

 Promiscuity as key to drug repurposing, off-target prediction, polypharmacology: What can be learned based on the comparison of binding sites and the description of their properties? Herein, we discuss binding site similarities with a special focus on medicinal chemistry.
Promiscuity as key to drug repurposing, off-target prediction, polypharmacology: What can be learned based on the comparison of binding sites and the description of their properties? Herein, we discuss binding site similarities with a special focus on medicinal chemistry.
Abstract
The elucidation of non-obvious binding site similarities has provided useful indications for the establishment of polypharmacology, the identification of potential off-targets, or the repurposing of known drugs. The concept underlying all of these approaches is promiscuous binding which can be analyzed from a ligand-based or a binding site-based perspective. Herein, we applied methods for the automated analysis and comparison of protein binding sites to study promiscuous binding on a novel dataset of sites in complex with ligands sharing common shape and physicochemical properties. We show the suitability of this dataset for the benchmarking of novel binding site comparison methods. Our investigations also reveal promising directions for further in-depth analyses of promiscuity and druggability in a pocket-centered manner. Drawbacks concerning binding site similarity assessment and druggability prediction are outlined, enabling researchers to avoid the typical pitfalls of binding site analyses.
Introduction
In the context of modern rational drug discovery and design, “druggability” and “promiscuity” are often occurring terms in the literature and have to be considered to ensure the success of drug development and the safety of the resulting compounds.
In general, promiscuous binding is defined as the ability of a small molecule to bind to and to modulate multiple targets.1 In the context of drugs, the term “polypharmacology” describes the beneficial and intentional modulation of multiple targets by one compound leading to additive or synergistic effects or improved efficacy.2 The prerequisite for polypharmacology is promiscuous binding which is mediated by distinct interactions to the different targets3 and which is often an interesting starting point for drug repurposing.4 However, promiscuous binding is also the basis for the binding to unwanted targets (off-targets) resulting in adverse drug reactions. In the case of drugs that also address antitargets, e.g., hERG or the 5-HT2B receptor, or a broad spectrum of unrelated targets,5 the term “promiscuity” often carries a negative connotation (“harmful promiscuity”).6 However, it was shown that ligand promiscuity across different target families is a rare phenomenon as compared to the binding of ligands to multiple targets of one family.1 Irrespective of the molecular and therapeutic consequences of the multi-target activity of a compound, we will use the word “promiscuity” herein. This term is not to be confused with the non-specific binding of small molecules as our analyses are based on known protein–ligand complexes.
As previously shown, one key to promiscuity is binding site similarity.4 However, the definitions of site similarity are manifold and based on different fundamentals. Whereas many binding site comparison methods rely on residue similarities, some of the approaches that exploit surface properties and interaction patterns are apparently better suited to detect similarities between unrelated proteins binding to similar or even identical ligands.5,6 In contrast, Sturm and co-workers could show that binding site similarity does not always account for binding sites complexed with promiscuous compounds.7 An obvious reason is the flexibility of otherwise identical ligands.4 Furthermore, the interactions to the target sites can be mediated by different functional groups of identical ligands.8
The counterpart to promiscuous binders is promiscuous proteins.9 Promiscuous protein binding sites often contain structural motifs that bind to many different ligands which are characterized by one or a small set of common functional groups. For example, positively charged compounds with two or more aromatic rings were identified to be promiscuous in an analysis of the available bioactivity data.10 Aminergic G protein-coupled receptors (GPCRs) and amine transporters contain a complementary structural motif responsible for the binding of ligands with these characteristics.
However, compounds forming aggregates, non-specifically inhibiting a large spectrum of proteins,11,12 and “frequent hitters”, like Pan-Assay Interference Compounds (PAINS),13,14 can lead to misleading results in the analysis of bioactivity data. As it is difficult to exclude such compounds from datasets for ligand-based bioactivity analyses of promiscuity, a protein-focused analysis as presented here might provide better insights into promiscuous binding.
We focus on binding site similarity and description for the analysis of promiscuity. Several tools are available to elucidate common binding site properties,15 but their performance highly relies on the applied datasets for validation, the analyzed properties, and the representation of these properties.16 The question “How to measure binding site similarity?”17 remains elusive and can only be answered by considering the complexity and flexibility of binding sites. Here, we use the structures of the small molecule-protein complexes as stored in the sc-PDB18 to elucidate similar binding sites of distant proteins starting at a ligand-based perspective and then re-evaluating the results with a number of successfully applied binding site comparison tools.
The second part of our analyses focuses on binding site descriptors, as they were successfully applied for druggability assessment. The term druggability in the context of binding sites refers to their ability to accommodate compounds with drug-like properties leading to a modulation of protein function.19 The prediction of druggable and non-druggable binding sites by means of machine-learning techniques based on binding site descriptors is nowadays possible on the fly and the actual challenge is to choose the appropriate tool.20 The ambiguity of pocket definition and protein flexibility additionally hamper the predictiveness and robustness of these prediction methods. Moreover, several contradictory guidelines with respect to binding site druggability were defined in the past.21 Our analysis of different methods based on the sc-PDB database of VolSite22-defined druggable binding sites underlines the necessity of a cautious application and evaluation of druggability prediction approaches. Nevertheless, we conclude our analysis by showing how binding site descriptors can be exploited to elucidate potential reasons for promiscuity, not from a ligand-based, but from a pocket-based point of view.
The overall outcome of this analysis highlights the tightrope that a medicinal chemist must walk when analyzing the similarity, promiscuity, and druggability of binding sites. Nevertheless, our results provide rationals for the choice of appropriate methods and elucidate potential pitfalls that should be considered to ensure the reliability of the conclusions drawn from binding site similarity and property analyses.
Experimental
Dataset preparation
The basic hypothesis that physicochemically similar ligands that bind in comparable conformations indicate related binding sites or similar protein–ligand interaction patterns led to the development of our so-called ROCS dataset. It was generated to evaluate whether binding site comparison tools enable the researcher to discriminate between binding sites of unrelated proteins in complex with highly similar small molecules (the active site pairs) and those of dissimilar ligands (the decoy site pairs). Fig. 1 summarizes the steps that led to the final dataset. The OpenEye tool ROCS (Rapid Overlay of Chemical Structures)23 was applied to screen for shape and physicochemical similarities between the ligands in the sc-PDB.18 The complete structure library was downloaded (04/2017) and the ligand MOL2 files were used for an all-against-all comparison with ROCS. The results were filtered with respect to the TanimotoCombo similarity score to obtain similar and dissimilar ligand pairs. Active (similar) pairs show a TanimotoCombo of at least 1.4, while decoy (dissimilar) pairs are characterized by a TanimotoCombo below 0.2. Pairs of sites in proteins with identical UniProt24 accession codes were excluded. The remaining protein structure pairs of the active and decoy pairs were used as input for TM-align25 to exclude protein pairs with high overall similarity. To this end, all chains involved in ligand binding as documented for the sc-PDB were used as input structures. Protein pairs with a TM-score above 0.3 were excluded. The resulting dataset comprises 15 339 active site pairs of dissimilar proteins binding to similar ligands and 56 179 decoy site pairs of unrelated proteins binding to dissimilar ligands.
Fig. 1. The workflow for the generation of the ROCS dataset.

The datasets of protein structures with identical sequences (X-ray dataset) and NMR ensemble models (NMR dataset) and the preparation of the dataset structures are described in an earlier publication.16
Dataset analyses
The ligands of the unrelated structures in complex with conformationally and physicochemically similar ligands were extracted from the sc-PDB, saved to an SDF file, and processed with KNIME26 as follows: the ECFP4 fingerprints27 of all molecules were calculated using the CDK toolkit.28 The Tanimoto coefficient-based similarity matrix was calculated and the k-Medoids algorithm was applied to cluster the molecules. After a visual inspection of the resulting clusters with varying partition counts k, a partition count of 10 was chosen for the clustering of all molecules in the dataset. Descriptors for the clustered molecules were calculated with the RDKit29 descriptor calculation node. The RDKit MCS node was applied to extract the common SMARTS patterns of the molecules in the individual clusters.
The ligands of the similar site pairs were searched in the PDB and the ChEMBL database30 to calculate the number of targets per compound. With respect to the ChEMBL search, a protein was assumed to be a target of the ligand if either the IC50 or the Ki or the Kd value was at least 10 μM.
The analysis of the target types was realized by annotating the structures in the PDB with the following target families: protein kinase (GO-ID 3672), protease (GO-ID 8322), transcription factor (GO-ID 3700), GPCR (GO-ID 4930), ion channel (GO-ID 5216), transmembrane transporter (GO-ID 22857), and nuclear receptor (GO-ID 4878). If the protein could not be assigned to any of these classes, it was annotated as an enzyme if its EC Number was available from the PDB. Otherwise, it was classified as “other”. Enzymes were further classified by the first two digits of their EC number.
The protein structures of the PDB, the sc-PDB, and the ROCS dataset (the complete set, and the structures of the active and decoy pairs) were sequence-culled using the PISCES server.31 The sequence identity threshold was set to 25% and the number of protein chains in the sequence-culled sets was counted.
The search for typical antitargets as previously defined in different publications32–34 was performed based on the corresponding UniProt accession codes.
Benchmark analyses
The binding site comparison methods were selected based on their successful applications in projects related to different questions arising in medicinal chemistry.35 They were grouped according to the underlying approach: residue-based (Cavbase,36,37 FuzCav,38 PocketMatch,39 RAPMAD,40 SiteAlign,41 SMAP,42 TM-align25), surface-based (ProBiS,43 VolSite/Shaper,22 SiteEngine,44,45 SiteHopper46), and interaction-based (Grim,47 IsoMIF,48 KRIPO,49 TIFP47). Details regarding their application and usage can be found elsewhere.16 In contrast to these earlier studies, the recent IChem version 5.2.850 was applied herein (necessary for the comparisons with FuzCav, VolSite/Shaper, Grim, and TIFP) to ensure the reproducibility of the results. With respect to TM-align, it is worthwhile mentioning that only the residues in a 10 Å environment of the ligand atoms were used for the binding site comparison.
The ROC curves were generated using KNIME.26 The AUC values, the statistical significance of the area under the ROC curve (AUC) values and their differences for the methods can be found in the ESI† (Tables S3 and S4). They were calculated according to DeLong and colleagues51 using the pROC package52 in R.53
Binding site analyses
VolSite,22 DoGSite,54 and dpocket (as included in the second version of the fpocket55 software) were applied with default settings to derive the descriptors and druggability scores for the binding sites under investigation. The ligands utilized for the binding site comparisons were used to define the corresponding sites. For VolSite, the sum of the number of hydrogen bond and charged site points divided by the number of all site points was used as the measure of the site polarity. The number of hydrophobic site points divided by the number of all site points was used as the measure of the hydrophobicity. For DoGSite, the number of hydrogen bond donor and acceptor atoms divided by the number of all site atoms was used as the measure of the site polarity. All pocket descriptors were rescaled using a min-max normalization based on all sites of the respective datasets to ensure comparability.
A more detailed analysis was performed for the active (similar) site pairs of the ROCS dataset. We calculated the SiteHopper scores for these pairs and split the pairs into two categories. The first group consists of site structures with a predominant shape similarity. In this group, sites were included whose ShapeTanimoto was at least two times as high as the ColorTanimoto. The remaining sites were assigned to the second group. We excluded sites which were found in both groups and calculated the dpocket descriptors for the remaining pockets in both groups to highlight differences between both types of site pairs.
An additional analysis focused on the ligands of active site pairs with a PatchScore below 0.82 (dissimilar site pairs). The properties of their ligand binding sites as stored in the sc-PDB were calculated with VolSite, DoGSite, and dpocket. Ligand-specific sites with a median DoGSite-derived polarity below 0.4 and a median hydrophobicity above 0.5 were analysed separately. For comparison purposes, these descriptors were also calculated for all structures in the sc-PDB.
Results and discussion
The ligand-based elucidation of binding site similarities
The starting point of the analyses presented herein was the sc-PDB18 – a database of binding sites predicted as being druggable. The general advantage of restricting all analyses to this structural subspace can be found in the absence of biologically irrelevant ligands as well as the availability of unique site identifiers for proteins with multiple binding sites. An earlier analysis of the corresponding ligands shows that a number of promiscuous ligands can be found.7 Nevertheless, identical ligands do not necessarily bind with comparable conformations.4,56 Moreover, their relative orientation within the binding site has a significant impact on the definition of promiscuity. While the latter point is not addressed within this study but was analyzed elsewhere,57,58 we tried to tackle the first one.
Given the ligand coordinates as stored in the sc-PDB, a ROCS23-based small molecule similarity search was performed to identify ligands with a similar shape and similar physicochemical properties indicating a similar binding mode to the corresponding sites. Altogether, we found 149 032 pairs of binding sites with ligands showing a TanimotoCombo (Tcombo) of at least 1.4. An analysis of the ShapeTanimoto (Tshape) and ColorTanimoto (Tcolor) with respect to the binned Tcombo values shows that especially for lower Tcombo values, the shape similarity dominates the overall similarity (see Fig. S1, ESI†).
The focus of this study was on sequentially and structurally unrelated proteins binding to similar ligands. Therefore, the binding site pairs corresponding to the ligand similarities were analyzed with respect to the relationship of the corresponding proteins. We excluded binding site pairs of proteins with identical UniProt24 accession codes leading to 139 378 remaining binding site pairs with similar ligands, but dissimilar proteins. The complete protein structures of these pairs were compared using TM-align to exclude pairs with a high overall similarity. Taking a maximum TM-score threshold of 0.3 into account,59 only 15 339 binding site pairs of structurally unrelated proteins binding to similar ligands remained. The contributions of the similarity measures Tshape and Tcolor to their overall similarity was also analyzed for this filtered dataset (Fig. S1, ESI†). Intriguingly, the exclusion of structurally related protein pairs by means of TM-align comparisons did not lead to a significantly different distribution of the shape and physicochemical similarities.
We then analyzed the dataset of active site pairs with respect to the chemical space and ligand properties. An ECFP4 fingerprint-based similarity analysis and subsequent clustering of the corresponding ligands (Fig. 2 and S2, ESI†) shows distinct clusters of small molecules which include mainly cofactors, but also fragments (cluster 1), a class of aromatic, condensed, and heterocyclic ring systems which can in most cases be assigned to the classes of flavonoids and isoflavonoids (cluster 4), steroids and lipids (clusters 2, 3, and 6), and a group which mainly includes flavone scaffolds (cluster 8). Within the groups of cofactors, guanosine-related compounds (cluster 7), adenosine-related compounds (cluster 9), and phosphorylated adenosine-related compounds (including NAD and FAD, cluster 10), as well as thymidine-related compounds (cluster 5), were found. This diversity underlines that the dataset is not biased toward one specific compound class, although cofactor-related compounds are clearly overrepresented. Many of the compounds were already analyzed in a previous study that focused on promiscuous ligands in the sc-PDB.7 In that study, the compounds with the following PDB-IDs were characterized as “super-promiscuous” ligands of distant binding sites: AE2, ASD, STR, CHD, RTL, RBF, QUE, and DES. These compounds except for AE2, RBF, and RTL are also included in our filtered dataset. The name “super-promiscuous” is derived from the finding that they bind with identical chemical moieties to unrelated sites without significant conformational changes. This suggests that they show unique properties when compared to other ligands.
Fig. 2. SMARTS patterns of maximum common substructures within the clusters derived from the ligands of the active site pairs in the ROCS dataset. The percentage of dataset compounds per cluster is given in parentheses.

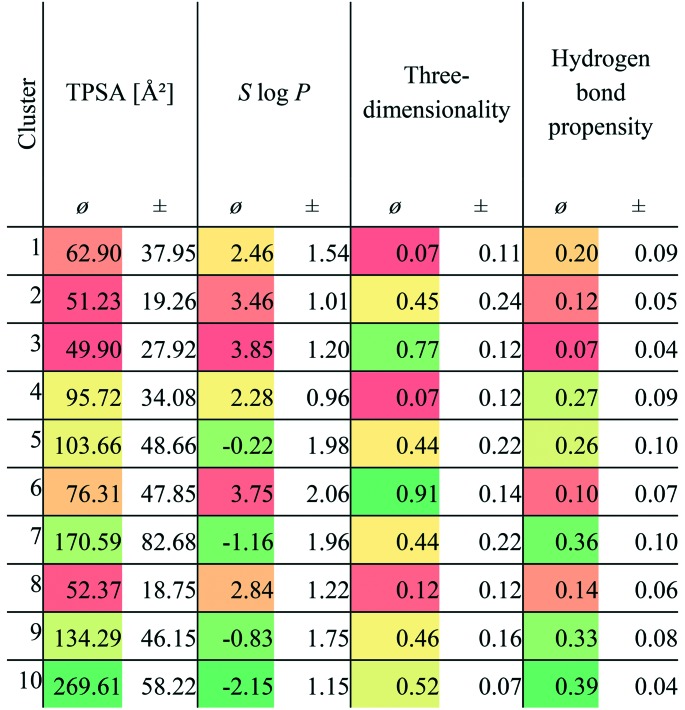
The question arises of whether this ligand type predominates the active ligands (i.e., the ligands corresponding to the active site pairs) as their similarities evolve from their “promiscuous” properties. In the previous study,7 the identified compounds binding to multiple targets were characterized by their three-dimensionality (number of carbon atoms with sp3 hybridization divided by the total number of carbon atoms) and hydrogen bond propensity (number of hydrogen bond donors or acceptors divided by the total number of atoms). We analyzed these properties for the compounds of our dataset (Table 1). A group of high three-dimensionality and low or high hydrogen bond propensities was identified in the analysis of Sturm and co-workers which corresponds to clusters 2, 3, and 6 (mainly lipids) and cluster 4 (mainly flavonoids) in our dataset, respectively. Another group was characterized by an extraordinarily low three-dimensionality and low hydrogen bond propensities. Members of clusters 1 and 8 can be assigned to this category, although their hydrogen bond propensities is high. In the case of cofactors, a medium three-dimensionality as well as a high hydrogen bond propensity was observed in the earlier study which also holds true in the case of our dataset (clusters 5, 7, 9 and 10). The “super-promiscuous” ligands are found spread across clusters 1, 3, 4, 5, and 6.
Table 1. Mean (ø) TPSA, S log P, three-dimensionality, and hydrogen bond propensities of the ligands of the similar binding site pairs (active pairs). The standard deviation (±) for all descriptors is also given. A color gradient ranging from green to yellow to red was applied. Green is used for the highest values in the case of TPSA, three-dimensionality, and hydrogen bond propensity. For the S log P descriptors, the lowest values are colored green.
|
The compounds of clusters 7 and 10 show the highest mean number of targets per ligand as calculated from the data in the PDB (Fig. S3, ESI†). This can be attributed to the overrepresentation of cofactors in these compound groups. However, the analysis of the ChEMBL30 data shows a considerably high average number of targets for the compounds in clusters 3 and 4. The targets of the compounds of cluster 3 are mainly members of the cytochrome P450 family, the nuclear hormone receptor family, the GPCR family, and the family of transmembrane transporters. In contrast, the targets of the compounds of cluster 4 are more broadly distributed, including enzymes such as protein kinases, carbonic anhydrases, peptidases, lipoxygenases, but also GPCRs, nuclear hormone receptors, and members of the cytochrome P450 family.
Lipophilicity and the correlation with promiscuity is another property that is controversially discussed in the literature.60,61 The different results hint at a dependency on the dataset and a general infeasibility to derive general trends. Table 1 shows that the retrieved clusters exhibit highly different mean TPSA and S log P values and there is even a high variation observed within the clusters. The mean S log P values range from –2.15 to 3.85 and the mean TPSA from 49.9 to 269.61 Å2. A general correlation between lipophilicity and promiscuity in this dataset can be excluded. However, for clusters 1, 2, 3, 6, and 8, this relationship was verified. For cluster 4, the missing three-dimensionality might explain the promiscuity of this compound class. In contrast, the compounds in clusters 5, 7, 9, and 10 show a high three-dimensionality and polarity. Their binding to unrelated proteins is explicable by their conserved function in nature (mainly cofactors and cofactor-related compounds).
From a target-based viewpoint, the distribution of enzymes in general, protein kinases, proteases, transcription factors, GPCRs, ion channels, transmembrane transporters, and nuclear receptors in the ROCS dataset is similar to that of the complete sc-PDB (Fig. S3, ESI†). As compared to the distribution in the PDB, we find a significantly higher percentage of enzymes in general and protein kinases in particular, whereas the percentage of ion channels and transmembrane transporters is even lower than in the PDB. Glycosylases, peptidases, and C–O lyases are underrepresented in the similar site pairs as compared to the sc-PDB, while enzymes that transfer one-carbon groups, phosphatases and C–N bond synthetases are overrepresented (Table S1, ESI†). This overrepresentation results from the high occurrence of the compounds SAM, SAH, and ADP in the structures of these enzymes.
Of the 54 234 unique proteins in the PDB (in terms of their UniProt24 accession codes), 3701 are included in the sc-PDB. The ROCS dataset consists of 2930 unique protein structures and the subsets of proteins with similar and dissimilar sites include 1254 and 2381 unique proteins. The diversity of the targets was assessed by a sequence culling of the datasets with PISCES.31 The sequence-culled sets of the PDB, the sc-PDB, and the ROCS dataset contain 12 225, 1429, and 846 unique entries. This reduction in diversity can be attributed to the necessity of ligand-occupied and druggable sites for the ROCS dataset and the restricted number of protein structures for certain protein classes, e.g., transmembrane proteins.
An analysis of the proteins with similar sites reveals that most pairs are composed of proteins from different target families. Thus, our dataset reflects difficult cases of cross-target family binding which are not readily detectable based on already available knowledge.
Concluding this analysis, we looked for pharmacological interesting similarities within the dataset. Several typical antitargets32–34 were found in the dataset of similar site pairs. They include several sulfotransferases and nuclear receptors in complex with different steroid hormones or related compounds as well as cAMP-specific 3′,5′-cyclic phosphodiesterase 4D (see Table S2 in the ESI† for some examples).
From a ligand-based point of view, several similarities between the sites of unrelated proteins were identified. A search with DrugBank62 molecules that are known as ligands in the PDB (; https://www.rcsb.org/pdb/ligand/drugMapping.do) revealed several intriguing binding site similarities. Fig. S4 and S5 in the ESI† present the ligand-based superimpositions of the protein binding sites and the structures of the ligands. Fludrocortisone, which is known in complex with a mutant human androgen receptor (PDB-ID ; 1gs4), shows high shape and physicochemical similarities to dexamethasone-21-phosphate bound to the Pin1 substrate binding domain (PDB-ID ; 3tc5). Nevertheless, the Kd of 4.4 mM illustrates the low affinity of the ligand toward Pin1 which mainly interacts via its phosphate moiety with the enzyme. Another similarity was found between sulfathiazole in sepiapterin reductase (PDB-ID ; 4j7u) and a weak thumb pocket 2-binding inhibitor of the HCV NS5B polymerase (PDB-ID ; 4iz0). This compound (2,4,5-trichloro-N-(5-methyl-1,2-oxazol-3-yl)benzenesulfonamide, IC50 = 100 μM) resulted from a fragment-based screening approach. The binding mode of chlorzoxazone bound to nitric oxide synthase (PDB-ID ; 1m8d) shares high shape and physicochemical similarities with 3-methyl-3,4-dihydroquinazolin-2(1H)-one in complex with the N-terminal bromodomain of BRD2 (PDB-ID ; 4a9e). Moreover, a similarity between a peptide-mimetic ligand bound to Escherichia coli peptide deformylase (PDB-ID ; 3k6l) and marimastat bound to ADAMTS-1 (PDB-ID ; 2jih) was identified.
We also find known similarities, as for example the binding of flavone-derived compounds to enzymes of the flavonoid metabolism and protein kinases: QUE in the dihydroflavonol 4-reductase structures with the PDB-IDs 2nnl and ; 3bxx, respectively, shows similarities to ERD in the Ser/Thr-protein kinase 17B structure (PDB-ID ; 3lm5), ERD in the structure with the PDB-ID ; 2nnl, and AGI in a casein kinase II subunit alpha structure (PDB-ID ; 4dgm). Ligand-based similarities can also be found for flavoenzymes and tankyrase 2 (ERD in the structure with the PDB-ID ; 2nnl and LU2 in the tankyrase structure with the PDB-ID ; 4hkn, see also Fig. S5, ESI†). Correspondingly, similarities between compounds binding to tankyrase and kinases were found (F94 in the tankyrase structure with the PDB-ID ; 4hmh and AGI in the casein kinase II structure with the PDB-ID ; 4dgm, and 15W in the tankyrase structure with the PDB-ID ; 4hl5 and FSE in the CDK6 structure with the PDB-ID ; 1xo2). The question arises of whether these small molecule similarities can be accounted for by binding site similarities.
The comparison of sites binding to similar ligands
We compared the active and decoy site pars of the ROCS dataset with different methods to identify potential relationships between the binding sites of unrelated proteins in complex with similar ligands. As it was shown before,7,8,63 ligands with a high 2D similarity might bind in highly different conformations to their target proteins. This can be partially attributed to a high degree of flexibility of the ligand of interest. The application of an initial ROCS 3D similarity analysis circumvents this problem.
Interaction-based binding site comparison tools were shown to be in general less robust with respect to the bound ligand and the flexibility of the binding site.16 However, this dataset might disclose the benefits of these comparison tools. This is exemplified by the complex structures in Fig. 3. Both proteins bind the same ligand S-adenosyl-l-homocysteine (SAH) in a highly similar conformation and both ligands share a common interaction pattern. Nonetheless, the cavity environment is different and residue- and surface-based comparison methods will probably not assign high similarity scores for this binding site pair, although it represents an interesting match.
Fig. 3. Non-obvious binding site similarities. On the left, the binding site alignment of the human enzyme arginine N-methyltransferase 6 (PDB-ID ; 4hc4 chain A, green) and N-4 cytosine-specific methyltransferase from Proteus vulgaris (PDB-ID ; 1boo chain A, orange) is presented with SAH in ball-and-stick representation. The corresponding binding site residues are colored according to the structure. The figure was generated with UCSF Chimera.64 The interaction patterns of SAH in the binding sites of the complexes with the PDB-IDs ; 4hc4 and ; 1boo are shown on the right (the schemes were generated with LigandScout 4.065).

This basic assumption cannot be completely verified by the outcome of the benchmark analysis presented in Fig. 4 (see Table S3, ESI† for the AUC differences and their significance). Although IsoMIF,48 KRIPO,49 and Grim47 show a high early enrichment, TIFP47 only follows this trend if the Hamming distance is used as the distance measure. The early enrichment of IsoMIF and KRIPO is comparable. Both methods are appropriate to correctly identify close relationships between the binding sites of similar ligands. Despite these promising findings, the overall performance in terms of the AUC of all tools except for IsoMIF is worse than that of SiteHopper.46
Fig. 4. Evaluation of different binding site comparison tools with respect to the dataset of ROCS structures. Top: ROC curves for different binding site comparison methods for the initial purely ligand-based dataset. Bottom: ROC curves for the methods when only matches with a high SiteHopper PatchScore are taken into account as active site pairs. The name of the tool is colored according to its corresponding ROC curve. The binding site comparison tools are sorted in descending order with respect to their overall AUC. The scoring measures that yielded the highest AUC for both datasets were distance score d1, the similarity score TC, the SVA, and the DistanceScore for SiteAlign (thin red line), SMAP (thin blue line), ProBiS (thin purple line), and SiteEngine (thin orange line), respectively. TIFP AUC values significantly improved when using the hamming distance as the scoring measure in both datasets. Top: Shaper(PDB), VolSite/Shaper, and VolSite/Shaper(PDB) showed the highest AUC values when using the Tanimoto (fit) as the similarity score. Bottom: The SiteHopper AUC value improved slightly upon using the ColorTanimoto.

Intriguingly, different scoring measures lead to highly different performance for some tools of this analysis. TIFP performs best if the Hamming distance is used to differentiate between similar and dissimilar sites. This metric mainly distinguishes based on size and complexity.66 While the decoy site pairs of the dataset are in complex with differently sized ligands, the size and complexity of the ligands of the active site pairs are comparable. Therefore, the Hamming distance allows a good differentiation between the active and decoy site pairs. The performance of SiteAlign41 notably improves if the d1 distance is applied. The underlying scoring scheme uses a distance measure that detects global binding site similarities. In contrast, the distance measure d3, which was the most suitable one for the datasets in our previous analysis,16 allows for the identification of local binding site similarities. Additionally, it uses the distance between the centroid and the center of the side chain (Ccentroid) as opposed to the distance of the centroid and to the residue's Cβ atom which is used for the calculation of d3. In the case of SiteEngine,44,45 the use of the DistanceScore instead of the CurvatureScore seems to be most appropriate to reveal similarities between pockets of unrelated proteins.
These findings underline a peculiarity of this dataset. In comparison to most datasets used for binding site comparison evaluation, the ligands of the active site pairs overlap with respect to shape and physicochemical properties. Therefore, the binding site definition allows for comparably sized binding site sections whose global similarity will be high. The applicability of TIFP, SiteAlign, and SiteEngine for the detection of non-obvious similarities for proteins that bind similar ligands is restricted to these scoring schemes and similarly excised binding sites.
The poor performance of FuzCav38 can be explained when having a look at Fig. 3. The side chains of the hydrophobic residues Met166 and Phe273 are located in a similar manner with respect to the ligand. Nevertheless, their backbone atoms are part of different backbone structures in opposite locations of the binding site. In contrast to the previously investigated datasets for the benchmarking of site comparison methods,16 this is a unique feature of this set of active site pairs and hampers the success of residue-based methods that make use of Cα atom positions for the assignment of binding site features. One exception to this rule is PocketMatch39 which showed a mediocre performance when compared to the other residue-based methods in our previous studies.
An even more striking observation for this dataset is the high early enrichment for TM-align25 which also uses Cα atom coordinates to superimpose structures. This is discussed in more detail in Text S1, ESI.† However, most residue-based methods did not correctly differentiate between similar and dissimilar site pairs. The same holds true for the surface-based methods with the exception of SiteHopper which clearly outperforms all residue- and surface-based methods.
Overall, IsoMIF and SiteHopper lead to AUC values of nearly 1, which suggests that the binding of similar ligands to the sites of unrelated proteins can be explained based on the underlying site similarities. However, there might be other explanations for this superior performance which will be further discussed below.
Similar ligands–similar binding sites?
The outcome of the analysis was unanticipated as nearly all analyzed tools performed satisfactorily on different datasets of interesting binding site similarities.16 The clear superiority of IsoMIF and SiteHopper was not observed before. However, both tools reliably scored similarities and dissimilarities of binding sites. To shed light on the question of whether the ligand-based similarities indeed correspond to similar binding sites, we analyzed the similarity scores for the binding site pairs obtained with SiteHopper. This method reports a measure for both, the shape and the physicochemical similarity. For most site pairs, the shape similarity was much higher than the Tanimoto for common physicochemical properties. This is in accordance with our analyses of the ligand-based similarities. In general, only one third of the binding site similarities show a ColorTanimoto above 0.15. Binding sites of similar ligands that share mainly common shape properties are characterized by a high fpocket hydrophobicity and low fpocket polarity score when compared to those that bind to similar ligands and show high physicochemical similarity (see Fig. S6, ESI†). This suggests that the overall superiority of IsoMIF and SiteHopper results from scores derived from a high shape, but not physicochemical similarity. However, this observation does not controvert their general ability to detect physicochemical similarities and score them appropriately.16 For IsoMIF, it was shown that the number of hydrophobic and aromatic probes is considerably higher in promiscuous sites leading to a smaller number of interactions that enable a clear differentiation.67 This partially explains the superiority of the method for this dataset. Predominantly hydrophobic and aromatic interactions fields enable a purely shape-based matching which will nonetheless lead to a high similarity score (see also Table 2).
Table 2. SiteHopper similarity scores and the percentages of matched hydrophobic and aromatic interaction field grid points by IsoMIF for selected active site pairs.
| Site 1 (PDB-ID, ligand-ID) | Site 2 (PDB-ID, ligand-ID) | SiteHopper scores |
IsoMIF | ||
| PatchScore | Shape-Tanimoto | Color-Tanimoto | Matched % hyd, % aro | ||
| 1gs4, ZK5 | 3tc5, 3T5 | 0.56 | 0.24 | 0.10 | 62.5, 18.8 |
| 4j7u, YTZ | 4iz0, 2BI | 0.69 | 0.35 | 0.11 | 45.2, 35.5 |
| 1m8d, H4B | 4a9e, 3PF | 0.69 | 0.36 | 0.11 | 65.0, 10.0 |
| 3k6l, 2BB | 2jih, 097 | 0.93 | 0.32 | 0.20 | 50.0, 0.0 |
| 4hkn, LU2 | 2nnl, ERD | 0.87 | 0.33 | 0.10 | 56.8, 29.5 |
| 3lm5, QUE | 2nnl, ERD | 0.68 | 0.36 | 0.10 | 64.3, 0.0 |
| 4dgm, AGI | 2nnl, ERD | 0.75 | 0.33 | 0.14 | 73.0, 10.8 |
| 4dgm, AGI | 4hmh, F94 | 0.75 | 0.38 | 0.12 | 63.6, 21.2 |
| 4hl5, 15W | 1xo2, FSE | 0.74 | 0.36 | 0.13 | 53.3, 16.7 |
We exemplified the impact of pocket hydrophobicity using the pairs of interesting similarities between enzymes based on the ligand similarities of therapeutically interesting targets discussed in the first section (Table 2). Most of them show a comparatively low binding site similarity. Their SiteHopper scores result from a very high shape, but comparably low physicochemical similarity.
For binding sites complexed with flavone-scaffolds, the overall similarity between the binding sites is low and most similarities fall below the threshold for significant binding site similarities. For binding sites of related flavone-scaffolds, distinct interaction patterns can be found that convey selectivity. This is shown in Fig. 5 for the similarity between tankyrase 2 (PDB-ID ; 4hkn) and dihydroflavonol-4-reductase (PDB-ID ; 2nnl). Although, the ROC curve for SiteHopper given in Fig. 4 suggests that binding site similarity can reveal reasons for promiscuous binding, the analysis of the similarity scores is inevitable for further investigations.16 A visualization of the binding site alignment, together with the corresponding ligand-based alignment, underlines the differences in the interactions of both molecules with their respective target sites. In contrast, the similarity between Escherichia coli peptide deformylase and ADAMTS-1 (both metalloproteases) is higher.
Fig. 5. Examples of dissimilar and similar binding sites bound to ligands showing a high similarity in terms of the SiteHopper-derived PatchScore. Left panel: SiteHopper- (top) and ligand-based (bottom) alignment of the binding sites of tankyrase 2 (PDB-ID 4hkn, ligand-ID LU2) and dihydroflavonol-4-reductase (PDB-ID ; 2nnl, ligand-ID ERD). Despite their common shape and physicochemical properties, both ligands interact in a unique way with their respective targets. Right panel: Alignment of the metalloproteases E. coli peptide deformylase (PDB-ID ; 3k6l, ligand-ID 2BB) and human ADAMTS-1 (PDB-ID ; 2jih, ligand-ID 097) together with bound peptidomimetic compounds (top). The crucial common interactions are depicted separately with the corresponding ligand 2D representations. Blue and red arrows indicate hydrogen bond donor and acceptor functionalities of the protein residues.

A closer examination of all similarities to ADAMTS-1 (PDB-ID 2jih) in complex with marimastat shows pronounced similarities to peptide deformylase enzymes of different pathogenic organisms (e.g., Pseudomonas aeruginosa, PDB-ID ; 1ix1, Fig. 5) and the human enzyme. This example highlights the potential of binding site comparison to elucidate similarities between functionally related enzymes which show no obvious protein structure similarities.
Taking these examples into account, we can derive two important issues, which have to be considered when comparing binding sites. First, the researcher has to be aware of the score distributions. For tools which do not provide measures to highlight the significance of found matches, defined score ranges that correspond to similar binding sites have to be considered. These findings are in agreement with our previous analyses.15 Secondly, care has to be taken if an indicated binding site similarity is solely based on shape similarity. Both common physicochemical and shape features are necessary to identify meaningful similarities.
Next, we excluded binding site pairs with overall low similarities in a biased manner as described in the following. Given the scores obtained with SiteHopper for highly similar binding sites, we applied a threshold to exclude binding sites with a low overall binding site similarity as defined by the PatchScore. This led to a decrease from 15 339 to only 4487 similar binding site pairs. We re-evaluated the performance of all methods on this reduced dataset (Fig. 4, see Table S3 in the ESI† for the AUC differences and their significance). Intriguingly, the performance of some tools that were previously characterized by a poor retrieval of active site pairs improved with respect to the AUC. Obviously, a predominant shape similarity leads to a low score with these tools.
Overcoming this thin line between sufficiently high shape similarity, but also significant physicochemical similarity is a crucial step when applying binding site comparison tools for drug repositioning or polypharmacology purposes. While the overall similarity has to be high enough to allow for repurposing, the binding site properties should be characterized by minor dissimilarities as a promising starting point to establish selectivity. Nonetheless, high shape similarity and a low number of hydrogen bond donor and acceptor features of the binding site might hint at a higher number of potential off-targets of the binding ligands. We investigated the meaning of the similarity scores for the binding sites of typical off-targets in the similar cavity pairs of the ROCS dataset. We searched the ChEMBL for common inhibitors for binding site matches between the antitargets and unrelated proteins. For 63 out of all 209 ligand-based binding site similarity pairs, we found at least one common inhibitor in the ChEMBL database (see Table S2, ESI†). However, more than 50% of those cavity pairs showed SiteHopper PatchScores below 0.82. The mean ShapeTanimoto for all pairs with common inhibitors in the ChEMBL database was 0.39 and the mean ColorTanimoto was 0.14. This finding indicates that even a high ShapeTanimoto-based similarity might hint at potential off-target effects and should not be neglected.
However, with respect to the establishment of polypharmacology or drug repurposing studies, we hypothesize that the meaning of binding site similarities with respect to shape is of minor interest as compared to physicochemical similarities, which can be readily identified most of the tools used within this study.16 In line with recent studies,67 the number of potential interaction hot spots in very hydrophobic pockets is small, rendering a differentiation between such sites a challenging issue.
The description of binding sites and druggability assessment
As the ROCS dataset was restricted to druggable binding sites which are often characterized by a high hydrophobicity,68 we investigated the properties of the sites in complex with similar ligands in the view of a druggability assessment. Different methods exist to predict druggable pockets.20 The underlying approaches for binding site definition and different assumptions concerning druggability restrict the applicability domains of these methods. To re-evaluate the druggability in our ROCS dataset, the binding sites were processed with DoGSite54 (see Fig. S7, ESI†). Although the binding sites of the ligands of the ten clusters (Fig. 2) show mean druggability scores above 0.5 (which corresponds to the minimum threshold that was recommended to identify druggable sites with DoGSite), the druggability scores for the structures range between 0 and 1.
This is in line with an earlier finding with respect to druggability assessment. Sets of identical binding sites that were derived from NMR ensembles were not consistently predicted as being druggable.16 This can be partially attributed to differences in the applied training sets (applicability domain) and discrepancies in the pocket definition. A closer analysis of three publicly available prediction tools (DoGSite, fpocket,55 VolSite22) hints at the overwhelming importance of comprehensive structural knowledge to reliably predict binding site druggability and is outlined in the following paragraphs.
A description of the binding site properties and a subsequent training based on known druggable and non-druggable sites is usually performed to establish predictive druggability models. As previously shown, the binding site definition has a considerable impact on the reliability of druggability predictions. Therefore, we investigated the robustness of not only the druggability scores but also the binding site properties with respect to binding site flexibility. To this end, we used two datasets from our ProSPECCTs dataset collection. The first dataset is derived from the sequence-culled subset of the sc-PDB. We searched for structures with 100% atom-based sequence identity in the PDB and built a dataset of 12 groups of structural ensembles of proteins (X-ray dataset). The second dataset was generated based on ligand-bound NMR ensemble structures from the sc-PDB (15 structural ensembles, NMR dataset). In comparison to the first dataset, this set is characterized by higher root-mean-square deviation (RMSD) values and a lower quality of the underlying structures.
Fig. 6 illustrates the assessment of the binding site properties for all structures of the X-ray dataset with the corresponding protein UniProt accession code and protein name. Some general trends are predicted consistently, e.g., the high hydrophobicity and low polarity of the bromodomain-containing enzyme 4 (BRD4) and the androgen receptor (AR) binding site or the low hydrophobicity of the site in the Drosophila homolog of the mammalian angiotensin-converting enzyme (AnCE). However, striking differences in the pocket descriptors hydrophobicity and polarity for the models within the structural ensembles can be observed. This trend very likely manifests itself in the derived druggability scores. For fpocket, the ligand-based binding site definition and druggability prediction were not possible. Therefore, we used the druggability scores of the automatically detected pockets with the highest overlap with the reference ligand. This might contribute to the high fluctuations of the pocket descriptors for some binding sites. Despite the generally high fluctuations of the two analyzed binding site descriptors, it can be observed that very hydrophobic binding sites with low polarity are also characterized by high druggability scores. Results based on an experimental assessment of druggability are in line with this trend.69,70
Fig. 6. Box plots of the scaled hydrophobicity (top) and polarity (center) descriptors derived from the analyses with fpocket45 (dark green boxes), VolSite21 (light green boxes), and DoGSite44 (yellow boxes) together with the obtained druggability scores (bottom) for the X-ray dataset. The cut-offs to distinguish between druggable and non-druggable sites are given as gray lines. The dashed line represents a stricter threshold for druggability predictions with fpocket.

The major drawback of the X-ray dataset is the differing nature of the ligands. To evaluate whether the discussed trend can also be found for structural ensembles of proteins binding to identical ligands, the dataset of NMR structures was used. Fig. S8 in the ESI† gives the binding site descriptors, together with the druggability assessment, and shows that binding site definition, as well as flexibility, hamper a unique prediction of druggability.
Finally, we compared the robustness of two druggability scoring models implemented in DoGSite. The SimpleScore is the more robust scoring scheme with respect to sensitivity to binding site flexibility (Fig. S9, ESI†). In contrast to the Support Vector Machine-based DrugScore, it is derived from a simple regression model that takes only three site descriptors into account: volume, the relative number of solvent-exposed hull grid points, and the relative number of lipophilic site interaction points. If little structural knowledge of the protein of interest exists, this score might be the more appropriate way to assess the druggability of the site of interest.
Overall, the high fluctuations of the druggability scores and physicochemical site descriptors for similar ligand binding sites highlight the necessity of structural ensembles (e.g., multiple crystal structures with sites in complex with different ligands, molecular dynamics simulation data, NMR ensembles) for a rigorous and reliable calculation of binding site descriptors and finally a robust assessment of druggability and binding site properties. A prediction for novel binding sites with only one structural representative might be misleading. Nonetheless, distinct trends can be visualized with the help of binding site descriptors, such as the extraordinarily high hydrophobicity of BRD4 binding sites, the low one of AnCE, and a high polarity of the thrombin binding sites. Consequently, we used different descriptors to analyze the binding sites of the ROCS dataset in more detail.
The use of binding site descriptors to identify promiscuous binding sites
We tried to tackle the question of promiscuous binding within our sc-PDB-derived dataset. Recent studies of ligand promiscuity highlighted some crucial small molecule descriptors which might reveal promiscuous binders, e.g., hydrophobicity, the number of aromatic rings etc.60 As a matter of fact, we should find complementary properties within the binding sites of proteins in complex with promiscuous ligands. As a basis for our analysis, the “super-promiscuous” ligands described by Sturm et al. were considered. The binding sites of ASD, STR, QUE, DES, and CHD (RTL, RBF, and AE2 were not found in the final dataset) are characterized by a low physicochemical and overall similarity. They belong to the group of ligands binding to dissimilar enzymes with distant cavities. Therefore, we strived to elucidate why those ligands bind to different sites of structurally dissimilar proteins. For ASD, we also find binding sites that show a PatchScore above the previously defined threshold. Nonetheless, the corresponding alignment shows that both sites are dissimilar with respect to the bound ligands.
Analyzing the pocket properties hydrophobicity and polarity as reported by DoGSite, we can derive a distinct trend for the corresponding binding sites (Fig. 7, highlighted). Most of them are characterized by a high hydrophobicity and a low polarity. An extension of this analysis to the ligands that were characterized by similar shape and physicochemical properties, but bind to dissimilar sites, led to the trend that was as also observed for the “super-promiscuous” ligands (Fig. 7). Similar outcomes could be observed when using fpocket and VolSite (Fig. S10, ESI†). Therefore, the question arises of whether we can define typical “side-effect” targets6 based on easily accessible binding site descriptors. Going a step backward and looking for targets in the sc-PDB that bind to these molecules further underlines the results. Among them are human serum albumin,71 PETN reductase,72 and the mineralocorticoid receptor,73 besides GPCRs, ion channels, and transporters.
Fig. 7. Box plots of the DoGSite44-derived scaled hydrophobicity (solid filled boxes) and polarity (hatch pattern-filled boxes) scores of the binding sites of ligands that bind to multiple binding sites which do not show a high similarity using SiteHopper (PatchScore < 0.82, mainly hydrophobic). For comparison purposes, the box plot of these properties is also given for all ligands of the sc-PDB. “Super-promiscuous” ligands as defined by an earlier study are highlighted in gray.

Besides those compound types, cofactor-related compounds (e.g., 5GP, AMP, APC, APR, CDP, DG3, DTP, IBM, IMP, RBF, and XMP) and flavone-like molecules (AGI and MYC) were identified to bind to dissimilar binding sites. In contrast, their corresponding binding site properties followed an opposite trend, i.e., the binding sites are characterized by a high polarity (see Fig. S11, ESI†). Accordingly, these molecules are characterized by a high number of hydrogen bond donors and acceptors, negatively charged atoms, and aromatic rings. These characteristics enable a multitude of unique selective interactions with multiple targets. However, the ratio of hydrophobicity to polarity for the pockets of the flavone-like compounds AGI and MYC tends to that of the promiscuous binders. The same holds true for the site of the compound with the PDB-based ligand-ID DXC.
Conclusion
Several conclusions can be drawn from the presented analyses, especially with respect to binding site similarity and property analyses in chemical biology and medicinal chemistry. The results presented herein provide some useful hints at potential pitfalls and chances of binding site analysis and comparison.
In view of the different potential applications of binding site comparison, we can assign tools that are of special interest. A small group of approaches has major difficulties to identify similarities between binding sites of distant proteins (FuzCav, RAPMAD, VolSite/Shaper, TIFP) and should be predominantly used for the comparison of binding site of related proteins, e.g., for the analysis of evolutionary relationships or the elucidation of minor dissimilarities that might assist in the process of drug development. Other comparison algorithms are especially successful in the identification of physicochemical similarities with a focus on similar interaction patterns (PocketMatch, SMAP, Cavbase, SiteAlign, SiteEngine, ProBiS, KRIPO, Grim) and seem to be best suited for applications such as drug repurposing or polypharmacology analyses. For our ROCS-based dataset of similar binding sites, they showed a high early enrichment of similar sites. In contrast, the tools SiteHopper and IsoMIF not only allow for the identification of similar binding sites with respect to the chemical functionalities, but also focus on the shape similarities between the sites. Although they might be misleading for searching for similarities which can be exploited for ligand discovery, they are highly useful for off-target prediction. For drug repurposing, both approaches can bridge the gap between similar sites and sufficiently dissimilar sites to ensure the safety of repositioning strategies. The ROCS dataset which was used for our analyses will be made available as part of the ProSPECCTs dataset collection for the evaluation of further binding site comparison methods (download site: ; http://dx.doi.org/10.17877/DE290M-2).
Intriguingly, we can identify binding site pairs with pronounced shape similarities, but a small number of common physicochemical properties. This purely geometry-based similarity is reflected by similarly shaped ligands that bind to these sites without necessarily exhibiting similar interaction patterns. Especially some of the previously described “super-promiscuous” ligands7 fall into this category. The overall outcome shows that we have to adjust our view on promiscuity. Descriptors of ligands that were previously characterized as promiscuous binders include hydrophobicity, a high aromatic ring count, or positive ionization.10 Correspondingly, we can find binding site descriptors that highlight protein pockets which are able to accommodate a varying number of different, probably unselectively binding ligands due to their high hydrophobicity. Therefore, the analysis of promiscuity should not only take the ligand-based analysis into account, but should also utilize the structural knowledge of protein cavities, comparison tools, and the available pocket descriptors to prevent the targeting of binding sites with properties that hint at an increased promiscuity.10
The second focus of this article was the assessment of binding site druggability. Although the number of tested tools is low when compared to the number of the available ones, we want to underline the necessity of an appropriate application of binding site description as well as druggability prediction methods. Given the complexity and flexibility of binding sites, the question arises of whether we will ever be able to assess robust binding site properties with respect to one target binding site. Nonetheless, the overall results show a general consensus in druggability assessment. The prediction of druggable and non-druggable binding sites should never be overrated if only a small number of structures is available. With increasing structural knowledge, an overall assessment becomes more meaningful by taking multiple structures into account. The same holds true for the underlying site descriptors. Additionally, we show that we should focus our attention not only on promiscuous binders, but also binding sites with respect to their properties. Besides other factors that explain promiscuous binding,9 we find that binding site hydrophobicity is one of the major determinants of missing selectivity from a binding site-based point of view. This is in line with a recent study based on molecular interaction fields.67
Overall, we can declare that binding site comparison in combination with binding site characterization is a potential key to the phenomenon of promiscuous binding. Special care has to be taken with respect to druggability. While a high hydrophobicity is a commonly accepted indicator of druggability, a predominance of hydrophobic properties might also indicate a promiscuous binding site that accommodates varying ligand types in an unselective manner. However, further in-depth analyses of a larger set of tools and comprehensive benchmark analyses of methods for the characterization of binding sites are a major prerequisite to substantiate these general conclusions. Altogether, we should avoid over-optimism with respect to results of tools for binding site comparison and druggability assessment. Nonetheless, knowing their pitfalls, we can establish suitable protocols to cope with minor drawbacks and apply these tools in medicinal chemistry projects.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support from the German Federal Ministry for Education and Research (BMBF, Medizinische Chemie in Dortmund, Grant BMBF 1316053) and from the Chemical Industry Fund (Kekulé Mobility Fellowship). We also wish to thank D. Rognan (IChem) and BioSolveIT (DoGSiteScorer) for providing access to these tools. We acknowledge OpenEye for the supply of an academic license to use ROCS and SiteHopper.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00102f
References
- Hu Y., Bajorath J. Drug Discov. Today. 2013;18(13–14):644–650. doi: 10.1016/j.drudis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Proschak E., Stark H., Merk D. J. Med. Chem. 2018;62(2):420–444. doi: 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- Anighoro A., Bajorath J., Rastelli G. J. Med. Chem. 2014;57(19):7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Haupt V. J., Daminelli S., Schroeder M. PLoS One. 2013;8(6):e65894. doi: 10.1371/journal.pone.0065894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saqib U., Kelley T. T., Panguluri S. K., Liu D., Savai R., Baig M. S. Front. Pharmacol. 2018;9:1201. doi: 10.3389/fphar.2018.01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lounkine E., Keiser M. J., Whitebread S., Mikhailov D., Hamon J., Jenkins J. L. Nature. 2012;486(7403):361–367. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm N., Desaphy J., Quinn R. J., Rognan D., Kellenberger E. J. Chem. Inf. Model. 2012;52(9):2410–2421. doi: 10.1021/ci300196g. [DOI] [PubMed] [Google Scholar]
- Barelier S., Sterling T., O'Meara M. J., Shoichet B. K. ACS Chem. Biol. 2015;10(12):2772–2784. doi: 10.1021/acschembio.5b00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobeli I., Favia A. D., Thornton J. M. Nat. Biotechnol. 2009;27(2):157–167. doi: 10.1038/nbt1519. [DOI] [PubMed] [Google Scholar]
- Peters J.-U., Hert J., Bissantz C., Hillebrecht A., Gerebtzoff G., Bendels S. Drug Discov. Today. 2012;17(7–8):325–335. doi: 10.1016/j.drudis.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Seidler J., McGovern S. L., Doman T. N., Shoichet B. K. J. Med. Chem. 2003;46(21):4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- Feng B. Y., Simeonov A., Jadhav A., Babaoglu K., Inglese J., Shoichet B. K. J. Med. Chem. 2007;50(10):2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- Baell J. B., Holloway G. A. J. Med. Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell J. B., Nissink J. W. M. ACS Chem. Biol. 2018;13(1):36–44. doi: 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalencas X., Mestres J. Mol. Inf. 2013;32(11–12):976–990. doi: 10.1002/minf.201300082. [DOI] [PubMed] [Google Scholar]
- Ehrt C., Brinkjost T., Koch O. PLoS Comput. Biol. 2018;14(11):e1006483. doi: 10.1371/journal.pcbi.1006483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger E., Schalon C., Rognan D. Curr. Comput.-Aided Drug Des. 2008;4(3):209–220. [Google Scholar]
- Kellenberger E., Muller P., Schalon C., Bret G., Foata N., Rognan D. J. Chem. Inf. Model. 2006;46(2):717–727. doi: 10.1021/ci050372x. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L., Groom C. R. Nat. Rev. Drug Discovery. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Abi Hussein H., Geneix C., Petitjean M., Borrel A., Flatters D., Camproux A.-C. Drug Discov. Today. 2017;22(2):404–415. doi: 10.1016/j.drudis.2016.11.021. [DOI] [PubMed] [Google Scholar]
- Barril X. WIREs Comput. Mol. Sci. 2013;3(4):327–338. [Google Scholar]
- Desaphy J., Azdimousa K., Kellenberger E., Rognan D. J. Chem. Inf. Model. 2012;52(8):2287–2299. doi: 10.1021/ci300184x. [DOI] [PubMed] [Google Scholar]
- ROCS, OpenEye Scientific Software, Santa Fe, NM, Available from: URL: http://www.eyesopen.com.
- The UniProt Consortium Nucleic Acids Res. 2019;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Skolnick J. Nucleic Acids Res. 2005;33(7):2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold M. R., Cebron N., Dill F., Gabriel T. R., Kötter T. and Meinl T., et al., KNIME: The Konstanz Information Miner, in Studies in Classification, Data Analysis, and Knowledge Organization (GfKL 2007), Springer, 2007. [Google Scholar]
- Rogers D., Hahn M. J. Chem. Inf. Model. 2010;50(5):742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Beisken S., Meinl T., Wiswedel B., de Figueiredo L. F., Berthold M., Steinbeck C. BMC Bioinf. 2013;14:257. doi: 10.1186/1471-2105-14-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RDKit: Open-source cheminformatics, 2016, Available from: URL: http://www.rdkit.org.
- Bento A. P., Gaulton A., Hersey A., Bellis L. J., Chambers J., Davies M. Nucleic Acids Res. 2014;42(Database issue):D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Dunbrack Jr. R. L. Bioinformatics. 2003;19(12):1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- Bowes J., Brown A. J., Hamon J., Jarolimek W., Sridhar A., Waldron G. Nat. Rev. Drug Discovery. 2012;11(12):909–922. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- Peters J.-U. J. Med. Chem. 2013;56(22):8955–8971. doi: 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]
- Tanaka E. J. Clin. Pharm. Ther. 1998;23(6):403–416. doi: 10.1046/j.1365-2710.1998.00086.x. [DOI] [PubMed] [Google Scholar]
- Ehrt C., Brinkjost T., Koch O. J. Med. Chem. 2016;59(9):4121–4151. doi: 10.1021/acs.jmedchem.6b00078. [DOI] [PubMed] [Google Scholar]
- Schmitt S., Hendlich M., Klebe G. Angew. Chem., Int. Ed. 2001;40(17):3141–3144. doi: 10.1002/1521-3773(20010903)40:17<3141::AID-ANIE3141>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Schmitt S., Kuhn D., Klebe G. J. Mol. Biol. 2002;323(2):387–406. doi: 10.1016/s0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- Weill N., Rognan D. J. Chem. Inf. Model. 2010;50(1):123–135. doi: 10.1021/ci900349y. [DOI] [PubMed] [Google Scholar]
- Yeturu K., Chandra N. BMC Bioinf. 2008;9:543. doi: 10.1186/1471-2105-9-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krotzky T., Grunwald C., Egerland U., Klebe G. J. Chem. Inf. Model. 2015;55(1):165–179. doi: 10.1021/ci5005898. [DOI] [PubMed] [Google Scholar]
- Schalon C., Surgand J.-S., Kellenberger E., Rognan D. Proteins. 2008;71(4):1755–1778. doi: 10.1002/prot.21858. [DOI] [PubMed] [Google Scholar]
- Xie L., Xie L., Bourne P. E. Bioinformatics. 2009;25(12):i305–i312. doi: 10.1093/bioinformatics/btp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J., Janezic D. Bioinformatics. 2010;26(9):1160–1168. doi: 10.1093/bioinformatics/btq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman-Peleg A., Nussinov R., Wolfson H. J. J. Mol. Biol. 2004;339(3):607–633. doi: 10.1016/j.jmb.2004.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman-Peleg A., Nussinov R., Wolfson H. J. Nucleic Acids Res. 2005;33(Web Server issue):W337–W341. doi: 10.1093/nar/gki482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista J., Hawkins P. C. D., Tolbert R., Geballe M. T. Aust. J. Chem. 2014;6(Suppl 1):P57. [Google Scholar]
- Desaphy J., Raimbaud E., Ducrot P., Rognan D. J. Chem. Inf. Model. 2013;53(3):623–637. doi: 10.1021/ci300566n. [DOI] [PubMed] [Google Scholar]
- Chartier M., Najmanovich R. J. Chem. Inf. Model. 2015;55(8):1600–1615. doi: 10.1021/acs.jcim.5b00333. [DOI] [PubMed] [Google Scholar]
- Wood D. J., de Vlieg J., Wagener M., Ritschel T. J. Chem. Inf. Model. 2012;52(8):2031–2043. doi: 10.1021/ci3000776. [DOI] [PubMed] [Google Scholar]
- Da Silva F., Desaphy J., Rognan D. ChemMedChem. 2018;13(6):507–510. doi: 10.1002/cmdc.201700505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong E. R., DeLong D. M., Clarke-Pearson D. L. Biometrics. 1988;44(3):837–845. [PubMed] [Google Scholar]
- Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2015, Available from: URL: http://www.R-project.org/.
- Volkamer A., Kuhn D., Grombacher T., Rippmann F., Rarey M. J. Chem. Inf. Model. 2012;52(2):360–372. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- Schmidtke P., Barril X. J. Med. Chem. 2010;53(15):5858–5867. doi: 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- Stegemann B., Klebe G. Proteins. 2012;80(2):626–648. doi: 10.1002/prot.23226. [DOI] [PubMed] [Google Scholar]
- Gao M., Skolnick J. Bioinformatics. 2013;29(5):597–604. doi: 10.1093/bioinformatics/btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Skolnick J. PLoS Comput. Biol. 2013;9(10):e1003302. doi: 10.1371/journal.pcbi.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Zhang Y. Bioinformatics. 2010;26(7):889–895. doi: 10.1093/bioinformatics/btq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. J., Green D. V. S., Luscombe C. N., Hill A. P. Drug Discov. Today. 2011;16(17–18):822–830. doi: 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Korkuć P., Walther D. Front. Mol. Biosci. 2015;2:51. doi: 10.3389/fmolb.2015.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S., Knox C., Guo A. C., Shrivastava S., Hassanali M., Stothard P. Nucleic Acids Res. 2006;34(Database issue):D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahraman A., Morris R. J., Laskowski R. A., Favia A. D., Thornton J. M. Proteins. 2010;78(5):1120–1136. doi: 10.1002/prot.22633. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Wolber G., Langer T. J. Chem. Inf. Model. 2005;45(1):160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Martin E., Cao E. J. Comput.-Aided Mol. Des. 2015;29(5):387–395. doi: 10.1007/s10822-014-9819-y. [DOI] [PubMed] [Google Scholar]
- Chartier M., Morency L.-P., Zylber M. I., Najmanovich R. J. BMC Pharmacol. Toxicol. 2017;18(1):18. doi: 10.1186/s40360-017-0128-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Pei J., Lai L. Curr. Pharm. Des. 2013;19(12):2326–2333. doi: 10.2174/1381612811319120019. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J., Huth J. R., Fesik S. W. J. Med. Chem. 2005;48(7):2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J., Huth J. R., Tse C. Drug Discov. Today. 2005;10(23–24):1675–1682. doi: 10.1016/S1359-6446(05)03624-X. [DOI] [PubMed] [Google Scholar]
- Ghuman J., Zunszain P. A., Petitpas I., Bhattacharya A. A., Otagiri M., Curry S. J. Mol. Biol. 2005;353(1):38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- Khan H., Harris R. J., Barna T., Craig D. H., Bruce N. C., Munro A. W. J. Biol. Chem. 2002;277(24):21906–21912. doi: 10.1074/jbc.M200637200. [DOI] [PubMed] [Google Scholar]
- Funder J. W. Cardiovasc. Res. 1995;30(2):177–180. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.