

Abstract
The electrochemical systems of both grafted catechol as a pH-responsive electrophore and immobilized cytochrome c as a model redox protein are detected by cyclic voltammetry at an optimized lipid deposit-modified glassy carbon electrode. The catechol covalent grafting is successfully performed by the one-pot/three-step electrochemical reduction of 3,4-dihydroxybenzenediazonium salts generated in situ from 4-nitrocatechol. The resulting glassy carbon electrode electrochemically modified by grafted catechol species is evaluated as an efficient electrochemical pH sensor. The optimized molar ratio for the lipid deposit, promoting cytochrome c electrochemical activity in solution onto glassy carbon electrode, is reached for the lipid mixture composed of 75% 1,2-dioleoyl-sn-glycero-3-phosphocholine and 25% cardiolipin. Cytochrome c immobilization into the optimized supported lipid deposit is efficiently achieved by cyclic voltammetry (10 cycles) recorded at the modified glassy carbon electrode in a cytochrome c solution. The pH-dependent redox response of the grafted catechol and that of the immobilized cytochrome c are finally detected at the same lipid-modified glassy carbon electrode without alteration of their structure and electrochemical properties in the pH range 5–9.
Introduction
Electroactive bacteria are living microorganisms that are able to directly connect their respiratory metabolism to their extracellular environment by transferring electrons across biological membranes to or from solids like metal oxides or electrodes.1−3 These bacteria are organized at conducting surfaces as biofilms and can indeed shuttle electrons via periplasmic and membrane proteins to/from electrodes.3,4 Hence, electroactive bacteria represent living, stable, self-replicating, and low-cost electrode catalysts. This unique property leads to potential “green” biotechnology applications, such as microbial fuel cells, microbial electrosynthesis cells, wastewater treatment, desalination, and biosensors.1,2,4,5 To permit the advent of these promising microbial electrochemical technologies, it is crucial to progress toward the fundamental knowledge of these electroactive biofilms. In particular, it is important to understand the role and function of membrane proteins in electroactive bacteria. Indeed, little is known on the coupling of extracellular electron and proton transfers in electroactive bacteria and these phenomena are key factors for optimizing and designing relevant applications.
Electroactive microorganisms such as Gram-negative Geobacter sulfurreducens directly connect and transfer electrons to anodes via outer-membrane c-type cytochrome redox proteins as ultimate redox relays.3,6 In the absence of an organic substrate like acetate, at least four redox proteins may be detected electrochemically in the biofilm, whereas in the presence of an organic substrate, extracellular electron release from bacterial catabolism is easily followed by recording a catalytic anodic current.7 The anolyte then becomes more acidic, consistent with the oxidation of the organic substrate to carbon dioxide concomitantly with proton release. Acidification of the anolyte, of the biofilm, and/or of the biofilm/electrode interface compromises the performance and viability of the catalytic biofilms.8−10 In fact, the extracellular electron transfer may be coupled to proton transfer in outer-membrane c-type cytochromes or, alternatively, nonredox proton transport proteins may be responsible for the acidification of the anolyte. Although extracellular electron and proton transfers are known to occur in electroactive bacteria,9,11,12 the fundamental understanding of these processes is still in its infancy and precludes the optimization of microbial electrochemical technologies.
The overall objective of our research is to develop an efficient and versatile electrochemical platform for probing electroactive bacteria membrane proteins incorporated in artificial lipid deposits supported on modified carbon electrode. In a first approach, the electrodes are modified with pH-responsive electrophores such as quinone units and then covered by lipid layers or deposits, creating an artificial supported lipid membrane in which membrane proteins are incorporated and electrochemically probed (Figure 1). Indeed, the intrinsic electroactivity of the protein (if any) can be probed directly with the lipid-modified electrode and possible proton transport occurring across the lipid layer by the membrane protein can in principle be detected with the grafted pH-responsive redox probe (Figure 1).13,14 The first experimental challenge of this work is to immobilize and to analyze the electrochemical activity of the pH-responsive redox probe and that of the membrane protein at the same electrode without alteration of their native and electrochemical properties.15
Figure 1.

Conceptual scheme of the target electrochemical platform: pH-responsive electrophores (here quinones) grafted on electrodes supporting lipid deposits allow the study of the charge transfer properties of membrane-associated proteins (electron or proton transfer or both). Note that the selected proteins in this scheme are for an illustrative purpose only.
Here, a proof of concept of the electrochemical platform is presented using surface-grafted catechol moieties as pH-responsive electrophores and cytochrome c as the model membrane-associated redox protein. Cytochrome c is immobilized into the supported lipid deposit at the catechol-modified glassy carbon electrode surface. We focus on the detection of both the pH-dependent electrophore and the redox protein reversible redox systems at glassy carbon successively modified by covalent catechol grafting, then by an optimized lipid deposit, and finally by immobilization of cytochrome c. First, the grafting of catechol onto the glassy carbon electrode is performed by a one-pot/three-step electrochemical procedure that consists of the cathodic reduction of 4-nitrocatechol in acidic conditions in the presence of sodium nitrite. The electrode functionalization and the properties of the grafted catechol moieties are evaluated by cyclic voltammetry in phosphate buffer aqueous solution at different pH. Then, optimization of the lipid deposit promoting cytochrome c electrochemical activity at glassy carbon leads to a 75% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 25% cardiolipin (CL) ratio. Cytochrome c immobilization is achieved by cyclic voltammetry of either a lipid-modified glassy carbon electrode or a catechol/lipid-modified glassy carbon electrode, and the electrochemical properties of the modified electrodes are studied by cyclic voltammetry in phosphate buffer aqueous electrolyte at various pH values.
Results and Discussion
Catechol Grafted onto Glassy Carbon Electrode
In this section, the grafting of pH-responsive catechol moieties and the electrochemical properties of the modified carbon electrodes are discussed. First, a bare glassy carbon electrode was modified by electrochemical reduction of aryldiazonium salts in situ generated from reduction of the nitro precursor to the arylamine (Figure 2). Then, the catechol-modified glassy carbon electrode was studied by cyclic voltammetry in phosphate buffer aqueous solution at different pH values (Figure 3).
Figure 2.

Catechol electrografting by one-pot/three-step electrochemical reduction of 3,4-dihydroxybenzenediazonium salts in situ generated from 4-nitrocatechol. Top: Schematic of the reaction. Bottom: Cyclic voltammograms (2 cycles) recorded at 50 mV s–1 at a bare glassy carbon electrode in 1 mM 4-nitrocatechol + 3 mM NaNO2 with 0.1 M HCl as aqueous electrolyte under inert atmosphere (Ar).
Figure 3.
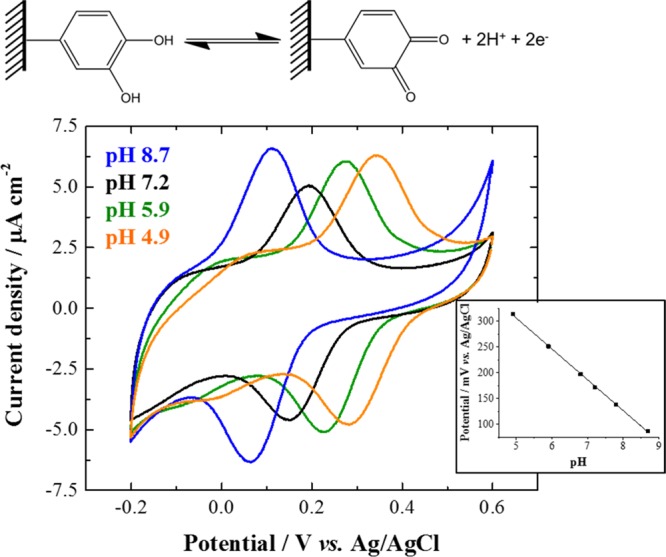
Grafted catechol redox probe. Top: Schematic of the grafted quinone/hydroquinone redox couple. Bottom: Cyclic voltammograms (third cycle shown) recorded at 20 mV s–1 on catechol-modified glassy carbon electrode in 10 mM phosphate buffer aqueous electrolyte successively at pH 7.2 (black line), pH 8.7 (blue line), pH 5.9 (green line), and pH 4.9 (orange line) under inert atmosphere (Ar). Inset: pH dependence of the apparent normal potential of the grafted catechol with the corresponding linear regression analysis: y = −59.8x + 605, R2 = 0.9994.
The covalent electrografting is a powerful method to functionalize a wide range of electrode materials’ surfaces for many applications.16 Especially, the reductive electrografting of aryldiazonium cations is very efficient for surface modification because of its simplicity, versatility, and robustness.16−18 Here, catechol species were grafted on glassy carbon electrode following a surface derivatization strategy initially reported by Cougnon et al. and based on the one-pot/three-step electrochemical reduction of aryldiazonium cations in situ generated from the nitro precursor (Figure 2).19,20 For our work, the main advantage to use this one-pot/three-step procedure is to limit the surface coverage (to less than a monolayer equivalent)20 to retain fast electron transfer kinetics between the redox protein and the modified electrode. Indeed, this procedure allows to produce a source of arylamine “on demand” and only in the vicinity of the working electrode surface.19,20 This grafting method is based on the cathodic reduction of 4-nitrocatechol to the corresponding amino derivative in the presence of sodium nitrite in acidic aqueous solution (0.1 M HCl). On the first cyclic voltammogram (Figure 2), the broad reduction wave is divided into two peaks located at −0.5 and −0.7 V. The first reduction process at Epc = −0.5 V is assigned to the reduction of the 4-nitrocatechol to 4-aminocatechol, whereas the reduction at Epc = −0.7 V is tentatively assigned to species originating from the reaction of grafted catechol moieties with nucleophilic species such as amine (Figures S1 and 2).20,21 In addition, the presence of only one reduction peak in the second cyclic voltammogram at Epc = −0.6 V is consistent with the presence of a first grafted catechol “layer” (Figure 2). The current intensity of the reduction peak is also lower on the second cycle, suggesting a partial passivation of the electrode surface consistent with glassy carbon functionalization.20 The aryldiazonium reduction peak is expected at ca. +0.2 V.20 However, since the diazotization of the arylamine is fast,22 the aryldiazonium is generated at an electrode potential where it is immediately reduced. Hence, the electrochemical response associated to the reduction of the in situ generated aryldiazonium cannot be detected on the cyclic voltammogram. In this electrochemical reaction mechanism, 4-aminocatechol is electrogenerated at the electrode from 4-nitrocatechol and immediately converted into 3,4-dihydroxybenzenediazonium, which is then reduced at the electrode in a subsequent step, leading to the surface covalent derivatization (Figure 2).
Catechol grafting onto glassy carbon was confirmed by sonicating the modified electrode for 2 min in ultrapure water to desorb any material not covalently attached to its surface, followed by the electrochemical detection of the grafted catechol at different pH values (Figure 3). All apparent normal potentials of redox species immobilized onto glassy carbon electrodes and discussed in the manuscript are reported in Table 1. In Figure 3, a pH-dependent reversible redox system is detected (Table 1) and ascribed to the grafted quinone/hydroquinone redox couple. Figure S2 shows the cyclic voltammograms recorded at the glassy carbon electrode before and after surface modification, which confirms the effectiveness of the grafting procedure. In addition, the linear relationship between the oxidative peak currents and the scan rate (Figure S3) indicates that the catechol groups are confined to the electrode surface.19,20,23 The surface coverage of the grafted catechol species onto the modified glassy carbon calculated from integration of the faradaic current on the cyclic voltammograms and based on the geometrical area of the electrode (0.0707 cm2) is (1.5 ± 0.5) × 10–10 mol cm–2. This corresponds to a submonolayer modification and is consistent with the results reported previously.20 Indeed, the grafting of catechol units through the one-pot/three-step electrochemical reduction procedure from the nitro precursor allows a fine control of the grafting yield. In addition, and as expected for this pH-responsive electrophore, the apparent normal potential of the grafted catechol is linearly proportional to the pH with a slope of 60 mV per pH unit over a pH range from 5 to 9 in the phosphate buffer electrolyte used here (Figure 3, inset). These results show that the glassy carbon electrode is electrochemically modified by grafted catechol species from a 4-nitrocatechol precursor and the resulting modified electrode is an efficient electrochemical pH sensor.
Table 1. Apparent Normal Potentials of the Grafted Catechol and Immobilized Cytochrome c Detected by Cyclic Voltammetry at the Modified Glassy Carbon Electrodes in 10 mM Phosphate Buffer Aqueous Electrolyte.
| glassy carbon electrode modified by | apparent normal potential of the grafted catechol (V vs Ag/AgCl) | apparent normal potential of the immobilized cytochrome c (V vs Ag/AgCl) |
|---|---|---|
| catechol | +0.314 V at pH 4.9 | |
| +0.251 V at pH 5.9 | ||
| +0.172 V at pH 7.2 | ||
| +0.087 V at pH 8.7 | ||
| 75% DOPC/25% CL lipid deposit/cytochrome c | +0.005 V (at pH range 5–9) | |
| catechol/75% DOPC/25% CL lipid deposit/cytochrome c | +0.293 V at pH 5.5 | +0.012 V (at pH range 5–9) |
| +0.267 V at pH 6.0 | ||
| +0.232 V at pH 7.0 | ||
| +0.181 V at pH 8.2 | ||
| +0.137 V at pH 9.1 |
Cytochrome c Electroactivity in Solution and Immobilized onto Lipid-Modified Glassy Carbon Electrode
In this section, we discuss the electrochemical study and immobilization of cytochrome c at a glassy carbon electrode modified by an optimized supported lipid mixture deposit. The optimum lipid deposit ratio of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and cardiolipin (CL) was first determined by cyclic voltammetry recorded at the different lipid-modified glassy carbon electrodes in a cytochrome c solution (Figure 4). In a second step, the immobilization of cytochrome c onto the glassy carbon electrode modified by the optimized lipid film was performed by recurrent cyclic voltammetry (10 cycles) and then the resulting modified electrode was transferred to a protein-free phosphate buffer aqueous electrolyte for studying the immobilized protein electroactivity (Figure 5).
Figure 4.
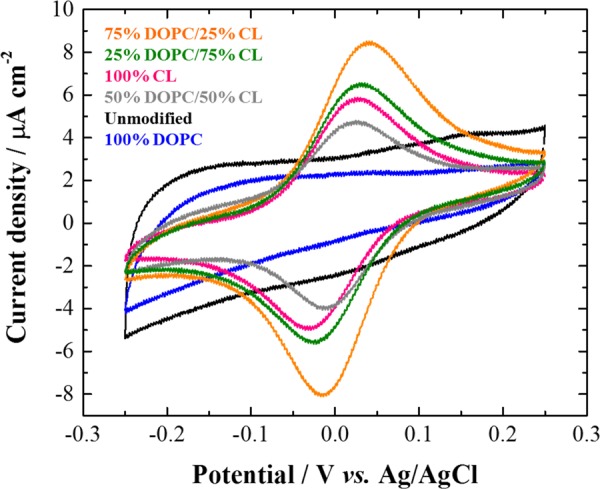
Cyclic voltammograms (10th cycle shown) recorded at 20 mV s–1 on glassy carbon electrode before (black) and after modification with a lipid deposit comprising 100% DOPC (blue), 100% CL (pink), 75% DOPC/25% CL (orange), 50% DOPC/50% CL (gray), and 25% DOPC/75% CL (green) in a 0.05 mM cytochrome c solution in 10 mM phosphate buffer aqueous electrolyte at pH 7.2 under inert atmosphere (Ar).
Figure 5.

(A) Cyclic voltammograms (10 cycles) recorded at 20 mV s–1 on glassy carbon electrode modified by a 75% DOPC/25% CL lipid deposit in 0.15 mM cytochrome c solution 10 mM phosphate buffer aqueous electrolyte at pH 7.2 under inert atmosphere (Ar). (B) Cyclic voltammogram (third cycle shown) recorded at 20 mV s–1 in 10 mM protein-free phosphate buffer aqueous electrolyte at pH 7.2 under Ar with a glassy carbon electrode modified by a 75% DOPC/25% CL lipid deposit and previously cycled in a cytochrome c solution for protein immobilization.
In a previous work, we have investigated the effect of pure cardiolipin (CL) deposit to promote the electrochemical activity of cytochrome c at a glassy carbon electrode since it is known that this lipid is crucial for the physiological function of this redox protein.24 Here, the dilution of cardiolipin within a 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) matrix has been optimized. DOPC is a phospholipid commonly used in supported lipid bilayers but inefficient to promote the cytochrome c electroactivity (vide infra). To optimize the lipids mixture deposited on glassy carbon electrode surface, the best ratio of DOPC/CL for promoting the cytochrome c electroactivity was determined by cyclic voltammetry with different lipid deposits supported on glassy carbon electrode in a cytochrome c aqueous solution (Figure 4). Each lipid deposit was carried out by dipping for a few seconds the electrode in a 3.4 mM lipid solution in ethanol with different molar ratios of DOPC and CL and by subsequently letting the electrode dry upside down under air. As expected, the cyclic voltammogram recorded at the bare glassy carbon electrode in the cytochrome c solution does not display the protein redox signal because of denaturation or unfavorable orientation.25,26 The same result is also observed in the cyclic voltammogram recorded at the 100% DOPC lipid deposit-modified glassy carbon electrode although with a lower capacitive current in comparison to the unmodified electrode. This observation confirms that cardiolipin and its specific interaction with cytochrome c is an essential parameter for promoting the protein electrochemical activity.24 Indeed, a reversible redox system at +0.010 V (ΔEp = 47 ± 8 mV at 20 mV s–1) corresponding to the typical electrochemical response of cytochrome c in solution is observed in the cyclic voltammograms recorded at glassy carbon electrodes modified by a lipid deposit containing cardiolipin.25,27−29 One can also note a slight variation of the apparent normal potential of cytochrome c (from −0.002 to +0.013 V, Table S1) depending on the cardiolipin fraction in the lipid deposit on the electrodes. In fact, cardiolipin interaction with cytochrome c in systems that do not mimic the native negative curvature and length scale of the inner mitochondrial membrane relies on electrostatic interactions involving lysines and histidines in the so called A- and L-sites.30 This is likely the case at the present modified electrodes. A-site lysines are also involved in the alkaline transition and play an important role in the stabilization of the native heme axial coordination in the physiological pH range.31 Changes in the population fraction of cytochrome c destabilized upon cardiolipin binding for the different lipid ratios may be at the core of the slight variation of the cytochrome c voltammetric signal with lipid composition. Furthermore, the highest current intensity of the protein redox signal is reached for the glassy carbon electrode modified by a 75% DOPC 25% CL lipid deposit. Hence, for promoting cytochrome c electroactivity by a supported lipid deposit, the optimized lipid molar ratio of 75% DOPC/25% CL is used for all further experiments.
A simple and efficient strategy to immobilize cytochrome c on the lipid-modified electrode consists in performing consecutive cyclic voltammetry (here 10 cycles were performed) in a cytochrome c solution until a steady-state current is reached (Figures 5A and S4) and then to transfer the modified electrode into a protein-free phosphate buffer where the electroactivity of the immobilized protein can be studied (Figure 5B).24,25,27Figure 5A shows the increase of the cytochrome c redox peak current intensity during continuous cyclic voltammetry in a cytochrome c solution at a glassy carbon electrode modified by a 75% DOPC/25% CL lipid deposit. The redox peak current intensity increases gradually until a steady-state current is reached at the 10th cycle (Figure S4). This result suggests that the lipid-modified glassy carbon electrode is saturated with electroactive surface-confined cytochrome c captured from solution thanks to the specific interaction between this redox protein and cardiolipin contained in the supported lipid deposit.24,25,27
In Figure 5B, the cyclic voltammogram recorded in a protein-free aqueous electrolyte on 75% DOPC/25% CL lipid deposit-modified glassy carbon electrode and previously cycled in cytochrome c solution displays a redox system at +0.005 V and is assigned to immobilized cytochrome c. The protein apparent potential (+0.005 V) is only slighty shifted compared to that in solution (+0.013 V), indicating that cytochrome c immobilized in the lipid film is not denaturated.24,27,32 The electrochemical signal of the immobilized protein corresponds to an electrochemically quasi-reversible electron transfer reaction since the peak-to-peak potential separation is ΔEp = 90 ± 5 mV at 20 mV s–1.33−35 The surface coverage of the immobilized electroactive cytochrome c estimated from faradaic charge integration of the cyclic voltammogram is ca. (1.7 ± 0.5) × 10–10 mol cm–2. This value is at least 10 times higher than that expected for cytochrome c immobilization onto a supported lipid deposit,24,25,27 because here the modified electrode is directly transferred from the cytochrome c solution to the protein-free buffer aqueous electrolyte after a simple gentle wash in phosphate buffer.35 Indeed, the fraction of the immobilized protein that is only loosely adsorbed at the modified electrode quickly desorbs during consecutive cyclic voltammograms (Figure S5). The redox potential of immobilized cytochrome c is not dependent on the pH of the solution in the range of 5–9 (Figure S5).36,37 These results show that cytochrome c immobilization is easily and efficiently carried out by cyclic voltammetry at an optimized DOPC/CL lipid deposit-modified glassy carbon electrode in a cytochrome c solution and the electrochemical properties of the redox protein are kept intact in these conditions.
Grafted Catechol, Optimized Lipid Deposit, and Immobilized Cytochrome c onto Glassy Carbon Electrode
In this section, we report the electrochemical properties of a glassy carbon electrode successively modified by catechol following the one-pot/three-step electrochemical reduction grafting procedure, then by the optimized 75% DOPC/25% CL lipid deposit (solvent evaporation), and finally by the immobilization of cytochrome c through cyclic voltammetry in a solution of the redox protein (Figure S6). The catechol/lipid deposit/cytochrome c-modified glassy carbon electrode was transferred and studied by cyclic voltammetry in a phosphate buffer aqueous electrolyte at different pH values (Figure 6).
Figure 6.

(A) Cyclic voltammograms (third cycle shown) at 20 mV s–1 at a glassy carbon electrode modified by catechol/lipid deposit/cytochrome c in 10 mM phosphate buffer aqueous electrolyte successively recorded at pH 7 (solid line), pH 6 (dotted line), pH 8.2 (dashed line), pH 9.1 (not shown), and pH 5.5 (not shown) under inert atmosphere (Ar) (see Figure S7 for a plot of catechol redox potential vs all five pH values). (B) Faradaic signal of interest obtained by subtracting the capacitive current from the raw cyclic voltammograms.
The cyclic voltammograms successively recorded at the catechol/lipid deposit/cytochrome c-modified glassy carbon electrode in phosphate buffer aqueous electrolyte at various pH values (pH 7, 6, and 8.2 in the chronological order of measurements) are presented in Figure 6A. Figure 6B shows the faradaic contribution38 in the cyclic voltammograms of Figure 6A obtained by capacitive current subtraction using the QSoas software.39 The cyclic voltammograms clearly show two reversible redox systems located at +0.232 V (ΔEp = 50 ± 3 mV at 20 mV s–1) and at +0.012 V (ΔEp = 43 ± 2 mV at 20 mV s–1) in phosphate buffer aqueous electrolyte at pH 7, corresponding to the grafted catechol and immobilized cytochrome c electrochemical signals, respectively. In addition, the presence of the redox protein in the methanol/ethanol/water washing solution of modified electrodes has been confirmed by UV–visible spectroscopy measurements (Figure S8). Electrochemical impedance spectroscopy measurements at each modification step of the glassy carbon electrode (Figure S9) do not evidence any significant changes after the initial catechol grafting, indicating that the subsequent lipid/protein deposit still contains defects and remains permeable. As previously observed for the immobilized redox protein onto the lipid-modified glassy carbon electrode (Figure S5), the electrochemical response of cytochrome c presented in Figure 6 is independent of the pH in the range studied (pH 5–9). A decreasing of the redox peaks’ current intensity is also observed upon consecutive cyclic voltammograms at different pH values, indicating a partial desorption of the protein during cyclic voltammetry. In contrast, the redox signal of grafted catechol shows a better stability because of the robustness of the covalent bond with the electrode surface. Importantly, after the lipid/protein adduct onto the electrode, the grafted catechol kept its dependence of pH-responsive properties with a redox potential linearly proportional to the pH (Table 1 and Figure S7) and a slope of 42 mV per pH unit in the phosphate buffer electrolyte used here. This work demonstrates that grafted catechol as pH-responsive electrophore and immobilized cytochrome c as model redox protein can be detected by cyclic voltammetry at a catechol/lipid deposit/redox protein-modified glassy carbon electrode, while keeping intact their electrochemical properties without alteration of their native structure.
Conclusions
In summary, the electrochemical detection of both grafted catechol and immobilized cytochrome c onto lipid deposit-modified glassy carbon electrode has been achieved at the same electrode. First, a glassy carbon electrode has been functionalized by a catechol covalent grafting procedure following the one-pot/three-step electrochemical reduction of 3,4-dihydroxybenzenediazonium salts in situ generated from 4-nitrocatechol. Then, an optimized lipid film composed of 75% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 25% cardiolipin (CL) has been deposited by solvent evaporation on the catechol-modified glassy carbon electrode surface for promoting the cytochrome c electrochemical activity. The cytochrome c immobilization onto the catechol/lipid deposit-modified electrode has been achieved following a simple and efficient strategy consisting of recurrent cyclic voltammetry in a cytochrome c solution until a steady-state current is reached. The resulting catechol/lipid deposit/cytochrome c-modified glassy carbon electrode has been finally transferred into a protein-free phosphate buffer where its electrochemical behavior has been studied by cyclic voltammetry. The electrochemical detection of both grafted catechol and immobilized cytochrome c reversible redox systems has been achieved while maintaining intact their native electrochemical properties, namely, the pH-dependent reversible system of the grafted quinone/catechol redox couple and the pH-independent electroactivity of the redox protein (in the pH range studied). This work is a first proof of concept for an electrochemical platform aimed at probing charge transfer properties of membrane-associated proteins. The pH range (ca. pH 5–9) studied here is physiologically relevant for a wide range of proteins, and the associated potential shift of the grafted catechol (namely, 293–137 mV vs Ag/AgCl at pH 5.5 and 9.1, respectively) does not overlap with any of the known outer-membrane c-type cytochromes of electroactive bacteria.40 Current work in our laboratory is devoted to the application of this platform to membrane and periplasmic proteins from electroactive bacteria.
Experimental Section
Reagents
Milli-Q water (18.2 MΩ cm) was used to prepare all solutions. Cardiolipin (CL) solution from bovine heart (∼4.7–5.3 mg mL–1 in ethanol, ≥97%) and cytochrome c from equine heart (≥95%) were obtained from Sigma-Aldrich and stored in a freezer (−18 °C). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) powder was purchased from Avanti Polar Lipids and stored in a freezer. The 10 mM phosphate buffer aqueous solution at pH 7.2 was prepared by using sodium hydrogen phosphate anhydrous ACS (99.0% min) and potassium dihydrogen phosphate ACS (99.0% min) from Alfa Aesar. Anhydrous absolute ethanol from Carlo Erba Reagents was used to prepare DOPC/CL lipid mixture solutions. Concentrated hydrochloric acid (12 M, 37%, ACS Reagent) and pellets for making sodium hydroxide solution (ACS) were obtained from VWR and were used to adjust the pH of the phosphate buffer solution. Sodium nitrite (>99%) was obtained from Sigma-Aldrich, and 4-nitrocatechol (>98%) was obtained from Alfa Aesar.
Electrochemical Measurements
A three-electrode cell with a glassy carbon disk electrode (3 mm diameter) obtained from BASi as the working electrode, a platinum wire as the counter electrode, and an Ag/AgCl, KCl 3 M reference electrode were used to perform all electrochemical measurements. The working electrode was mirror-polished under a flux of ultrapure water on a fine grid silicon carbide paper (4000-grid SiC paper, Struers) mounted on a DAP-V Struers polishing equipment rotating at 75 rpm, then rinsed and sonicated in ultrapure water for 5 min before each experiment. Electrochemical experiments were carried out at room temperature (21 ± 3 °C) with an Autolab PGSTAT204 potentiostat/galvanostat (Eco Chemie B.V., The Netherlands) using Nova as the electrochemical software (Metrohm). Before each measurement, all solutions were deaerated by bubbling argon for at least 10 min. Electrochemical impedance spectroscopy measurements were carried out at open circuit potential in the frequency range from 100 kHz down to 50 mHz, with a signal amplitude of 10 mV in 10 mM phosphate buffer aqueous solution at pH 7.2.
Surface Modification Procedures
The catechol grafting on glassy carbon surfaces was performed by cyclic voltammetry in aqueous acidic solution (0.1 M HCl) containing 1 mM 4-nitrocatechol and 3 mM NaNO2 by recording 2 cycles between +0.1 and −0.9 V versus Ag/AgCl, KCl 3 M at a scan rate of 50 mV s–1. Then, the modified glassy carbon electrode was sonicated in ultrapure water for 2 min. The supported lipid deposit was obtained by dipping the glassy carbon electrodes in DOPC/CL lipid mixture solutions (3.4 mM lipids dissolved in ethanol) for 5 s and then let dry under air (less than 30 s for complete evaporation of ethanol). After the complete evaporation of ethanol, the dry electrode was immediately dipped into the 10 mM aqueous phosphate buffer pH 7.2. Cytochrome c immobilization on the deposited lipid film was carried out by immersing the electrode in a 0.15 mM cytochrome c solution and by recording cyclic voltammetry (10 cycles) between 0.3 and −0.2 V versus Ag/AgCl, KCl 3 M at a scan rate of 20 mV s–1. Then, the protein-modified electrode was gently washed with phosphate buffer solution and transferred to a cytochrome-free phosphate buffer electrolyte for electrochemical experiments.
Spectrophotometry Measurements
UV–visible absorption spectra were recorded using a UV–visible spectrophotometer Shimadzu UV-1605 and 1 cm optical length cuvettes. A 0.15 mM cytochrome c solution in 10 mM phosphate buffer at pH 7.2 was diluted in a methanol/ethanol/water mixture (1 mL of each solvent) used as blank, to obtain a protein concentration of 25 nmol L–1. Six modified glassy carbon electrodes were washed with the same solvent mixture (1 mL methanol + 1 mL ethanol + 1 mL ultrapure water), and the presence of cytochrome c was checked by UV–visible spectroscopy in this washing solution.
Acknowledgments
E.L. is supported by a Marie Skłodowska Curie Individual Fellowship through funding from the European Union’s Horizon 2020 research and innovation program, grant agreement no. 745689. The authors also acknowledge the France-Portugal PHC PESSOA program for support, project 40814ZE. Dr. Vincent Fourmond is warmly thanked for providing the QSoas software.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01425.
Cyclic voltammograms and electrochemical impedance spectroscopy measurements recorded at different modified glassy carbon electrodes in phosphate buffer aqueous electrolyte; UV–visible spectra of cytochrome c in a solvent mixture solution (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Logan B. E.; Rabaey K. Conversion of Wastes into Bioelectricity and Chemicals by Using Microbial Electrochemical Technologies. Science 2012, 337, 686–690. 10.1126/science.1217412. [DOI] [PubMed] [Google Scholar]
- Sydow A.; Krieg T.; Mayer F.; Schrader J.; Holtmann D. Electroactive bacteria—molecular mechanisms and genetic tools. Appl. Microbiol. Biotechnol. 2014, 98, 8481–8495. 10.1007/s00253-014-6005-z. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Hsu L. H.-H.; Kavanagh P.; Barrière F.; Lens P. N. L.; Lapinsonnière L.; Lienhard V J. H.; Schröder U.; Jiang X.; Leech D. The ins and outs of microorganism–electrode electron transfer reactions. Nat. Rev. Chem. 2017, 1, 0024 10.1038/s41570-017-0024. [DOI] [Google Scholar]
- Smida H.; Lebègue E.; Bergamini J.-F.; Barrière F.; Lagrost C. Reductive electrografting of in situ produced diazopyridinium cations: Tailoring the interface between carbon electrodes and electroactive bacterial films. Bioelectrochemistry 2018, 120, 157–165. 10.1016/j.bioelechem.2017.12.006. [DOI] [PubMed] [Google Scholar]
- Smida H.; Flinois T.; Lebègue E.; Lagrost C.; Barrière F.. Microbial Fuel Cells—Wastewater Utilization. In Encyclopedia of Interfacial Chemistry; Wandelt K., Ed.; Elsevier: Oxford, 2018; pp 328–336. [Google Scholar]
- Alves A.; Ly H. K.; Hildebrandt P.; Louro R. O.; Millo D. Nature of the Surface-Exposed Cytochrome–Electrode Interactions in Electroactive Biofilms of Desulfuromonas acetoxidans. J. Phys. Chem. B 2015, 119, 7968–7974. 10.1021/acs.jpcb.5b03419. [DOI] [PubMed] [Google Scholar]
- Picot M.; Lapinsonnière L.; Rothballer M.; Barrière F. Graphite anode surface modification with controlled reduction of specific aryl diazonium salts for improved microbial fuel cells power output. Biosens. Bioelectron. 2011, 28, 181–188. 10.1016/j.bios.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Blanchet E.; Pécastaings S.; Erable B.; Roques C.; Bergel A. Protons accumulation during anodic phase turned to advantage for oxygen reduction during cathodic phase in reversible bioelectrodes. Bioresour. Technol. 2014, 173, 224–230. 10.1016/j.biortech.2014.09.076. [DOI] [PubMed] [Google Scholar]
- Torres C. I.; Marcus A. K.; Rittmann B. E. Proton transport inside the biofilm limits electrical current generation by anode-respiring bacteria. Biotechnol. Bioeng. 2008, 100, 872–881. 10.1002/bit.21821. [DOI] [PubMed] [Google Scholar]
- Harnisch F.; Schröder U. Selectivity versus Mobility: Separation of Anode and Cathode in Microbial Bioelectrochemical Systems. ChemSusChem 2009, 2, 921–926. 10.1002/cssc.200900111. [DOI] [PubMed] [Google Scholar]
- Babauta J. T.; Nguyen H. D.; Harrington T. D.; Renslow R.; Beyenal H. pH, redox potential and local biofilm potential microenvironments within Geobacter sulfurreducens biofilms and their roles in electron transfer. Biotechnol. Bioeng. 2012, 109, 2651–2662. 10.1002/bit.24538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto A.; Yoshihide T.; Shafeer K.; Kazuhito H. Proton Transport in the Outer-Membrane Flavocytochrome Complex Limits the Rate of Extracellular Electron Transport. Angew. Chem., Int. Ed. 2017, 56, 9082–9086. 10.1002/anie.201704241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-khouri R. J.; Bricarello D. A.; Watkins E. B.; Kim C. Y.; Miller C. E.; Patten T. E.; Parikh A. N.; Kuhl T. L. pH Responsive Polymer Cushions for Probing Membrane Environment Interactions. Nano Lett. 2011, 11, 2169–2172. 10.1021/nl200832c. [DOI] [PubMed] [Google Scholar]
- Jürmann G.; Schiffrin D. J.; Tammeveski K. The pH-dependence of oxygen reduction on quinone-modified glassy carbon electrodes. Electrochim. Acta 2007, 53, 390–399. 10.1016/j.electacta.2007.03.053. [DOI] [Google Scholar]
- Vacek J.; Zatloukalova M.; Novak D.. Electrochemistry of membrane proteins and protein–lipid assemblies. Curr. Opin. Electrochem. 2018. 10.1016/j.coelec.2018.04.012. [DOI] [Google Scholar]
- Bélanger D.; Pinson J. Electrografting: a powerful method for surface modification. Chem. Soc. Rev. 2011, 40, 3995–4048. 10.1039/c0cs00149j. [DOI] [PubMed] [Google Scholar]
- Delamar M.; Hitmi R.; Pinson J.; Saveant J. M. Covalent Modification of Carbon Surfaces by Grafting of Functionalized Aryl Radicals Produced from Electrochemical Reduction of Diazonium Salts. J. Am. Chem. Soc. 1992, 114, 5883–5884. 10.1021/ja00040a074. [DOI] [Google Scholar]
- Allongue P.; Delamar M.; Desbat B.; Fagebaume O.; Hitmi R.; Pinson J.; Saveant J. M. Covalent modification of carbon surfaces by aryl radicals generated from the electrochemical reduction of diazonium salts. J. Am. Chem. Soc. 1997, 119, 201–207. 10.1021/ja963354s. [DOI] [Google Scholar]
- Cougnon C.; Gohier F.; Belanger D.; Mauzeroll J. In Situ Formation of Diazonium Salts from Nitro Precursors for Scanning Electrochemical Microscopy Patterning of Surfaces. Angew. Chem., Int. Ed. 2009, 48, 4006–4008. 10.1002/anie.200900498. [DOI] [PubMed] [Google Scholar]
- Cougnon C.; Nguyen N. H.; Dabos-Seignon S.; Mauzeroll J.; Bélanger D. Carbon surface derivatization by electrochemical reduction of a diazonium salt in situ produced from the nitro precursor. J. Electroanal. Chem. 2011, 661, 13–19. 10.1016/j.jelechem.2011.07.002. [DOI] [Google Scholar]
- Blurton K. F. An electrochemical investigation of graphite surfaces. Electrochim. Acta 1973, 18, 869–875. 10.1016/0013-4686(73)85040-6. [DOI] [Google Scholar]
- Baranton S.; Belanger D. In situ generation of diazonium cations in organic electrolyte for electrochemical modification of electrode surface. Electrochim. Acta 2008, 53, 6961–6967. 10.1016/j.electacta.2008.02.105. [DOI] [Google Scholar]
- Nguyen N. H.; Esnault C.; Gohier F.; Belanger D.; Cougnon C. Electrochemistry and Reactivity of Surface-Confined Catechol Groups Derived from Diazonium Reduction. Bias-Assisted Michael Addition at the Solid/Liquid Interface. Langmuir 2009, 25, 3504–3508. 10.1021/la804205d. [DOI] [PubMed] [Google Scholar]
- Lebègue E.; Smida H.; Flinois T.; Vié V.; Lagrost C.; Barrière F. An optimal surface concentration of pure cardiolipin deposited onto glassy carbon electrode promoting the direct electron transfer of cytochrome-c. J. Electroanal. Chem. 2018, 808, 286–292. 10.1016/j.jelechem.2017.12.024. [DOI] [Google Scholar]
- Park H.; Park J.-S.; Shim Y.-B. Electrochemical and in situ UV–visible spectroscopic behavior of cytochrome c at a cardiolipin-modified electrode. J. Electroanal. Chem. 2001, 514, 67–74. 10.1016/S0022-0728(01)00627-1. [DOI] [Google Scholar]
- Wang J.; Li M.; Shi Z.; Li N.; Gu Z. Direct Electrochemistry of Cytochrome c at a Glassy Carbon Electrode Modified with Single-Wall Carbon Nanotubes. Anal. Chem. 2002, 74, 1993–1997. 10.1021/ac010978u. [DOI] [PubMed] [Google Scholar]
- Perhirin A.; Kraffe E.; Marty Y.; Quentel F.; Elies P.; Gloaguen F. Electrochemistry of cytochrome c immobilized on cardiolipin-modified electrodes: A probe for protein–lipid interactions. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 2798–2803. 10.1016/j.bbagen.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Liu L.; Shi C.; Huang J.; Li G. Electrochemical analysis of the effect of Ca2+ on cardiolipin–cytochrome c interaction. Biochim. Biophys. Acta, Gen. Subj. 2006, 1760, 1827–1830. 10.1016/j.bbagen.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Salamon Z.; Tollin G. Interaction of Horse Heart Cytochrome c with Lipid Bilayer Membranes: Effects on Redox Potentials. J. Bioenerg. Biomembr. 1997, 29, 211–221. 10.1023/A:1022401825287. [DOI] [PubMed] [Google Scholar]
- O’Brien E. S.; Nucci N. V.; Fuglestad B.; Tommos C.; Wand A. J. Defining the Apoptotic Trigger: The interaction of cytochrome c and cardiolipin. J. Biol. Chem. 2015, 290, 30879–30887. 10.1074/jbc.M115.689406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi F.; Howes B. D.; Droghetti E.; Polticelli F.; Piro M. C.; Di Pierro D.; Fiorucci L.; Coletta M.; Smulevich G.; Santucci R. Role of Lysines in Cytochrome c–Cardiolipin Interaction. Biochemistry 2013, 52, 4578–4588. 10.1021/bi400324c. [DOI] [PubMed] [Google Scholar]
- Tarlov M. J.; Bowden E. F. Electron-transfer reaction of cytochrome c adsorbed on carboxylic acid terminated alkanethiol monolayer electrodes. J. Am. Chem. Soc. 1991, 113, 1847–1849. 10.1021/ja00005a068. [DOI] [Google Scholar]
- Lee K.-S.; Won M.-S.; Noh H.-B.; Shim Y.-B. Triggering the redox reaction of cytochrome c on a biomimetic layer and elimination of interferences for NADH detection. Biomaterials 2010, 31, 7827–7835. 10.1016/j.biomaterials.2010.06.052. [DOI] [PubMed] [Google Scholar]
- Liu L.; Zeng L.; Wu L.; Jiang X. Label-Free Surface-Enhanced Infrared Spectroelectrochemistry Studies the Interaction of Cytochrome c with Cardiolipin-Containing Membranes. J. Phys. Chem. C 2015, 119, 3990–3999. 10.1021/jp507225m. [DOI] [Google Scholar]
- Yue H.; Waldeck D. H.; Petrović J.; Clark R. A. The Effect of Ionic Strength on the Electron-Transfer Rate of Surface Immobilized Cytochrome c. J. Phys. Chem. B 2006, 110, 5062–5072. 10.1021/jp055768q. [DOI] [PubMed] [Google Scholar]
- Hagen W. R. Direct electron transfer of redox proteins at the bare glassy carbon electrode. Eur. J. Biochem. 1989, 182, 523–530. 10.1111/j.1432-1033.1989.tb14859.x. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Borsari M.; Loschi L.; Martinelli A.; Sola M. Thermodynamics of the Alkaline Transition of Cytochrome c. Biochemistry 1999, 38, 7900–7907. 10.1021/bi983060e. [DOI] [PubMed] [Google Scholar]
- Léger C.; Elliott S. J.; Hoke K. R.; Jeuken L. J. C.; Jones A. K.; Armstrong F. A. Enzyme Electrokinetics: Using Protein Film Voltammetry To Investigate Redox Enzymes and Their Mechanisms. Biochemistry 2003, 42, 8653–8662. 10.1021/bi034789c. [DOI] [PubMed] [Google Scholar]
- Fourmond V. QSoas: A Versatile Software for Data Analysis. Anal. Chem. 2016, 88, 5050–5052. 10.1021/acs.analchem.6b00224. [DOI] [PubMed] [Google Scholar]
- Paquete C. M.; Fonseca B. M.; Cruz D. R.; Pereira T. M.; Pacheco I.; Soares C. M.; Louro R. O. Exploring the molecular mechanisms of electron shuttling across the microbe/metal space. Front. Microbiol. 2014, 5, 318 10.3389/fmicb.2014.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.