

Abstract
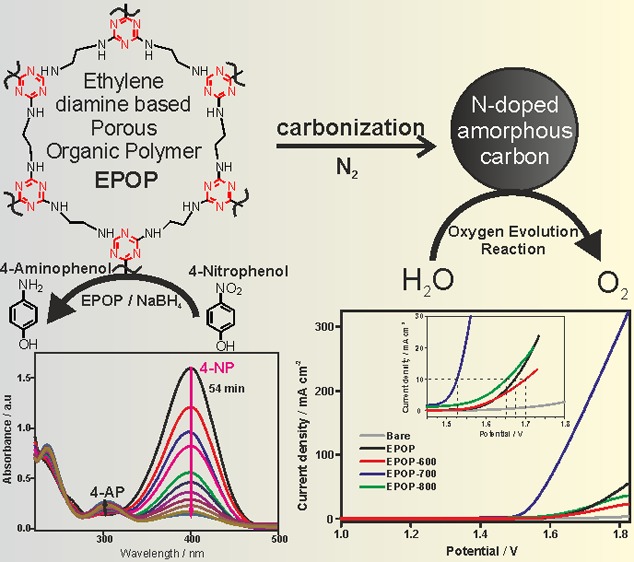
Ethylene diamine-based porous organic polymer (EPOP) was synthesized, carbonized at different temperatures, and characterized. The successful formation of the triazine polymer was confirmed by Fourier-transform infrared spectroscopy, 13C, and 15N cross-polarization magic angle spinning solid-state NMR. The two-dimensional layered architecture and graphitic nature of the samples resembled that of nitrogen-doped amorphous carbon, as confirmed by Raman, powder X-ray diffraction, and transmission electron microscopy measurements. The catalytic activity of these materials toward nitrophenol reduction and electrocatalytic activity toward oxygen evolution reaction (OER) were systematically evaluated in detail. Electrocatalytic activity toward oxygen evolution reaction was systematically evaluated by chronoamperometry and linear sweep voltammetry. Results clearly demonstrate that all of these catalysts exhibit good OER activity and excellent stability. Among all catalysts, EPOP-700 showed better OER activity, as reflected by its onset potential and current density, comparable with that of the metal-based OER catalysts and better than that of metal-free catalysts. Further, their catalytic activity toward the reduction of 4-nitrophenol to 4-aminophenol was tested with NaBH4; although all of these catalysts showed good catalytic activity; EPOP-800 displayed better catalytic activity.
Introduction
Porous organic polymers (POPs), a subclass of organic polymers/organic materials, show great advantage over conventional polymers in the area of catalysis due to their high surface area.1,2 They are highly cross-linked and are amorphous in nature.1,3 Proper tuning of the porosity in these materials can be achieved with desired functional groups or linking units. It is anticipated that porous organic polymers with high nitrogen content are desirable for catalytic applications.4 It is also advantageous that the “N” atom can coordinate to metal atoms;5 hence, their surface can be modified with desired metals to further improve their catalytic properties.6 Among the porous organic polymers, covalent triazine frameworks constitute an important class because of their high nitrogen content, high surface area, high thermal and chemical stability, easy preparation on a large scale, and use in catalytic applications.6−9 Thomas and co-workers developed a novel ionothermal process for the synthesis of covalent triazine frameworks from aromatic nitriles using ZnCl2 at elevated temperatures (≥400 °C) in molten state.10 Such a high-temperature procedure usually yields highly porous framework due to the partial decomposition of organic moiety, although it was reported that nitrile trimers are stable up to 400 °C.6,10,11 POPs can also be synthesized from melamine3 and cyanuric chloride (C-Cl) under mild conditions;8,12,13 however, POPs prepared under mild conditions exhibit very low surface area.14,15 It is further shown that these syntheses are solvent dependent. The condensation reaction in dimethylacetamide and N-methyl pyrrolidine yielded 4 times high surface area material than the one obtained using dioxane as a solvent, indicating the impact of solvent on porosity in such reactions. Fifteen POPs are electron rich and they exhibit good π-electron mobility along the triazine ring and if conjugated with an aromatic linker, they exhibit good electronic conductivity, which enhances their chemical, electrochemical, and photocatalytic activity.16 Altogether, POPs find application in energy storage devices, gas storage/separation, photocatalysis, etc. wherein the triazine nitrogen is shown to play a dominant role.16−21 Recent applications of POPs focus on the heteroatom doping, multilayer assembling, and metal cocatalysts aimed toward further enhancement of their catalytic properties.22−25
Oxygen evolution reaction (OER) is one of the most promising sustainable systems to generate clean and effective energy. OER is a complex multistep reaction as it involves several surface-adsorbed intermediates, and this reaction usually requires large overpotential for the actual process, which distinctly reduces the process efficiency even if the benchmark catalysts are applied.26 Numerous electrocatalysts have been developed till date to improve the efficiency of oxygen evolution.27 Ru- and Ir-based noble metal catalysts were frequently employed as benchmark catalysts due to their efficiency in oxygen evolution reaction. Further, nanoscale catalysts with reduced noble metal content and high surface area were developed to reduce the cost. Later, metal nanocatalysts on carbon matrices were developed. In this scenario, metal-free catalysts have attracted wide attention owing to their analogous performance with noble metal catalysts. Frequently studied metal-free catalysts include graphene-based materials, carbon nanotubes (CNTs), and so on, which are usually expensive. Metal-free catalysts were also developed from inexpensive carbon-based materials by high-temperature treatment under inert conditions.
On the other hand, 4-nitrophenol (4-NP) is an important organic pollutant obtained from agricultural and industrial sources.28 Metal-catalyzed reduction of 4-NP with excess NaBH4 yields 4-aminophenol (4-AP), a key intermediate in the production of paracetamol and dyeing industry. Hence, development of an efficient catalyst for the reduction of 4-NP to 4-AP will be of broad interest owing to its industrial value. In general, metal or metal-based nanocatalysts were employed for the catalytic reduction of 4-NP to 4-AP with excess NaBH4. Till date, very few metal-free catalysts based on graphene materials are known for the reduction of 4-NP to 4-AP, which are usually expensive.28,29 In this context, development of a catalyst with dual performance in OER and nitrophenol reduction would be interesting, provided they display excellent catalytic activities in both the reactions, as they follow a different pathway.
Recently, we reported on the synthesis of phenylenediamine-based POP network and its electrocatalytic activity toward the nitrobenzene reduction and oxygen evolution reaction (OER).14 Further, the carbonized sample was tested for its energy storage performance.30 There are very few reports available on OER and nitrophenol reduction using metal-free catalysts derived from POPs.
Herein, we report the synthesis, carbonization, and characterization of ethylene diamine-based porous organic polymer (EPOP) and their catalytic activity toward oxygen evolution reaction and nitrophenol reduction.
Results and Discussion
The reported procedure was followed to obtain EPOP (Scheme 1).14 EPOP-600, EPOP-700, and EPOP-800 were obtained by the carbonization of EPOP sample at 600, 700, and 800 °C under inert atmosphere (3 h) in a tubular furnace, respectively.
Scheme 1. Synthesis of EPOP and Its Carbon Composites.

The successful formation of the porous organic polymer is confirmed by Fourier-transform infrared spectroscopy (FT-IR) analysis. The absence of cyanuric chloride (C-Cl) stretching vibration at 850 cm–1 and a broad peak at 3400 cm–1 corresponding to “NH2” of ethylene diamine confirms the absence of both the starting materials (Figure S1). Apart from this, key peaks such as the stretching frequencies of triazine rings were observed at 1347 and 1571 cm–1, bending and stretching vibration of sp3 “CH2” moiety were observed at 1447 and 2940 cm–1 respectively. The peak at 804 cm–1 corresponds to the breathing mode of vibration of triazine unit (Figure S2). All of these characteristic peaks indicate the successful formation of triazine polymer, which is further confirmed by solid-state 13C and 15N cross-polarization magic angle spinning (CP-MAS) spectroscopy. The FT-IR of the carbonized sample (EPOP-800) showed key features of the triazine ring to some extent; however, the characteristic peaks corresponding to CH2 group were absent and the pattern looked very similar to that of the nitrogen-doped graphene.31
The successful formation and the nature of the formed porous organic polymers can be confirmed by cross-polarization magic angle spinning (CP-MAS) solid-state 13C and 15N NMR spectroscopy. 13C CP-MAS measurement of EPOP sample (Figure S3a) confirmed the coexistence of triazine and ethylene diamine moieties in the polymer; the aromatic sp2 carbon of triazine was observed at δ 166 ppm, and the aliphatic sp3 carbon of ethylene diamine was observed at δ 41 ppm, indicating that these two individual moieties are covalently linked during the synthesis [starting materials are soluble in organic solvents; upon completion of the reaction, the product EPOP was washed several times with organic solvents to remove unreacted starting materials]. Similarly, 15N CP-MAS measurement of EPOP sample (Figure S3b) showed two broad peaks at δ −172 and −90 ppm, corresponding to the triazine N and linker ethylene diamine N, respectively. This confirms the presence of two different N (triazine N and ethylene diamine N) atoms in the sample.
Raman spectroscopy is an important tool that specifies the defects and disorderly nature of carbon materials. Figure 1 shows the Raman spectra of carbon composites of EPOP, and the Raman spectra of EPOP is given in Figure S4a. It is clear that after carbonization at different temperatures, we observe only two peaks at 1330 (D-band) and 1600 cm–1 (G-band). This clearly proves that EPOP has undergone chemical and structural changes. The observed peaks lie in the D- and G-band region; usually, D-band arises due to the disorders and imperfection present in the carbon lattice. G-band is assigned to one of the two E2g modes corresponding to stretching vibrations in the basal-plane (sp2 domains) of carbon lattice. The D- and G-band peaks were observed at 1346, 1358, and 1342 cm–1 and 1542, 1550, and 1567 cm–1, EPOP-600, 700, 800 respectively. The G peaks of all samples shift to higher frequencies as the pyrolyzing temperature increased. The quantitative amount of blueshift for EPOP-700 and EPOP-800 is 8 and 25 cm–1, respectively, when compared to that of EPOP-600. It is obvious that the shift in Raman peaks may be attributed to the effect of doping and strain. Usually, the doping of nitrogen in carbon-based materials, such as graphene and CNTs, the blueshift of G peak is apparent, which we observed in our case. Typically, it is known that the compressive/tensile strain in carbon materials (graphene and CNTs) may induce a blue/redshift of Raman peaks. Doping of nitrogen atoms in carbon lattice may lead to the defect pinning and distortion of lattice. The doping may be associated with bond formation and this produces deformation and stress fields. The formation of pyrrolic nitrogen by doping of nitrogen will have a C–N bond length of 1.37 Å, which shortens compared to that of C–C bond (1.42 Å). The extent of structural disorder present in the carbonized POP can be considered from the ID/IG ratio.32 The ID/IG ratios of all three carbonized samples were found to be 1.47 (EPOP-600), 1.41 (EPOP-700), and 0.87 (EPOP-800). The increase in ID/IG ratio indicates more structural disorder; if the value is high, it indicates a low degree of graphitization. The order of graphitization for all three carbonized samples can be given as EPOP-800 > EPOP-700 > EPOP-600.
Figure 1.
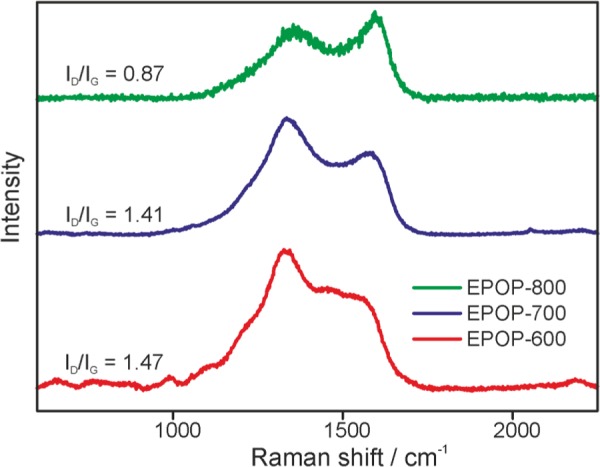
Raman spectrum of carbon composites of EPOP.
Figure 2a–d shows the powder X-ray diffraction (PXRD) pattern of EPOP and its carbon composites. All samples displayed a broad peak, and hence deduction of any structural information of the POP becomes feeble. EPOP showed a broad peak around the 2θ value of 23.7°, ascribed to the graphitic carbon like structure, with a d-spacing of 3.72 Å,32 whereas EPOP-600 and EPOP-800 showed a broad peak with a spacing of 3.68 and 3.59 Å, respectively, suggesting the presence of graphitic structure.32,33 EPOP-700 showed a broad peak around the 2θ value of 26.1° close to that of graphitic carbon with d-spacing of 3.47 Å.33 From the PXRD, we infer that all samples were found to resemble graphitic carbon. The reason for the resemblance to graphitic carbon could be attributed to the carbonization process; a similar kind of observation was made for room-temperature analogues.6 Further, these results were supported by Transmission electron microscopy (TEM) analysis.
Figure 2.

XRD profile of (a) EPOP, (b) EPOP-600, (c) EPOP-700, and (d) EPOP-800.
TEM image of the EPOP showed stacked sheetlike network, whereas the high-resolution transmission electron microscopy (HR-TEM) images of the carbonized samples EPOP-600 and EPOP-700 showed exfoliated sheetlike structure. EPOP-800 displayed carbon sheetlike morphology, which was crumpled to spherelike morphology on the topographical view34 (Figure 3). Selected area electron diffraction patterns of all of these samples showed amorphous nature of the material (Figures S5–S8). These results are highly consistent with XRD analyses.
Figure 3.

(a) TEM image of EPOP, HR-TEM images of (b) EPOP-600, (c) EPOP-700, and (d) EPOP-800.
Field emission scanning electron microscopy (FE-SEM) images of EPOP and the carbonized samples were shown in Figures S9–S12. EPOP sample exhibited irregularly agglomerated particle-like morphology. In the case of carbonized samples, change in morphology was observed. As the carbonization temperature increased from 600 to 800 °C, the morphology changed from irregularly agglomerated network to highly agglomerated network, which could be attributed to the high pyrolysis temperature.4
The oxygen evolution activities of the prepared carbon sample (EPOP, EPOP-600, EPOP-700, and EPOP-800) were studied in 1 M KOH using linear sweep voltammetry (LSV). The prepared samples were coated on carbon paper and used as working electrode. Figure 4 shows the polarization curves of the prepared carbon sample. Among these samples, EPOP-700 showed an onset potential of 1.527 V, which is lower than that of the other carbon samples (EPOP: 1.673 V, EPOP-600: 1.70 V, EPOP-800: 1.65 V). EPOP-700 sample exhibited overpotential of 297 mV to attain a current density of 10 mA/cm2; however, other carbon samples showed relatively high overpotential, as shown in Table 1. Also, at high current density of 300 mA/cm2, the EPOP-700 sample showed a low overpotential of 580 mV, whereas other samples exhibited high overpotential. The obtained results with high current density are found to be better than the metal-based OER catalysts.35 The influence of pH on the OER activity has been studied for EPOP-700 sample (Figure S21). Among the four catalysts used in this study, EPOP-700 showed high electrocatalytic activity, which could be ascribed to the presence of nitrogen atoms present on the surface/edges of the sample. It is evident from the elemental analysis that the N content in the sample EPOP-700 has relatively high nitrogen content than other samples as a result of high nitrogen atom doping during carbonization process (Supporting Information (SI), Table 1). From N %, it may possibly be concluded that 700 °C is the ideal temperature to attain high nitrogen doping with respect to EPOP samples. The presence of nitrogen atoms on the surface impacts the polarity and hydrophilicity of the sample, which is known to improve mass transfer properties at the electrode–electrolyte interface. The OER kinetics of the samples EPOP, EPOP-600, EPOP-700, and EPOP-800 were examined using a Tafel plot. From figure 4b, it is apparent that favorable reaction kinetics and a small Tafel slope of 76 mV/dec was found for EPOP-700 among all studied catalysts (SI, Table 2). The Tafel slope value implies that a lower electrochemical polarization occurred at the interface. The enhanced OER activity of EPOP-700 sample can be explained on the basis of N % (SI, Table 1). A high N % of 16.87 was obtained for EPOP-700 in comparison to other samples. The morphology analysis of EPOP-700 reveals the high stability of the carbon after prolonged OER for 10 h (Figure S19). EPOP-700 sample displayed better electrocatalytic performance among the studied catalysts (EPOP, EPOP-600, and EPOP-800), and this improved performance might be related to the increased electron transfer at the electrode interface accompanied by high ionic conductivity. To elucidate this, electrochemical impedance spectroscopy (EIS) measurements for all four catalysts were carried out (Figure S17). Nyquist plots obtained from the EIS measurement showed a lower resistance of 90 Ω for EPOP-700 compared to that of other catalysts (EPOP = 293 Ω, EPOP-600 = 238 Ω, EPOP-800 = 102 Ω). This lower charge-transfer resistance arises from the efficient interfacial contact of porous carbon network with electrolyte. EPOP-700 catalyst holds a smaller Rct, which facilitates the faster charge-transfer rate and hence better OER performance. Also, we anticipate that such OER performance might be concomitantly related to the intrinsic activity of the catalyst, high electrochemical surface area (ECSA), and efficient charge-transfer kinetics prevailing at the electrode surface. The mechanistic aspect of the EPOP-700 can be explained as follows: the nitrogen atom present in the network of EPOP-700 might be negatively charged because of the electron withdrawing nature, and hence the adjacent carbon atoms might become positively charged (Figure S20). The positively charged carbon adsorbs OH– from the electrolyte, which leads to the accumulation of OH– on the surface of the catalyst, and this may have positive influence on the catalytic reaction. For the rate-determining step of OER, the adjacent positively charged carbon atoms should undergo facile recombination of the two adsorbed species. The electron density localized on the nitrogen atoms in the carbon network may reside near the Fermi level, and so they can participate in the electrocatalytic reaction.36
Figure 4.

Electrochemical performance of EPOP and its carbon composites toward OER. (a) LSV; (b) Tafel plots in 0.1 M KOH solution at 2 mV/s.
Table 1. Electrochemical Data of the Catalysts.
| s. no | catalyst | onset (V) | current density (mA) | Tafel slope (mV/dec) | overpotential (mV) | active surface area ECSA (cm–2) |
|---|---|---|---|---|---|---|
| 1 | EPOP | 1.673 | 53 | 169 | 440 | 0.0568 |
| 2 | EPOP-600 | 1.70 | 22 | 177 | 470 | 0.0016 |
| 3 | EPOP-700 | 1.527 | 322 | 76 | 297 | 0.233 |
| 4 | EPOP-800 | 1.65 | 37 | 106 | 420 | 0.0598 |
To evaluate the catalytic performance of EPOP and its carbon composites toward the reduction of nitro group to amino group, the catalytic reduction of 4-NP was carried out in excess NaBH4 in aqueous medium. The decrease and increase in absorption intensity of 4-NP or 4-AP is measured in the presence of different catalysts as a function of time, as shown in Figure 5a–d. As the time increases, absorbance at 400 nm corresponding to 4-nitrophenolate anion decreases and the absorbance at 298 nm corresponding to 4-aminophenolate anion increases, indicating the catalytic activity of all four catalysts. An identical experiment was carried out with excess NaBH4 without catalyst. It is remarkable that even after a day, no change in intensity of 4-nitrophenolate peak was observed, indicating that the catalyst is necessary for this conversion. The difference in the degree of catalytic activity can be correlated with the complete disappearance of the peak at 400 nm vs function of time. Although all catalysts displayed good catalytic activity toward the reduction of 4-NP to 4-AP, the time taken for the complete reduction of 4-NP to 4-AP varied. It is evident that EPOP-800 sample took 35 min for the complete conversion of 4-NP to 4-AP, displaying the best catalytic activity among all. The order of catalytic activity can be derived as EPOP-800 > EPOP-700 > EPOP-600 > EPOP. From these observations, it can be concluded that the catalyst with a high degree of graphitization showed better catalytic activity in 4-NP reduction, whereas the same is not in the case of OER catalysis that follows a different mechanism. The reaction followed pseudo first-order kinetics. The improved catalytic performance of EPOP-800 can be ascribed to the facile adsorption of 4-NP on the catalyst’s surface. The concept of facile adsorption of 4-NP on the carbon surface has been reported using density functional theory calculation in the literature.37 The rate constant for 4-NP reduction of all catalysts was tabulated (SI, Table 3), and the activity factors for all catalysts were found to be 63.3, 90, 113, 150 min–1/g, respectively. The observed rate constant and activity factor values are better than those of the reported nonmetallic catalysts.29,37
Figure 5.

UV–vis absorption spectra for the catalytic reduction of 4-nitrophenol by NaBH4 over (a) EPOP, (b) EPOP-600, (c) EPOP-700, and (d) EPOP-800.
Conclusions
In conclusion, porous organic polymers based on ethylene diamine and triazine were successfully synthesized, carbonized at different temperatures, and characterized. The as-synthesized materials were studied for the catalytic activity toward OER and nitrophenol reduction. Among the catalysts employed in this study, EPOP-700 sample displayed excellent OER activity with an overpotential of 580 mV at a current density of 300 mA/cm2, with a prolonged stability over 10 h. These results were further complimented by Tafel slope and EIS measurements. A small Tafel slope of 76 mV/dec and lower resistance of 90 Ω were obtained for EPOP-700 compared to that of other catalysts employed in this study. The excellent activity of EPOP-700 could be attributed to high N doping, as apparent from elemental analysis. The obtained results were found to be better than those of the metal-free catalysts and are comparable with those of metal-based OER catalysts. In addition, these catalysts showed excellent catalytic activity toward the reduction of 4-NP to 4-AP. EPOP-800 sample displayed excellent catalytic activity by reducing 4-NP to 4-AP in 35 min, whereas other catalysts took relatively longer times. The observed rate constant and activity factor values are better than those of the reported nonmetallic catalysts.
Experimental Section
Materials and Methods
All reagents and solvents were purchased from Sigma-Aldrich/Alfa Aesar and used without further purification.
13C and 15N CP-MAS measurements were carried out on a Bruker Avance 400 spectrometer, operating at 100.6 MHz for 13C and 40.53 MHz for 15N using a Bruker 4 mm double resonance probe-head at a spinning rate of 10 kHz. The X-ray diffraction (XRD) patterns were measured at room temperature (RT) using a Bruker D8 ADVANCE instrument using Cu Kα radiation with a wavelength of 1.5418 Å. The powder diffraction covered the angle ranges from 5–65°, with a step angle of 0.02°/min. The morphological structures of the prepared samples were captured using a scanning electron microscope (SEM) of TESCAN, VEGA 3 with Bruker detector. Thermogravimetric analysis (TGA) was used to study the thermal degradation of synthesized materials using a TGA/SDT Q600, TA instruments at a scanning rate of 5 °C/min, from room temperature to 1000 °C under nitrogen atmosphere. FT-IR analysis was carried out on a Bruker Tensor 27 (Optik GmbH) using RT DLaTGS (Varian) detector. Transmission electron microscope (Tecnai 20 G2 (FEI make), Netherlands) was used to analyze the surface morphology. Brunauer–Emmett–Teller surface area and porosity analysis was carried out on an Accelerated Surface Area and Porosimetry system (ASAP-2020 V4.03 (V4.03 H)) at 77 K. Raman spectra were recorded using a high-resolution Renishaw Raman microscope employing a He–Ne laser of 18 mW at 633 nm.
Cyclic voltammetry was carried on an autolab PGSTAT 302N workstation at room temperature in a standard three-electrode cell. The working electrode was a carbon paper (area = 1 cm2, mass loading of 2 mg/cm2) and modified glassy carbon, and the counter electrode was a Pt wire. The reference electrode was Ag/AgCl/KCl (3 M) electrode. The oxygen evolution reaction was studied by using linear sweep voltammetry carried out at room temperature using 1 M KOH as the electrolyte at a scan rate of 1 mV/s. For the ease of comparison of results, the potentials were converted to the reversible hydrogen electrode scale.
Ink Preparation
The ink was prepared by equimolar preparation 1:1:1 (water/isopropyl alcohol/Nafion), and the solution was sonicated for 3 h. A small volume (100 μL) of this solution was added to 5 mg of the catalyst to prepare the slurry and was sonicated for 30 min. The catalyst ink was coated on the surface of tory carbon by manual brush coating. The modified tory carbon was allowed to dry at room temperature.
Nitrophenol Reduction
Reduction of 4-NP to 4-AP was carried out in the presence of excess NaBH4, which is an important organic catalytic reaction. This reaction was carried out to evaluate the catalytic performance of EPOP catalyst. Two milligrams of 4-NP was dissolved in 40 mL of deionized water and sonicated for half an hour; the solution was light yellow. Then, NaBH4 (120 mg) was added to the 4-NP solution. In the absence of EPOP catalysts, a small amount of bubbles was observed because of the hydrogen generation by the reduction reaction between NaBH4 and water. However, in the presence of EPOP (3 mg) catalysts, a large amount of bubbles was observed and the gas release rate became much faster.
Synthesis of EPOP
Under inert conditions, ethylene diamine (1.4 g, 24.4 mmol) was dissolved in anhydrous 1,4-dioxane (100 mL) under constant stirring at RT. To this solution, K2CO3 (4.5 g, 32.5 mmol) was added and stirred for 30 min. Then, the solution was cooled to 10 °C and cyanuric chloride (3 g, 16.27 mmol) dissolved in 100 mL anhydrous 1,4-dioxane was added dropwise over 8 h. The mixture was allowed to warm to RT and refluxed for 3 days (during the course of the reaction, color changed from dark brown to pale brown). The solution was cooled to RT; the mixture was filtered, washed with 1,4-dioxane and methanol to remove unreacted SM, and dried under vacuum to yield EPOP (yield = 90%).
Acknowledgments
Dr. M.K. thanks DST-INSPIRE for the faculty award and DST-SERB Start-up Research grant for research funding. Director and support staffs of CSIR-CECRI are gratefully acknowledged for their constant encouragement and support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00574.
FT-IR, 13C and 15N NMR, HR-TEM, FE-SEM, TGA, electrochemical active surface area calculation, SEM image of EPOP-700 modified electrode after OER measurements, EIS, chronoamperometry, XPS (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang Y.; Riduan S. N. Functional porous organic polymers for heterogeneous catalysis. Chem. Soc. Rev. 2012, 41, 2083–2094. 10.1039/c1cs15227k. [DOI] [PubMed] [Google Scholar]
- Patra B. C.; Khilari S.; Manna R. N.; Mondal S.; Pradhan D.; Pradhan A.; Bhaumik A. A Metal-Free Covalent Organic Polymer for Electrocatalytic Hydrogen Evolution. ACS Catal. 2017, 7, 6120–6127. 10.1021/acscatal.7b01067. [DOI] [Google Scholar]
- Schwab M. G.; Fassbender B.; Spiess H. W.; Thomas A.; Feng X.; Müllen K. Catalyst-free Preparation of Melamine-Based Microporous Polymer Networks through Schiff Base Chemistry. J. Am. Chem. Soc. 2009, 131, 7216–7217. 10.1021/ja902116f. [DOI] [PubMed] [Google Scholar]
- Liu J.; Hu Y.; Cao J. Covalent triazine-based frameworks as efficient metal-free electrocatalysts for oxygen reduction reaction in alkaline media. Catal. Commun. 2015, 66, 91–94. 10.1016/j.catcom.2015.03.027. [DOI] [Google Scholar]
- Iwase K.; Yoshioka T.; Nakanishi S.; Hashimoto K.; Kamiya K. Copper-Modified Covalent Triazine Frameworks as Non-Noble-Metal Electrocatalysts for Oxygen Reduction. Angew. Chem., Int. Ed. 2015, 54, 11068–11072. 10.1002/anie.201503637. [DOI] [PubMed] [Google Scholar]
- Ren S.; Bojdys M. J.; Dawson R.; Laybourn A.; Khimyak Y. Z.; Adams D. J.; Cooper A. I. Porous, Fluorescent, Covalent Triazine-Based Frameworks Via Room-Temperature and Microwave-Assisted Synthesis. Adv. Mater. 2012, 24, 2357–2361. 10.1002/adma.201200751. [DOI] [PubMed] [Google Scholar]
- Tao L.; Niu F.; Liu J.; Wang T.; Wang Q. Troger’s base functionalized covalent triazine frameworks for CO2 capture. RSC Adv. 2016, 6, 94365–94372. 10.1039/C6RA21196H. [DOI] [Google Scholar]
- Puthiaraj P.; Lee Y.-R.; Zhang S.; Ahn W.-S. Triazine-based covalent organic polymers: design, synthesis and applications in heterogeneous catalysis. J. Mater. Chem. A 2016, 4, 16288–16311. 10.1039/C6TA06089G. [DOI] [Google Scholar]
- Buyukcakir O.; Je S. H.; Talapaneni S. N.; Kim D.; Coskun A. Charged Covalent Triazine Frameworks for CO2 Capture and Conversion. ACS Appl. Mater. Interfaces 2017, 9, 7209–7216. 10.1021/acsami.6b16769. [DOI] [PubMed] [Google Scholar]
- Kuhn P.; Antonietti M.; Thomas A. Porous, Covalent Triazine-Based Frameworks Prepared by Ionothermal Synthesis. Angew. Chem., Int. Ed. 2008, 47, 3450–3453. 10.1002/anie.200705710. [DOI] [PubMed] [Google Scholar]
- Hug S.; Stegbauer L.; Oh H.; Hirscher M.; Lotsch B. V. Nitrogen-Rich Covalent Triazine Frameworks as High-Performance Platforms for Selective Carbon Capture and Storage. Chem. Mater. 2015, 27, 8001–8010. 10.1021/acs.chemmater.5b03330. [DOI] [Google Scholar]
- Zhao H.; Jin Z.; Su H.; Jing X.; Sun F.; Zhu G. Targeted synthesis of a 2D ordered porous organic framework for drug release. Chem. Commun. 2011, 47, 6389–6391. 10.1039/c1cc00084e. [DOI] [PubMed] [Google Scholar]
- Kundu S. K.; Bhaumik A. A triazine-based porous organic polymer: a novel heterogeneous basic organocatalyst for facile one-pot synthesis of 2-amino-4H-chromenes. RSC Adv. 2015, 5, 32730–32739. 10.1039/C5RA00951K. [DOI] [Google Scholar]
- Gopi S.; Kathiresan M. 1,4-Phenylenediamine based covalent triazine framework as an electro catalyst. Polymer 2017, 109, 315–320. 10.1016/j.polymer.2016.12.052. [DOI] [Google Scholar]
- Zulfiqar S.; Sarwar M. I.; Yavuz C. T. Melamine based porous organic amide polymers for CO2 capture. RSC Adv. 2014, 4, 52263–52269. 10.1039/C4RA11442F. [DOI] [Google Scholar]
- Niu F.; Tao L.; Deng Y.; Gao H.; Liu J.; Song W. A covalent triazine framework as an efficient catalyst for photodegradation of methylene blue under visible light illumination. New J. Chem. 2014, 38, 5695–5699. 10.1039/C4NJ01534G. [DOI] [Google Scholar]
- Hao L.; Ning J.; Luo B.; Wang B.; Zhang Y.; Tang Z.; Yang J.; Thomas A.; Zhi L. Structural Evolution of 2D Microporous Covalent Triazine-Based Framework toward the Study of High-Performance Supercapacitors. J. Am. Chem. Soc. 2015, 137, 219–225. 10.1021/ja508693y. [DOI] [PubMed] [Google Scholar]
- Bhanja P.; Bhunia K.; Das S. K.; Pradhan D.; Kimura R.; Hijikata Y.; Irle S.; Bhaumik A. A New Triazine-Based Covalent Organic Framework for High-Performance Capacitive Energy Storage. ChemSusChem 2017, 10, 921–929. 10.1002/cssc.201601571. [DOI] [PubMed] [Google Scholar]
- Bhunia A.; Esquivel D.; Dey S.; Fernandez-Teran R.; Goto Y.; Inagaki S.; Van Der Voort P.; Janiak C. A photoluminescent covalent triazine framework: CO2 adsorption, light-driven hydrogen evolution and sensing of nitroaromatics. J. Mater. Chem. A 2016, 4, 13450–13457. 10.1039/C6TA04623A. [DOI] [Google Scholar]
- Jiang X.; Wang P.; Zhao J. 2D covalent triazine framework: a new class of organic photocatalyst for water splitting. J. Mater. Chem. A 2015, 3, 7750–7758. 10.1039/C4TA03438D. [DOI] [Google Scholar]
- Wang Y.; Li J.; Yang Q.; Zhong C. Two-Dimensional Covalent Triazine Framework Membrane for Helium Separation and Hydrogen Purification. ACS Appl. Mater. Interfaces 2016, 8, 8694–8701. 10.1021/acsami.6b00657. [DOI] [PubMed] [Google Scholar]
- Liu G.; Niu P.; Sun C.; Smith S. C.; Chen Z.; Lu G. Q.; Cheng H.-M. Unique Electronic Structure Induced High Photoreactivity of Sulfur-Doped Graphitic C3N4. J. Am. Chem. Soc. 2010, 132, 11642–11648. 10.1021/ja103798k. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Sun J.; Maeda K.; Domen K.; Liu P.; Antonietti M.; Fu X.; Wang X. Sulfur-mediated synthesis of carbon nitride: Band-gap engineering and improved functions for photocatalysis. Energy Environ. Sci. 2011, 4, 675–678. 10.1039/C0EE00418A. [DOI] [Google Scholar]
- Zhang X.; Xie X.; Wang H.; Zhang J.; Pan B.; Xie Y. Enhanced Photoresponsive Ultrathin Graphitic-Phase C3N4 Nanosheets for Bioimaging. J. Am. Chem. Soc. 2013, 135, 18–21. 10.1021/ja308249k. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Wang X.; Nishihara Y.; Lu D.; Antonietti M.; Domen K. Photocatalytic Activities of Graphitic Carbon Nitride Powder for Water Reduction and Oxidation under Visible Light. J. Phys. Chem. C 2009, 113, 4940–4947. 10.1021/jp809119m. [DOI] [Google Scholar]
- Dau H.; Limberg C.; Reier T.; Risch M.; Roggan S.; Strasser P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. 10.1002/cctc.201000126. [DOI] [Google Scholar]
- Suen N.-T.; Hung S.-F.; Quan Q.; Zhang N.; Xu Y.-J.; Chen H. M. Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem. Soc. Rev. 2017, 46, 337–365. 10.1039/C6CS00328A. [DOI] [PubMed] [Google Scholar]
- Kong X.-k.; Chen Q.-w.; Lun Z.-y. Probing the influence of different oxygenated groups on graphene oxide’s catalytic performance. J. Mater. Chem. A 2014, 2, 610–613. 10.1039/C3TA13946H. [DOI] [Google Scholar]
- Wang Z.; Chen Q. Metal-Free Catalytic Reduction of 4-Nitrophenol by MOFs-Derived N-Doped Carbon. ChemistrySelect 2018, 3, 1108–1112. 10.1002/slct.201702721. [DOI] [Google Scholar]
- Selvamani V.; Gopi S.; Rajagopal V.; Kathiresan M.; Vembu S.; Velayutham D.; Gopukumar S. High Rate Performing in Situ Nitrogen Enriched Spherical Carbon Particles for Li/Na-Ion Cells. ACS Appl. Mater. Interfaces 2017, 9, 39326–39335. 10.1021/acsami.7b10882. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Sun Z.; Wang H.; Wang Y.; Liang M.; Xue S. Nitrogen-doped graphene as a cathode material for dye-sensitized solar cells: effects of hydrothermal reaction and annealing on electrocatalytic performance. RSC Adv. 2015, 5, 10430–10439. 10.1039/C4RA13224F. [DOI] [Google Scholar]
- Panomsuwan G.; Saito N.; Ishizaki T. Nitrogen-Doped Carbon Nanoparticle–Carbon Nanofiber Composite as an Efficient Metal-Free Cathode Catalyst for Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2016, 8, 6962–6971. 10.1021/acsami.5b10493. [DOI] [PubMed] [Google Scholar]
- Wu X.; Yu X.; Lin Z.; Huang J.; Cao L.; Zhang B.; Zhan Y.; Meng H.; Zhu Y.; Zhang Y. Nitrogen doped graphitic carbon ribbons from cellulose as non noble metal catalyst for oxygen reduction reaction. Int. J. Hydrogen Energy 2016, 41, 14111–14122. 10.1016/j.ijhydene.2016.05.275. [DOI] [Google Scholar]
- Li D.; Chen H.; Liu G.; Wei M.; Ding L.-x.; Wang S.; Wang H. Porous nitrogen doped carbon sphere as high performance anode of sodium-ion battery. Carbon 2015, 94, 888–894. 10.1016/j.carbon.2015.07.067. [DOI] [Google Scholar]
- Han L.; Dong S.; Wang E. Transition-Metal (Co, Ni, and Fe)-Based Electrocatalysts for the Water Oxidation Reaction. Adv. Mater. 2016, 28, 9266–9291. 10.1002/adma.201602270. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Nakamura R.; Kamiya K.; Nakanishi S.; Hashimoto K. Nitrogen-doped carbon nanomaterials as non-metal electrocatalysts for water oxidation. Nat. Commun. 2013, 4, 2390 10.1038/ncomms3390. [DOI] [PubMed] [Google Scholar]
- Kong X.-k.; Sun Z.-y.; Chen M.; Chen C.-l.; Chen Q.-w. Metal-free catalytic reduction of 4-nitrophenol to 4-aminophenol by N-doped graphene. Energy Environ. Sci. 2013, 6, 3260–3266. 10.1039/c3ee40918j. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.