

Abstract
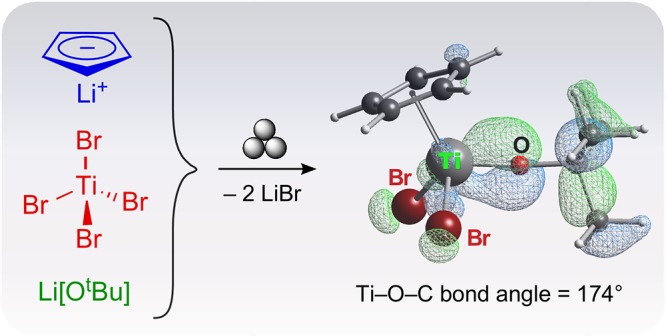
Titanium tert-butoxy halides of the formula CpxTiXy(OtBu)4–(x+y) (x, y = 1, 2; X = Cl, Br) have been prepared thorough milling the reagents without solvent. In the case of the chloride derivatives, Cp2TiCl2 is used as a starting material; in the case of the bromides, a mixture of LiCp, TiBr4, and Li[OtBu] is used. The stoichiometric ratios of the starting materials are reflected in the major products of the reactions. Single-crystal X-ray structures are reported for Cp2TiCl(OtBu), Cp2TiBr(OtBu), and CpTiBr2(OtBu), as well as for Cp2TiCl(OiPr) and a redetermination of Cp2TiCl(OMe). The tert-butoxy derivatives are notable for their nearly linear Ti–O–C angles (>170°) that reflect Ti–O π-bonding, an interpretation supported with density functional theory calculations.
1. Introduction
The neutral d0 Cpx′MLyL4–(x+y) framework comprises an enormous number of complexes in early transition-metal and actinide chemistry.1 This is the result of the combinatorial possibilities within the basic metallocene structure, coupled with an extended range of compatible L and L′ ligands. Many applications have been developed for such complexes, mainly in catalysis (e.g., syndiotactic polymerization of styrene,2 alkyne hydroamination,3 olefin polymerization,4−13 including copolymerization, e.g., of ethylene/norbornene,14,15 polymerization of lactide monomers and ε-caprolactone16−18), but there are also uses in organic synthesis19−22 and for the chemical vapor deposition production of thin-film oxides23,24 and carbides.25
The richness of the synthetic landscape of these complexes can be appreciated by considering a subset of complexes in which the cyclopentadienyl ligand is the unsubstituted C5H5, L′ is a halide, and L″ is an alkoxide. Not counting the homoleptic complexes MX4, M(OR)4, and MCp4, 12 general compositions can satisfy the CpxMXy(OR)4–(x+y) formula (Figure 1). The very flexibility that makes the metallocene framework so versatile, however, can also complicate the synthesis of specific compositions. Remarkably, for the specific set with M = Ti, X = Cl, and R = tBu, all of the possible compounds with the exception of Cp2TiCl(OtBu) are known.26 This is one of the most complete such families, however. With the sole change of Cl to Br, for example, none of the heteroleptic CpxTiBry(OtBu)4–(x+y) or TiBrx(OtBu)4–x complexes has been described. Even when known, the assembly of particular ligand sets in a Cp2MLL′ complex is not always straightforward. The dichlorides Cp2MCl2 are commonly used starting materials for bis(cyclopentadienyl) derivatives, but the reaction of Cp2TiCl2 and 2 equiv Li[OtBu] in tetrahydrofuran (THF) yields not only Cp2Ti(OtBu)2 (61%) but also CpTi(OtBu)3 (26%) and Ti(OtBu)4 (13%).26f Similarly, the reaction of Cp2ZrCl2 and (Li or Na)[OtBu] yields an inseparable mixture of Cp2ZrCl(OtBu) and Cp2Zr(OtBu)2, even with the use of more than 2 equiv of the alkoxide.23
Figure 1.
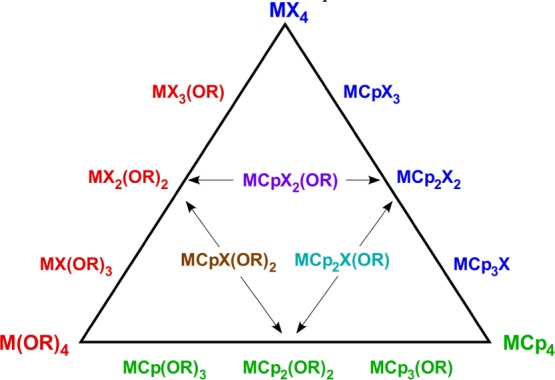
Combinatorial possibilities of mixed group 4 CpxMXy(OR)4–(x+y) complexes, starting from the homoleptic species MX4, M(OR)4, and MCp4. Complexes in the interior represent conceptual blends of the compounds on the edges.
The formation and disruption of Ti–O bonds has been of particular interest because although such bonds are typically rather robust (e.g., ca. 90 kcal mol–1 in various Cp2Ti(OR)2 complexes),28 their strength depends markedly on the metal oxidation state and the identity of other ligands on the metal center.29−31 In general, increased electron density on the metal center is associated with weaker Ti–O bond strength. Thus, the Ti–O energy in the Ti(IV) complex Ti(OtBu)4 has been placed at 108 kcal mol–1, whereas in the Ti(III) species Cp2TiCl(TEMPO) [TEMPO = (2,2,6,6-tetramethylpiperidine-N-oxyl)], the bond strength is calculated at 17 kcal mol–1.31 The ready scission of the Ti(III)–TEMPO bond has been exploited synthetically for the formation of CpTiCl(OR)2 (R = complexes).32
In contrast to the solution-based methods described above, we recently explored the use of mechanochemical methods in the preparation of CpxMCly(OtBu)4–(x+y) (M = Ti, Zr, and Hf) complexes.26f Mechanochemical synthesis, which typically employs grinding or milling, usually under solvent-free conditions, has been investigated for the preparation of organic,33 inorganic,34−37 and organometallic compounds.38 Such reactions can occur on a much faster time scale than equivalent solution methods.39 Part of the reason for this is the much higher concentration of reagents in the solid state than is the case in solution. Solvents can also coordinate to reagents and interfere with their subsequent reactivity or stabilize intermediates that require more time to form products.40 Mechanochemistry may yield unique products that cannot be isolated from solution-based approaches.41,42 The distribution of products and isomers can also be tuned.26f,43,44 In addition, the solvent-free approach provides flexibility to use reagents that are not compatible with typical organic solvents, especially ethers. As demonstrated below, this proves to be valuable in work with titanium halides. We report here mechanochemical routes to CpxTiCly(OtBu)4–(x+y) (X = Cl, Br) complexes, with solution syntheses of methoxy45 and isopropoxy derivatives added for comparison.
2. Results and Discussion
2.1. Synthesis
Preliminary attempts to prepare Cp2TiCl(OMe)45 (1) and the previously unknown Cp2TiCl(OiPr) (2) through grinding Cp2TiCl2 with the appropriate alkali metal alkoxide did not produce clean results, and hence, for these two compounds, we turned to alcoholysis reactions in THF with an added base.46 This approach has been used with both bis(cyclopentadienyl)47 and mono(cyclopentadienyl)titanium26h complexes and provides control over substitution, as indicated in eq 1 (n = 2 or 3; R = Me, Et, and iPr) for the latter compounds.26h For this study, we used Cp2TiCl2 as a starting material, which is inexpensive and its chloride ligands are substitutable.
| 1 |
The methoxy derivative 1 has been described before45 but its synthesis was given without details. The new isoproxy derivative 2 is an orange solid with the expected 1H and 13C NMR spectra. The ethoxy derivative has been previously synthesized,47,49 but it was useful for comparative studies (see below) and was also prepared via the literature solution method.
These solution reactions are slow (ca. 16 h) and even then may be incomplete,47 and for the synthesis of the tert-butoxy derivative Cp2TiCl(OtBu) (3), we investigated halide metathesis instead. This approach has been used to form Cp2ZrCl(OtBu) in THF,23 although the solution route fails for Cp2HfCl(OtBu).26f Mechanochemical activation works successfully with both of the heavier group 4 metals and it was adopted for the preparation of 3. At the outset, we were not confident that the reaction would be selective, as our previous investigation into tert-butoxide substitution with Cp2TiCl2 demonstrated that at ratios of [OtBu]−/Cp2TiCl2 ≥ 2, mixtures of products were obtained whose composition depended on whether or not solvent was used in the reaction and if so, whether it was an alkane or an ether.26f Nevertheless, we proceeded with the assumption that the composition of the desired product could be generated from the corresponding ratio of starting materials, that is, 1 equivalent of [OtBu]− would replace one Cl– or Br–. With the use of an equimolar ratio of K[OtBu] to Cp2TiCl2, in fact, grinding in a planetary mill for 15 min at 600 rpm did yield solid orange 3 exclusively. This result parallels those with Zr and Hf. The formation of 3, which completes the set of CpxTiXy(OtBu)4–(x+y) compounds for X = Cl and R = tBu (Figure 1), has been proposed as an intermediary in the formation of Cp3Ti(OtBu) from Cp2TiCl2 and K[OtBu] and LiCp (eq 2).26f Its isolation raises the reasonableness of this route.
| 2 |
The scarcity of organotitanium bromides stems from a lack of readily accessible starting materials. The solid TiBr4 would be a logical choice but it reacts with ethereal solvents at room temperature (in the case of THF, with ring cleavage).50 The adduct TiBr4(thf)2 has been used in the preparation of some titanium complexes, but its preparation requires a low-temperature (−78 °C) reaction between TiBr4 and THF followed by several days of vacuum drying to obtain a solid product. The resulting material is not soluble in THF, CHCl3, or CCl4.50 Even if it were, such solvents are problematic from an environmental viewpoint and move the traditional route even farther away from a “green” approach to synthesis.51 Thus, a mechanochemical route seemed especially suitable for the preparation of alkoxybromo derivatives. Rather than starting with Cp2TiBr2, which itself can be made from Cp2TiCl2 and LiBr,52 the one-pot synthesis of Cp2TiBr(OtBu) (4) was accomplished by milling TiBr4, Li[OtBu], and LiCp in a 1:1:2 M ratio for 15 min (eq 3)
 |
3 |
Adjusting the reaction stoichiometry of TiBr4, Li[OtBu], and LiCp to 1:1:1 and 1:2:1 ratios forms CpTiBr2(OtBu) (5) and CpTiBr(OtBu)2 (6), respectively. Perhaps not surprisingly, compound 5, formed from the simplest ratio of reagents, is the bromo complex that is consistently formed most cleanly. In contrast, the formation of 4 is also accompanied by 6 (ca. 56%); crystals 4 can be obtained from hexanes. Compound 6, in turn, which is produced with a larger amount of the tert-butoxide than is 5, is usually accompanied by the homoleptic alkoxide Ti(OtBu)4 (ca. 30%). Such differences reflect the role that kinetic factors play in mechanochemical synthesis, as grinding and milling environments are often far from equilibrium,53−56 and it is not uncommon to observe mixtures of the kinetically preferred products from solid-state reactions.
All of the compounds 1–6 are air- and moisture-sensitive and are soluble in hydrocarbons.
2.2. Crystal Structures
Crystals of 1 and 2 that were suitable for X-ray diffraction were grown from toluene; those for 3–5 were grown from hexanes. Comparative listings of selected bond lengths and angles are found in Table 1.
Table 1. Crystallographic and Calculateda Structures for CpxTiXy(OR)4–(x+y) Complexes.
| Ti–Cp (cent) (Å) | Ti–X (Å) | Ti–O (Å) | Ti–O–R (deg) | refs | |
|---|---|---|---|---|---|
| Cp2TiCl(OMe) (1) | 2.091 (ave) | 2.405(1) | 1.839(2) | 141.4(3) | (45) |
| Cp2TiCl(OMe) | 2.083 (ave) | 2.4215(3) | 1.8300(19) | 138.53(13) | this work |
| Cp2TiCl(OMe) | 2.111 (ave) | 2.379 | 1.844 | 136.8 | calc |
| Cp2TiCl(OEt) | 2.09 (ave) | 2.405(1) | 1.855(2) | 133.2(2) | (49) |
| Cp2TiCl(OEt) | 2.084 (ave) | 2.4044(12) | 1.858(3) | 133.3(2) | this work |
| Cp2TiCl(OEt) | 2.113 | 2.378 | 1.840 | 140.2 | calc |
| Cp2TiCl(OiPr) (2) | 2.052 (ave) | 2.4031(16) | 1.802(3) | 160.0(3) | this work |
| Cp2TiCl(OiPr) | 2.116 (ave) | 2.387 | 1.831 | 147.7 | calc |
| Cp2TiCl(OtBu) (3) | 2.103 (ave) | 2.4101(4) | 1.7864(9) | 172.48(8) | this work |
| Cp2TiCl(OtBu) | 2.130 (ave) | 2.394 | 1.800 | 168.0 | calc |
| Cp3Ti(OtBu) | 2.112 (η5) | 2.304(1) (η1-Cp) | 1.794(1) | 170.88(9) | (26f) |
| Cp3Ti(OtBu) | 2.142 (ave) | 2.366 (η1-Cp) | 1.792 | 175.5 | calc |
| Cp2TiBr(OtBu) (4) | 2.096 (ave) | 2.534(2) | 1.774(3) | 175.8(3) | this work |
| Cp2TiBr(OtBu) | 2.126 (ave) | 2.585 | 1.797 | 170.6 | calc |
| CpTiBr2(OtBu) (5) | 2.024 | 2.433(7) (ave) | 1.7274(18) | 173.95(19) | this work |
| CpTiBr2(OtBu) | 2.024 | 2.430 | 1.755 | 178.9 | calc |
B2PLYP/def2TZVP(Ti,Br,Cl); def2SVP(C,O,H).
2.2.1. Cp2TiCl(OMe) (1)
An X-ray structure determination of 1 has been reported.45 The room-temperature (295 K) structure was affected by disorder in the Cp rings that could not be satisfactorily modeled. Interestingly, in the present low-temperature version, which crystallizes in the same space group (Pbca), the Cp rings are not disordered but there is a second chlorine position possibly arising from cocrystallized Cp2TiCl2; its occupancy is refined (9%) with a Ti–Cl bond similarity restraint. Its presence may be the result of incomplete alcoholysis, a difficulty that has been observed before with this synthetic method.47 Nevertheless, both the original and our low-temperature redetermination agree on the pseudotetrahedral nature of the molecule, defined by the centroids of the two Cp rings, the chloride, and the oxygen of the methyl group (Figure 2). The Ti–Cl, Ti–O, and Ti-ring centroid distances in the two structures differ by less than 0.01 Å and the Cl–Ti–O angles differ by only 0.19°. The major difference involves the methoxy ligand; the O1–C11 bond distance is 0.05 Å longer in the present structure and the Ti–O–C bond angle has contracted by 2.9°. The low-temperature structure parameters are more in line with those of Cp2TiCl(OEt).49 For example, the C–O distance in the ethoxy derivative [1.415(4) Å] is not statistically distinguishable from that in the low-temperature 1 and the Ti–O–C angles, although differing by 5.3°, are closer than the ethoxy/room-temp methoxy values (Δ = 8.2°).
Figure 2.
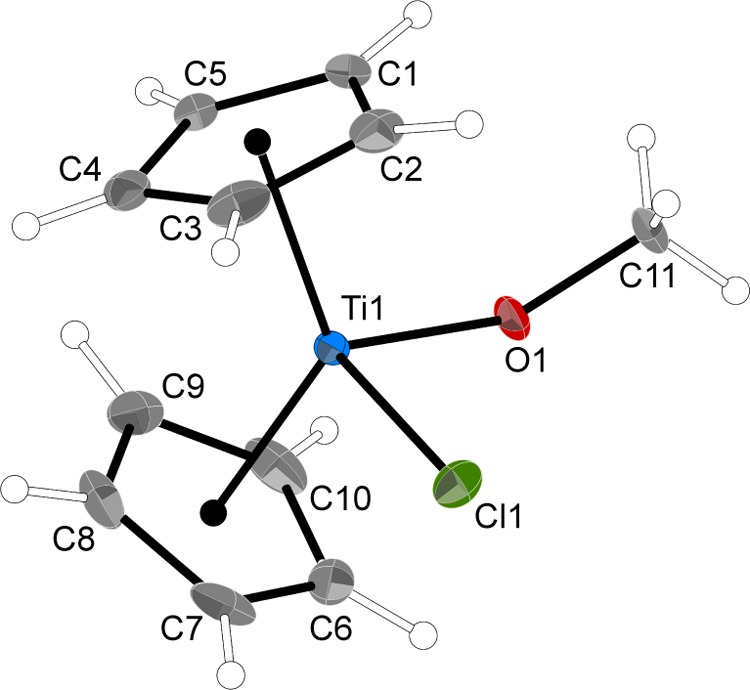
Thermal ellipsoid plot of 1 (50% level); hydrogens have arbitrary radii. A second chloride (2.326 Å from Ti, see text) has been removed for clarity. Selected bond distances (Å) and angles (deg): Ti1–O1, 1.8300(19); Ti1–Cl1, 2.4215(3); O1–C11, 1.419(2); Ti–Cp′ (ring centroid), 2.083 (ave); O1–Ti1–Cl1, 93.61(5); Ti1–O1–C11, 138.53(13); and Cp–Ti–Cp′, 130.9.
As a result of the unusual ordering of geometric parameters in the previously reported Cp2TiCl(OEt) structure relative to 1 and 2 (see below), Cp2TiCl(OEt) was resynthesized (see Experimental Section) and its structure was redetermined (see Supporting Information). Crystals were grown in view of the possibility that a polymorph might exist with a different Ti–O length and/or Ti–O–C angle. Although the new crystals were twinned, unlike those used for the earlier determination, all of the relevant structural parameters of the two structures agree to within 1σ (see Table 1).
2.2.2. Cp2TiCl(OiPr) (2)
Compound 2 shares the same pseudotetrahedral structure as 1 and Cp2TiCl(OEt); in the case of 2, both the Cp rings and OiPr ligand are disordered. One set of these is depicted in Figure 3. In particular, the alkoxide ligand is disordered across a crystallographically imposed mirror plane, that is, Cl1, Ti1, O1, C7, and C8 all lie in the plane and C6 is disordered above and below it (50:50). The general details of 2 mimic those of 1, in that the Ti-ring centroid distances differ only by 0.03 Å and the Ti–Cl bond lengths are within 0.02 Å of each other. Some details of the geometry of the isopropoxy ligand suggest a change in the metal ligand bonding compared to that in 1 and Cp2TiCl(OEt). The Ti–O bond has shortened to 1.802(3) Å and the Ti–O–C angle has widened to 160.0(3)°, features that could be consistent with some π-bonding character between Ti and the isopropoxy oxygen. The C–O distance of 1.309(6) Å, however, although not an unprecedented value for such a bond,57 is nearly 0.1 Å shorter than that found in any of the other complexes in this study. It also is not supported by computational modeling (see below), suggesting that the ligand disorder has unrealistically shortened it. Other features may have been altered by the disorder as well.
Figure 3.
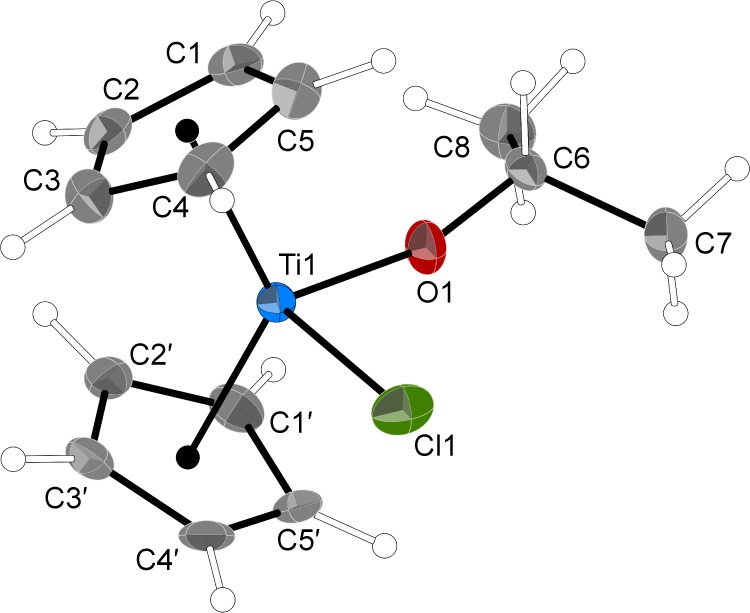
Thermal ellipsoid plot of 2 (30% level); hydrogens have arbitrary radii. Only one conformation of the disordered isopropyl and cyclopentadienyl ligands is shown. Selected bond distances (Å) and angles (deg): Ti1–O1, 1.802(3); Ti1–Cl1, 2.4031(16); O1–C6, 1.309(6); Ti–Cp′ (ring centroid), 2.052 (ave); O1–Ti1–Cl1, 94.87(13); Ti1–O1–C6, 160.0(3); and Cp–Ti–Cp′, 129.0.
2.2.3. Cp2TiCl(OtBu) (3)
Compound 3 is not affected by the disorder problems in 1 and 2 and interpretation of its structural features is more straightforward (Figure 4). The molecule has the same pseudotetrahedral geometry displayed by 1 and 2, but 3 can probably be compared most directly with the [Cp2Ti(OtBu)(thf)]+ cation;58 the Ti–O bond of 1.806(4) Å and Ti–O–C angle of 166.9(3)° of the latter differ by 0.02 Å and 5.6°, respectively, from that of 3. Comparisons are also possible with the structure of Cp2TiCl(OEt), which has been suggested as representing a sterically unencumbered Cp2TiCl(OR) complex.49 The Ti–Cp (centroid) distances and Cp–Ti–Cp′ angles in the two complexes differ by only 0.016 Å and 1.0°, respectively. The Ti–Cl distances are also nearly identical (0.005 Å difference) and are ca. 0.04 Å longer than those in Cp2TiCl2.59
Figure 4.
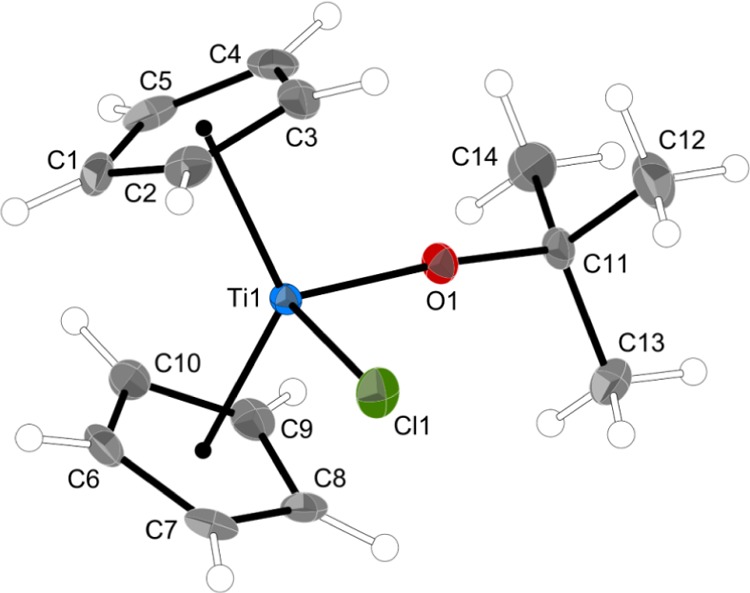
Thermal ellipsoid plot of 3 (50% level); hydrogens have arbitrary radii. Selected bond distances (Å) and angles (deg): Ti1–O1, 1.7864(9); Ti1–Cl1, 2.4101(4); O1–C11, 1.4204(14); Ti–Cp′ (ring centroid), 2.103 (ave); O1–Ti1–Cl1, 94.40(3); Ti1–O1–C11, 172.48(8); and Cp–M–Cp′, 129.5.
The differences between 3 and Cp2TiCl(OEt), however, are the most revealing. The Ti–O–C angle in 3 is 172.5°, 39.3° wider than that in Cp2TiCl(OEt), and which to our knowledge is the widest such angle reported for a Cp2TiCl(OR) complex.60 It might be thought that, given the bulk of the OtBu ligand, there could be some steric influence on this value, but the closest intramolecular contact between a methyl carbon atom of the OtBu group and a Cp carbon is at 3.49 Å (C8···C13). Although the comparison is not exact, in Cp2TiCl(OEt), the methylene carbon displays nearest contacts at 3.15 and 3.23 Å to Cp carbon atoms. More telling is the short (1.786 Å) Ti–O bond distance in 3, compared to the 1.855 Å distance in the ethoxide. There is clearly no steric impediment to adopting the very short Ti–O distance in 3 and it would seem that the OtBu group is serving as a strong π donor in this complex.
2.2.4. Cp2TiBr(OtBu) (4)
There are two crystallographically independent but closely similar molecules in the unit cell; only molecule “A” will be discussed here. Not unexpectedly, the bromoalkoxy derivative 4 (Figure 5) is isostructural with the chloroalkoxy derivative 3. They share the same pseudotetrahedral framework, with closely similar Ti–Cp (centroid) distances (<0.01 Å difference). The Ti–Br bond length in 4 matches that in Cp2TiBr2 exactly61 and is 0.12 Å longer than the Ti–Cl distance in 3, which is slightly less than the difference in covalent radii (0.18 Å).62 The tert-butoxy ligand bonding is also similar to that in 3, as measured by the Ti–O and O–C distances and the Ti–O–C angle (0.012, 0.006 Å, and 3.3° difference, respectively). The differences, although small, are in a direction that indicates that the bromide ligand provides less competition for π-bonding to the metal than does the more electronegative chloride. This is reflected in the contraction of the Ti–O bond and the widening of the Ti–O–C angle in 4 compared to 3. There is even less reason to posit any steric influence on the wide Ti–O–C angle in 4 (175.8°), as the closest intramolecular contact between a carbon atom of the OtBu group and a Cp carbon is now at 3.68 Å (C1···C14).
Figure 5.
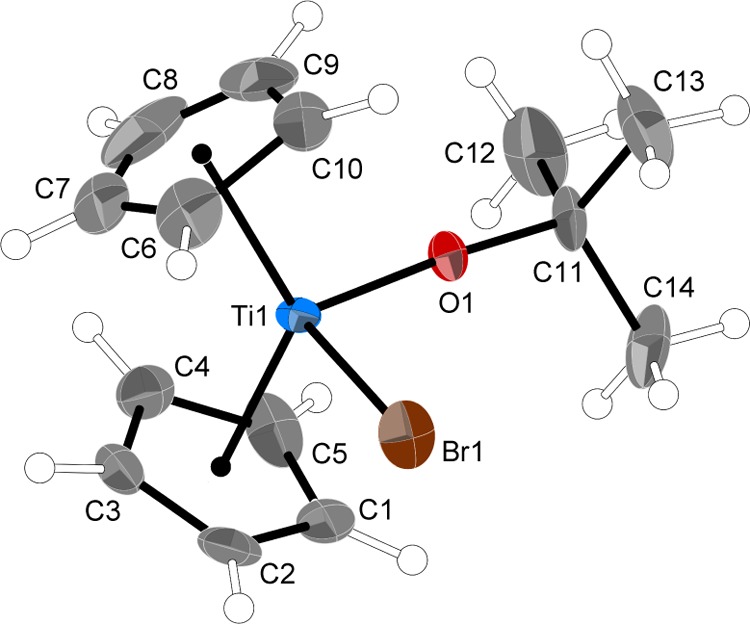
Thermal ellipsoid plot of 4 (50% level); hydrogens have arbitrary radii. Selected bond distances (Å) and angles (deg): Ti1–O1, 1.774(3); Ti1–Br1, 2.534(2); O1–C11, 1.414(5); Ti–Cp′ (ring centroid), 2.096 (ave); O1–Ti1–Br1, 84.67(16); Ti1–O1–C11, 175.8(3); and Cp–Ti–Cp′, 128.8.
2.2.5. CpTiBr2(OtBu) (5)
Similar to 4, there are two crystallographically independent but closely similar molecules in the unit cell of 5; only molecule “A” will be discussed here. Also similar to 4, compound 5 has a pseudotetrahedral geometry, with one of the Cp rings of 4 replaced with another Br (Figure 6). The molecule possesses approximate, although not crystallographically imposed, Cs symmetry; the atoms C5, Ti1, O1, C6, and C8 are all within 0.03 Å of a least-squares plane drawn through their centers. Consistent with the reduced coordination number, all of the distances to the metal have decreased relative to 4. The Ti–Cp (centroid) distance is smaller in 5 by 0.067 Å and the Ti–Br distances are 0.10 Å shorter. The already short Ti–O distance in 4 is now reduced by 0.047 Å to 1.727 Å. The Ti–O–C angle in 5 is 173.9°, slightly but not appreciably different from the value in 4.
Figure 6.
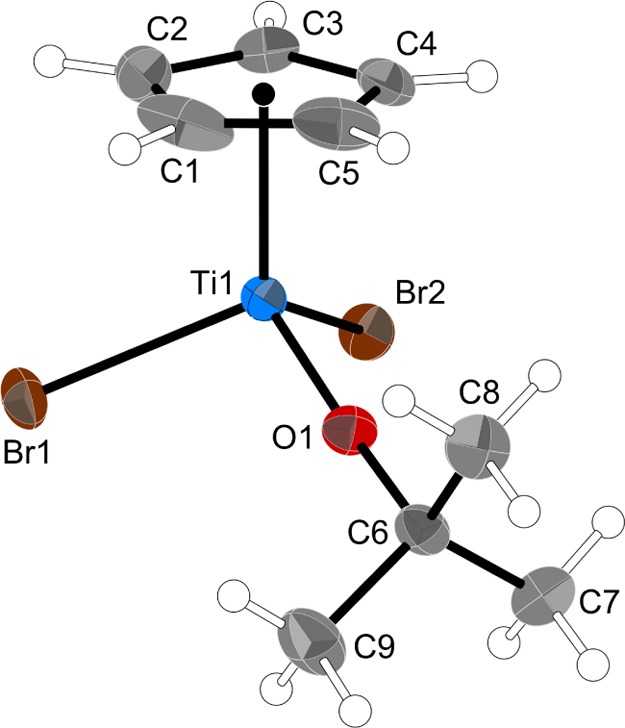
Thermal ellipsoid plot of 5 (50% level); hydrogens have arbitrary radii. Selected bond distances (Å) and angles (deg): Ti1–O1, 1.7274(18); Ti1–Br1, 2.4333(5); Ti1–Br2, 2.4326(5); O1–C6, 1.445(3); Ti–Cp (ring centroid), 2.024 (ave); Br1–Ti1–Br2, 103.05(2); O1–Ti1–Br1, 101.69(7); O1–Ti1–Br2, 101.88(7); and Ti1–O1–C6, 173.95(19).
2.3. Metal–Alkoxide Bonding
The new complexes described here provide a basis for re-examining the long-standing issue of the unusual geometries displayed by terminal transition-metal alkoxides.63 It was noted over 20 years ago that many such alkoxides of the second- and third-row early transition metals and the lanthanides (e.g., Zr, Nb, Ta, and Sm) possess nearly linear M–O–R angles and short M–O distances, although there is no direct correlation between the distances and angles.64,65 Several sources for the linear structures have been proposed, including steric crowding (bulkier alkoxides tend to display more linear M–O–R angles), π-bonding between occupied p orbitals of the oxygen and empty metal orbitals, or conversely, ionic interactions (i.e., Mδ+ORδ−).65 A role has also been assigned to electrostatic repulsion between the α-carbon of the alkoxide and the metal centers.66 Confounding these explanations are difficulties with solid-state artifacts in crystallographic structures, that is, crystal packing effects and disordered ligands, which can obscure inferences made about bonding arrangements.
Even if a largely ionic interaction is a primary bonding contributor for the heavier metal alkoxides, the situation with the early first-row metals is not necessarily the same. A broad range of M–O–R angles has been observed in first-row alkoxide complexes, although as with the heavier metals, correlation between M–O distances and M–O–C angles is essentially nonexistent. For example, even when restricting examples to complexes comparable to those in the present study, that is, mononuclear Ti(IV) species with terminal tert-butoxide ligands, a plot of Ti–O distances versus Ti–O–C angles is effectively a random scatter (Figure 7).
Figure 7.
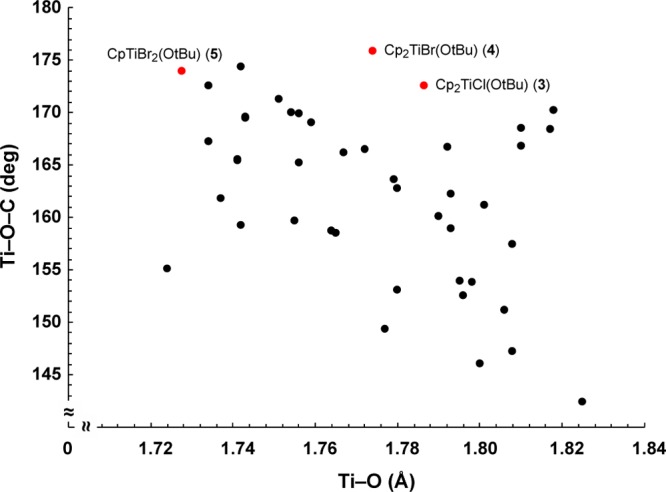
Ti–O distances vs Ti–O–C angles in mononuclear Ti(IV) tert-butoxide complexes. Compounds from the present study are marked in red and are detailed in Table 1. Data are from the Cambridge Crystallographic Database.67
Such randomness affects the plausibility of certain explanations for the observed Ti–O–C angles. For example, the steric bulk of the −OtBu ligand, which is obviously a fixed value, is not always accompanied by wide (ca. >160°) Ti–O–C bond angles. In addition, even the longest Ti–O bond in the set of complexes [1.825(4) Å] in (OPy)2Ti(OtBu)2 (OPy = 2-pyridylmethoxy68) is well below the sum of the covalent radii of Ti and O (2.09 Å), making an ionic contribution to the bonding reasonable for all of the complexes.
The just-mentioned (OPy)2Ti(OtBu)2 complex, which possesses two independent (nonsymmetry related) tert-butoxide ligands, also illustrates the importance of packing effects on experimentally determined bond distances and angles. Specifically, there are two independent molecules in its asymmetric unit68 and between the four alkoxide ligands, the Ti–O bonds range from 1.790 Å to the previously noted 1.825 Å; the Ti–O–C angles vary from 142.4° to 162.2°. The large difference in angles (Δ = 19.8°) for the small change in bond lengths (Δ = 0.035 Å) underscores why correlation between the two variables cannot be expected to be strong, if it exists at all.
Even apart from such experimental difficulties, the alkoxide ligand is notoriously ambivalent in its bonding.69,70 Depending on how the lone pairs on the oxygen interact with the metal, single, double, and (weak) triple covalent bonding can be envisaged or a purely ionic representation can be drawn as well; the latter two forms are consistent with wide M–O–R angles (Figure 8).
Figure 8.

Bonding arrangements for the alkoxide ligand. Forms (a) and (b) represent single and double bonds, respectively; forms (c) and (d) are both compatible with near linear M–O–R bonds.
We were interested in the opportunity that the present set of complexes offered for illuminating the bonding involved in the CpxTiXy(OR)4–(x+y) series. Calculations on isolated molecules remove the ambiguities of crystal packing effects, of course, and also provide the opportunity to look for orbital evidence of M–L π-bonding.
2.4. Computational Results
Even considering the vagaries of crystal packing effects, preliminary density functional theory (DFT) calculations using the M0671 and APF-D72 global hybrid functionals provided somewhat disappointing reproductions of the crystallographic structures. Tests with the double hybrid B2PLYP functional,73 which incorporates perturbative second-order correlation (PT2) obtained from the Kohn–Sham (GGA) orbitals and eigenvalues, were more encouraging and were used for subsequent studies. Not unexpectedly, reproducing angles is more difficult than bond distances (Table 1). Specific points about individual molecules are listed below.
2.4.1. Cp2TiCl(OMe) (1)
Although the calculated Ti–O–C angle of 136.8° is 4.6° smaller than that in the original room-temperature structure determination, it is only 1.7° smaller than that in the present redetermined low-temperature geometry, providing confidence that the latter is the more reliable figure.
2.4.2. Cp2TiCl(OiPr) (2)
The wide Ti–O–C angle of 160.0° found in the crystal structure cannot be reproduced by the B2PLYP calculations, which underestimates it by 12.3° [the underestimation is larger (15.4°) with the M06 functional], but the large difference supports the conclusion that the crystallographically observed value, suffering as it does from disorder both in the isopropoxy group and cyclopentadienyl rings, cannot be used uncritically. Other features of the molecule not directly involved in the disorder may be affected as well. The calculated Ti–Cl bond, for example, is 0.16 Å shorter than the crystallographically observed value.
2.4.3. Cp2TiCl(OtBu) (3)
The calculated Ti–O–C angle is smaller than the crystallographic value by 4.5° and the Ti–Cl and Ti–O bonds are in good agreement, with a mismatch of only 0.016 Å (under) and 0.014 Å (over), respectively.
2.4.4. Cp2TiBr(OtBu) (4)
The calculated Ti–O–C angle underestimates the crystallographic value by 5.2°; the Ti–Br and Ti–O bonds are slightly overestimated by 0.051 and 0.023 Å, respectively.
2.4.5. CpTiBr2(OtBu) (5)
The calculated Ti–O–C angle is larger than the crystallographic value by 5.0°; the Ti–Br and Ti–O bonds display good agreement, with a difference of only 0.003 Å (under) and 0.028 Å (over), respectively.
2.5. π-Bonding in Cyclopentadienyl Alkoxide Complexes
When there is no evidence for steric crowding or crystallographic disorder, a wide M–O–C angle accompanied by a shortened M–O bond is consistent with metal–ligand π-bonding.74 Three of the new compounds reported here, 3, 4, and 5, meet these structural criteria, as they have Ti–O–C angles >170° and Ti–O bonds of <1.80 Å. The bonding in 3 and 5 is discussed in detail.
An inspection of selected orbitals of 3 makes it obvious that two of them display evidence of π-bonding between Ti and O (Figure 9; the x axis is coincident with the Ti–O bond; the Cl lies in the xz plane). In MO#69 [highest occupied molecular orbital (HOMO) – 6, Figure 9a], Ti is involved in sigma bonding to Cl as well as π-type bonding to the oxygen. A natural bond orbital (NBO) analysis of the composition indicates that for oxygen, 98% of its contribution to the MO is from its 2pz orbital. For Ti, both its 3dz2 and 3dxz orbitals are involved, in a ratio of roughly 3:5. In MO#68 (HOMO – 7, Figure 9b), Ti is again involved in π-type bonding to the oxygen. The contribution from oxygen is 30.5% of the total MO and 99% of this is from the 2py orbital. Almost 84% of the contribution of titanium is from the 3dxy orbital, with smaller amounts from 3dyz. Much lower in energy (MO#48) are orbitals that represent Ti–O sigma bonding (Figure 9c) and the C–H bonds in the OtBu ligand. More than 95% of the contribution of oxygen to the MO is from the 2px orbital. Most of the contribution of titanium is from the 3dx2–y2 orbital.
Figure 9.
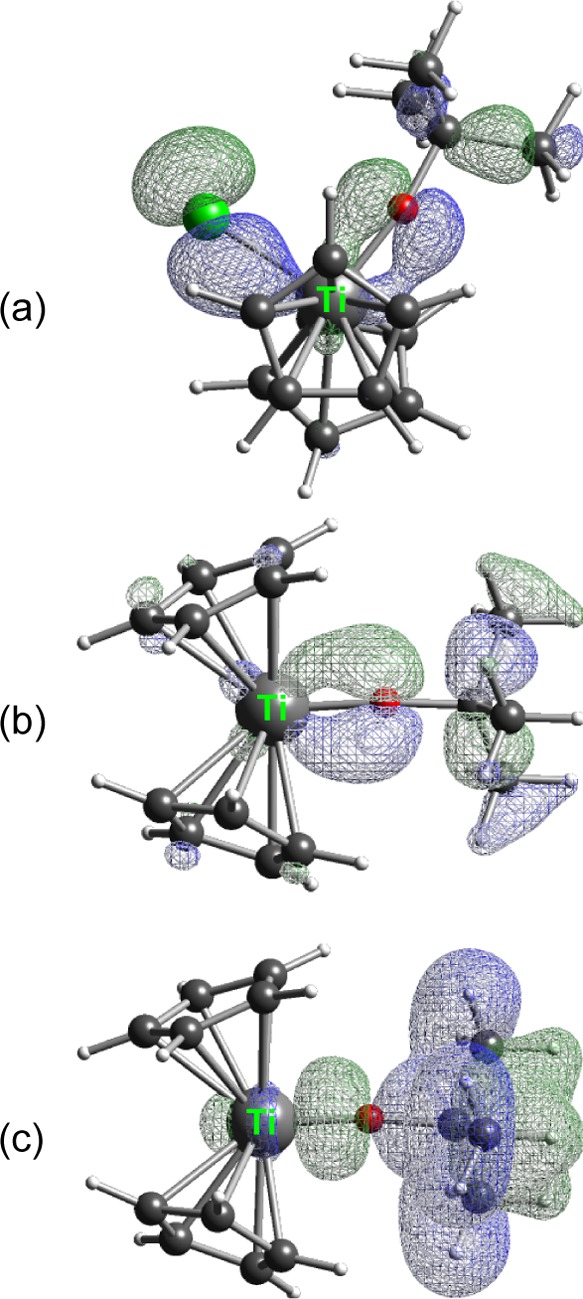
(a) MO#69 of Cp2TiCl(OtBu). (b) MO#68 of Cp2TiCl(OtBu). The isodensity surface for the two orbitals is 0.050. (c) MO#48 of Cp2TiCl(OtBu); isodensity surface = 0.025.
A similar arrangement of orbitals exists for 5. In MO#76 (HOMO – 8, Figure 10a), Ti is involved in σ-bonding to Cl as well as π-type bonding to the oxygen. NBO analysis of the composition indicates that for oxygen, over 99% of its contribution to MO is from its 2py orbital. For Ti, 88% of its contribution is from the 3dxy orbital. Complementary Ti–O π-bonding is found in MO#75 (HOMO – 9, Figure 10b), in which over 99% of the contribution of oxygen is from its 2pz orbital; almost 85% of the contribution of oxygen is from the 3dxz orbital, with 10.5% from the 4pz. As is the case with 3, the Ti–O σ interaction is substantially lower in energy (Figure 10c). In MO#57, the overlap is again primarily between the oxygen 2px orbital and the Ti 3dx2–y2 orbital.
Figure 10.
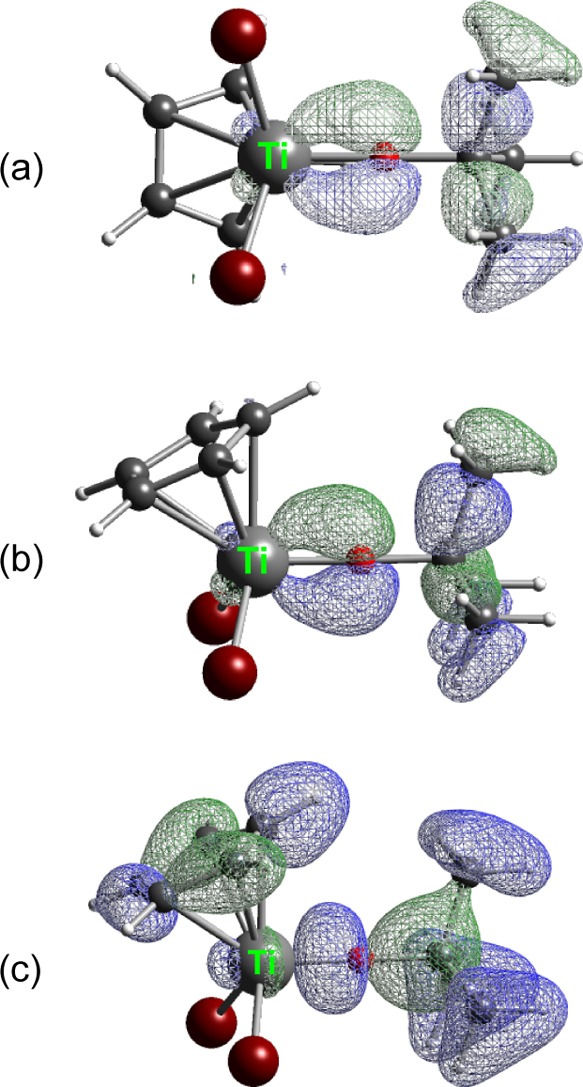
(a) MO#76 of CpTiBr2(OtBu). (b) MO#75 of CpTiBr2(OtBu). The isodensity surface for the two orbitals is 0.050. (c) MO#57 of CpTiBr2(OtBu); isodensity surface = 0.035.
The bonding picture presented here is closely related to that developed for the monocyclopentadienyl CpTiMe2((O,S)Ar) complexes.75 For the arylsulfide CpTiMe2(SAr), a straightforward Lewis structure based on an sp3-hybridized sulfur atom was sufficient to explain the bonding and geometrical arrangement of the Ti–SAr interaction. For the aryloxide analogue, in contrast, it was not possible to draw a satisfactory bonding picture in terms of a single classical Lewis structure and a resonance blend of forms c (with an sp-hybridized oxygen) and d (Figure 8) was proposed instead. It seems that the same situation largely applies to the present tert-butoxide complexes (Figure 11).
Figure 11.

Limiting bonding arrangements for Cp2TiCl(OtBu). Both extremes support a linear Ti–O–C bond angle.
Estimation of the covalency/ionicity in the Ti–O interaction in the alkoxide complexes is not straightforward. Natural population charges are relatively insensitive to basis set composition76 and that on Ti is 0.56 and 0.49 for 3 and 5, respectively. These are higher than the values of 0.12 and 0.06 for Cp2TiCl2 and Cp2TiBr2, respectively, and reflect the electron-withdrawing effect of the more electronegative oxygen and thus a greater ionic contribution to a resonance blend. Although as noted above, the Ti–O bond distances (1.73–1.79 for 3–5) are far shorter than the sum of single-bond covalent radii (2.09 Å), they are close to the sum of double-bond radii (1.74 Å).77 The Mayer bond order (MBO)78,79 for the Ti–O bond of 3 and 5 is 1.08 and 1.23, respectively, suggestive of at least a full single bond. In contrast, the “fuzzy” bond order (FBO)80 (numerically the same as the delocalization index calculated in “fuzzy” atomic space81 and intended to reflect the number of electron pairs delocalized (shared) between two atomic spaces) of 3 and 5 is 1.80 and 2.07, respectively. As the MBO can tend to underestimate the total bond order and the FBO can overestimate it,80 assigning an approximate bond order of ∼1.5 seems reasonable.
3. Conclusions
Mechanochemical synthesis can be used to produce CpxTiXy(OtBu)4–(x+y) (X = Cl, Br) complexes from the mixture of two or three starting materials, depending on whether bis(cyclopentadienyl) or mono(cyclopentadienyl) compounds are desired. Adjustment of the stoichiometric ratios of the reagents is reflected in the composition of the major products, although the outcomes are cleanest if the ratios are 1:1 (as in the production of 3) or 1:1:1 (as in the formation of 5). One noticeable advantage of the mechanochemical approach is that TiBr4, which reacts at room temperature with ethereal solvents, can be used in the solid state without modification. This probably applies to solid TiI4 as well and should make the synthesis of titanium-bromo and -iodo complexes more accessible, while also minimizing the use of solvents with environmental concerns.
All of the tert-butoxide complexes display the hallmarks of π-bonding between the titanium and alkoxide ligand, that is, short Ti–O bonds and wide (>170°) Ti–O–C angles. DFT calculations support this interpretation of the bonding, with a total bond order between 1 and 2.
It is notable that the calculations support an increasingly linear Ti–O–C angle in the order Me < Et < OiPr < OtBu, which comports with the increasing π-donor ability of the ligands. The X-ray crystal structures in general support this ordering, with the notable exception of the ethoxy complex Cp2TiCl(OEt), whose Ti–O–C angle is the smallest of those studied here. We are at present unsure of the reason for this difference, unless crystal packing effects have a larger than expected influence on the geometry.
4. Experimental Section
4.1. General Considerations
All manipulations were performed with the exclusion of air and moisture using high vacuum, Schlenk, or glovebox techniques. All of the solid reagents are slightly to highly moisture-sensitive and/or air-sensitive and should be handled accordingly. Proton and carbon (13C(1H)) NMR spectra were obtained on a DRX-400 spectrometer at 400 (1H) and 100.1 (13C) MHz and were referenced to the residual proton and 13C resonances of C6D6. Elemental analysis was performed by ALS, Tucson, AZ.
4.2. Materials
TiBr4, LiOtBu, KOtBu, LiCp, and Cp2TiCl2 were purchased from commercial suppliers and used as received. Toluene and hexanes were distilled under nitrogen from potassium benzophenone ketyl.82 Anhydrous THF was stored over molecular sieves. MeOH was obtained from MB-SPS. Isopropyl alcohol and triethylamine (TEA) were distilled and dried over CaH2. C6D6 was vacuum-distilled from Na/K (22/78) alloy and stored over type 4A molecular sieves prior to use. Cp2TiCl(OEt) was prepared according to the literature procedure.49
4.3. Mechanochemical Protocol
Ball-milling reactions used 50 stainless steel (440 grade) ball bearings [3/16 in. (5 mm), 0.44 g] that were thoroughly cleaned with hexanes and acetone prior to use. Planetary milling was performed with a Retsch PM100 mill, 50 mL stainless steel grinding jar type C, and a safety clamp for air-sensitive grinding. A typical reaction involved 300 mg total sample weight, sealed under an inert atmosphere. The ground mixture was extracted with minimal hexanes (<100 mL) and filtered through a medium porosity ground glass frit. The extraction is designed to dissolve the complex and the filtration removes traces of KCl or KBr. The filtrate was then dried under vacuum prior to NMR analysis. In the case of TiBr4 reactions, TiBr4 was added to the grinding jar first, followed by LiCp and then Li[OtBu]; this is to prevent the solid-state reaction of the alkoxide with TiBr4.
4.3.1. Cp2TiCl(OMe) (1)
This was prepared following the literature procedure.45 Cp2TiCl2 (0.502 g, 2.01 mmol) was added to a Schlenk flask containing 40 mL of THF and a magnetic stirrer bar. This was stirred at room temperature under N2. To this mixture, TEA (0.52 mL, 3.7 mmol) and MeOH (0.16 mL, 3.9 mmol) were added. The reaction mixture was allowed to stir at room temperature for 16 h. THF was removed in vacuo. The resulting product was then extracted with toluene to yield an orange filtrate. Toluene was removed to afford 0.441 g (90% yield) of an orange solid. 1H NMR (400 MHz, C6D6, 298 K): δ 4.06 ppm (s, 3H, CH3), δ 5.88 (s, 10H, C5H5). 13C NMR (100 MHz, C6D6, 298 K): δ 70.7 (s, OCH3), δ 117.0 (s, C5H5).
4.3.2. Cp2TiCl(OiPr) (2)
Cp2TiCl2 (0.500 g, 2.01 mmol) was added to a Schlenk flask containing 40 mL of THF and a magnetic stirrer bar. This was stirred at room temperature under N2. To this mixture, TEA (0.29 mL, 2.08 mmol) and iPrOH (0.16 mL, 2.08 mmol) were added. The reaction mixture was allowed to stir at room temperature for 16 h. THF was removed in vacuo. The resulting product was then extracted with hexanes and filtered through a medium porosity glass-fritted glass filter. Hexane was removed, leaving an orange solid (0.241 g, 44%). 1H NMR (400 MHz, C6D6, 298 K): δ 1.03 (d, 6H, CH(CH3)2), δ 4.48 (sept, 1H, J = 6 Hz, CH(CH3)2), δ 5.88 (s, 10H, C5H5). 13C NMR (100 MHz, C6D6, 298 K): δ 25.5 (s, CH(CH3)2), δ 84.1 (s, CH(CH3)2), 116.5 (s, C5H5). Anal. Calcd for C13H17ClOTi: C, 57.3; H, 6.3; Ti, 17.6. Found: C, 56.2; H, 5.9; Ti, 17.8.
4.3.3. Cp2TiCl(OtBu) (3)
Cp2TiCl2 (0.249 g, 1.00 mmol), K[OtBu] (0.113 g, 1.01 mmol), and 50 ball bearings were added to a grinding jar. The jar was sealed under an inert atmosphere and the reaction mixture was ground at 600 rpm for 15 min. Upon completion, the jar was opened under an inert atmosphere to reveal an orange solid which was then extracted with minimal hexanes and filtered through a fritted glass filter. The resulting orange filtrate was then dried, resulting in an orange solid (0.181 g, 63%). 1H NMR (400 MHz, C6D6, 298 K): δ 1.12 (s, 9H, OC(CH3)3), δ 5.94 (s, 10H, C5H5). 13C NMR (100 MHz, C6D6, 298 K): δ 31.3 (s, CH3), δ 87.8 (s OC(CH3)3), 116.6 (s, C5H5). Anal. Calcd for C14H19ClOTi: C, 58.7; H, 6.7; Ti, 16.7. Found: C, 58.5; H, 6.6; Ti, 16.9.
4.3.4. Cp2TiBr(OtBu) (4)
TiBr4 (0.457 g, 1.24 mmol), Li[OtBu] (0.101 g, 1.26 mmol), LiCp (0.177 g, 2.46 mmol), and 50 ball bearings were added to a grinding jar. The jar was sealed under an inert atmosphere and the reaction mixture was ground at 600 rpm for 15 min. Upon completion, the jar was opened under an inert atmosphere to reveal an orange solid which was then extracted with minimal hexanes and filtered through a fritted glass filter. The resulting filtrate was then dried, resulting in an orange solid (0.214 g, 52%). The product also contains CpTiBr(OtBu)2; crystals of 4 can be obtained from hexanes. 1H NMR of 4 (400 MHz, C6D6, 298 K): δ 1.10 (s, 9H OC(CH3)3), δ 5.92 (s, 10H, C5H5). 13C NMR (100 MHz, C6D6, 298 K); δ 31.2 (s, CH(CH3)2), δ 88.4 (s, CH(CH3)2), 116.2 (s, C5H5).
4.3.5. CpTiBr2(OtBu) (5)
TiBr4 (0.576 g, 1.57 mmol), Li[OtBu] (0.127 g, 1.59 mmol), LiCp (0.114 g, 1.58 mmol), and 50 ball bearings were added to a grinding jar. The jar was sealed under an inert atmosphere and the reaction mixture was ground at 600 rpm for 15 min. Upon completion, the jar was opened under an inert atmosphere to reveal a yellow-brown solid which was then extracted with minimal hexanes and filtered through a fritted glass filter. The resulting yellow filtrate was then dried, resulting in a yellow solid (0.241 g, 44%). 1H NMR (400 MHz, C6D6, 298 K): δ 1.14 (s, 9H C4H9), δ 6.16 (s, 10H, C5H5). 13C NMR (100 MHz, C6D6, 298 K): δ 30.1 (s, CH3), δ 92.6 (s OC(CH3)3), 118.0 (s, C5H5). Anal. Calcd for C9H14Br2OTi: C, 31.25; H, 4.08. Found: C, 31.42; H, 3.96.
4.3.6. CpTiBr(OtBu)2 (6)
TiBr4 (0.382 g, 1.04 mmol), Li[OtBu] (0.167 g, 2.09 mmol), LiCp (0.0747 g, 1.04 mmol), and 50 ball bearings were added to a grinding jar. The jar was sealed under an inert atmosphere and the reaction mixture was ground at 600 rpm for 15 min. Upon completion, the jar was opened under an inert atmosphere to reveal an orange paste that was then extracted with minimal hexanes and filtered through a fritted glass filter. The resulting yellow filtrate was then placed under vacuum, resulting in an orange oil (0.217 g, 62%). Analysis with 1H NMR indicates that ca. 31% of the sample consisted of Ti(OtBu)4. 1H NMR of 6 (400 MHz, C6D6, 298 K): δ 1.19 (s, 18H, C4H9), δ 6.22 (s, 5H, C5H5). 13C (100 MHz, C6D6, 298 K): δ 31.8 (s, CH(CH3)2), δ 86.8 (s, CH(CH3)2), 115.0 (s, C5H5).
4.4. General Procedures for X-ray Crystallography
A suitable crystal of each sample was located, mounted in a polyimide loop, and mounted on an Agilent SuperNova (Dual, Cu at zero, EosS2) diffractometer. The crystals were maintained at 100 K (223 K for 2) during data collection. Under Olex2,83 the structure was solved with the SHELXT84 structure solution program using direct methods and refined with the SHELXL85 refinement package using least squares minimization. All nonhydrogen atoms were refined with anisotropic displacement parameters.
4.5. General Procedures for Calculations
All calculations were performed with the Gaussian 09W suite of programs.86 The double hybrid B2PLYP functional,73 which incorporates perturbative second-order correlation (PT2) that is obtained from the Kohn–Sham (GGA) orbitals and eigenvalues, was used for geometry optimization. The def2TZVP basis set was used on Ti, Cl, and Br; the def2SVP basis was used on all other atoms.87 An ultrafine grid was used for all calculations (Gaussian keyword: int = ultrafine).
Acknowledgments
Financial support by the National Science Foundation (CHE-1112181 and CHE-1665327), The American Chemical Society–Petroleum Research Fund (56027-ND3), and a Discovery Grant of Vanderbilt University is gratefully acknowledged. Professor Nathan D. Schley is thanked for providing the crystal structures of 1–5 and Cp2TiCl(OEt).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00943.
Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as 1815544 (CpTiBr2(OtBu)), 1815545 (Cp2TiCl(OtBu)), 1815546 (Cp2TiCl(OMe)), 1815547 (Cp2TiCl(OiPr)), 1815548 (Cp2TiCl(OEt)), and 1815549 (Cp2TiBr(OtBu)). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44)1223-336-033; e-mail: deposit@ccdc.cam.ac.uk) (ZIP)
Optimized coordinates of all structures (XYZ)
Crystal data and summary of X-ray data collection for compounds 1–5; thermal ellipsoid plot of Cp2TiCl(OEt); and 1H NMR spectra of 4 and 6 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Rabaa H.; Saillard J. Y.; Hoffmann R. Hydrogen-hydrogen and carbon-hydrogen activation reactions at d0 metal centers. J. Am. Chem. Soc. 1986, 108, 4327–4333. 10.1021/ja00275a015. [DOI] [Google Scholar]
- Tsai J.-C.; Kuo J. C.; Chen Y.-C. Novel catalyst compositions for the syndiospecific polymerization of styrene prepared by the combination of cyclopentadienyl complexes of group IIA or group IIIA elements with titanium alkoxides. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 2304–2315. 10.1002/pola.20715. [DOI] [Google Scholar]
- Buil M. L.; Esteruelas M. A.; López A. M.; Mateo A. C.; Oñate E. Preparation and X-ray Structures of Alkyl–Titanium(IV) Complexes Stabilized by Indenyl Ligands with a Pendant Ether or Amine Substituent and Their Use in the Catalytic Hydroamination of Alkynes. Organometallics 2007, 26, 554–565. 10.1021/om060909b. [DOI] [Google Scholar]
- Hsiao T.-J.; Tsai J.-C. Novel monocyclopentadienyl zirconium and hafnium trialkoxide complexes: Syntheses and catalytic properties for olefin polymerization. J. Appl. Polym. Sci. 2010, 116, 2040–2049. 10.1002/app.31731. [DOI] [Google Scholar]
- Grafov A. V.; Firme C. L.; Grafova I. A.; Benetollo F.; Dias M. L.; Abadie M. J. M. Olefin polymerisation with hafnocenes: A bridged alicyclic alcohol as a ligand and as the hafnocene modifier. Polymer 2005, 46, 9626–9631. 10.1016/j.polymer.2005.07.076. [DOI] [Google Scholar]
- Suzuki N.Stereospecific Olefin Polymerization Catalyzed by Metallocene Complexes. In Metallocenes in Regio- and Stereoselective Synthesis; Takahashi T., Ed.; Topics in Organometallic Chemistry 8; Springer-Verlag: Berlin, 2005; pp 177–216. [Google Scholar]
- Collins R. A.; Russell A. F.; Mountford P. Group 4 metal complexes for homogeneous olefin polymerisation: a short tutorial review. Appl. Petrochem. Res. 2015, 5, 153–171. 10.1007/s13203-015-0105-2. [DOI] [Google Scholar]
- Nomura K.; Naga N.; Miki M.; Yanagi K.; Imai A. Synthesis of Various Nonbridged Titanium(IV) Cyclopentadienyl–Aryloxy Complexes of the Type CpTi(OAr)X2 and Their Use in the Catalysis of Alkene Polymerization. Important Roles of Substituents on both Aryloxy and Cyclopentadienyl Groups. Organometallics 1998, 17, 2152–2154. 10.1021/om980106r. [DOI] [Google Scholar]
- Phomphrai K.; Fenwick A. E.; Sharma S.; Fanwick P. E.; Caruthers J. M.; Delgass W. N.; Abu-Omar M. M.; Rothwell I. P. Diverse Pathways of Activation and Deactivation of Half-Sandwich Aryloxide Titanium Polymerization Catalysts. Organometallics 2006, 25, 214–220. 10.1021/om0507272. [DOI] [Google Scholar]
- Manz T. A.; Phomphrai K.; Medvedev G.; Krishnamurthy B. B.; Sharma S.; Haq J.; Novstrup K. A.; Thomson K. T.; Delgass W. N.; Caruthers J. M.; Abu-Omar M. M. Structure–Activity Correlation in Titanium Single-Site Olefin Polymerization Catalysts Containing Mixed Cyclopentadienyl/Aryloxide Ligation. J. Am. Chem. Soc. 2007, 129, 3776–3777. 10.1021/ja0640849. [DOI] [PubMed] [Google Scholar]
- Manz T. A.; Sharma S.; Phomphrai K.; Novstrup K. A.; Fenwick A. E.; Fanwick P. E.; Medvedev G. A.; Abu-Omar M. M.; Delgass W. N.; Thomson K. T.; Caruthers J. M. Quantitative Effects of Ion Pairing and Sterics on Chain Propagation Kinetics for 1-Hexene Polymerization Catalyzed by Mixed Cp′/ArO Complexes. Organometallics 2008, 27, 5504–5520. 10.1021/om8004993. [DOI] [Google Scholar]
- Dove A. P.; Kiesewetter E. T.; Ottenwaelder X.; Waymouth R. M. Propylene Polymerization with Cyclopentadienyltitanium(IV) Hydroxylaminato Complexes. Organometallics 2009, 28, 405–412. 10.1021/om800571j. [DOI] [Google Scholar]
- Fraser D. A. X.; Turner Z. R.; Buffet J.-C.; O’Hare D. Titanium and Zirconium Permethylpentalene Complexes, Pn*MCpRX, as Ethylene Polymerization Catalysts. Organometallics 2016, 35, 2664–2674. 10.1021/acs.organomet.6b00417. [DOI] [Google Scholar]
- Nomura K.; Fukuda H.; Katao S.; Fujiki M.; Kim H. J.; Kim D.-H.; Saeed I. Olefin Polymerization by Half-Titanocenes Containing η2-Pyrazolato Ligands–MAO Catalyst Systems. Macromolecules 2011, 44, 1986–1998. 10.1021/ma200018z. [DOI] [Google Scholar]
- Zhao W.; Yan Q.; Tsutsumi K.; Nomura K. Efficient Norbornene (NBE) Incorporation in Ethylene/NBE Copolymerization by Half-Titanocene Catalysts Containing Chlorinated Aryloxo Ligands. Organometallics 2016, 35, 1895–1905. 10.1021/acs.organomet.6b00242. [DOI] [Google Scholar]
- Turner Z. R.; Buffet J.-C.; O’Hare D. Chiral Group 4 Cyclopentadienyl Complexes and Their Use in Polymerization of Lactide Monomers. Organometallics 2014, 33, 3891–3903. 10.1021/om500634a. [DOI] [Google Scholar]
- Buffet J.-C.; Harris G. R.; Coward J. J.; Arnold T. A. Q.; Turner Z. R.; O’Hare D. Zirconocene alkoxides and aryloxides for the polymerization of L- and rac-lactide. J. Organomet. Chem. 2016, 801, 87–95. 10.1016/j.jorganchem.2015.10.024. [DOI] [Google Scholar]
- Ning Y.; Zhang Y.; Rodriguez-Delgado A.; Chen E. Y.-X. Neutral Metallocene Ester Enolate and Non-Metallocene Alkoxy Complexes of Zirconium for Catalytic Ring-Opening Polymerization of Cyclic Esters. Organometallics 2008, 27, 5632–5640. 10.1021/om800602s. [DOI] [Google Scholar]
- Lee C. B.; Wu Z.; Zhang F.; Chappell M. D.; Stachel S. J.; Chou T.-C.; Guan Y.; Danishefsky S. J. Insights into Long-Range Structural Effects on the Stereochemistry of Aldol Condensations: A Practical Total Synthesis of Desoxyepothilone F. J. Am. Chem. Soc. 2001, 123, 5249–5259. 10.1021/ja010039j. [DOI] [PubMed] [Google Scholar]
- Sturla S. J.; Buchwald S. L. Monocyclopentadienyltitanium Aryloxide Complexes: Preparation, Characterization, and Application in Cyclization Reactions. Organometallics 2002, 21, 739–748. 10.1021/om010755u. [DOI] [Google Scholar]
- Schetter B.; Mahrwald R. Modern Aldol Methods for the Total Synthesis of Polyketides. Angew. Chem., Int. Ed. 2006, 45, 7506–7525. 10.1002/anie.200602780. [DOI] [PubMed] [Google Scholar]
- Allred T. K.; Manoni F.; Harran P. G. Exploring the Boundaries of “Practical”: De Novo Syntheses of Complex Natural Product-Based Drug Candidates. Chem. Rev. 2017, 117, 11994–12051. 10.1021/acs.chemrev.7b00126. [DOI] [PubMed] [Google Scholar]
- Sartori A.; El Habra N.; De Zorzi C.; Sitran S.; Casarin M.; Cavinato G.; Sada C.; Gerbasi R.; Rossetto G. Zirconocene Alkoxides, Promising Precursors for MOCVD of Zirconium Dioxide Thin Films. Chem. Vap. Deposition 2012, 18, 151–158. 10.1002/cvde.201106950. [DOI] [Google Scholar]
- Niinistö J.; Putkonen M.; Niinistö L.; Song F.; Williams P.; Heys P. N.; Odedra R. Atomic Layer Deposition of HfO2 Thin Films Exploiting Novel Cyclopentadienyl Precursors at High Temperatures. Chem. Mater. 2007, 19, 3319–3324. 10.1021/cm0626583. [DOI] [Google Scholar]
- Slifirski J.; Teyssandier F. Thermodynamic approach to the OMCVD of titanium carbide from titanocene dichloride. Chem. Vap. Deposition 1996, 2, 247–251. 10.1002/cvde.19960020608. [DOI] [Google Scholar]
- a (TiCl4)Vigoroux E.; Arrivant G. The Preparation of Titanium Tetrachloride. Bull. Soc. Chim. Fr. 1907, 1, 19. [Google Scholar]; b (CpTiCl3)Masnadi M.; Jamjah R.; Ahmadjo S.; Nekoomanesh M. Synthesis and Characterization of Cyclopentadienyl Titanium Trichloride and Indenyltitanium Trichloride; Monocyclictitanium Trihalide Complexes. Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2006, 36, 543–547. 10.1080/15533170600862481. [DOI] [Google Scholar]; c (Cp2TiCl2)Hunt C. C.; Doyle J. R. Synthesis of cyclopentadienide derivatives. Inorg. Nucl. Chem. Lett. 1966, 2, 283–288. 10.1016/0020-1650(66)80043-0. [DOI] [Google Scholar]; d (Cp3TiCl)Cotton F. A.; Calderon J. L.; Takats J. Stereochemically nonrigid organometallic molecules. XXVII. Fluxional behavior of tetra(cyclopentadienyl)titanium. J. Am. Chem. Soc. 1971, 93, 3587–3591. 10.1021/ja00744a008. [DOI] [Google Scholar]; e (Cp4Ti)Siegert F. W.; Meijer H. J. D. M. Tetracyclopentadienyltitanium(IV) and tricyclopentadienyltitanium(III). J. Organomet. Chem. 1969, 20, 141–145. 10.1016/s0022-328x(00)80098-4. [DOI] [Google Scholar]; f (Cp3Ti(OtBu))Boyde N. C.; Rightmire N. R.; Bierschenk E. J.; Steelman G. W.; Hanusa T. P.; Brennessel W. W. Reaction environment and ligand lability in group 4 Cp2MXY (X, Y = Cl, OtBu) complexes. Dalton Trans. 2016, 45, 18635–18642. 10.1039/c6dt03199d. [DOI] [PubMed] [Google Scholar]; g (Cp2Ti(OtBu)2)Ott K. C.; De Boer E. J. M.; Grubbs R. H. An investigation of the reaction of bis(cyclopentadienyl)titanium dichlorides with trimethylaluminum. Mechanism of an α-hydrogen abstraction reaction. Organometallics 1984, 3, 223–230. 10.1021/om00080a010. [DOI] [Google Scholar]; h (CpTi(OtBu)3)Wang Q.; Quyoum R.; Gillis D. J.; Tudoret M.-J.; Jeremic D.; Hunter B. K.; Baird M. C. Ethylene, Styrene, and α-Methylstyrene Polymerization by Mono(pentamethylcyclopentadienyl) (Cp*) Complexes of Titanium, Zirconium, and Hafnium: Roles of Cationic Complexes of the Type [Cp*MR2]+(R = Alkyl) as both Coordination Polymerization Catalysts and Carbocationic Polymerization Initiators. Organometallics 1996, 15, 693–703. 10.1021/om9501945. [DOI] [Google Scholar]; i (Ti(OtBu)4)Deluzarche A. Orthotitanates. Ann. Chim. (Paris) 1961, 6, 661–676. [Google Scholar]; j (TiCl(OtBu)3)Razuvaev G. A.; Vyshinskaya L. I.; Samarina T. P.; Drobotenko V. V. Preparation and reaction of titanium tert-butoxo complexes with triisobutylaluminum. Koord. Khim. 1987, 13, 1035–1038. [Google Scholar]; k (TiCl2(OtBu)2)Vyshinskaya L. I.; Timoshenko S. Ya.; Vishnyakova T. A. Preparation and decomposition of compoundscontaining a titanium-tin bond. Zh. Obshch. Khim. 1990, 60, 1937–1942. [Google Scholar]; l (TiCl3(OtBu))Razuvaez G. A.; Bobinova L. M.; Etlis V. S. Gewinnung und eigenschaften einiger titanorganischer verbindungen. Tetrahedron 1959, 6, 154–160. 10.1016/0040-4020(59)85009-2. [DOI] [Google Scholar]; m (CpTiCl2(OtBu)26h)Wang Q.; Quyoum R.; Gillis D. J.; Tudoret M.-J.; Jeremic D.; Hunter B. K.; Baird M. C. Ethylene, Styrene, and α-Methylstyrene Polymerization by Mono(pentamethylcyclopentadienyl) (Cp*) Complexes of Titanium, Zirconium, and Hafnium: Roles of Cationic Complexes of the Type [Cp*MR2]+(R = Alkyl) as both Coordination Polymerization Catalysts and Carbocationic Polymerization Initiators. Organometallics 1996, 15, 693–703. 10.1021/om9501945. [DOI] [Google Scholar]; n (CpTiCl(OtBu)226h)Wang Q.; Quyoum R.; Gillis D. J.; Tudoret M.-J.; Jeremic D.; Hunter B. K.; Baird M. C. Ethylene, Styrene, and α-Methylstyrene Polymerization by Mono(pentamethylcyclopentadienyl) (Cp*) Complexes of Titanium, Zirconium, and Hafnium: Roles of Cationic Complexes of the Type [Cp*MR2]+ (R = Alkyl) as both Coordination Polymerization Catalysts and Carbocationic Polymerization Initiators. Organometallics 1996, 15, 693–703. 10.1021/om9501945. [DOI] [Google Scholar]
- Calhorda M. J.; Carrondo M. A. A. F. d. C. T.; Dias A. R.; Domingos A. M. T. S.; Simoes J. A. M.; Teixeira C. Energetics of transition-metal-sulfur and -oxygen bonds in M(η5-C5H5)2L2 complexes (M = Ti, Mo, W). Molecular structure of Mo(η5-C5H5)2(SO4). Organometallics 1986, 5, 660–667. 10.1021/om00135a007. [DOI] [Google Scholar]
- Huang K.-W.; Waymouth R. M. Coordination Chemistry of Stable Radicals: Homolysis of a Titanium–Oxygen Bond. J. Am. Chem. Soc. 2002, 124, 8200–8201. 10.1021/ja0264854. [DOI] [PubMed] [Google Scholar]
- Huang K.-W.; Han J. H.; Cole A. P.; Musgrave C. B.; Waymouth R. M. Homolysis of Weak Ti–O Bonds: Experimental and Theoretical Studies of Titanium Oxygen Bonds Derived from Stable Nitroxyl Radicals. J. Am. Chem. Soc. 2005, 127, 3807–3816. 10.1021/ja044512f. [DOI] [PubMed] [Google Scholar]
- Huang K.-W.; Han J. H.; Musgrave C. B.; Waymouth R. M. Density Functional Theory Calculations of Ti–TEMPO Complexes: Influence of Ancillary Ligation on the Strength of the Ti–O Bond. Organometallics 2006, 25, 3317–3323. 10.1021/om060148c. [DOI] [Google Scholar]
- Huang K.-W.; Waymouth R. M. Hydrolysis of CpTiCl2(TEMPO) and its application on one-pot syntheses of CpTiCl(OR)2 complexes. Dalton Trans. 2004, 354–356. 10.1039/b314027j. [DOI] [PubMed] [Google Scholar]
- Margetić D.; Štrukil V.. Mechanochemical Organic Synthesis, 1st ed.; Elsevier: Amsterdam, 2016; p 386. [Google Scholar]
- Jobbágy C.; Tunyogi T.; Pálinkás G.; Deák A. Versatile Solvent-Free Mechanochemical Route to the Synthesis of Heterometallic Dicyanoaurate-Based Coordination Polymers. Inorg. Chem. 2011, 50, 7301–7308. 10.1021/ic200893n. [DOI] [PubMed] [Google Scholar]
- Jobbágy C.; Molnár M.; Baranyai P.; Deák A. Mechanochemical synthesis of crystalline and amorphous digold(I) helicates exhibiting anion- and phase-switchable luminescence properties. Dalton Trans. 2014, 43, 11807–11810. 10.1039/c4dt01214c. [DOI] [PubMed] [Google Scholar]
- Bowmaker G. A.; Hanna J. V.; Hart R. D.; Healy P. C.; King S. P.; Marchetti F.; Pettinari C.; Skelton B. W.; Tabacaru A.; White A. H. Mechanochemical and solution synthesis, X-ray structure and IR and 31P solid state NMR spectroscopic studies of copper(I) thiocyanate adducts with bulky monodentate tertiary phosphine ligands. Dalton Trans. 2012, 41, 7513–7525. 10.1039/c2dt30579h. [DOI] [PubMed] [Google Scholar]
- Garay A. L.; Pichon A.; James S. L. Solvent-free synthesis of metal complexes. Chem. Soc. Rev. 2007, 36, 846–855. 10.1039/b600363j. [DOI] [PubMed] [Google Scholar]
- Rightmire N. R.; Hanusa T. P. Advances in Organometallic Synthesis with Mechanochemical Methods. Dalton Trans. 2016, 45, 2352–2362. 10.1039/c5dt03866a. [DOI] [PubMed] [Google Scholar]
- Orita A.; Jiang L.; Nakano T.; Ma N.; Otera J. Solventless reaction dramatically accelerates supramolecular self-assembly. Chem. Commun. 2002, 1362–1363. 10.1039/b203651g. [DOI] [Google Scholar]
- Garci A.; Castor K. J.; Fakhoury J.; Do J.-L.; Di Trani J.; Chidchob P.; Stein R. S.; Mittermaier A. K.; Friščić T.; Sleiman H. Efficient and Rapid Mechanochemical Assembly of Platinum(II) Squares for Guanine Quadruplex Targeting. J. Am. Chem. Soc. 2017, 139, 16913–16922. 10.1021/jacs.7b09819. [DOI] [PubMed] [Google Scholar]
- Rightmire N. R.; Hanusa T. P.; Rheingold A. L. Mechanochemical Synthesis of [1,3-(SiMe3)2C3H3]3(Al,Sc), a Base-Free Tris(allyl)aluminum Complex and Its Scandium Analogue. Organometallics 2014, 33, 5952–5955. 10.1021/om5009204. [DOI] [Google Scholar]
- Shi Y. X.; Xu K.; Clegg J. K.; Ganguly R.; Hirao H.; Friščić T.; García F. The First Synthesis of the Sterically Encumbered Adamantoid Phosphazane P4(NtBu)6 : Enabled by Mechanochemistry. Angew. Chem., Int. Ed. 2016, 55, 12736–12740. 10.1002/anie.201605936. [DOI] [PubMed] [Google Scholar]
- Hernández J. G.; Macdonald N. A. J.; Mottillo C.; Butler I. S.; Friščić T. A mechanochemical strategy for oxidative addition: remarkable yields and stereoselectivity in the halogenation of organometallic Re(I) complexes. Green Chem. 2014, 16, 1087–1092. 10.1039/c3gc42104j. [DOI] [Google Scholar]
- Rightmire N. R.; Bruns D. L.; Hanusa T. P.; Brennessel W. W. Mechanochemical Influence on the Stereoselectivity of Halide Metathesis: Synthesis of Group 15 Tris(allyl) Complexes. Organometallics 2016, 35, 1698–1706. 10.1021/acs.organomet.6b00151. [DOI] [Google Scholar]
- Gibson D. H.; Ding Y.; Mashuta M. S.; Richardson J. F. Chlorobis(η5-cyclopentadienyl)methoxytitanium. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1996, 52, 559–560. 10.1107/s0108270195012297. [DOI] [Google Scholar]
- Nesmeyanov A. N.; Nogina O. V.; Lazareva N. A.; Dubovitskii V. A. Inverse disproportionation reaction and ester interchange of monocyclopentadienylitaniums. Izvest. Akad. Nauk SSSR, Ser. Khim. 1967, 4, 808–814. [Google Scholar]
- Nesmeyanov A. N.; Nogina O. V.; Berlin A. M.; Girshovich A. S.; Shatalov G. V. Acyl and alkoxy derivatives of dicyclopentadienyltitanium and the refraction increment of the π-C5H5Ti group. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1961, 10, 2008. 10.1007/bf01182943. [DOI] [Google Scholar]
- Huffman J. C.; Moloy K. G.; Marsella J. A.; Caulton K. G. Molecular structure of (η5-C5H5)2Ti(OC2H5)Cl and [(η5-C5H5)Cl2Ti]2O2C2(CH3)4. A structural basis for deoxygenation using titanium. J. Am. Chem. Soc. 1980, 102, 3009–3014. 10.1021/ja00529a022. [DOI] [Google Scholar]
- Rolsten R. F.; Sisler H. H. Molecular Addition Compounds of Titanium Tetrabromide and Titanium Tetraiodide with Several Ethers. J. Am. Chem. Soc. 1957, 79, 1068–1070. 10.1021/ja01562a016. [DOI] [Google Scholar]
- Anastas P. T.; Warner J. C.. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998; p 30. [Google Scholar]
- Klouras N.; Nastopoulos V.; Demakopoulos I.; Leban I. Molecular and crystal structure of Bis(η5-cyclopentadienyl)titanium(IV) Dibromide, Ti(η5-C5H5)2Br2. Z. Anorg. Allg. Chem. 1993, 619, 1927–1930. 10.1002/zaac.19936191117. [DOI] [Google Scholar]
- Hernández J. G.; Friščić T. Metal-catalyzed organic reactions using mechanochemistry. Tetrahedron Lett. 2015, 56, 4253–4265. 10.1016/j.tetlet.2015.03.135. [DOI] [Google Scholar]
- Boldyreva E. Mechanochemistry of inorganic and organic systems: what is similar, what is different?. Chem. Soc. Rev. 2013, 42, 7719–7738. 10.1039/c3cs60052a. [DOI] [PubMed] [Google Scholar]
- Wang G.-W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. 10.1039/c3cs35526h. [DOI] [PubMed] [Google Scholar]
- Waddell D. C.; Thiel I.; Clark T. D.; Marcum S. T.; Mack J. Making kinetic and thermodynamic enolates via solvent-free high speed ball milling. Green Chem. 2010, 12, 209–211. 10.1039/b922108p. [DOI] [Google Scholar]
- Among many examples that could be cited is the 1.277 Å C–O length in Cp2Ti(CHO2)2 (Gibson D. H.; Ding Y.; Richardson J. F.; Mashuta M. S. Bis(η5-cyclopentadienyl)bis(formato-O)titanium. Acta Crystallogr. Sect. C 1996, 52, 1614–1616. 10.1107/s0108270196002715. [DOI] [Google Scholar]; ). The electronic properties of the acyloxy group contribute to the bond shortening.
- Niibayashi S.; Mitsui K.; Matsubara K.; Nagashima H. Novel Titanium–Cobalt Complexes Formed by Reductive Cleavage of a Co–Co Bond in Co2(CO)8 by Titanocene tert-Butoxides: Synthesis, Characterization, and Mechanistic Aspects for Metal–Metal Bond Recombination. Organometallics 2003, 22, 4885–4892. 10.1021/om0340701. [DOI] [Google Scholar]
- Clearfield A.; Warner D. K.; Saldarriaga-Molina C. H.; Ropal R.; Bernal I. Structural Studies of (π-C5H5)2MX2 Complexes and their Derivatives. The Structure of Bis(π-cyclopentadienyl)titanium Dichloride. Can. J. Chem. 1975, 53, 1622–1629. 10.1139/v75-228. [DOI] [Google Scholar]
- The next widest angle is 152.9(5)°, found in the Cp2TiCl(10,10-dimethyl-3,3-dioxo-3-thia-4-azatricyclo[5. 2. 1. 01, 5]decan-6-olato) complex. The Ti–O bond length is 1.824(5) Å, shorter than the distance in Cp2TiCl(OEt), but 0.38 Å longer than the distance in 3.Carvalho M. F. N. N.; Galvão A. M.; Kredatusová J.; Merna J.; Pinheiro P. F.; Salema M. M. Synthesis and catalytic activity of camphor titanium complexes. Inorg. Chim. Acta 2012, 383, 244–249. 10.1016/j.ica.2011.11.019. [DOI] [Google Scholar]
- Jones P. G.; Kienitz C.; Thöne C. Crystal structure of dibromobis(cyclopentadienyl)titanium(IV), C10H10Br2Ti. Z. für Kristallogr.—Cryst. Mater. 1994, 209, 85. 10.1524/zkri.1994.209.1.85. [DOI] [Google Scholar]
- Cordero B.; Gómez V.; Platero-Prats A. E.; Revés M.; Echeverría J.; Cremades E.; Barragán F.; Alvarez S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- Bradley D.; Mehrotra R. C.; Rothwell I.; Singh A.. X-Ray Crystal Structures of Alkoxo Metal Compounds. In Alkoxo and Aryloxo Derivatives of Metals; Academic: New York, 2001; pp 229–382. [Google Scholar]
- Steffey B. D.; Fanwick P. E.; Rothwell I. P. Solid state structure of the tantalum bis-aryl compounds Ta(OAr-2,6R2)3(C6H5)2 (R = CH3, Pri; OAr-2,6R2 = 2,6-dialkylphenoxide): Observation of a lack of correlation of MOAr distances and MOAr angles for aryloxide derivatives of niobium(V) and tantalum(V). Polyhedron 1990, 9, 963–968. 10.1016/s0277-5387(00)84298-9. [DOI] [Google Scholar]
- Howard W. A.; Trnka T. M.; Parkin G. Syntheses of the Phenylchalcogenolate Complexes (η5-C5Me5)2Zr(EPh)2 (E = O, S, Se, Te) and (η5-C5H5)2Zr(OPh)2: Structural Comparisons within a Series of Complexes Containing Zirconium-Chalcogen Single Bonds. Inorg. Chem. 1995, 34, 5900–5909. 10.1021/ic00127a031. [DOI] [Google Scholar]
- Russo M. R.; Kaltsoyannis N.; Sella A. Are metal alkoxides linear owing to electrostatic repulsion?. Chem. Commun. 2002, 2458–2459. 10.1039/b207435d. [DOI] [PubMed] [Google Scholar]
- Data from Cambridge Crystallographic Database. Release of Nov. 2016. Complexes with disordered alkoxides were excluded from the compilation.
- Boyle T. J.; Sewell R. M.; Ottley L. A. M.; Pratt H. D.; Quintana C. J.; Bunge S. D. Controlled Synthesis of a Structurally Characterized Family of Sterically Constrained Heterocyclic Alkoxy-Modified Titanium Alkoxides. Inorg. Chem. 2007, 46, 1825–1835. 10.1021/ic0618798. [DOI] [PubMed] [Google Scholar]
- Mingos D. M. P. A review of complexes of ambivalent and ambiphilic Lewis acid/bases with symmetry signatures and an alternative notation for these non-innocent ligands. J. Organomet. Chem. 2014, 751, 153–173. 10.1016/j.jorganchem.2013.08.033. [DOI] [Google Scholar]
- Mingos D. M. P. A theoretical analysis of ambivalent and ambiphilic Lewis acid/bases with symmetry signatures. Coord. Chem. Rev. 2015, 293–294, 2–18. 10.1016/j.ccr.2014.11.009. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Austin A.; Petersson G. A.; Frisch M. J.; Dobek F. J.; Scalmani G.; Throssell K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. 10.1021/ct300778e. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Niibayashi S.; Mitsui K.; Motoyama Y.; Nagashima H. The effect of titanium alkoxides in the synthesis of heterobimetallic complexes by titanocene(III) alkoxide-induced metal-metal bond cleavage of metal carbonyl dimers. J. Organomet. Chem. 2005, 690, 276–285. 10.1016/j.jorganchem.2004.09.075. [DOI] [Google Scholar]
- Manz T. A.; Fenwick A. E.; Phomphrai K.; Rothwell I. P.; Thomson K. T. The nature of aryloxide and arylsulfide ligand bonding in dimethyltitanium complexes containing cyclopentadienyl ligation. Dalton Trans. 2005, 668–674. 10.1039/b412455c. [DOI] [PubMed] [Google Scholar]
- Reed A. E.; Weinstock R. B.; Weinhold F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. 10.1063/1.449486. [DOI] [Google Scholar]
- Pyykkö P.; Atsumi M. Molecular Double-Bond Covalent Radii for Elements Li–E112. Chem.—Eur. J. 2009, 15, 12770–12779. 10.1002/chem.200901472. [DOI] [PubMed] [Google Scholar]
- Mayer I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. 10.1016/0009-2614(83)80005-0. [DOI] [Google Scholar]
- Bridgeman A. J.; Cavigliasso G.; Ireland L. R.; Rothery J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc., Dalton Trans. 2001, 2095–2108. 10.1039/b102094n. [DOI] [Google Scholar]
- Mayer I.; Salvador P. Overlap populations, bond orders and valences for “fuzzy” atoms. Chem. Phys. Lett. 2004, 383, 368–375. 10.1016/j.cplett.2003.11.048. [DOI] [Google Scholar]
- Matito E.; Poater J.; Solà M.; Duran M.; Salvador P. Comparison of the AIM Delocalization Index and the Mayer and Fuzzy Atom Bond Orders. J. Phys. Chem. A 2005, 109, 9904–9910. 10.1021/jp0538464. [DOI] [PubMed] [Google Scholar]
- Armarego W. L. F., Perrin D. D.. Purification of Laboratory Chemicals, 4th ed.; Pergamon: Oxford, 1997; p 16. [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/s0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT- Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/s2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09W, Revision D.01; Gaussian, Inc.: Wallingford CT, 2009.
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.