

Abstract
H-bonds between neutral tyrosine and arginine in nonpolar environments are modeled by small-molecule phenol/guanidine complexes. From the temperature and concentration dependence of UV spectra, a value of ΔH° = −74 ± 4 kJ mol–1 is deduced for the formation of H-bonded p-cresol/dodecylguanidine in hexane. ΔE = −71 kJ mol–1 is computed with density functional theory (in vacuo). In dimethyl sulfoxide or crystals, (p-phenolyl)alkylguanidines form head-to-tail homodimers with two strong H-bonding interactions, as evidenced by UV, IR, and NMR spectral shifts, strong IR continuum absorbance bands, and short O···N distances in X-ray crystal structures. Phenol/alkylguanidine H-bonded complexes consist of polarizable rapidly interconverting tautomers, with the proton shift from phenol to guanidine increasing with increase in the polarity of the aprotic solvent. As measured by NMR, both groups in these strongly H-bonded neutral complexes can simultaneously appear to be predominantly protonated. These systems serve as models for the hypothetical hydrogen-Bonded Uncharged (aRginine + tYrosine), or “BU(RY)”, motifs in membrane proteins.
Introduction
There has been little experimental modeling of H-bonding interactions between the guanidine group of arginine and the phenolic group of tyrosine, especially with the guanidine in a deprotonated state. Geometries suitable for H-bonding between tyrosine and arginine were for a long time quite uncommon in observed crystal structures of proteins, making such H-bonds a relatively uninteresting field of study. (See section titled “Possible New Motif for Membrane Protein Structures”, for more details on the relative infrequency of such groupings in the crystallographic database.)
However, this has changed recently with the publication of a number of crystal structures of active intermediates of membrane proteins, in which conserved arg–tyr pairs approach each other within a hydrophobic region of the protein interior, in a geometry that would permit H-bonding between them. Examples were seen first in the M state of bacteriorhodopsin (bR), a microbial rhodopsin,1 and subsequently in bovine opsin, a G-protein-coupled receptor (GPCR).2 (For an explanation of the distinction between these the two major superfamilies of heptahelical transmembrane (7-TM) proteins, see a recent review of microbial rhodopsins.3) In examples from both these superfamilies,1,2 the conformational change that forms the active intermediate involves significant displacement of a highly conserved arginine in the third transmembrane helix, away from an asp or glu salt-bridge partner and into a less-polar protein environment that includes a conserved tyr.
Evidence for arg–tyr interaction can be seen in greater detail in more recent crystal structures of metarhodopsin II (Meta II), with and without the C-terminal binding portion of the α subunit of Gt, GαCT (see Figure 1). These structures show that one of the key interactions in Meta II involves the protrusion of GαCT deep into the protein interior, allowing a direct H-bonded interaction of the backbone of GαCT with the highly conserved arginine (R135 in rhodopsin).4
Figure 1.

H-bonding interactions of the conserved arginine–tyrosine grouping in activated rhodopsin (Meta II), without (left) and with (right) the peptide backbone of the bound C-terminal portion of transducin (GαCT, with a backbone shown in green in the right panel). Coordinates were obtained from data published with 3.0 Å (3XPO) and 2.85 Å (3PQR) resolutions, respectively.4 In Meta II without GαCT (at left), one of terminal nitrogens of Arg135 from TM3 is only 2.7 Å from the Tyr223 oxygen atom. The other two nitrogens are not within the H-bond distance to donor or acceptor group detected in the crystal structure. Upon binding of Meta II with GαCT (right panel), the latter protein’s Val347 backbone C=O group serves as an H-bond acceptor for the other terminal nitrogen of Arg135, as well as for the intrachain nitrogen, with N–O distances of 3.0 and 3.1 Å, respectively. (Only the former distance is indicated by a dashed line in this figure.) Furthermore, the C-terminal carboxylate of GαCT moves in to become the closest anion, ∼9 Å away from Arg135. Formation of these ionic and H-bonding interactions between GαCT and Arg135 apparently weakens the H-bonding interaction of the arginine with tyr223, as evidenced by an increased N–O distance (3.3 Å in the right panel).
Each time a Gt binds to a Meta II in this fashion, the Gt nucleotide-binding site opens up, permitting the spontaneous release of GDP, and replacement with GTP from solution. This cyclical, catalytic G-protein activation process clearly depends on the specificity, tightness, and reversibility of the binding of Gt to Meta II. Many aspects of the binding interaction are conserved in other GPCR systems.
Both arginine and tyrosine side chains are generally thought to be protonated (cationic and neutral, respectively) in most biological environments, including the highly conserved arginine–tyrosine grouping within Meta II (Figure 1), as well as other activated GPCRs.6−9 However, interpretation of these side chains’ protonation and H-bonding state in these structures is complicated by their unusually aprotic environment. In the published crystal structure of Meta II in the absence of GαCT (3XPO, Figure 1), numerous structural water molecules are observable in the protein interior, but the closest is at a distance of ∼8.5 Å from arg135.4,5 In fact, the environment within 5 Å of the arg135–tyr223 H-bond consists almost exclusively of hydrocarbon side chains. Such an environment is very different from those that have previously been used to model the arginine side chain spectroscopically.10,11 Deprotonated arginine side chains, and H-bonded arginine–tyrosine dyads, should be spectroscopically modeled in aprotic environments similar to these proteins’ interiors. The current study, and the accompanying paper,12 provides some key steps toward achieving this.
Scheme 1 summarizes three generalized proton configurations corresponding to H-bonded arginine–tyrosine dyads that would most likely be observable in nonpolar environments. The first (A) involves neutral tyrosine acting as an H-bond acceptor for protonated arginine, which additionally participates in a stabilizing Coulombic interaction with a nearby counterion (X–). The second (B) involves a zwitterion comprising deprotonated tyrosine and protonated arginine, in which the tyrosine oxygen again acts as an H-bond acceptor. The third (C) is fully neutral, comprising neutral tyrosine serving as an H-bond donor, as well as neutral (deprotonated) arginine taking on the new role of H-bond acceptor (right).
Scheme 1. Three Possible Protomeric Configurations for Arg−Tyr Dyads in Nonpolar Environments.

Scheme 1B,C are the proton-limiting structures of an overall-neutral H-bonded pair. That is, they differ only by a very small movement of a proton within an H-bond. Therefore, if either is present, the other is expected also to be present, in rapid equilibrium.
We modeled structures B/C computationally; and A–C experimentally by synthesizing model compounds that exhibited these three structures in properly selected aprotic solvents, based on spectroscopic evidence. Three common aprotic solvents, and one protic solvent used as a control, were chosen to span a wide range of dielectric constants (ε): hexane (ε = 1.8), CCl4 (ε = 2.2), dimethyl sulfoxide (DMSO, ε = 47), and methanol (ε = 33). This span includes the smaller ranges generally assumed for protein and membrane interiors. These solvents were also selected for several other important properties, including ready availability with needed isotope variations for NMR; maintaining chemical stability of our samples; and allowing sufficient solubility of them to obtain UV, IR, and/or NMR spectra with suitable signal/noise ratios. Water itself is not generally a suitable solvent for spectroscopy of any of the deprotonated guanidine compounds because hydroxide-catalyzed hydrolysis occurs within minutes at high pH. We were also able to crystallize several of the model compounds in the forms corresponding to both A and B.
Using our model systems, we demonstrate that the net-neutral pair (structures B and C in Scheme 1) can indeed be formed with well-defined stoichiometry in the three aprotic solvents examined, and structure B can be observed in the crystalline state. The H-bond between guanidine and phenol in such overall-neutral systems is not only unusually strong but also highly polarizable. In the least-polar environments, both protomers participate nearly equally.
These results allow more accurate spectroscopic modeling of membrane-buried arg–tyr dyads that might undergo deprotonation, that is, with overall loss of a proton shared between arginine and a nearby tyrosine. We term this an H-Bonded Uncharged (aRginine–tYrosine), or “BU(RY)” dyad. We also present a specific hypothesis for the role such a BU(RY) dyad might play in G-protein activation by GPCRs.
Results
Computational Studies of the Phenol–Guanidine H-Bond
The complex between p-cresol and monoalkylguanidines was modeled by using density functional theory (DFT). Rather than dodecylamine itself, the computational modeling used the shorter alkyl chain of ethylguanidine. The energy-optimized structure of the 1:1 complex of p-cresol with ethylguanidine in vacuo is shown in Figure 2A (see Supporting Information for details). The computed binding energy is −71 kJ mol–1, which corresponds to the energy of the optimized geometry (Figure 2A), relative to the sum of the energies of the same two molecules optimized individually. Even when a larger basis set was used (e.g., 6-311G** as compared to 6-31G**), the computed binding energy remained unchanged to within ∼1%.
Figure 2.
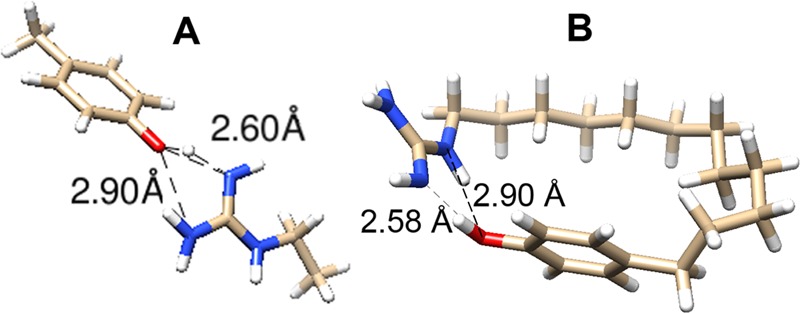
DFT-computed H-bonding geometries. (A) p-Cresol/ethylguanidine complex. (B) Internally H-bonded p-phenol-dodecylguanidine. See Supporting Information for computational details and energy-optimized Cartesian coordinates.
In the optimized geometry (Figure 2A), both H-bonds are significantly nonlinear. For example, there is a 158° O–H–N angle for the primary H-bond, which has a 2.60 Å distance from the phenolic oxygen to the imino nitrogen. There is also a more distorted secondary (2.90 Å) H-bond, from an amine nitrogen back to the oxygen. Formation of the H-bond(s) resulted in significant lengthening of the O–H covalent bond length, to 1.03 Å (from 0.98 Å in isolated p-cresol).
A similar H-bonding geometry was obtained when the ethylguanidine and cresol were linked together with a dodecyl chain, as shown in Figure 2B. One interesting difference is the involvement of the intrachain NH group in the H-bond, instead of only the two terminal nitrogens. Despite the overall similar bond lengths and angles for the H-bonding groups as in Figure 2A, the binding energy in Figure 2B is only −39 kJ mol–1. In this case, the reference state was the optimized local energy minimum, with an extended dodecyl chain in an all-s-trans conformation. Presumably, the H-bond energy in Figure 2B, which by itself would not be expected to be greatly different than that in Figure 2A, was significantly canceled by unfavorable strain introduced in the alkane chain.
UV Spectroscopic Detection of Phenol–Guanidine H-Bonding in Hexane
The formation of p-cresol/alkylguanidine complexes in nonpolar solvents was confirmed experimentally by UV spectral measurements. The ∼280 nm absorption band of the phenol group is strongly affected by the formation of H-bonded complexes, as has been shown previously using amines as the H-bond acceptor groups.13,14 Similar effects are clearly seen in the UV spectra shown in Figure 3, which demonstrates the interaction between p-cresol and dodecylguanidine in hexane. From these data, we determined a value for the enthalpy of complex formation in hexane (ΔH° = −74 ± 4 kJ mol–1) that closely matched the computed in vacuo ΔE = −71 kJ mol–1. (The closeness of this match is fortuitous; see Discussion.) Details of this experimental determination are as follows.
Figure 3.

UV absorption spectra demonstrating the reversible complexation of p-cresol with equimolar dodecylguanidine in hexane. Spectra were obtained as a function of total concentration of p-cresol (A, in which all seven spectra were measured at 58 °C; the inset shows a log–log plot of A308 vs total concentration); or as a function of temperature (B, in which all eight spectra were measured with 0.50 mM total concentrations of p-cresol and dodecylguanidine; the inset shows a van’t Hoff plot). Temperatures in (B) are indicated by trace colors: blue, 58 °C; green, 60 °C; orange, 62 °C; red, 64 °C. Two nearly superimposed traces of each color are plotted, measured during up-then-down temperature steps. Spectral traces in black (overlapping curves near 0 absorbance) in (A) and (B) represent the residuals after subtracting the best-fitted linear combination of the spectra of free p-cresol and complexed p-cresol/dodecylguanidine (C). The contribution of free p-cresol was computed as the measured extinction-coefficient spectrum of pure p-cresol in hexane (C, black trace), multiplied by its measured total concentration and the best-fit mole fraction for it. These 11 mole fractions of free p-cresol, 1 for each of the 11 measured spectra, were calculated as 1 – Xi, where the values of Xi were 11 of the 512 adjustable parameters during the least-squares fit. These Xi were the mole fractions of the p-cresol present as a 1:1 complex with dodecylguanidine. All 501 values of the extinction-coefficient spectrum of this 1:1 complex (C, red trace) were also optimized during the fit. See Supporting Information for further details.
First, absorption spectra of the unassociated species were determined. When dissolved by itself in hexane, p-cresol gives a strong UV absorption with a maximum at 278 nm as well as vibronic structure (Figure 3C, black trace). This spectrum shows no sign of concentration dependence below ∼10 mM. Similarly, dilute dodecylguanidine free base in hexane shows no measurable extinction coefficient (<1 M –1 cm–1) anywhere in this 250–350 nm range. However, at dodecylguanidine concentrations above ∼5 mM, significant light scattering occurs (data not shown), indicating self-association as reverse micelles. Unassociated dodecylguanidinium cation was presumed also not to have any significant absorption in hexane, based on the low absorbance (<0.001) measured for ∼10 mM solutions of dodecylguanidinium bromide in other solvents (DMSO, CHCl3; data not shown). However, dodecylguanidine–HBr was not sufficiently soluble in hexane to measure its extinction-coefficient spectrum directly.
Combining ethylguanidine and p-cresol in hexane clearly results in an H-bonded complex. This is evident from the concentration and temperature dependence of the p-cresol absorption band in the presence of an equimolar amount of dodecylguanidine (Figure 3). Exact stoichiometric equivalence is easily obtained by mixing together ∼1 mM hexane solutions of the two components, and then collecting the resulting crystalline precipitate (“dodecylguanidinium p-cresolate”).
This precipitated salt has a 1:1 fixed stoichiometry regardless of relative initial concentrations, as shown by relative 1H NMR peak areas after the salt is collected and redissolved in DMSO. The solid dodecylguanidinium p-cresolate salt has very low solubility in hexane at room temperature, but it can easily be redissolved to a concentration of ∼10–3 M in hexane at temperatures >55 °C. UV absorbance spectra were measured as function of further dilution with hexane (Figure 3A), or further temperature elevation (Figure 3B). These spectra clearly show a broad absorption band centered near 310 nm. This species likely has a structure related to phenolate anion, because pure sodium p-cresolate also gives a broad absorption band near 310 nm in DMSO15 (see also Supporting Information).
It is a priori unlikely that such a low-dielectric solvent as hexane (ε = 1.8) would contain independently solvated dodecylguanidinium and p-cresolate ions. This hypothesis is supported experimentally by the observed dilution-dependent increases in the A280/A310 spectral absorbance ratio in Figure 3A. This is not what is expected, if a simple Brønsted–Lowry acid–base equilibrium prevails, that is, ϕ-OH + :N-guan ⇄ ϕ-O– + HN+-guan. For such an equilibrium, the ratio of [ϕ-OH]/[ϕ-O–] should be unaffected by dilution with additional solvent. (As a control, such Brønsted–Lowry behavior was shown to occur when methanol was used as a solvent for the same equilibrium; see below.)
Instead, we determined that concentration- and temperature-dependent spectral changes in hexane could be better modeled by the equilibrium, ϕ-OH + :N-guan ⇄ ϕ-OH:N-guan, which describes the formation of a Lewis acid–base complex. Note that what is written as a unitary complexation product on the right-hand side probably involves multiple interconverting protomers, such as the ones shown in Scheme 1B,C.
The simplest demonstration of this is a log–log plot of A308 versus dissolved concentration of dodecylguanidinium p-cresolate (Figure 3A inset). This shows a limiting slope of ∼2 at low concentrations, a clear sign that the 310 nm absorption band results from the formation of a binary complex. A higher-order complex would be expected to show a higher-power concentration dependence.
It was possible to determine more precise values of equilibrium concentrations by fitting a wider range of wavelengths simultaneously. At a total concentration near 1 mM and temperatures near 60 °C, the 1:1 mixture of p-cresol and dodecylguanidine shows a broad spectral shape with a variable plateau from 298 to 310 nm (Figure 3A,B). The relative size of this plateau decreases either upon overall dilution with hexane (Figure 3A) or upon heating (Figure 3B). The reversibility of the complexation reaction is demonstrated by the nearly perfect superposition of spectra in Figure 3B obtained during heating (from 58 to 64 °C) and then cooling (from 64 to 58 °C), at intervals of 2 °C There was evidence for ∼1% overall decrease in p-cresol content over the course of the entire heating–cooling cycle, as evidenced by a uniform decrease in the entire absorption spectrum in the final spectrum at 58 °C, as compared with the initial spectrum at the same temperature. This continual slow loss of sample was corrected (to first order) during the subsequent data analysis by averaging the identical-temperature spectra pairwise from the heating–cooling cycle.
By subtracting out differently weighted amounts of the pure p-cresol spectrum in hexane (Figure 3C, black trace), it required only a simple least-squares fitting procedure to compute the shape of the UV spectrum of the single additional component (Figure 3C, red trace) that could best account for all the measured spectra in Figure 3A,B. The fitting procedure utilized singular value decomposition (SVD) to minimize the sum-of-squares in the residual absorbance spectra (thin black traces in Figure 3A,B) that could not be accounted for by linear combinations of just p-cresol and one other species.
This SVD-based fitting procedure gave a unique spectral shape, but not its vertical scale. That is, based solely on the SVD procedure, the fitted spectral bandshape (Figure 3C, red trace) could be rescaled vertically by any arbitrary factor. Choosing the scale for the red trace in Figure 3C required one additional assumption, namely, that the eight different concentrations of the 1:1 complex, deduced from the properly scaled extinction-coefficient spectrum, should give the same equilibrium constant for 1:1 complex formation, Kcomplex, for all eight dilutions measured at 58 °C (seven measurements in Figure 3A, plus one in Figure 3B). In fact, the scale was optimized to minimize the standard deviation among eight resultant values of ΔG°, rather than among the values of Kcomplex itself (see Table 2-S in Supporting Information for details).
The optimization gave a value of ΔG° = −23.5 ± 0.8 kJ mol–1 at 58 °C, corresponding to Kcomplex = 5100 ± 1500 M –1. The reciprocal of this Kcomplex represents the dissociation constant in hexane at 58 °C, Kd = 0.195 ± 0.06 mM. The moderate standard deviation is generally consistent with the initial hypothesis that 1:1 complexation is the main reaction occurring over the 12-fold concentration range used in these experiments, although clearly this is not the whole story.
The best-fit extinction-coefficient spectrum (Figure 3C, red trace) can be most easily interpreted as involving two species in rapid internal proton-transfer equilibrium, corresponding to structures B and C in Scheme 1. These structures give rise, respectively, to “guanidinium/phenolate” absorbance near 310 nm and “guanidine/phenol” absorbance near 280 nm in Figure 3C. However, even the latter subcomponent shows a significantly different vibronic substructure than the isolated p-cresol in hexane (Figure 3C, black trace).
The scaled spectrum (Figure 3C, red trace) was also used to determine the temperature dependence of the concentrations in Figure 3B, and thus of the equilibrium constant. A linear least-squares fit of the resulting van’t Hoff plot (Figure 3B, inset) gave a value of ΔH° = −74 ± 4 kJ mol–1 for the formation of the 1:1 complex. (The stated uncertainty here is simply R times the standard error of the slope, obtained from the linear regression of ln K vs 1/T. Concentration errors from the SVD fit contribute to errors in the four individual values for Kcomplex, but these end up being insignificant compared with 4 kJ mol–1, as can be judged by smaller size of the error in ΔG°.) Combining this value of ΔH° with the value of ΔG° at 58 °C, we obtain the value of ΔS° = −152 ± 12 J mol–1 K–1 at 58 °C. We can further extrapolate these results to physiological temperature (37 °C), obtaining a value of ΔG° = −26.7 ± 0.8 kJ mol–1 and a corresponding Kd = 0.03 ± 0.01 mM for the formation of a heterodimer complex in hexane.
UV Measurements in More Polar Solvents
DMSO offers distinct advantages for studying the interactions between phenol and guanidine groups. The most important is significantly greater solubility, for example, for the covalently linked free bases (p-phenolyl)-dodecylguanidine and (p-phenolyl)ethylguanidine and their HBr salts, as well as for potassium p-cresolate. None of these compounds dissolved sufficiently in alkanes to permit measurement of an interpretable UV spectrum. Furthermore, dodecylguanidine itself could be dissolved to a much higher concentration in DMSO (∼50 mM), without producing strongly scattering micelles as was consistently observed above ∼2 mM in hexane. Similar high concentrations could also be obtained in the protic solvent MeOH, which served as an important control.
UV spectral measurements in DMSO (Figure 4A–D) also provide evidence for Lewis acid–base complexation rather than Brønsted–Lowry H+ transfer. The 1:1 dodecylguanidinium p-cresolate salt, obtained as above, when redissolved at 0.1 and 1 mM in DMSO (Figure 4B, black and red traces) gave extinction-coefficient spectra superimposable on those of pure p-cresol (Figure 4A, black and red traces). Thus, in DMSO, the presence of <1 mM concentrations of the very strong base, dodecylguanidine, was insufficient to deprotonate a similar concentration of p-cresol, a weak acid, contrary to expectation if Brønsted–Lowry acid–base equilibrium predominated.
Figure 4.

UV spectral detection of phenol–guanidine interactions, measured using DMSO (A–D) or MeOH (E–H) as solvent. Each column represents spectra obtained on the solute molecule shown above (or two solutes, in the case of the second column). Spectra are presented as extinction-coefficient data, to correct for varying path lengths and concentrations used during measurements, and are color-coded according to solute concentration(s). Every panel (A–H) includes one spectrum of the indicated phenol-containing compound at 0.1 mM (black), and one at 1.0 mM (red); in (B) and (F), those red and black traces also include 0.1 or 1.0 mM dodecylguanidine free base, respectively. (B) and (F) each also include three additional traces in green, blue, and purple, taken respectively with 5, 10, and 50 mM total concentration of dodecylguanidine free base; all of these still have only 1.0 mM p-cresol. All spectra were measured at 25 °C with 1 cm pathlength sample and reference cuvettes on a Shimadzu UV-265 spectrometer. Absolute vertical scales of the spectra are accurate only to ±20% due to weighing and dilution imprecision and were adjusted within this range to facilitate comparisons.
We did not obtain measurements of the 1:1 salt in DMSO at concentrations above 1 mM, due to too high p-cresol absorption for the 1 cm pathlength cuvette we were using. However, when additional crystalline dodecylguanidine free base was added to the 1 mM salt, a new absorption maximum could be seen at ∼318 nm (Figure 4B). This increased in intensity as the total dodecylguanidine concentration was raised successively to 5, 10, and 50 mM (Figure 4B, green, blue, and violet traces, respectively). This 318 nm peak is consistent with Lewis acid–base complex formation, analogous to that seen in hexane (Figure 3A). However, the appearance of a new band at 272 nm at the highest (50 mM) dodecylguanidine concentration (Figure 4B) makes clear that, compared with hexane, it is less reasonable in DMSO to analyze the equilibrium based on the assumption of only two species (isolated p-cresol and its 1:1 complex with dodecylguanidine). From the spectra in Figure 4B, we can only estimate an apparent Kd for the p-cresol/dodecylguanidine complex of 10–50 mM in DMSO at 25 °C. This is the dodecylguanidine concentration for which 50% of the maximum possible 318 nm cresolate absorbance is achieved. This apparent Kd is at least ∼100-fold greater than in hexane (extrapolated to a similar temperature), indicating a weaker H-bonding interaction in DMSO.
Complexation in DMSO clearly occurs at a lower concentration for covalently linked (p-phenolyl)alkylguanidines (Figure 4C,D) than is required for p-cresol/dodecylguanidine (Figure 4B). That is, the spectra of (p-phenolyl)ethylguanidine and (p-phenolyl)dodecylguanidine in DMSO (Figure 4C,D) show clear evidence of Lewis acid–base complexation, as evidenced by the appearance of a 318 nm band even at concentrations of ∼1 mM but not at 0.1 mM. This complexation is probably intermolecular rather than intramolecular, given the absence of the 318 nm peak at the lower (0.1 mM) concentration, and its similar size at 1 mM, for both alkyl chain lengths. The short ethyl linker clearly cannot permit the formation of an intramolecular H-bond. The dodecyl linker also does not appear to favor intramolecular H-bonding in DMSO solvent, despite computations (Figure 2B), indicating that a conformational rearrangement to permit intramolecular H-bond formation is energetically favored in vacuo. Empirically, the extended chain form appears to allow much more favorable external interactions in condensed phases, that is, both in solid state (as shown in crystal structures below) and for either dimers or monomers solvated in DMSO, as suggested by Figure 4C,D. There is a barely perceptible 318 nm absorbance above the baseline for 0.1 M (p-phenolyl)dodecylguanidine in DMSO (Figure 4D, black trace), which is not present for the ethyl-linked compound (Figure 4C, black trace). This could be a sign of a small amount of intramolecular H-bonding in the former. However, we cannot rule out that this could simply be due to a minor difference in light scattering.
Free-energy and enthalpy changes for (p-phenolyl)ethylguanidine dimerization in DMSO were determined by using concentration- and temperature-dependent UV spectral analyses, as described above for the p-cresol/dodecylguanidine system in hexane. Due to considerably higher solubility, the spectra in DMSO could be measured over a wider temperature range (19–45 °C), as well as a wider concentration range (0.1–10 mM; Figure 4C and Supporting Information). The thermodynamic values obtained for (p-phenolyl)ethylguanidine dimerization in DMSO are ΔG°298 = −8.4 ± 0.6 kJ mol–1 and ΔH° = −22 ± 3 kJ mol–1. Because there are two H-bonding interactions per dimer, the enthalpy change for forming each phenol/guanidine H-bond is only −11 ± 2 kJ mol–1, that is, barely one-sixth of the value in hexane.
In contrast to the complexation behavior in hexane and DMSO, the same molecules in MeOH simply undergo Brønsted–Lowry acid–base equilibria. This is seen in Figure 4F, where sufficiently high relative concentrations of the base (dodecylguanidine) fully deprotonate the acid (p-cresol), as evidenced by the complete replacement of the 280 nm absorption band by the 296 nm band characteristic of cresolate in MeOH. That is, the purple trace in Figure 4F, corresponding to 1 mM p-cresol + 50 mM dodecylguanidine, matches almost perfectly the spectrum of 1 mM potassium p-cresolate in MeOH (data not shown). There is a fairly clean isosbestic point in Figure 4F, indicating that only two species (solvated p-cresol and solvated p-cresolate) contribute to the spectra in this panel.
Likewise, the lack of concentration dependence in the spectra in Figure 4G,H is indicative of the presence of buffering concentrations of conjugate acid and base forms of both groups within a single amphiprotic solute. The concentration-independent acid–base equilibrium of these (p-phenolyl)alkylguandine compounds is analogous to other amphiprotic zwitterions, such as amino acids. When present as the only solute in water or other protic solvents, such zwitterions buffer the pH at the midpoint between the two pKa values and show almost no concentration dependence for the extent of H+ transfer.
The presence of both phenol and phenolate forms in Figure 4F–H indicates that in methanol, the pKa value of the phenol group in this solvent must be quite similar to the pKa of the guanidinium. In the limit of phenol and guanidinium groups sitting on separate molecules in MeOH, ΔpKa ≈1.6. This is seen in Figure 4F, where 50% conversion of p-cresol to p-cresolate (corresponding roughly to the green trace), requires approximately a 20-fold excess of total guanidine (10 mM) over total p-cresol (0.5 mM). Thus, surprisingly, in MeOH, which is often thought of as only a little less polar than water, the relative acidity of these two groups is already swapped, with guanidinium clearly more acidic than phenol.
However, as the two groups are held progressively closer, for example, by a dodecyl or an ethyl linker, the phenol becomes progressively more acidic, eventually becoming about as acidic as guanidine, as seen by the nearly 50% contribution of phenolate to the spectrum (compare 296 nm peaks for black traces in Figure 4F–H). That is, the zwitterionic form is favored by progressively closer distances between the charges, a clear sign of electrostatic perturbation of the pKa values. This is analogous to the well-known phenomenon in amino acids, where the pKa values of amino and carboxylic acid groups are pushed farther apart, favoring the zwitterionic form, as a result of these groups’ proximity and mutual electrostatic stabilization.
IR Spectroscopy
Direct IR evidence for 1:1 complexation was not as strong for IR as for UV spectra, because measurement of the concentration dependence of the IR solution spectra was not possible. In nonpolar solvents (hexane, CCl4), the maximum room-temperature solubility of (p-phenolyl)alkylguanidine compounds was too low (<0.5 mM) to measure spectra accurately. In DMSO, the background solvent absorbances were too high to permit precise quantitative measurements even at saturating concentrations (∼150 mM). Nevertheless, IR spectra are consistent with the presence of H-bonded complexes of phenol with guanidine free bases. Such spectra show clearly that the H-bond between these groups is polarizable, that is, the equilibrium position of the proton shifts moving increasingly toward the guanidine group as the polarity of the environment increases from CCl4 to DMSO to MeOH to tightly packed crystals.
Nonpolar Solvents
Due to its low solubility, we failed to obtain adequate-quality IR spectra of p-cresol-dodecylguanidine 1:1 in hexane, which would be directly analogous to the UV spectra in Figure 3. The closest solvent we could use is CCl4 (Figure 5). This spectrum supports formation of a binary H-bonded complex, as indicated by characteristic bands due to both p-cresol (1514 and 1255 cm–1) and p-cresolate (1498 and 1272 cm–1).16,18 The characteristic C–N stretch region of the guanidino group, between 1500 and 1700 cm–1, also shows features characteristic of both deprotonated guanidine (1629 and 1555 cm–1) and guanindinium ion (1665 cm–1). A key feature observed is the strong broad absorption band between 3500 and 2500 cm–1, a “continuum” absorbance characteristic of polarizable H-bonded systems.
Figure 5.

IR absorbance spectrum of p-cresol/dodecylguanidine 1:1 dissolved in CCl4. The strong continuum absorbance from 3500 to 2500 cm–1 is a clear evidence of the formation of a strongly H-bonded complex, rather than a simple Brønsted–Lowry proton transfer equilibrium. The exact position and intensity of the most characteristic peak of the deprotonated guanidine group (1555 cm–1) are difficult to quantify due to interference by a strong solvent absorption band at this frequency. Spectral region near 2350 cm–1 is blanked due to interference by CO2 absorption.
DMSO and MeOH Solutions
It was possible to obtain spectra of (p-phenolyl)dodecylguanidine in DMSO (Figure 6A) and in MeOH (Figure 6C), but only at ∼100-fold higher concentrations than for the corresponding UV spectra (Figure 4D,H). The strongest characteristic IR bands of the protonated forms of both guanidinium (∼1670 cm–1 shoulder) and phenol (1514 cm–1) are present in DMSO (Figure 6A). The additional bands at 1642 and (especially) 1548 cm–1 are characteristic of deprotonated guanidine in either DMSO12,17 or chloroform.19 Meanwhile, characterisitic phenolate bands are observed at 1599 and 1494 cm–1 (Figure 6A), that is, shifted somewhat relative to this compound in MeOH (Figure 6B, 1602 and 1501 cm–1). The latter wavenumber values are identical with those seen for high-pH aqueous phenolate18 and tyrosine.16
Figure 6.
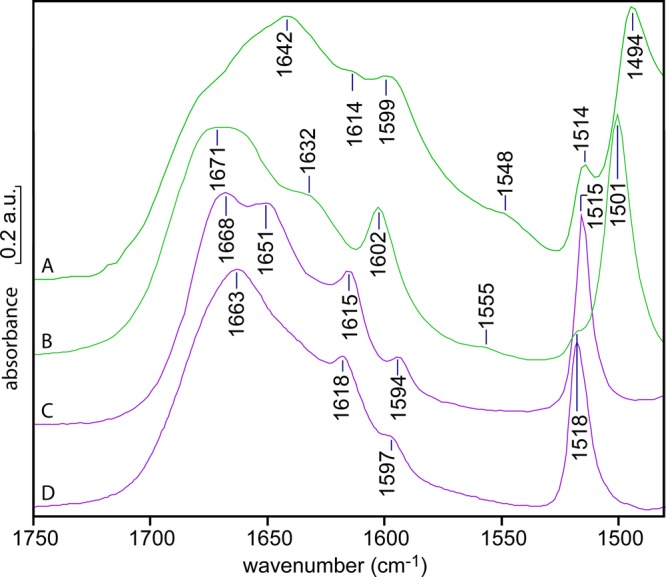
IR spectra of (p-phenolyl)dodecylguanidine, as the “free base” (green; A, B) or as the HBr salt (purple; C, D), with the former two showing clear evidence of H-bonding and/or proton transfer from the phenol. Each sample was recrystallized from methanol, dried, and then redissolved to a concentration of ∼100 mM in DMSO (A, C) or methanol (B, D). The absorbance scale on the y axis is approximate; the measured spectra had somewhat different concentrations and path lengths and were rescaled for optimum visual comparison.
Thus, these IR spectra confirm that in DMSO, this free base compound exists in an equilibrium between a guanidinium/phenolate zwitterion (Scheme 1B) and a fully neutral guanidine/phenol form (Scheme 1C). The relative sizes of the bands suggest that the zwitterion accounts for some 50–75% of the total concentration, more than seen in the UV absorption spectra in Figure 4D. This can be explained by the presence of a higher proportion of the H-bonded complex, as a result of a ∼100-fold higher overall concentration in the IR samples.
An even greater contribution from the zwitterionic form is observed in MeOH (Figure 6B) (e.g., larger characteristic guanidinium peak near 1670 cm–1 in Figure 6B compared with an unlabeled shoulder in Figure 6A). This vibration is characteristic of guanidinium; it is clearly seen in the spectra of the corresponding HBr salt in either DMSO or MeOH (Figure 6C,D). Likewise, in MeOH, phenolate peaks are stronger (1602 and 1501 cm–1 in Figure 6B). Similar features were observed in the spectrum of (p-phenolyl)ethylguanidine free base dissolved in methanol (data not shown). These spectra also indicate that the zwitterion contribution to the spectrum is increased significantly by the use of >100-fold higher concentration for the IR spectrum (Figure 6B), as compared with the <1 mM concentrations used to measure UV spectra (Figure 4H). This might contradict somewhat the hypothesis of simple Brønsted–Lowry acid–base equilibrium. Alternatively, it could simply result from favorable long-range electrostatic (dipole–dipole) interactions coming into play at the highest concentrations.
Very different IR results are obtained with the salt (p-phenolyl) dodecylguanidinium bromide, which corresponds to the structure in Scheme 1A, with X– = Br–. This HBr salt has a much higher solubility in most solvents compared with the free-base compound, especially in DMSO. This makes it easier to measure accurately over a wider spectral range (Figure 7). In both DMSO (Figure 6C; and Figure 7, purple trace) and MeOH (Figure 6D), this salt contains a solvated fully protonated guanidinium, with no evidence of interaction with the phenolic group. The monoalkylguanidinium C–N stretch bands at 1668 and 1651 cm–1 in DMSO (Figure 6C; Figure 7, purple trace) are largely unchanged from their appearance in dodecylguanidinium bromide in the same solvent (Figure 7, red trace).5,17
Figure 7.
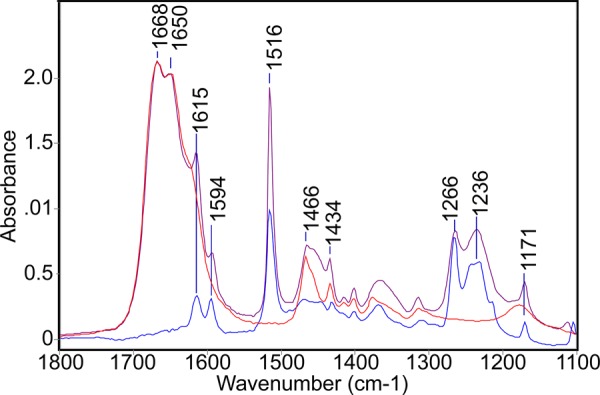
IR evidence that the phenol and guanidinium groups in (p-phenolyl)dodecylguanidinium bromide are independently solvated by DMSO, that is, there is no significant interaction between them. The IR spectrum of this salt (purple) resembles a simple sum of absorbances due to separate samples of dodecylguanidinium bromide (red) and p-cresol (blue). All spectra were obtained in DMSO.
Likewise, characteristic phenol peaks are observed near 1615, 1594, 1515, 1266, and 1236 cm–1 in (p-phenolyl)dodecylguanidinium bromide (Figure 6C,D; Figure 7, purple trace), virtually unchanged from p-cresol (Figure 7, blue trace). In fact, these characteristic phenol peaks are generally very insensitive to the environment. This is shown in greater detail in Figure 8. The C–O stretch vibration is consistently observed as a strong IR absorbance band for p-alkylated phenols in a fairly narrow range between 1245 and 1275 cm–1. The lowest frequencies in this range have been reported for poly-tyrosine in dried films or aqueous solvents.18 In Figures 7 and 8A–C, which are limited to crystalline samples and solution measurements in methanol and DMSO, the variation is within a narrower range of 1265–1268 cm–1.
Figure 8.

Phenolic C–O stretch region (1250–1310 cm–1) is less sensitive to solvent environment when the phenolic group is protonated (A–C) than when it is unprotonated (D–G). (A) p-Cresol in DMSO; (B) (p-phenolyl)dodecylguanidinium bromide in DMSO, continuation of spectrum in Figure 6C; (C) (p-phenolyl)ethylguanidinium bromide in MeOH, continuation of Figure 6D; (D) (p-phenolyl)ethylguanidine free base in methanol, continuation of Figure 6B; (E) (p-phenolyl)dodecylguanidine free base in DMSO, continuation of Figure 6A; (F) p-cresol and dodecylguanidine free base (1:1) in CCl4, expanded region from Figure 5; (G) potassium p-cresolate in DMSO.
In contrast, deprotonation of the phenolic group leads to a much larger range of C–O stretch frequencies, as seen in Figure 8D–G. The lowest observed value, 1264 cm–1, is for (p-phenolyl)ethylguanidine free base in MeOH (Figure 8D). This value overlaps with the range seen for protonated phenol (Figure 8A–C), yet it was measured under conditions where the phenolate form predominates, as shown by the C=C ring modes (1602, 1501 cm–1 in Figure 6B). At the same time, the structurally similar compound, (p-phenolyl)dodecylguanidine in DMSO, appears to show a 40 cm–1 higher frequency for its phenolate C–O stretch (1309 cm–1, Figure 8E).
These big differences in C–O stretch frequency for phenolate in various environments are likely attributable to a strong dependence of the C–O bond order on solvation and H-bond strength. These also produce changes in the degree of delocalization of negative charge into the ring, weakly altering other characteristic vibrational frequencies. For example, the phenolate C=C ring stretching vibrations are lower in frequency for the H-bonded complex of (p-phenolyl)dodecylguanidine in DMSO (1599 and 1494 cm–1, Figure 6A) than in MeOH (1602 and 1501 cm–1, Figure 6B). The corresponding frequencies for tyrosinate in water at a high pH16,18 are intermediate, that is, 1600 and 1498 cm–1, suggesting that the H-bonding interactions of phenolate with water are stronger than with MeOH, but that neither type of solvent produces an H-bonding perturbation on phenol that is as strong overall as a single H-bond to guanidine in an aprotic solvent such as DMSO. The protonated phenol group again serves as a useful control; its solvent interactions are weaker than those of phenolate, that is, only small solvent-dependent effects are observed for the phenol ring vibrations (near 1615, 1595, and 1515 cm–1 in Figures 6A,C,D and 7).
Solid-State IR Spectra
An important question is, what do Fourier transform infrared (FTIR) spectra indicate for the protomeric form(s) present in the crystalline state, specifically in crystals that are suitable for determining high-resolution X-ray structures? In such crystals, the polar ends of neighboring dimers are packed close to each other, and also within the H-bonding distance of structural MeOH molecules (see below). Thus, the environment around the guanidine–phenol H-bond is quite polar.
Indeed, in the IR spectrum of these crystals (green trace in Figure 9), only the phenolate peak at 1498 cm–1 is observed; the peak at 1516 cm–1, characteristic of protonated (neutral) phenol, is completely missing. Additionally, in the crystalline forms of both the free base (Figure 9, green trace) and the corresponding HBr salt (Figure 9, purple trace), the strongest C–N stretch bands are very similar in frequency, near 1655 cm–1. This is a lower wavenumber than is typically seen for monoalkylguanidinium salts in most solvents (cf. Figures 6 and 7). Nonetheless, it agrees with the measurements in the crystalline state of the highest-frequency C–N stretch vibration for dodecylguanidinium bromide, anisolyl dodecylguanidinium bromide, phenolyl ethylguanidine free base, and phenolyl ethylguanidinium bromide, which are consistently observed between 1659 and 1669 cm–1 (Figure 6-S). In all these crystals, therefore, the free base exists mostly as the zwitterionic form, with a guanidinium group (Scheme 1B). In contrast, the highest C–N stretch frequencies for the crystalline states of authentic monoalkylguanidine free bases, such as dodecylguanidine or anisolyldodecylguanidine, are 1637 and 1636 cm–1, respectively (see spectra C and E in Figure 6-S).
Figure 9.

Broad IR continuum absorbance 3500–2500 cm–1 in FTIR spectrum of a crystalline sample of p-phenol-dodecylguanidine free base (green) is the evidence that it is more strongly H-bonded than the corresponding crystalline p-phenol-dodecylguanidinium bromide salt (purple). Both samples were measured in KBr disks. Blanked regions had errors due to background correction, for example, CO2 absorbance near 2350 cm–1.
Nevertheless, the IR spectrum of the p-phenolyl-dodecylguanidine free base (Figure 9, green trace) may be consistent with the presence of small amounts of the strongly H-bonded but fully neutral structure (Scheme 1C). The new peaks, which are observed only in the free base crystals (Figure 8, green trace), and not the HBr salt (Figure 9, purple trace), include a characteristic deprotonated guanidine frequency (1554 cm–1), as well as a broad continuum absorption band from 2500 to 3500 cm–1. Both of these features can also be discerned in the crystalline samples of either (p-phenolyl)ethylguanidine, or (less prominently) dodecylguanidinium p-cresolate salt (see Supporting Information). Because of the absence of characteristic vibrational bands of MeOH, at most, a small portion of the continuum bands (above ∼3200 cm–1) can be attributed to O–H stretch vibrations from the cocrystallized MeOH in the phenolyl alkylguanidine free bases (see crystal structures below). Instead, these broad absorption bands are likely indicative of proton polarizability,20 which is associated with a strong H-bonding.21,22
1H and 13C NMR Measurements of the Phenol–Guanidine Interaction
Clear support for the formation of an H-bonding interaction between phenol and guanidine groups was also observed in the solution-state 1H and 13C NMR spectra. Figure 10 shows that at a low concentration of (p-phenolyl)ethylguanidine, the chemical shifts of both the guanidine group and the phenol group are much the same as when measured individually in dodecylguanidine or p-cresol, respectively. However, as the concentration is increased above ∼30 mM, there is a significant change, especially in the value for the guanidine protons.
Figure 10.

Concentration dependence of the 1H chemical shifts of the phenolic ring (gray solid circle, orange solid circle) and of the guanidine group (blue solid circle), in (p-phenolyl)ethylguanidine. Average values are shown for peaks split by j–j coupling. Likewise, for the monoalkylguanidinium bromide salts, there were two to three partially resolved resonances for the guanidino group protons in the range of 6.9–7.5; these were averaged with weightings of 1:2:2 to give the plotted value of 7.25 ppm (blue line next to the right axis). Well below ∼30 mM, the chemical shifts in (p-phenolyl)ethylguanidine are nearly identical with those seen for isolated phenol and guanidine groups, indicated by lines next to the left vertical axis. The changing values over the 20–150 mM concentration range are consistent with the formation of an H-bonded dimer complex. That is, at the highest concentrations, the averaged chemical shift of the guanidine protons approaches that of dodecylguanidinium cation, whereas the phenolic ring protons move somewhat toward the values for phenolate. The latter are represented by the chemical shifts of Br– or K+ salt solutions in DMSO, respectively, as indicated next to the right axis.
Figure 11 shows additional 13C NMR results in DMSO solution, which also demonstrate an H-bonding interaction in (p-phenolyl)ethylguanidine dimers at elevated concentrations. The chemical shift of the phenol ring carbons is clearly dependent on concentration. The dependence is strongest for the ipso carbon, which shifts from 157.0 ppm at 3 mM (Figure 11A) to 158.7 ppm at 200 mM (Figure 11C), and for the para carbon (128.1–126.3 ppm; same spectra).
Figure 11.

13C NMR spectra (112–172 ppm portion) of the indicated samples, calibrated to the chemical shift value for the solvent (d6-DMSO, 39.5 ppm). (A)–(G) were measured with a 600 MHz Bruker NMR and (H)–(K), with a 300 MHz instrument. The chemical shift values labeled in blue are assigned to the guanidine carbon; in black, to the six phenolic ring carbons. The two largest phenolic peaks always correspond to ortho and meta positions, each representing 2 equiv carbons. Horizontally expanded and horizontally shifted insets for (D), (F), and (G) aid in visualizing spectral details near 157 ppm. Spectrum (F) was measured at 2× finer resolution than the others, the better to resolve the nearly overlapping phenolic ipso and guanidine resonances at 157.2 ppm.
Properly assigning the phenolic ipso carbons within their overlapping ranges with the guanidine carbon required careful measurements of a number of control spectra (Figure 11D–K). To minimize perturbations on the guanidine group from H2O impurities, which could lead to formation of guanidinium hydroxide rather than the free base, these were measured on samples that had been recrystallized from acetonitrile, protected from moisture during storage, weighed quickly, then redissolved at 100–200 mM by adding each directly to a freshly opened glass ampule of d6-DMSO. Comparisons between monoalkylguanidine free bases lacking other H-bond donor groups (Figure 11F,J), and their corresponding HBr salts (Figure 11G,K) show firstly that protonation of guanidine produces only a ∼0.5 ppm change in its 13C chemical shift. These control measurements made clear that regardless of the protonation state, the 13C chemical shifts for monoalkylguanidine compounds in DMSO can safely be assigned within the narrow range of 156.7–157.3 ppm.
The lowest concentration of (p-phenolyl)ethylguanidine (Figure 11A) showed closely spaced 13C resonances at 157.0 and 157.2 ppm. These could be definitively assigned based on a comparison with the similar compound (p-phenolyl)dodecylguanidine (Figure 11D). The latter was measured as a 50/50 mix of the natural-abundance compound with an otherwise-identical compound that had been prepared with both terminal nitrogens labeled with >97% 15N. Such labeling produced a triplet pattern due to j–j coupling between 13C and two attached 15N nuclei, which could easily be seen for the peak at 157.2 (Figure 11D inset). The fourth component peak at 157.2 ppm, which is the largest and nearest to the center of the triplet pattern, is due to the ∼50% of sample with 14N on all three of the guanidine nitrogens. The coupling pattern in this sample allows the 157.2 peak in Figure 11D to be assigned to the guanidine group, and by analogy also in Figure 11A. The minor differences in phenolic ipso-carbon chemical shifts between Figure 11A,D might be due to the difference in the H-binding geometries, as suggested by different X-ray crystal structures (see below).
As with the UV spectra in Figure 4D and the IR spectra in Figure 6A above, the concentration-dependent NMR results in both Figures 10 and 11 are consistent with the formation of an H-bonded Lewis acid–base dimer of (p-phenolyl)ethylguanidine, rather than the formation of solvated zwitterion monomers with complete H+ transfer. In particular, even at the highest concentrations, the 1H and 13C chemical shift values for the phenol group never come close to those seen for potassium p-cresolate. The concentration range over which the chemical shifts are seen to change is approximately consistent with the dissociation constant (34 mM) computed from the concentration dependence of the UV spectra of (p-phenolyl)ethylguanidine (see ΔG° value given above; and raw data in Supporting Information). Thus, the NMR results confirm that in DMSO, phenol and guanidine groups readily form strong H-bonded Lewis acid–base complexes, with partial H+ transfer, that is, a hybrid of the structures shown in Scheme 1B,C.
At the highest concentration for (p-phenolyl)ethylguanidine in Figure 10 (0.05 M), the average chemical shift of the guanidine protons (5.9) had risen just over 50% of the way from the value for dodecylguanidine free base (4.3) to the value for protonated dodecylguanidinium bromide (7.3). Overall, there is less sensitivity of the guanidine 13C NMR signal to its protonation state. Nevertheless, for (p-phenolyl)ethylguanidine at 0.05 M (Figure 11C), it is also shifted to 157.0, that is, about halfway from the ∼157.3 ppm value for deprotonated guanidine (Figure 11F,J) to the ∼156.7 ppm value seen for protonated guanidinium salts (Figure 11E,G,K). This change is consistent with the predicted dimer fraction of 55% (of the total mass) at 0.05 M (p-phenolyl)ethylguanidine, based on Kd = 34 mM. Together, these chemical shifts suggest that the guanidinium group in the dimer complex behaves as if it is fully protonated, that is, the proton transfer from phenol to guanidine is nearly complete in the dimer in DMSO.
However, at the same (p-phenolyl)ethylguanidine concentration, the phenolic ring protons moved only ∼20% of the way from their values for p-cresol to p-cresolate (Figure 10). The phenolic ipso carbon 13C shift in Figure 11C, at 158.7 ppm, is also only 25% of the way from its value in p-cresol (155.2 ppm, Figure 11G) to the value in p-cresolate (Figure 11H). Likewise, the 13C shifts for the other ring carbons in the (p-phenolyl)ethylguanidine dimer (Figure 11C) are very similar to those of p-cresol (Figure 11I), and quite different from those of the potassium salt of p-cresolate (Figure 11H). Based purely on an assumption of time-averaging of chemical shifts, this would suggest that the phenol group is less than 50% deprotonated in the dimer complex, a conclusion that would be somewhat at odds with the results from optical spectroscopy (both UV and IR).
In summary, the NMR chemical shifts in Figure 11A make the guanidine group look about 50% protonated, whereas the phenolic group looks nearly ∼75% protonated. That is, by NMR both groups participating in the H-bond look predominantly protonated, despite the lack of HBr or other possible external proton donor. This is not due to contamination of the NMR solvent (DMSO-d6) with water (or another acid), which could indeed cause both groups of the free base to appear protonated because of the formation of guanidinium hydroxide (or another salt). Contrary to what would be expected in such hypothetical cases, simple dilution with the same DMSO-d6 causes a breakup of the H-bonded complex and results in the guanidine group becoming less protonated, that is, both its 1H and 13C signals move closer to the values seen in guanidine free bases without strong H-bond donor groups.
15N NMR
We were unable to observe 15N chemical shifts of the terminal guanidine nitrogens for the phenolyl-dodecylguanidine free base in DMSO, despite the use of 50% 15N isotopic enrichment. This was true over a range of temperatures from 25 to 50 °C, even after sufficient signal-averaging that we could observe the 15N resonance from the natural-isotope-abundance intrachain nitrogen at 63 ppm (see Figure 11-S). However, we were able to obtain high-quality solid-state cross-polarized magic-angle-spinning (CP-MAS) signals from crystalline samples of this compound, as well as a number of control samples (Figure 12).
Figure 12.

15N solid-state CP-MAS NMR spectrum of the free bases of (p-anisolyl)-dodecylguanidine-[15N2] (A); (p-phenolyl)dodecylguanidine-[15N2] (B); (p-phenolyl)ethylguanidine-[15N2] (C); and the corresponding HBr salts (D-F, respectively). The x axis scale represents chemical shift, in ppm, relative to solid ammonium-[15N] sulfate.
The first control spectrum is noteworthy. Crystalline (p-anisolyl)-dodecylguanidine-[15N2] free base (Figure 12A) shows widely separated resonances for imine (110 ppm) and amine (52 and 46 ppm) nitrogens. A similar separation was also seen for the dodecylguanidine-[15N2] free base.12 However, the dodecylguanidine spectrum showed only a single amine resonance at 49 ppm; addition of the anisolyl group (Figure 12A) splits this into two peaks. Only the two terminal nitrogens in the (p-anisolyl)-dodecylguanidine were 15N-labeled, so it is not immediately obvious why these crystals should show three distinct resonances of similar intensity (Figure 12A). The likeliest explanation is the possible presence of crystalline polymorphs. Another explanation is that in the crystal unit cell, there could be two nonequivalent molecules with different environments for their amino nitrogen, as was seen for a number of crystalline arginine salts.25 A full explanation of the amine resonance splitting in Figure 12A is not possible in the absence of a crystal structure, which we have not yet been able to obtain for (p-anisolyl)-dodecylguanidine. Nevertheless, it is clear that the anisole group, which lacks an H-bond donor, produces only a minor perturbation on the 15N spectrum of the deprotonated dodecylguanidine.12
A very different situation prevails when a phenol group is present instead of anisole. The free bases of crystalline (p-phenolyl)dodecylguanidine-[15N2] (Figure 12B) and (p-phenolyl)-ethylguanidine-[15N2] (Figure 12C) each show only two distinct resonances, at ∼50 and ∼45 ppm. The 5 ppm chemical shift splittings likely correspond to the distinct H-bonding patterns for the two terminal nitrogens, as seen in the corresponding crystal structures (see below). The distinct 15N chemical shift ranges confirm the distinct chemical identities of the free bases and their HBr salts.
Among the three HBr salts (Figure 12D–F), there are varying degrees of separation for the two terminal −NH2 resonances. However, none show any sign of an imino-type nitrogen with a strongly downfield-shifted 15N resonance near 110 ppm. That is, the guanidine groups in these compounds behave almost fully protonated, as their 15N NMR spectra are generally similar to those of the corresponding HBr salts (Figure 12E,F). In (p-phenolyl)ethylguanidinium-[15N2] bromide (Figure 12F), the 6 ppm separation likely corresponds to the different H-bonding environment of the two −NH2 groups, which is clearly visible in the crystal structure (see below). Somewhat surprisingly, in the corresponding dodecyl-linked compounds (Figure 12E), the resonances are superimposed (Figure 12E). This indicates that H-bonding environments of the two terminal nitrogens are very similar. In the corresponding (p-anisolyl)dodecylguanidine-[15N2] compound (Figure 12D), the asymmetry in the environment of the two terminal nitrogens appears to be highest among the three HBr salts. Without the crystal structures for either of the dodecyl-linked HBr salts (Figure 12D,F), we cannot provide any detailed rationalization for these results.
All of the HBr salts (Figure 12D–F) give resonances near 54 ppm, that is, shifted a bit downfield from the amino resonances of the corresponding free bases (Figure 12A–C). However, the chemical shift of all the −NH2 resonances in Figure 12B–F, as well as the splittings between resonances within any one molecule, is well within the range of those seen previously seen among various arginine salts.25 We thus conclude that these five spectra represent fully protonated guanidinium groups. Only Figure 12A, as well as the similar result obtained for dodecylguanidine,12 represent good models for the 15N spectrum of a fully deprotonated monoalkylguanidine.
X-ray Crystallography
High-resolution crystal structures were obtained from (p-phenolyl)-dodecylguanidine free base (Figure 13A), (p-phenolyl)ethylguanidine free base (Figure 13B) and (p-phenolyl)guanidinium bromide (Figure 13C). These three compounds all crystallize from MeOH as head-to-tail dimers in the unit cell. Both free bases (Figure 13A,B) actually crystallize as guanidinium + phenolate zwitterions. In these, but not the HBr salt (Figure 13C), multiple solvent (MeOH) molecules per unit cell are clearly included, clustered about the phenol–guanidine interaction site. As a result of the presence of protic solvent, these zwitterion crystals have significantly weaker individual H-bonds between guanidinium and phenolate than are present in for dimer complexes formed in aprotic solvents (see Discussion). Thus, the X-ray crystal geometries are merely suggestive of the solution complexes.
Figure 13.

Crystal structures of compounds containing both phenol and guanidine groups, showing H-bond network and distances. (A) (p-phenolyl)dodecylguanidine free base. Formula: C21 H41N3O3. Space group: P – 1. Cell length (Å): a = 6.7626, b = 7.9144, c = 21.892. Cell angles: α = 94.407°, β = 98.175°, γ = 106.478°. R-factor (%): 4.27. (B) (p-Phenolyl)ethylguanidine free base. Formula: C10 H17N3O2. Space group: P21/c. Cell length (Å): a = 10.1822 (5), b = 10.0597 (5), c = 11.9084 (6). Cell angles: α = 90.00°, β = 109.8880° (10), γ = 90.00°. R-factor (%): 3.03. (C) (p-Phenolyl)ethylguanidinium bromide. Formula: C9H14BrN3O2. Space group: P21/c. Cell length (Å): a = 11.1931 (7), b = 10.0659 (6), c = 9.8327 (6). Cell angles: α = 90.00, β = 101.508(2), γ = 90.00. R-factor (%): 1.95.
These structures are even imperfect representations of the free-base samples that we measured using the IR spectra in KBr pellets and solid-state NMR, which did not include MeOH. That is, when crystals prepared for X-ray measurements were directly redissolved in DMSO, spectral signals from the cocrystallized MeOH could usually be seen (see, e.g., the 13C NMR signal from MeOH in Figure 10-S-E). However, even when the X-ray crystals were thoroughly ground with KBr to make pellets for IR measurements, no IR absorption bands from MeOH were ever detected. It appears the MeOH was lost during KBr grinding. Because stoichiometric MeOH could not be included in the IR samples, acetonitrile was used as the recrystallization solvent for those samples; and this choice was extended to solid-state NMR samples as well.
Nevertheless, these crystal structures (Figure 13) support the conclusion from solid-state 15N NMR (see above) that protons in the crystalline states of the (p-phenolyl)alkylguanidine free base compounds are essentially fully transferred from the phenol to form guanidinium. In all of them, the three C–N bonds of the guanidinium group are of equal length, lying in a narrow range of 1.33–1.34 Å. However, the free base crystal structures (Figure 13A,B) still show clear signs of stronger H-bonding than is present in the HBr salt (Figure 13C).
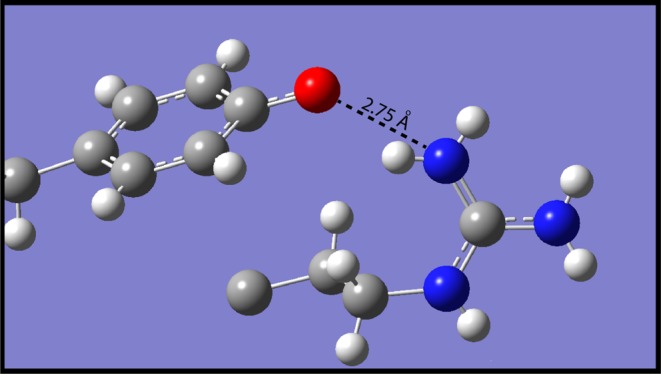
In (p-phenolyl)dodecylguanidine (Figure 13A), the two terminal guanidinium nitrogens form H-bonds with three different O atoms. The shortest, with an N–O distance of 2.75 Å, is with the phenolate of the partner molecule in the head-to-tail dimer. The others are to 2 distinct cocrystallized MeOH molecules. One MeOH accepts H-bonds from both terminal nitrogens, of very similar lengths (2.87 and 2.95 Å). The other MeOH accepts an even stronger H-bond (2.83 Å) from only one of the two terminal nitrogens. The two MeOH molecules also serve as H-bond donors to the phenolate oxygen, with O–O distances of 2.36 and 2.62 Å. Somewhat surprisingly, the intrachain nitrogen does not participate in any H-bonding in this crystal.
By contrast, in (p-phenolyl)ethylguanidine free base (Figure 13B), all of the three nitrogens participate in H-bonding. There are four H-bond acceptor groups for each guanidinium. Three of these are distinct MeOH oxygens, accepting H-bonds from the terminal nitrogens, with the O–N bond distances in the narrow range of 2.85–2.90 Å. The fourth H-bond acceptor, phenolate, shows an H-bonding pattern that is strikingly different from that in the dodecyl-linked compound (Figure 13A), in that it accepts moderately strong H-bonds of nearly equal length (2.87 and 2.88 Å) from two nitrogens of the same guanidinium group, one being the intrachain nitrogen. The phenolate in the ethyl-linked compound (Figure 13B) simultaneously accepts even stronger H-bonds from two other groups, as measured by their short lengths. One is MeOH (O–O distance of 1.69 Å); the other is a guanidine nitrogen from a neighboring dimer (O–N distance of 2.85 Å).
One desired control compound, the HBr salt of the dodecyl-linked compound in Figure 13A, has not yet been amenable to crystallography. However, the HBr salt of the corresponding ethyl-linked compound (Figure 13C) again clearly demonstrates head-to-tail dimer formation. In this case, Br– counterions are present, and no cocrystallized MeOH molecules. This results in somewhat different H-bonding patterns. As with the corresponding free base (Figure 13B), the intrachain nitrogen and one of the terminal nitrogens still form equal-length H-bonds with the phenolic oxygen of the dimer partner molecule. However, in the HBr salt (Figure 13C), the N–O bond distances are both larger than any N–O distances in the free bases. This indicates weaker H-bonding between guanidinium and protonated phenol, as compared to guanidinium + phenolate. Furthermore, as a result of complete proton transfer to the phenol group, the phenolic C–O bond length is also clearly 0.2–0.4 Å longer in the HBr salt than in the free bases.
Discussion
Phenol and Guanidine Free-Base Groups Form an Unusually Strong and Polarizable H-Bond in Nonpolar Environments
Computations in vacuo (Figure 2) and UV spectroscopic experiments in hexane (Figure 3) demonstrate that phenol and guanidine groups form an overall-neutral H-bonded complex in nonpolar environments, with a binding ΔE or ΔH of −70 kJ mol–1. The closeness of the agreement between the computational and experimental numbers is fortuitous, because the in vacuo computation did not take into account changes in solvent interfacial interactions that could easily contribute up to ±10–20 kJ mol–1 to the binding energy. Furthermore, Figure 2 presents only the single H-bonded structure with the very lowest energy. Several other local minima (e.g., involving H-bonding with a deprotonated intrachain nitrogen) were only a few kJ mol–1 higher in energy and would likely be represented in the physiological-temperature ensemble measured in the experimental work. Such alternative structures could also easily play a role inside proteins.
The experimental UV spectrum of the H-bonded complex in hexane (Figure 3C) was computed by a least-squares fitting procedure, based on the assumption of a simple complexation equilibrium that could be shifted by dilution or heating. This UV spectrum shows distinct features attributable to both phenol (∼280 nm) and phenolate (∼310 nm) structures, indicating a rapid protomeric equilibrium within the complex corresponding to the structures in Scheme 1B,C. There is no certain way to extract the individual extinction coefficients and individual concentrations of individual protomers. However, in hexane, they appear to provide roughly equal mole fractions on the basis of an assumption of roughly equal integrated extinction coefficients for the phenol (280 nm) and phenolate (310 nm) bands.
Even in a more polar aprotic solvent (DMSO), H-bonding is detectable in UV spectroscopic measurements, which show concentration-dependent formation of (p-phenolyl)alkylguanidine dimer complexes (Figure 4; additional data in Supporting Information). The experimental values of ΔG°298 = −4 kJ mol–1 and ΔH° = −11 kJ mol–1 per phenol–guanidine H-bonding pair are considerably smaller in DMSO than in hexane. This is attributable to the much stronger solvating power of DMSO than of hexane. As a result of solvation, formation of the dimer is expected to disrupt numerous energetically favorable interactions between DMSO molecules and each (p-phenolyl)ethylguanidine monomer. These include interactions in which DMSO oxygens act as H-bond acceptors from phenol, as well as other dipole–dipole interactions.
The UV spectrum of (p-phenolyl)ethylguanidine dimer in DMSO was also estimated (see Supporting Information). In this case, the phenolate-like absorbance band, with a maximum at ∼308 nm, was clearly predominant, and only much smaller features near 280 nm might be attributable to a contribution from phenol. Thus, shifting from hexane to a more polar solvent such as DMSO is sufficient to favor the zwitterionic protomer within the H-bonded dimer. Even taken by themselves, these UV spectral results demonstrate that the H-bond between phenol and guanidine is indeed highly polarizable. That is, the favored protomer can easily be changed by alterations in solvent environment. With widely differing dielectric constants, DMSO and hexane produce big changes in proton polarization.
The existence of phenol–guanidine complexes exhibiting rapid protomeric equilibria and large proton polarizability is also supported by IR spectroscopy in aprotic solvents. In both CCl4 (Figure 5) and DMSO (Figure 6), the phenol/guanidine H-bonded complexes simultaneously exhibit characteristic peaks of protonated and deprotonated phenol groups, as well as protonated and deprotonated monoalkylguanidine groups. In DMSO (Figure 6), the IR bands representative of the zwitterionic limiting structure (Scheme 1B) are more favored. However, in the less-polar CCl4 (Figure 5), there are nearly equal contributions of both forms (Scheme 1B,C).
Methylating the phenol, or protonating guanidine with a strong acid such as HBr, eliminates the possibility of this type of strong H-bonding interaction. Consistent with this, in crystals or in DMSO, the IR and NMR signals from the guanidine group of (p-anisolyl)dodecylguanidine free base are nearly the same as those from dodecylguanidine free base. The same similarities apply to the corresponding HBr salts, (p-anisolyl)dodecylguanidinium-Br and dodecylguanidinium bromide. In the latter case, the similarities also extend to (p-phenolyl)dodecylguanidinium bromide. This shows that a protonated (neutral) phenol group interacts only weakly with guanidinium. Spectral features of phenol are also reciprocally unaffected by the presence of guanidinium (Figure 7; see also Supporting Information). These control experiments show that the O of anisole or neutral phenol serves as only a very weak H-bond acceptor from guanidinium (and also, as shown in the case of anisole, from guanidine free base).
Phenolate Spectral Properties Are Strongly Perturbed When It Accepts an H-Bond from Guanidinium, but This Is Only Seen in Aprotic Environments
Strong perturbation of the phenolate group by H-bonding to a guanidinium counterion in aprotic solvents is detectable with UV, IR, and NMR spectroscopy.
UV
The phenolate component of the H-bonded complex (Scheme 1B) appears to have λmax near 310 nm in hexane (Figure 3) and double maxima at 293 and 310 nm in DMSO (see Figure 4-S). In contrast, cresolate salts in protic solvents show a single broad peak with λmax near ∼296 nm. This value is seen for MeOH in Figure 4F–H, and is nearly the same as that reported for tyrosinate in water at high pH (λmax ≈ 294 nm).18 That is, when combined in protic solvents, phenol and guanidine simply form solvated phenolate and guanidinium ions in the familiar Brønsted–Lowry type of acid–base equilibrium. Even at concentrations exceeding ∼50 mM (Figure 4), there is no evidence of significant formation of H-bonded complexes between these ions in protic solvents.
The neutral (phenol) species within the very same H-bonded complexes serves as a control. It shows considerably less solvent-dependent variability for λmax, which appears at 280–283 nm for both protic and aprotic solvents (Figures 3 and 4). Only the vibronic bandshape changes, with considerable sharpening as the solvent polarity is reduced.
IR
A clear IR demonstration of H-bonding perturbation on the phenolate is seen for the strongest C=C stretch mode. For (p-phenolyl)dodecylguanidine in DMSO, at a concentration expected to produce mainly the dimer complex, it is at 1494 cm–1 (Figure 6A), compared with 1501 cm–1 in MeOH (Figure 6B). The latter frequency is also observed in tyrosine solutions at high pH.18 This same vibration is at an intermediate frequency of 1498 cm–1 both in CCl4 (Figure 5) and in a solid state measurement (Figure 9).
In all these spectra, the phenolate peak at 1494–1501 cm–1 consistently has a larger half-width than the phenol peak at ∼1515 cm–1, likely due to inhomogeneous broadening. This is consistent with a greater degree of sensitivity of the phenolate vibrational frequency to the solvent environment as compared to phenol itself.
An even greater variability of phenolate vibrational frequencies is seen for the C–O stretch. This vibration ranges from 1264 to 1309 cm–1 depending on the solvent and counterion environments (Figure 8D–G). The lowest value, for (p-phenolyl)ethylguanidine in MeOH, is similar to the value of 1270 cm–1 reported for aqueous tyrosine and tyrosine-containing peptides at a high pH.18 The highest value of 1309 cm–1, for (p-phenolyl)ethylguanidine in DMSO, is probably related to the unusual H-bonding environment in the dimer complex. Once again, the corresponding C–O vibration in phenol itself is more constant (Figure 8A–C). It varies in intensity, sometimes resulting in the inability to resolve it from a nearby peak at 1235 cm–1, but appears to fall in a narrow (∼3-cm–1) range around 1246 cm–1. The latter is its value in MeOH (Figure 8C) and in water-solvated tyrosine.18 In summary, in the IR as in the UV region, phenolate spectral bands are much more sensitive to solvent environment than the corresponding features of phenol.
NMR
The phenol/phenolate ring 1H resonances in DMSO show a consistent dependence on the presence and concentration of guanidine/guanidinium H-bonding partners, but they always remain far from the values seen in potassium p-cresolate. This can be seen most clearly in Figure 10. The asymptotic values at low (<5 mM) concentration of (p-phenolyl)dodecylguanidine match those of p-cresol. The asymptotic values at high (>100 mM) concentration shift barely a quarter of the way to those seen for potassium p-cresolate in DMSO. Similarly, the spectra of dodecylguanidinium-phenolate salts in DMSO show ring 1H resonances (see Figures 9-S-B,C in Supporting Information) that are much closer to those of p-cresol than of potassium p-cresolate. This is seen with a dodecylguanidine/phenol ratio of either 1:1 or 1.6:1, either of which would be expected to fully deprotonate 0.06 M p-cresol in DMSO if Brønsted–Lowry acid–base equilibrium was present. That is, the 1H NMR spectra of phenolate when strongly H-bonded to a guanidinium counterion largely resemble those of p-cresol itself, and are very different than with a weakly H-bonded K+ counterion. Similar conclusions apply to the 13C resonance of the phenolate ring in aprotic solvents. With a guanidinium counterion, these phenolate salts remain closer to values seen for p-cresol, than to the corresponding potassium salt (see Figure 11).
Proton Polarizability
Based on UV, IR, and NMR spectroscopic measurements, the proton in the phenol–guanidine H-bond rapidly shifts position between the two bases (guanidine and phenolate), and is highly polarizable. That is, the fraction of time it spends on each of the two groups is strongly dependent on the solvent environment, with the zwitterionic form being favored in more polar solvents. Additionally, IR spectra of the H-bonded complexes in aprotic solvents (Figure 5), as well as in crystalline state (Figures 9 and 6-S-G) display a strong broad absorption band between 3500 and 2500 cm–1. Such a continuum absorption is characteristic of a polarizable H-bond. It is notably absent from the control spectra of the HBr salts of the same compounds in crystalline state (also shown in Figures 9 and 6-S-G in Supporting Information). This confirms that the phenol–guanidinium H-bonding interaction is much weaker than for phenolate–guanidinium.
The different degrees of polarization in various solvent environments can be correlated with solvent-dependent pKa values of the phenol and guanidine groups. In aprotic solvents less polar than DMSO, the acidity of guanidinium is expected to increase, whereas that of phenol is expected to decrease, effectively bringing the pKa values of guanidine and phenol closer together. This promotes the formation of an even stronger H-bond, with an even greater proton polarizability, because H-bonding is generally strongest when the pKa of the donor is closest to that of (the conjugate-acid form of) the acceptor.22 That is, the proton affinities of the groups competing for the H+ must be large to get the strongest H-bonds. This is precisely the case for guanidine and phenolate in our H-bonded complexes, most particularly in nonpolar solvents such as hexane and CCl4. A much weaker interaction is expected when guanidinium serves as the H-bond donor and phenol serves as the H-bond acceptor because of the much lower proton affinity of (neutral) phenol compared with phenolate.
No single parameter such as dielectric constant is a perfect predictor of solvent-dependent pKa, nor of whether phenol and guanidine are likely to participate in the H-bond interactions. Indeed, when considering such H-bonding in crystals and other solid states, including the interiors of any particular protein or lipid bilayer, only wide ranges are available for the dielectric constant. Furthermore, trying to predict the interactions of a highly directional and localized H-bond with its environment by using a continuum model is intrinsically unreliable. Therefore, we cannot claim to be modeling the arg–tyr system inside a protein or membrane with great precision by sampling their side chains’ behavior in only four homogeneous solvents. Observing our model compounds in a wider range of solvent environments will likely reveal interesting additional aspects of their interactions.
Additionally, the concept of pKa in nonaqueous environments is not rigorous and depends strongly on the particular counterions present. This lack of rigor explains some experimentally contradictory behavior. In water, guanidinium is unquestionably a weaker Brønsted–Lowry acid than phenol. However, we have shown quite directly (Figure 4F) that in MeOH, guanidinium is the stronger acid by about 1.3 pKa units. This contradicts the much lower published pKa value for p-cresol than for guanidinium in DMSO (18.523 and 28.5,24 respectively). These published values suggest that guanidine free base should be capable of completely deprotonating phenol in either MeOH or DMSO. Besides showing an opposite Brønsted–Lowry behavior in MeOH (Figure 4F), our spectra measured in DMSO (e.g., Figure 4B–D) indicate that Brønsted–Lowry acid–base equilibria do not occur with this pair of solutes. Instead, Lewis acid–base complexation predominates. Within such a complex, it is not easy to assign pKa values to the individual components.
Moderately Strong H-Bonding between Guanidine and Phenol Groups Is Retained in the Crystalline State
So far, we have only been able to obtain crystal structures of the zwitterion (Scheme 1B) in the presence of cocrystallized MeOH. Even in the absence of MeOH, the IR spectra of KBr pellets (Figure 8) support the conclusion that the zwitterion predominates in the solid state. These IR spectra nevertheless demonstrate that even this zwitterion form (predominantly Scheme 1B) retains a more polarizable H-bond than the phenol + guanidinium salt form (Scheme 1A), as evidenced by the stronger continuum absorption band (compare Figure 8 green and purple spectra). Thus, the phenolate + guanidinium H-bond remains much stronger than the phenol + guanidinium H-bond. This conclusion also finds support in the crystal structures, which show guanidinium-phenolate H-bonds that are shorter (2.75–2.9 Å, Figure 13A,B), than for guanidinium + phenol (2.96–3.00 Å; Figure 13C). The value of 2.75 Å is indicative of quite a strong H-bonding. Indeed, it is close to the range (<2.65 Å) for very strong N–H···O bonds that have been interpreted as having partial covalent character.26
Interpretation of 15N NMR Measurements
The solid-state 15N chemical shifts of the terminal nitrogens of (p-phenolyl)alkylguanidines (Figure 12B,C) are all within the range of 44–51 ppm. These are similar to those of the corresponding HBr salts (Figure 12E,F) and are also well within the 30–70 ppm range previously observed in 15N NMR chemical shifts of arginine and other guanidinium salts in solutions,17 as well as in the solid state.25 These results are consistent with the conclusion from X-ray crystallography and IR spectra that the zwitterion, that is, the guanidinium-phenolate protomer, predominates in the crystals of (p-phenolyl)alkylguanidine free bases.
In contrast, actual stoichiometric deprotonation of the guanidine group produces changes far outside the range for guanidinium. Thus, the ∼60 ppm separation between amino and imino 15N chemical shifts for crystalline (p-anisolyl)dodecylguanidine free base (Figure 12A) falls on a continuum with values previously published for alkylguanidine free bases in various solvent environments. The least H-bonded of these are the penta-alkylated guanidine free bases, which in aprotic solvents completely lack H-bond donors. In such solvents, the chemical shift splitting between the imino and amino nitrogens is very large, typically near 150 ppm.27 These splittings can be observed directly in the solution-state measurements in CHCl3 because there are no rapid H+ transfers that can make the terminal amino and imino nitrogens chemically equivalent. The same prior work showed that when even a single N–H bond is introduced, that is, in tetra-alkylated guanidine free bases in CHCl3, the 15N chemical shift splitting drops to 126 ppm. As we have now shown, with four N–H bonds per guanidine, specifically in monoalkylated guanidine free bases such as dodecylguanidine, the splitting drops further to 60 ppm. However, due to rapid tautomerization in a solution, this chemical shift splitting can only be observed with the solid-state measurements (see ref (12) and Figure 12A).
We conclude that large variations in 15N chemical shift splittings in deprotonated guanidines are produced by the presence or absence of H-bond donation to the imino nitrogen, mainly from neighboring amino nitrogens within the same alkylguanidine molecule. The presence of such H-bonding renders the chemical environment of the imino nitrogen more “amino-like”. Additionally, in the presence of a sufficiently strong proton donor(s) external to the guanidine group, the imino group becomes completely or nearly completely protonated, that is, nearly indistinguishable from the nearby amino group, leading to small splittings such as in Figure 12B,C.
The availability of internal and external H-bond donor(s) to the imino nitrogen thus can account for most of the enormous variation in 15N chemical-shift splittings seen in guanidine-free bases. The weaker the overall H+ donation to the imino group, the greater the splitting. If this hypothesis is true, then it should be possible to systematically adjust the H+-donating strength of the environment to obtain just about any value of splitting for the η-nitrogens of arginine, from 0 to 60 ppm. NMR spectroscopic modeling of possible arginine environments in biology cannot likely be considered complete until a good range of intermediate values have been observed experimentally.
In the (p-phenolyl)alkylguanidine model system we have developed, nearly complete H+ transfer prevails, and the splittings of only 5–6 ppm are obtained (Figure 12B,C). However, making the environment around the neutral phenol–guanidine H-bond less polar, so as to emphasize Scheme 1C more, should result in less H+ transfer to the imino group, and thus to greater splitting. The simplest way to accomplish this, while still allowing X-ray crystallography and solid-state 15N measurements, might be to crystallize our (p-phenolyl)alkylguanidine model compounds without including MeOH. Alternatively, it should be possible to resolve a large splitting of 15N chemical shifts of the two terminal nitrogens of the H-bonded dimer complex in DMSO, either in a liquid state or as a glassy solid. Our attempts at these measurements have not been successful to date (see Supporting Information for 15N NMR measurements in DMSO at 25 and 50 °C).
Possible New Motif for Membrane Protein Structures
The 70 kJ mol–1 binding enthalpy of the (phenol + guanidine) complex is quite large—nearly double that of (phenol + alkylamine) complexes, which have been measured by direct calorimetry,28 as well as by UV measurements similar to those shown in Figure 3 above.13,14
H-bonded dyads (e.g., arg–asp pairs with closest contacts commonly below 3.0 Å) have long been an identifiable motif in crystal structures, as shown in a statistical analysis of amino acid geometries in such structures.29 In this publication, no common H-bonded tyr–arg patterns were clearly discernable; however, no membrane proteins were included among the 62 proteins analyzed, as it was limited to structures determined prior to 1988. The database used for a more recently updated online version of this catalog30 now includes ∼40× as many structures. This probably includes a number of membrane proteins; however, the database is not organized according to protein type or localization. This recent database shows recurring geometric motifs that appear to be suitable for H-bonding not only for dyads of tyr–lys (“Cluster 6”) but also for tyr–arg (“Cluster 1” and “Cluster 5”). The H-bonded tyr–arg grouping in Figure 1 appears possibly to fit in the latter Cluster 5.
Still, few tyr–arg environments have been clearly identified in proteins that are sufficiently aprotic and nonpolar, for example, as in Meta II (Figure 1) that they might, based on our modeling, support deprotonation of arg; certainly not as readily as deprotonation of lys. Nevertheless, we hypothesize, based on our modeling, that the overall neutral form of the (arg + tyr) dyad may play a significant role in a small number of protein structures, specifically in very nonpolar aprotic environments. We designate this hypothetical grouping a Bonded Uncharged (aRginine + tYrosine) or BU(RY) motif, signifying any strongly H-bonded arg–tyr dyad that is overall neutral, that is, in zwitterionic (Scheme 1B), fully neutral (Scheme 1C), or intermediate protomeric states.
Our experimental modeling clearly shows this dyad is not likely allowed in protic environments. Thus, a functional BU(RY) grouping resembling our model compounds will probably occur only in biological environments deficient in H-bonding groups, predominantly membranes, where no proton acceptor for the tyrosine-OH is available, besides a deprotonated arginine; and no proton donor is available for that deprotonated arginine, besides the tyrosine-OH. A specific example of a sufficiently nonpolar aprotic environment is provided in the region surrounding the conserved R–Y grouping in Meta II (Figure 1).4 Conserved adjacent RY groupings in protein primary sequences are quite common in small-peptide hormones, such as members of the neuropeptide Y family. In fact, the powerful endogenous brain analgesic kyotorphin is simply Y–R dipeptide. The fact that kyotorphin and other RY-rich hormones tend to act at or within membranes suggests that the formation of a BU(RY) dyads within the nonpolar interior of a membrane bilayer should be considered as a possible physiological structure for such peptides.
Other candidates for BU(RY) motifs are in the activated intermediate states of heptahelical membrane proteins. X-ray crystallography shows a conserved arg135–tyr223 dyad in an aprotic environment in Meta II. When the G-protein transducin reversibly dissociates from this activated receptor (Figure 1A), there is no clear evidence of any counteranion for arg135 remaining behind or entering the binding pocket to substitute for transducin’s C-terminal carboxylate. In fact, the only protic or oxygen-containing group within 5 Å of the arg135 guanidine group is tyr227. Specifically, no structural water molecules are observable within this distance. Instead, nearby hydrocarbon side chains almost completely fill the space surrounding arg135 and tyr223. Their H-bonding distance of 2.7 Å matches well that seen in our best crystallographic model for a BU(RY) grouping, (p-phenolyl)dodecylguanidine dimer (2.75 Å, Figure 13).
Solid-state NMR measurements have previously been performed, which detected a clear perturbation of tyr227 during Meta II formation, but seemed to exclude the possibility that either it or arg135 underwent deprotonation.10 However, it may not be safe to rely on expectations based on prior model compounds,10,25,27,31,32 which did not include any with a guanidine–phenol pairing. In fact, (p-phenolyl)dodecylguanidine and (p-phenolyl)ethylguanidine in aprotic environments deviate significantly in their spectroscopic behavior from all of the earlier model compounds. Specifically, based only on earlier model compounds, deprotonation of tyr227 would be expected to result in a ∼15 ppm downfield shift for the 13C resonance of its ipso carbon.10 However, our new model compounds for such a BU(RY) dyad show 13C and 1H chemical shift values for the phenolic ring that are substantially closer to those of neutral p-cresol than potassium p-cresolate, while still showing the 13C and 1H chemical shifts closer to protonated guanidinium than to guanidine (see Figures 10 and 11). That is, when strongly H-bonded, both guanidine and phenol can appear spectroscopically to be mostly protonated, even though overall they are neutral, that is, there is a full stoichiometric deprotonation of the combined grouping.
Rather than NMR or IR, the best spectroscopic method to detect a BU(RY) grouping in a membrane protein may be UV absorption, using the spectral marker band for the phenolate component near 310–315 nm (Figures 3 and 4), or possibly UV resonance Raman. Unfortunately, the strong UV–visible absorption bands of retinylidene chromophores of rhodopsin and Meta II overlap this 310 nm region. As a result, Meta II-minus-rhodopsin UV–visible difference spectra provide no clear evidence, or lack of evidence, for a 310 nm difference band attributable to a BU(RY) dyad.33 However, substitution of a retinal analogue such as retinal2 could shift the absorption spectrum of both rhodopsin and Meta II chromophores to longer wavelengths, reducing spectral interference in the 310 nm region. Alternatively, many other GPCRs and their agonists have no competing absorptions in this region, so UV difference spectroscopy should provide a rather simple and unambiguous test of whether a BU(RY) dyad can form upon addition of agonist to receptor.
Materials and Methods
UV–Visible Spectra
Measurements were performed on a Shimadzu UV-265 dual-beam spectrometer, using 0.5 or 1 cm pathlength fused silica cuvettes as indicated.
IR Spectra
Spectra were obtained using a Nicolet Magna IR 860 spectrometer. For solution spectra, a refillable variable pathlength cell with 15 μm spacers and 2 mm thick CaF2 windows was used. For crystalline samples, the samples were prepared as KBr pellets.
UV/Visible/IR Spectral Manipulations and Fitting
Spectral manipulations (e.g., water vapor subtraction, smoothing, peak fitting) were performed using GRAMS32/AI software, except for SVD, which was performed by transferring the data as ASCII files and then manipulating them as matrices in Matlab. The SVD routine in Matlab was used to compute the best-fit spectrum of the 1:1 complex formed between p-cresol and n-dodecylguanidine in hexane, at varying dilutions of an initial total concentration of ∼700 μM; as well as the best-fit concentration of this species. The fitting was done in a least-squares fashion by fitting a set of 11 measured spectra of the sample at different concentrations and temperatures to weighted sums of two components: the separately measured spectrum of pure p-cresol in hexane and a single additional spectral component, which was allowed to vary. Further details are provided in Results and Supporting Information.
Computational Modeling
All computations were done by using GAUSSIAN09 implemented with the GAUSSVIEW 4.1 visualization interface. DFT energy computations used the B3LYP functional with 6-31G++ or 6-311G++ basis set.
Solution-State 1H and 13C NMR Spectra
Except where another solvent was indicated, all of the 1H and 13C NMR spectra were recorded in DMSO-d6 using a 400 MHz Bruker NMR spectrometer. Chemical shifts are presented in ppm and are referenced to the residual DMSO peak at 2.50 ppm for 1H and to the natural-abundance 13C-DMSO peak at 39.5 ppm peak.
15N Solid-State NMR Spectra
All solid-state 15N NMR cross-polarization magic-angle spinning (CP-MAS) measurements were performed at the Analytical and Technical services at the SUNY ESF using a 400 MHz Bruker spectrometer. The spinning rate was 4000 Hz, with a delay between pulse 5 s, 400 ppm sweep width (12 165.450 Hz), and 1H decoupling field of 47 200 Hz. Number of data points was 484, acquisition time was 0.02 s; processed with a line broadening of 40 Hz reference to ammonium-15N2 sulfate standard at 0 ppm.
Sources of Chemicals
p-Cresol, as well as all of the reagents and solvents used in the synthesis and analysis of other compounds were obtained from Sigma-Aldrich and used directly unless otherwise indicated. The solvents were dried by the addition of molecular sieves. KBr for solid-state IR samples was dried at 110 °C over several days before use.
Syntheses of Model Compounds
Detailed procedures for synthesis of p-phenol-dodecylguanidinium bromide and dodecylguanidinium bromide are described elsewhere.12 Analogous procedures were followed to prepare p-phenol-ethylguanidinium bromide, using commercially available p-phenol-ethylamine, that is, tyramine, as the starting point and converting the amine to a guanidine by reaction with S-methyl thiourea hydroiodide, just as was done to p-phenol-dodecylamine in the previously described synthesis of p-phenol-dodecylguanidinium bromide.12 The procedure for incorporating enriched (∼50 atom %) 15N at the guanidine terminal nitrogens, in this case to aid in obtaining solid-state 15N NMR spectra, was also described in the same publication.
Deprotonation of Alkylguanidine Compounds
A general method for deprotonating alkylguanidinium bromides to form alkylguanidine free bases is described in the accompanying paper.12 This procedure reproducibly gave crystalline samples, with elemental analyses that match the predicted values for the corresponding free bases. In brief, a dry crystalline alkylguanidinium bromide (or p-phenol alkylguanidinium bromide) was first recrystallized from acetonitrile and then redissolved in dry methanol (MeOH). Potassium tert-butoxide (1 mol equiv), also dissolved in dry methanol just before use, was added into the solution dropwise, causing the solution to become milky due to the precipitation of KBr. After the precipitation had stopped, the solution was heated briefly to boiling, then centrifuged while still hot (2 min, 3000g). The clear supernatant was transferred to a clean test tube and then concentrated ∼20-fold under a gentle stream of dry N2. The evaporative cooling aided in promoting a high yield of crystals. These were harvested while still cold by pipetting off the bulk of residual tert-butanol and MeOH and then drying under N2.
Sample Crystallization and X-ray Crystallography
Crystals of the free bases of (p-phenolyl)-dodecylguanidine and (p-phenolyl)ethylguanidine, as well as their HBr salts, were obtained by slowly evaporating and cooling concentrated solutions of them in dry MeOH, under a gentle stream of room-temperature N2. Making a sufficiently concentrated solution of (p-phenolyl)dodecylguanidine free base in dry MeOH required gentle heating, whereas the other compounds dissolved readily at room temperature. The resulting ∼0.1 mm size crystals were stored for up to ∼1 week in a minimal volume of MeOH at 4 °C before mounting in the diffractometer. Structures were recorded at 90 K.
Crystals of (p-phenolyl)-dodecylguanidinium bromide, (p-anisolyl)-dodecylguanidine free base and its HBr salt, as well as dodecylguanidine free base and its HBr salt could also be made by the approach described above, but were not of sufficient quality to produce good diffraction.
Acknowledgments
This work was supported by Syracuse University, and by NSF Instrumentation Grants for NMR and X-ray crystallography instruments. We thank Carlos Castaneda and Bruce Hudson for helpful suggestions, and Sharece Toner for assistance with GAUSSIAN computations.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00282.
Individual .cif files for the structures shown in Figure 13 (CIF) (CIF) (CIF)
(Tables 1-S–4-S) Gaussian input (.gjf) files for computed structures and energies as shown in Figure 2; tabulated raw UV absorbance spectral data on cresol + dodecylguanidine in hexane as shown in Figure 3; matrices obtained during the optimization used to compute thermodynamic parameters; and tabulated raw spectral absorbance for (p-phenolyl)ethylguanidine dimerization in DMSO; (Figures 1-S–4-S) computed UV spectral results for the dimerization of (p-phenolyl)ethylguanidine in DMSO; (Figures 5-S–6-S) additional IR spectra; (Figures 7-S–11-S) additional solution 1H, 13C, and 15N NMR spectra in MeOH and DMSO (PDF)
Author Present Address
∥ Department of Physics, University of Texas, El Paso, Texas 79968, United States (A.G.).
Author Present Address
§ Chemistry Department, University of Dodoma, Tanzania (A.T.B.).
The authors declare no competing financial interest.
Supplementary Material
References
- Sass H. J.; Büldt G.; Gessenich R.; Hehn D.; Neff D.; Schlesinger R.; Berendzen J.; Ormos P. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature 2000, 406, 649–653. 10.1038/35020607. [DOI] [PubMed] [Google Scholar]
- Choe H.-W.; Kim Y. J.; Park J. H.; Morizumi T.; Pai E. F.; Krauß N.; Hofmann K. P.; Scheerer P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455, 497–502. 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Govorunova E. G.; Sineshchekov O. A.; Li H.; Spudich J. L. Microbial rhodopsins: diversity, mechanisms, and optogenetic applications. Annu. Rev. Biochem. 2017, 86, 845. 10.1146/annurev-biochem-101910-144233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H. W.; Kim Y. J.; Park J. H.; Morizumi T.; Pai E. F.; Krauss N.; Hofmann K. P.; Scheerer P.; Ernst O. P. Crystal structure of metarhodopsin II. Nature 2011, 471, 651–655. 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- Braiman M. S.; Briercheck D. M.; Kriger K. M. Modeling vibrational spectra of amino acid side chains in proteins: Effects of protonation state, counterion, and solvent on arginine C–N stretch frequencies. J. Phys. Chem. B 1999, 103, 4744–4750. 10.1021/jp983011b. [DOI] [Google Scholar]
- Schneider E. H.; Schnell D.; Strasser A.; Dove S.; Seifert R. Impact of the DRY motif and the missing ionic lock on constitutive activity and G-protein coupling of the human histamine H4 receptor. J. Pharmacol. Exp. Ther. 2010, 333, 382–392. 10.1124/jpet.109.163220. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Jensen A. D.; Liapakis G.; Rasmussen S. G. F.; Shi L.; Gether U.; Javitch J. A. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001, 276, 29171–29177. 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- Valentin-Hansen L.; Groenen M.; Nygaard R.; Frimurer T. M.; Holliday N. D.; Schwartz T. W. The arginine of the DRY motif in transmembrane segment III functions as a balancing micro-switch in the activation of the β2-adrenergic receptor. J. Biol. Chem. 2012, 287, 31973–31982. 10.1074/jbc.M112.348565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J.; Khelashvili G.; Mondal S.; Weinstein H.; et al. Ligand-dependent conformations and dynamics of the serotonin 5-HT(2A) receptor determine its activation and membrane-driven oligomerization properties. PLoS Comput. Biol. 2012, 8, e1002473 10.1371/journal.pcbi.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves J. A.; South K.; Ahuja S.; Zaitseva E.; Opefi C. A.; Eilers M.; Vogel R.; Reeves P. J.; Smith S. O. Highly conserved tyrosine stabilizes the active state of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 19861–19866. 10.1073/pnas.1009405107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundel G. Hydrogen bonds with large proton polarizability in proteins—studies with model systems. Recent Res. Dev. Phys. Chem. 1998, 2, 501–532. [Google Scholar]
- Banyikwa A.; Miller S. E.; Gao T.; Krebs R. A.; Xiao Y.; Carney C.; Braiman M. S.. Small-molecule models for arginine side chains and arginine-tyrosine pairs in nonpolar environments ACS Omega (accompanying paper). [DOI] [PMC free article] [PubMed]
- Farah L.; Giles G.; Wilson D.; Ohno A.; Scott R. M. Hydrogen bonding interactions of aliphatic amines with ortho-substituted phenols. J. Phys. Chem. 1979, 83, 2455–2458. 10.1021/j100482a007. [DOI] [Google Scholar]
- Albrecht G.; Zundel G. Phenol-amine hydrogen bonds with large proton polarizabilities. Position of the OH···N ⇋ O···H+N equilibrium as a function of the donor and acceptor. J. Chem. Soc., Faraday Trans. 1 1984, 80, 553–561. 10.1039/f19848000553. [DOI] [Google Scholar]
- Bos M.; Ijpma S. T.; Dahmen E. A. M. F. The coulometric titration of acids and bases in dimethylsulfoxide media. Anal. Chim. Acta 1976, 83, 39–47. 10.1016/S0003-2670(01)84629-8. [DOI] [Google Scholar]
- DeLange F.; Klaassen C. H.; Wallace-Williams S. E.; Bovee-Geurts P. H.; Liu X. M.; DeGrip W. J.; Rothschild K. J. Tyrosine structural changes detected during the photoactivation of rhodopsin. J. Biol. Chem. 1998, 273, 23735–23739. 10.1074/jbc.273.37.23735. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Braiman M. Modeling amino acid side chains in proteins: 15N NMR spectra of guanidino groups in nonpolar environments. J. Phys. Chem. B 2005, 109, 16953–16958. 10.1021/jp051279e. [DOI] [PubMed] [Google Scholar]
- Rothschild K. J.; Roepe P.; Ahl P. L.; Earnest T. N.; Bogomolni R. A.; Das Gupta S. K.; Mulliken C. M.; Herzfeld J. Evidence for a tyrosine protonation change during the primary phototransition of bacteriorhodopsin at low temperature. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 347–351. 10.1073/pnas.83.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Hutson M. S.; Belenky M.; Herzfeld J.; Braiman M. S. Role of Arginine-82 in Fast Proton Release during the Bacteriorhodopsin Photocycle: A Time-Resolved FT-IR Study of Purple Membranes Containing 15N-Labeled Arginine. Biochemistry 2004, 43, 12809–12818. 10.1021/bi049238g. [DOI] [PubMed] [Google Scholar]
- Zundel G. Proton transfer in and proton polarizability of hydrogen bonds: IR and theoretical studies regarding mechanisms in biological systems. J. Mol. Struct. 1988, 177, 43–68. 10.1016/0022-2860(88)80078-4. [DOI] [Google Scholar]
- Zundel G.; Eckert M. IR continua of hydrogen bonds and hydrogen-bonded systems, calculated proton polarizabilities and line spectra. J. Mol. Struct.: THEOCHEM 1989, 200, 73–92. 10.1016/0166-1280(89)85045-6. [DOI] [Google Scholar]
- Brzezinski B.; Zundel G. Formation of hydrogen-bonded chains between a strong base with guanidine-like character and phenols with various pKa values—an FT-IR study. J. Mol. Struct.: THEOCHEM 1996, 380, 195–204. 10.1016/0022-2860(96)09221-6. [DOI] [Google Scholar]
- Bordwell F. G.; McCallum R. J.; Olmstead W. N. Acidities and hydrogen bonding of phenols in dimethyl sulfoxide. J. Org. Chem. 1984, 49, 1424–1427. 10.1021/jo00182a020. [DOI] [Google Scholar]
- Bordwell F. G.; Ji G. Z. Effects of structural changes on acidities and homolytic bond dissociation energies of the hydrogen-nitrogen bonds in amidines, carboxamides, and thiocarboxamides. J. Am. Chem. Soc. 1991, 113, 8398–8401. 10.1021/ja00022a029. [DOI] [Google Scholar]
- Petkova A. T.; Hu J. G.; Bizounok M.; Simpson M.; Griffin R. G.; Herzfeld J. Arginine Activity in the Proton-Motive Photocycle of Bacteriorhodopsin: Solid-State NMR Studies of the Wild-Type and D85N Proteins. Biochemistry 1999, 38, 1562–1572. 10.1021/bi981968z. [DOI] [PubMed] [Google Scholar]
- Gilli P.; Bertolasi V.; Ferretti V.; Gilli G. Evidence for intramolecular NH···O resonance-assisted hydrogen bonding in β-enaminones and related heterodienes: A combined crystal-structural, IR and NMR spectroscopic, and quantum-mechanical investivation. J. Am. Chem. Soc. 2000, 122, 10405–10407. 10.1021/ja000921+. [DOI] [Google Scholar]
- Kanamori K.; Roberts J. D. A nitrogen-15 NMR study of the barriers to isomerization about guanidinium and guanidino carbon-nitrogen bonds in L-arginine. J. Am. Chem. Soc. 1983, 105, 4698–4701. 10.1021/ja00352a028. [DOI] [Google Scholar]
- Kogowski G.; Scott R. M.; Filisko F. Enthalpy change of hydrogen bond formation between ortho-substituted phenols and aliphatic amines. J. Phys. Chem. 1980, 84, 2262–2265. 10.1021/j100455a009. [DOI] [Google Scholar]
- Singh J.; Thornton J. M.. Atlas of Protein Side-Chain Interactions; IRL Press: Oxford, 1992. [Google Scholar]
- Laskowski R. A. M. S.; Thornton J. M.. Atlas of Side-Chain Interactions. http://www.ebi.ac.uk/thornton-srv/databases/sidechains (accessed May 13, 2017).
- Pregosin P. S.; Randall E. W.; White A. I. Natural abundance nitrogen-15 nuclear magnetic resonance spectroscopy-amino-acid derivatives. J. Chem. Soc. D 1971, 1602–1603. 10.1039/C29710001602. [DOI] [Google Scholar]
- Blomberg F.; Maurer W.; Rüterjans H. 15N nuclear magnetic resonance investigations on amino acids. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 1409–1413. 10.1073/pnas.73.5.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto Y.; Kojima K.; Oka T.; Maeda R.; Schichida Y. Helical rearrangement of photoactivated rhodopsin in monomeric and dimeric forms probed by high-angle X-ray scattering. Photochem. Photobiol. Sci. 2015, 14, 1965–1973. 10.1039/C5PP00175G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.