

 A solid phase-based strategy gave access to DNA-tagged heterocycles by metal-mediated imine chemistry, exemplified by Cushman- and 1,3-dipolar cycloaddition reactions.
A solid phase-based strategy gave access to DNA-tagged heterocycles by metal-mediated imine chemistry, exemplified by Cushman- and 1,3-dipolar cycloaddition reactions.
Abstract
DNA-encoded libraries of chemically synthesized compounds are an important small molecule screening technology. The synthesis of encoded compounds in solution is currently restricted to a few DNA-compatible and water-tolerant reactions. Encoded compound synthesis of short DNA-barcodes covalently connected to solid supports benefits from a broad range of choices of organic solvents. Here, we show that this encoded chemistry approach allows for the synthesis of DNA-coupled isoquinolones by an Yb(iii)-mediated Castagnoli–Cushman reaction under anhydrous reaction conditions and for the synthesis of highly substituted pyrrolidines by Ag(i)-mediated 1,3-dipolar azomethine ylide cycloaddition. An encoding scheme for these DNA-barcoded compounds based on a DNA hairpin is demonstrated.
Introduction
Endowing chemically synthesized small molecules with barcoded information on their molecular structure 1 is a highly efficient strategy to handle large compound numbers for screening purposes (Fig. 1A). Not the least due to impressive technological advances in the efficiency to read massive genomic data sets, DNA is a highly attractive compound identifier. Genetically tagged collections of small molecules, commonly termed DELs (DNA-encoded libraries), have attracted much attention in recent years as an efficient technology for target-based screening.1–7 Unlike discrete compound libraries that require cost-intensive infrastructure for screening and biochemical or cell-based assays compatible with high-throughput experimentation,8 DELs are selected on minute amounts of target proteins either immobilized or captured on a solid support. Thus, through incorporating organic preparative small molecule synthesis for library preparation, as a technology, DELs are much more related to display libraries, such as phages, than to classical small molecule screening collections. Selection of encoded libraries on target proteins has led to the identification of a number of bioactive compounds with unique modes of action such as allosteric GPCR antagonists 2 and 3 and a protein homodimerizer 4 (Fig. 1B).9–11 Recently, the RIP1 kinase inhibitor 5 evolved from a DEL selection entering clinical phase 2, showing the potential of the technology for drug discovery projects.12 Encoded libraries are synthesized by alternating organic preparative synthesis and DNA ligation steps that track compound synthesis (Fig. 1C). Usually, DEL chemists are planning combinatorial strategies for compound synthesis to access high numbers of molecules efficiently.13–19
Fig. 1. DNA-encoded library technology. (A) Schematic presentation of a genetically tagged molecule. (B) Exemplary bioactive compounds developed from “hits” identified by DEL selection. (C) Encoded library synthesis may be initiated with a headpiece, 6, a hairpin-like DNA with a terminal aminolinker for attachment of chemical building blocks; or with a protected single-stranded DNA bound to a solid phase, 7.

Library synthesis can for instance be initiated by a hairpin-like “headpiece” structure that contains a linker moiety for attachment of small organic building blocks (Fig. 1C).14 For this strategy, any synthesis method applied to DEL synthesis needs to be DNA-compatible20 and it must tolerate water as a (co-)solvent too, requiring e.g. development of ligands for metal-mediated reactions.21–27 Initiating DEL synthesis with a solid support-based synthesis strategy is a viable alternative. The DNA oligonucleotide 7 modified with a linker moiety is synthesized by standard phosphoramidite chemistry on a controlled pore glass (CPG) solid phase. The solid phase material can directly be used for a chemical reaction, e.g. coupling of a building block by carbonyl chemistry or nucleophilic substitution reactions for encoded library synthesis or DNA labelling, as demonstrated by different research groups (Fig. 1C).28–33 In the context of DNA labelling, this methodology is called “post-synthetic” modification and comprised, for instance, Pd-mediated Sonogashira and Stille reactions using standard catalyst–ligand combinations.34–36 A disadvantage of this strategy is the need to deprotect and cleave oligonucleotide conjugates from the solid phase with concentrated ammonia solution which is an additional step and may damage target molecules by e.g. hydrolysis.37 This disadvantage must be weighed against the benefits of solid support chemistry. These are fully nucleobase-protected DNA which may display higher stability against reagents, and equally important, the choice of a broad range of (dry) organic solvents to perform reactions. Furthermore, the solid phase can be washed extensively to remove excess reactants and reagents such as metal catalysts which might otherwise contaminate the DNA oligomer.
We have a long-standing interest in developing encoding schemes and synthesis methods that give access to genetically tagged heterocycles from simple, readily available starting materials.38 This research direction in encoded chemistry is justified by the strong representation of heterocyclic structures among natural products and synthetic bioactive compounds including drugs.39–42 Following up on our systematic investigations in Au(i)-mediated reactions for heterocycle formation on different solid phase-coupled DNA oligonucleotides, among them the chemically highly stable adapter hexT, oligopyrimidine, A/T/C, and A/T/C/G DNA sequences,38,43 and especially the finding that an A/T/C-sequence tolerated a Au(i)-mediated spirocyclization reaction at room temperature,43 we decided to investigate further metal-mediated reactions that yield heterocyclic, drug-like structures under mild, ambient reaction conditions. Our focus was placed on imine chemistry. Imines are readily condensed from diverse aldehydes and amines giving rise to high appendage diversity, and they can be cyclized to different heterocyclic structures depending on the addition of further reactants and the mode of catalysis selected. Thus, imine chemistry is an attractive access to structural diversity.41 However, many imine-based reactions are likely incompatible with aqueous (co)-solvents due to competing hydrolysis of the imine. Here, we show the translation of the Yb(OTf)3-mediated Castagnoli–Cushman reaction into a DNA-tagged format giving rise to diverse substituted tetrahydroquinolones, and the translation of a Ag(i)-mediated [3+2]-cycloaddition yielding highly substituted pyrrolidines. Both reactions were successfully performed on short 10–14mer DNA strands coupled to a CPG solid phase. Following the reaction development, we demonstrate a preliminary scope of both reactions, the limitations of this strategy when applied to longer DNA oligonucleotides and to the acid-mediated Pictet–Spengler reaction, and finally a ligation strategy for short single-stranded 14mer coding strands based on a DNA hairpin sequence.
Yb(OTf)3-mediated Castagnoli–Cushman reaction on CPG-bound oligonucleotides
The Castagnoli–Cushman reaction is a well-established, versatile three component reaction yielding substituted lactams from diverse aldehydes, amines and cyclic anhydrides.44–48 Among the latter, homophthalic anhydride is particularly frequently used as it furnishes target tetrahydroisoquinolonic acids such as 8–10 under mild conditions.49–58 This scaffold can be found in numerous bioactive compounds (Fig. 2A). For instance, Floyd et al. reacted the acid further with anilines. The target tetrahydroisoquinolone carboxanilides showed potent antimalarial activity and compound 8 was selected as a preclinical candidate.59,60 Rothweiler et al. identified 9 as an inhibitor of the p53-MDM2 interaction.61,62 Humphries et al. characterized tetrahydroisoquinolonic acid 10 as a GPR40 antagonist.63
Fig. 2. Representative bioactive tetrahydroisoquinolonic acids (A) and pyrrolidines (B).

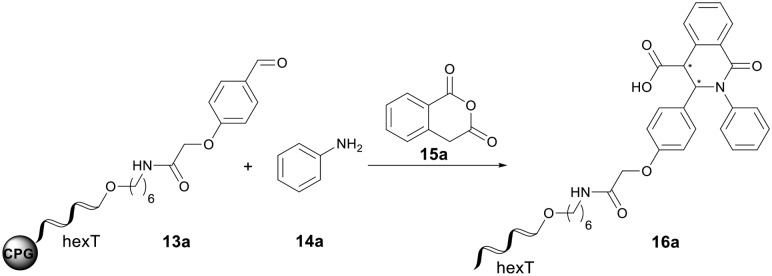
Attracted by its demonstrated potential for screening library synthesis, we were highly interested in translating the Castagnoli–Cushman reaction to a solid phase DEL synthesis. The mild reaction conditions reported by Wang et al. using Yb(OTf)3 for the Castagnoli–Cushman reaction were selected as a starting point for the reaction translation.53 We started our investigations with the CPG-bound adapter oligonucleotide “hexT” as previously described.38 In a first step, we coupled 2-(4-formylphenoxy)acetic acid to the C6-amino-linker of the hexT. This aldehyde conjugate 13a was treated in a two-step process first for 4 hours at room temperature with 500 equiv. of aniline 14a in dichloromethane/triethyl orthoformate to form the imine in situ, followed by 100 equiv. of Yb(OTf)3 and 500 equiv. of homophthalic anhydride 15a (Table 1) that were reacted with the imine for 2 hours at room temperature. Following the reaction, the hexT-conjugates were cleaved from the solid phase and analyzed by analytical HPLC and MALDI-MS. Product conversion was estimated by integration of the product and starting material peaks in the HPLC traces.
Table 1. Optimization of the Yb(OTf)3-mediated Castagnoli–Cushman reaction to the CPG-bound hexT-isoquinolonic acid conjugate 16a a .
| |||||
| Entry | Yb(OTf)3 [equiv.] | t 1 [h] | t 2 [h] | Solvent | Conversion b [%] |
| 1 c | 100 | 4 | 2 | CH2Cl2 | <5 |
| 2 | 100 | 4 | 2 | CH2Cl2 | 72 |
| 3 | 100 | 4 | 1 | CH2Cl2 | 73 |
| 4 | 100 | 4 | 0.5 | CH2Cl2 | 68 |
| 5 | 100 | 3 | 1 | CH2Cl2 | 69 |
| 6 | 100 | 2 | 1 | CH2Cl2 | 68 |
| 7 | 100 | 1 | 1 | CH2Cl2 | 65 |
| 8 | 100 | 16 | 1 | CH2Cl2 | 73 |
| 9 d | 100 | 4 | 1 | CH2Cl2 | 7 |
| 10 | 50 | 4 | 1 | CH2Cl2 | 70 |
| 11 | 25 | 4 | 1 | CH2Cl2 | 54 |
| 12 | 0 | 4 | 1 | CH2Cl2 | 38 |
| 13 e | 50 | 4 | 1 | CH2Cl2 | 65 |
| 14 f | 50 | 4 | 1 | CH2Cl2 | 63 |
| 15 g | 50 | 4 | 1 | CH2Cl2 | 51 |
| 16 | 50 | 4 | 1 | THF | 56 |
| 17 | 50 | 4 | 1 | DCE | 58 |
| 18 | 50 | 4 | 1 | ACN | 52 |
| 19 | 50 | 4 | 1 | MeOH | 68 |
| 20 | 50 | 4 | 1 | Toluene | 50 |
| 21 | 50 | 4 | 1 | 1,4-Dioxane | 52 |
| 22 | 50 | 4 | 1 | DMF | 31 |
| 23 | 50 | 4 | 1 | EtOAc | 40 |
aCPG-bound oligonucleotide conjugate 13a (20 nmol) and aniline 14a (500 equiv., 10 μmol) in 36 μL of solvent/triethyl orthoformate (2 : 1) at ambient temperature for t1, then Yb(OTf)3 (X equiv., 1 μmol) and anhydride 15a (500 equiv., 10 μmol) both suspended in 30 μL of solvent, at ambient temperature for t2. Afterwards, with AMA (30% aqueous ammonia/40% aqueous methylamine, 1 : 1 (vol/vol)) at ambient temperature for 0.5 h.
bDetermined by analytical RP-HPLC analysis.
cWashing of the CPG-bound hexT-conjugate after imine formation.
dImine formation performed without triethyl orthoformate.
e1000 equiv. of aniline 14a and anhydride 15 were used.
f1500 equiv. of aniline 14a and anhydride 15a were used.
g2000 equiv. of aniline 14a and anhydride 15 were used.
However, only traces of the desired product 16a were observed when the excess of aniline 14a was reduced by a washing step prior to addition of homophthalic anhydride 15a (Table 1, entry 1). To our delight, we obtained a 72% conversion into the hexT-lactam conjugate 16a by direct addition of Yb(OTf)3 and the homophthalic anhydride 15a suspensions to the in situ–formed DNA-imine conjugate, in spite of the high excess of aniline present in the reaction mixture (Table 1, entry 2).
Taking this as the starting point, the reaction conditions were systematically optimized. Decreasing the reaction time for the Castagnoli–Cushman reaction from two hours to one hour had no effect on the product conversion (Table 1, entries 2 and 3). Further decreasing to 0.5 hours led to a 5% lower conversion (Table 1, entry 4). Next, we reduced stepwise the time for the imine formation and observed a slight time-dependent decrease for the conversion too (Table 1, entries 3, 5–6). On the other hand, prolonging the time for imine formation to 16 hours did not improve the yield (Table 1, entry 8). Omitting triethyl orthoformate during imine formation led to a meagre 7% product formation (Table 1, entry 9) showing the need to promote the reaction by this condensing agent. Lowering the Yb(OTf)3 loading from 100 to 50 equiv. had a negligible effect on product formation (Table 1, entries 3 and 10). Yet, the usage of only 25 equiv. of the metal salt resulted in a lower conversion of 54% (Table 1, entry 11). Interestingly, the reaction also worked in the absence of Yb(OTf)3 but with a low conversion of 38%, probably driven by high reactant excess (Table 1, entry 12). Increasing the amounts of aniline and homophthalic anhydride had an opposite effect and led to a decrease in conversion (Table 1, entries 13–15). Finally, we explored the influence of different solvents on the reaction (Table 1, entries 16–23). Most of the tested solvents showed a lower starting material conversion compared to dichloromethane. Only methanol resulted in a similar outcome of the reaction with 68% product formation (Table 1, entries 19 and 10). In summary, the optimized reaction conditions for the Castagnoli–Cushman reaction on the CPG-bound hexT-aldehyde conjugate 16a are imine formation with 500 equiv. of aniline 14a in dichloromethane/triethyl orthoformate (2 : 1) for 4 hours at ambient temperature followed by the addition of suspensions of 50 equiv. of Yb(OTf)3 and 500 equiv. of homophthalic anhydride in dichloromethane for 1 hour at ambient temperature. The reaction proceeded reproducibly with 70% conversion to the target 16a (Table 1, entry 10).
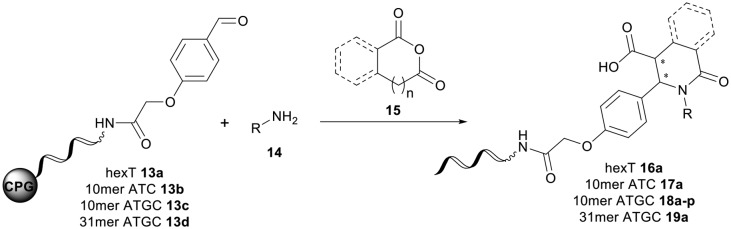
The next step was the transfer of the optimized reaction conditions from hexT to more complex 10mer ATC-43 and ATGC-oligonucleotides. Depending on the coding strategy these DNA oligonucleotides represent primer or barcode sequences, thus they enable a more efficient library synthesis than the hexT adapter. The required CPG-bound DNA-aldehyde conjugates 13b and 13c were synthesized in analogy to the hexT conjugate. To our delight, the treatment of the obtained CPG-bound 10mer ATC-aldehyde conjugate 13b with aniline 14a and homophthalic anhydride 15a under our optimized conditions furnished the desired Castagnoli–Cushman product 17a with 68% conversion and only minimal DNA decomposition (Table 2, entry 2). Fortunately, the reaction worked also with the CPG-bound 10mer ATGC-aldehyde conjugate 13c, with a similar conversion of 69% (Table 2, entry 3). To our surprise, the CPG-bound 31mer aldehyde conjugate 13d, i.e. an oligonucleotide containing both primer and code sequences, gave the target DNA-heterocycle product 19a as well (Table 2, entry 4). In all three cases, the Yb(OTf)3-promoted reactions proceeded without significant oligonucleotide degradation as determined by HPLC analysis of the crude. However, in the case of the 31mer and unlike the shorter DNA oligonucleotide conjugates, both the starting material and product were eluted close together on a standard RP-HPLC purification run.
Table 2. Scope of the Yb(OTf)3-mediated Castagnoli–Cushman reaction on CPG-bound oligonucleotides 13 using different amines 14 and anhydrides 15 a .
| ||||
| Entry | Product | Amine | Conversion b [%] | Masscalc. |
| Massfound c | ||||
| 1 | 16a |
|
70 | 2341.8 |
| 2342.3 | ||||
| 2 | 17a |

|
68 | 3540.6 |
| 3539.8 | ||||
| 3 | 18a |
|
69 | 3596.6 |
| 3596.1 | ||||
| 4 | 19a |
|
63 d | 9953.5 |
| 9942.6 | ||||
| 5 | 18b |
|
<5 | 3675.5 |
| n.d. | ||||
| 6 | 18c |
|
52 | 3675.5 |
| 3674.6 | ||||
| 7 | 18d |
|
46 | 3656.6 |
| 3654.1 | ||||
| 8 | 18e |
|
65 | 3624.6 |
| 3622.1 | ||||
| 9 | 18f |
|
72 | 3638.7 |
| 3640.0 | ||||
| 10 | 18g |
|
54 | 3654.6 |
| 3655.3 | ||||
| 11 | 18h |
|
50 | 3620.6 |
| 3619.7 | ||||
| 12 | 18i |
|
89 | 3562.6 |
| 3564.3 | ||||
| 13 | 18j |
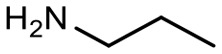
|
62 | 3610.6 |
| 3609.1 | ||||
| 14 | 18k |
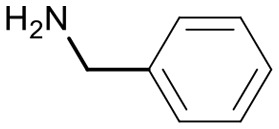
|
74 | 3736.5 |
| 3734.8 | ||||
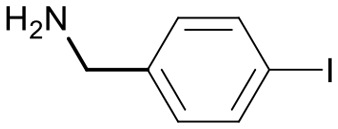
| 15 | 18l |
|
83 | 3640.6 |
| 3639.1 | ||||
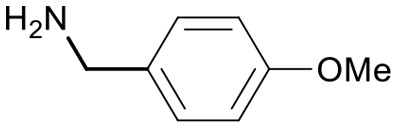
| 16 | 18m |
|
78 | 3663.7 |
| 3661.8 | ||||
| 17 e | 18n |
|
n.d. | 3548.5 |
| n.d. | ||||
| 18 f | 18o |
|
n.d. | 3520.5 |
| n.d. | ||||
| 19 f , g | 18o |
|
n.d. | 3548.5 |
| n.d. | ||||
aCPG-bound conjugate 13 (20 nmol) and amine 14 (500 equiv., 10 μmol) in 36 μL of CH2Cl2/triethyl orthoformate (2 : 1) at ambient temperature for 4 h, then Yb(OTf)3 (50 equiv., 1 μmol) and homophthalic anhydride 15a (500 equiv., 10 μmol) both suspended in 30 μL of CH2Cl2 at ambient temperature for 1 h. Afterwards in AMA (30% aqueous ammonia/40% aqueous methylamine, 1 : 1 (vol/vol)) at ambient temperature for 4 h.
bDetermined by HPLC analysis.
cMeasured by MALDI-MS.
dDetermined by preparative RP-HPLC.
eGlutaric anhydride 15b was used instead of homophthalic anhydride 15a.
fSuccinic anhydride 15c was used instead of homophthalic anhydride 15a.
gMeOH as the solvent, 50 °C in the second step. 10mer ATC = 5′-TTA CTA CCT A-3′, 10mer ATGC = 5′-GTC ATG ATC T-3′, 31mer ATGC = 5′-CAA ATC CGT TCA CAC CGA CCT GTC ATG ATC T-3′. n.d. = not detected.
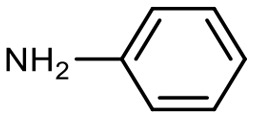
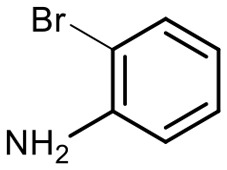
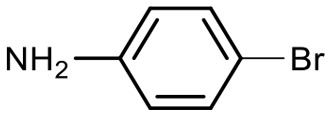
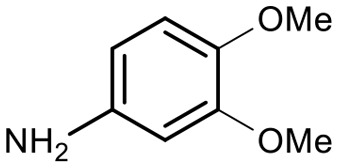
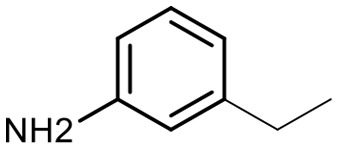
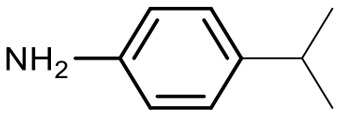
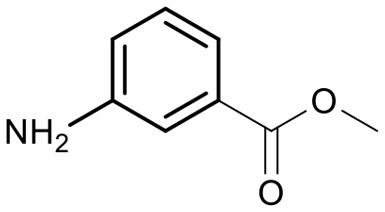
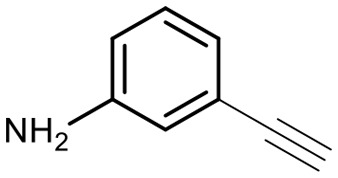
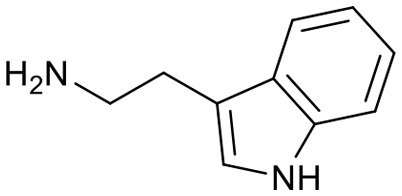
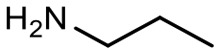
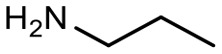
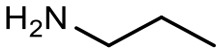
Encouraged by these results, we explored the substrate scope of the reaction on the CPG-bound ATGC-aldehyde conjugate 13c (Table 2, entries 5–18). First, diverse substituted anilines 14 were tested. Both electron withdrawing and electron donating substituents were tolerated with moderate to good conversions (46–72%). Electron donating substituents like a meta-ethyl or para-isopropyl group tended to show higher conversions (65% and 72%, Table 2, entries 8 and 9) compared to electron withdrawing substituents like para-bromo or meta-ethyl carboxylate groups (52% and 54%, Table 2, entries 6 and 10). Substituents in the meta-position led to slightly lower conversions than substituents in the para-position (Table 2, entries 8 and 9). No product formation was observed with a substituent in the ortho-position (Table 2, entry 5). Next, we investigated the reactivity of aliphatic amines 14 (Table 2, entries 12–16). Propylamine, benzylamine and tryptamine (Table 2, entries 12, 13 and 16) were well tolerated with even higher conversions (62–89%) than most of the tested anilines. All these products were isolated by preparative RP-HPLC with good purities. Unfortunately, the usage of different anhydrides like glutaric anhydride (Table 2, entry 17) or succinic anhydride (Table 2, entry 18) did not result in any product formation. Raising the temperature to 50 °C did not result in product formation in these cases (Table 2, entry 19). Monocyclic anhydrides typically demand high temperatures (>100 °C) that are likely not tolerated by DNA oligonucleotides.44–48
Finally, we explored if the Castagnoli–Cushman reaction can also be performed on oligonucleotides in solution (Scheme S1†). To this end, we performed the reaction in a mixture of water and methanol. However, the target heterocycle could not be detected, and the 10mer ATGC-aldehyde conjugate 13c was recovered intact. Instead, we noticed formation of DNA-ytterbium adducts by mass spectrometry and a marked shift of the retention time of 13c during ion-pair chromatography.
AgOAc-mediated 1,3-dipolar azomethine ylide-alkene cycloaddition on CPG-bound oligonucleotides
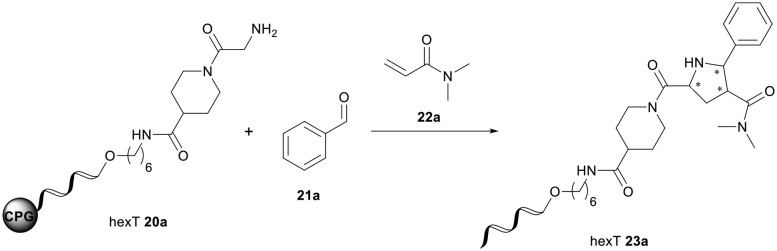
1,3-Dipolar cycloaddition (1,3-DC) reactions belong to the most powerful methods for the construction of five-membered heterocyclic compounds.64–66 Of particular interest is the 1,3-dipolar cycloaddition of azomethine ylides with electron-deficient alkenes that provides densely substituted pyrrolidines,67–69 which were previously used to populate screening libraries.70
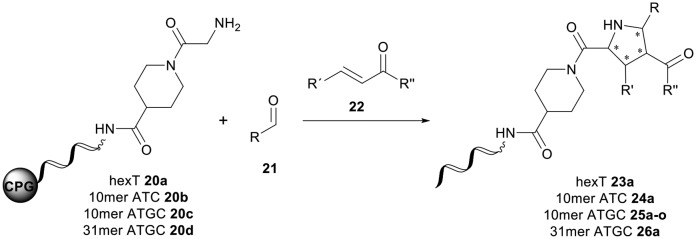
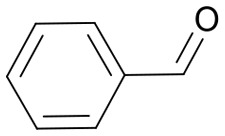
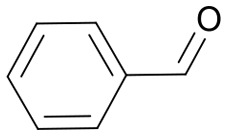
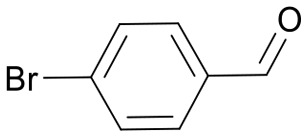
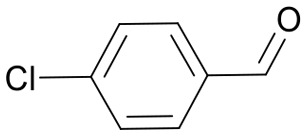
The pyrrolidine core is present in numerous natural products, biologically active molecules, and in drug candidates.71,72 One example of a drug candidate based on an N-acylpyrrolidine motif is compound 11 which was clinically evaluated for the treatment of hepatitis C (HCV, Fig. 2B).73 The core of this compound is accessible by 1,3-dipolar azomethine ylide-alkene cycloaddition. Wang et al. reported a series of MDM2-p53 interaction inhibitors, e.g.12, based on the 1,3-DC-accessible spirooxindole core (Fig. 2B).74 To establish the 1,3-dipolar cycloaddition on DNA oligonucleotides, we synthesized the CPG-bound hexT-glycine conjugate 20a by Fmoc peptide chemistry. The 1,3-DC reaction on the DNA was then performed by a two-step process. First, we treated hexT-20a with 1000 eq. of benzaldehyde 21a in acetonitrile to obtain the imine, this step was directly followed by addition of 100 equiv. of AgOAc, 1000 equiv. of N,N-dimethylacrylamide 22 and 1000 equiv. of triethylamine. In analogy to the Castagnoli–Cushman reaction, the CPG was washed following the reaction and the hexT conjugate was cleaved from the solid phase and analyzed by analytical HPLC and MALDI-MS. We observed 31% conversion to the desired product in this initial experiment (Table 3, entry 1). With this promising result in hand, the reaction was systematically optimized (Table 3). First, we explored the influence of the AgOAc loading at different temperatures (Table 3, entries 1–4). At ambient temperature, the use of 100 or 200 equiv. of AgOAc did not impact product formation (31% and 29%, Table 3, entries 1 and 2). At higher temperatures, a higher loading of the catalyst turned out to be detrimental (48% and 36%, Table 3, entries 3 and 4). With increasing amounts of reactants, the product conversion improved to 53% (Table 3, entries 4–6). In the next series of experiments, we changed the ratio of the reactants using 1000 equiv. of benzaldehyde 21a and 3000 or 4000 equiv. of N,N-dimethylacrylamide 22 and triethylamine. Changing the ratio of the reactants from a 1 : 1 : 1 to a 1 : 3 : 3 or 1 : 4 : 4 ratio in combination with elevated temperatures increased the conversion up to 70% (Table 3, entries 7 and 8). The addition of the condensing agent triethyl orthoformate was well tolerated by the reaction. The silver salts AgOTf, AgTFA and AgSbF6 that were previously reported in the 1,3-DC, reproduced the conversion obtained with AgOAc (66–68%, Table 3, entries 9–12). Finally, there were significant solvent effects on the reaction (Table 3, entries 9, 13–15). In particular, methanol, though pre-dried, turned out to be detrimental (Table 3, entry 13). In addition, numerous by-products were formed in this polar protic solvent. As suspected, the reaction did not give the target heterocycle conjugate on a deprotected oligonucleotide in aqueous solution (Scheme S2†). Of note, washing the CPG-bound oligonucleotide cycloadduct with EDTA was not sufficient to remove the silver ions in every experiment as we observed shifted HPLC retention times occasionally. An additional washing step by using Chelex 100 resin was therefore routinely done and sufficient to remove residual metal ions after oligonucleotide cleavage. Through systematic optimization of the reaction conditions, we arrived at a convenient one-pot two-step protocol for the 1,3-dipolar cycloaddition on the CPG-bound hexT-glycine conjugate 20a. It started with the addition of a 1000-fold excess of benzaldehyde 21a in acetonitrile/triethyl orthoformate (2 : 1) for 6 hours at room temperature for imine formation. This was followed by reacting the imine with 100 equiv. of AgOAc, 4000 equiv. of N,N-dimethylacrylamide 22 and 4000 equiv. of triethylamine for 16 hours at 50 °C to the target pyrrolidine (Table 3, entry 9). As described for the Castagnoli–Cushman reaction, we transferred the optimized reaction conditions for the silver-mediated 1,3-DC to the 10mer ATC- and ATGC-oligonucleotides. The required CPG-bound DNA-glycine conjugates 20b and 20c were prepared by Fmoc peptide chemistry. Gratifyingly, the transfer of the optimized 1,3-DC reaction conditions to the 10mer ATC- and ATGC-sequences succeeded as well (Table 4, entries 2 and 3). The desired CPG-bound 10mer ATC- and ATCG-conjugates 24a and 25a were both formed with a conversion of 50% (Table 4, entries 2 and 3). The 31mer oligonucleotide-aldehyde 20d could also be reacted with a similar conversion to the target pyrrolidine 26a (Table 4, entry 4). Next, we investigated the aldehyde scope of the 1,3-DC reaction on the CPG-bound ATGC-glycine conjugate 20c (Table 4, entries 5–16). Aromatic aldehydes having electron-withdrawing groups such as fluorine or nitrile in the para-position led to the formation of the target product with conversions ranging between 31% and 67% (Table 4, entries 5–7, 10). The highest conversions were obtained with a para-nitrile substituent (Table 4, entry 11). Electron-donating groups such as methoxy- or tert-butyl-groups in the para-position gave the product with lower conversions of 24–38% (Table 4, entries 8 and 9). Aromatic groups para-position, like phenyl or pyridyl, were better tolerated with conversions of 47% and 58% (Table 4, entries 12 and 13). Interestingly, a sterically hindered aromatic aldehyde substituted with a benzylether in the ortho-position gave the pyrrolidine with a conversion of 47% (Table 4, entry 14). The enolizable, aliphatic propylaldehyde was not productive at all (Table 4, entry 16). Standard yet highly reactive dipolarophiles methyl acrylate and dimethyl maleate gave only low product conversions of 10–20% of the target DNA conjugates. In both cases, a large number of by-products could be detected, thus they were not suitable starting materials for the solid phase strategy in our hands (Table 4, entries 17 and 18). All 1,3-DC products were isolated by preparative RP-HPLC with good purities.
Table 3. Optimization of the AgOAc-mediated (1,3)-cycloaddition on the CPG-bound oligonucleotides hexT conjugate 20a a .
| ||||||
| Entry | 21a [equiv.] | 22a [equiv.] | Ag(i) [equiv.] | T [°C] | Solvent | Conversion b [%] |
| 1 | 1000 | 1000 | 100 | 22 | ACN | 31 |
| 2 | 1000 | 1000 | 200 | 22 | ACN | 29 |
| 3 | 1000 | 1000 | 100 | 40 | ACN | 48 |
| 4 | 1000 | 1000 | 200 | 40 | ACN | 36 |
| 5 | 2000 | 2000 | 100 | 40 | ACN | 41 |
| 6 | 4000 | 4000 | 100 | 40 | ACN | 53 |
| 7 | 1000 | 3000 | 100 | 50 | ACN | 68 |
| 8 | 1000 | 4000 | 100 | 50 | ACN | 70 |
| 9 | 1000 | 4000 | 100 | 50 | ACN/TEOF (2 : 1) | 70 |
| 10 c | 1000 | 4000 | 100 | 50 | ACN/TEOF (2 : 1) | 66 |
| 11 d | 1000 | 4000 | 100 | 50 | ACN/TEOF (2 : 1) | 68 |
| 12 e | 1000 | 4000 | 100 | 50 | ACN/TEOF (2 : 1) | 68 |
| 13 | 1000 | 4000 | 100 | 50 | MeOH/TEOF (2 : 1) | 43 |
| 14 | 1000 | 4000 | 100 | 50 | THF/TEOF (2 : 1) | 70 |
| 15 | 1000 | 4000 | 100 | 50 | Toluene/TEOF (2 : 1) | 59 |
aCPG-bound oligonucleotide hexT-conjugate 20a (20 nmol) and aldehyde 21a (X equiv.) in 50 μL of solvent at ambient temperature for 6 h, then AgOAc (X equiv.), dissolved in 30 μL of ACN, triethylamine (X equiv.) and N,N-dimethylacrylamide 22 (X equiv.) at T overnight. Afterwards in AMA (30% aqueous ammonia/40% aqueous methylamine, 1 : 1 (vol/vol)) at ambient temperature for 4 h.
bDetermined by analytical RP-HPLC analysis.
cAgTFA was used instead of AgOAc.
dAgSbF6 was used instead of AgOAc.
eAgOTf was used instead of AgOAc. TEOF = triethyl orthoformate.
Table 4. Scope of the AgOAc-mediated (1,3)-Cycloaddition on CPG-bound oligonucleotides 20 using different aldehydes 21 and dipolarophiles 22 a .
| ||||
| Entry | Product | Aldehyde | Conversion b [%] | Masscalc. |
| Massfound c | ||||
| 1 | 23a |
|
70 | 2297.7 |
| 2301.1 | ||||
| 2 | 24a |
|
50 | 3496.6 |
| 3499.3 | ||||
| 3 | 25a |
|
50 | 3552.6 |
| 3553.3 | ||||
| 4 | 26a |
|
45 d | 9909.6 |
| 9908.6 | ||||
| 5 | 25b |
|
48 | 3631.5 |
| 3633.3 | ||||
| 6 | 25c |
|
41 | 3587.1 |
| 3589.8 | ||||
| 7 | 25d |
|
47 | 3570.6 |
| 3572.6 | ||||
| 8 | 25e |
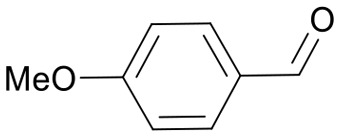
|
24 | 3582.6 |
| 3583.7 | ||||
| 9 | 25f |
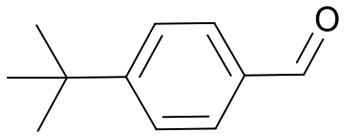
|
38 | 3608.7 |
| 3613.4 | ||||
| 10 | 25g |
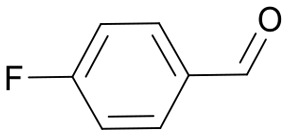
|
26 | 3576.6 |
| 3578.6 | ||||
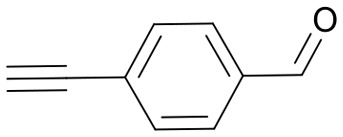
| 11 | 25h |
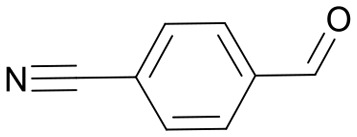
|
67 | 3577.6 |
| 3579.0 | ||||
| 12 | 25i |
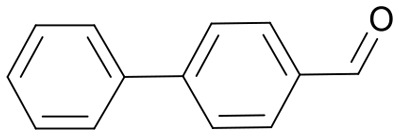
|
54 | 3628.7 |
| 3630.9 | ||||
| 13 | 25j |
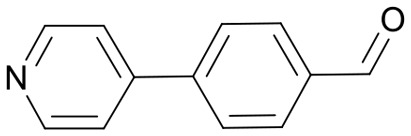
|
58 | 3629.7 |
| 3630.0 | ||||
| 14 | 25k |
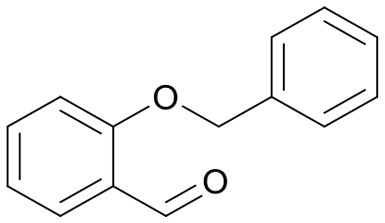
|
48 | 3658.7 |
| 3659.1 | ||||
| 15 | 25l |
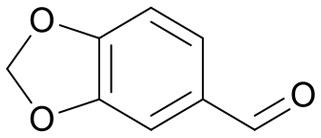
|
30 | 3596.6 |
| 3599.4 | ||||
| 16 | 25m |
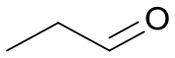
|
n.d. | 3504.6 |
| n.d. | ||||
| 17 e | 25n |
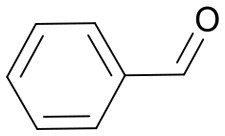
|
20 | 3538.6 |
| 3541.2 | ||||
| 18 f | 25o |
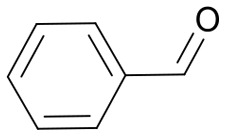
|
10 | 3595.6 |
| 3598.5 | ||||
aCPG-bound oligonucleotide conjugate 20 (20 nmol) and aldehyde 21 (1000 equiv., 20 μmol) in 50 μL of ACN/triethyl orthoformate (2 : 1) at ambient temperature for 6 h, then AgOAc (100 equiv., 2 μmol), dissolved in 30 μL of ACN/triethyl orthoformate (2 : 1), triethylamine (4000 equiv., 80 μmol) and N,N-dimethylacrylamide 22 (4000 equiv., 80 μmol) at 50 °C overnight. Afterwards, in AMA (30% aqueous ammonia/40% aqueous methylamine, 1 : 1 (vol/vol)) at ambient temperature for 4 h.
bDetermined by analytical RP-HPLC analysis.
cMeasured by MALDI-MS.
dDetermined by preparative RP-HPLC analysis.
eMethyl acrylate was used instead of N,N-dimethylacrylamide 22.
fDiemethyl maleate was used instead of N,N-dimethylacrylamide 22. 10mer ATC = 5′-TTA CTA CCT A-3′, 10mer ATGC = 5′-GTC ATG ATC T-3′, 31mer ATGC = 5′-CAA ATC CGT TCA CAC CGA CCT GTC ATG ATC T-3′, n.d. = not detected.
Investigation towards a Pictet–Spengler reaction on CPG-bound oligonucleotides
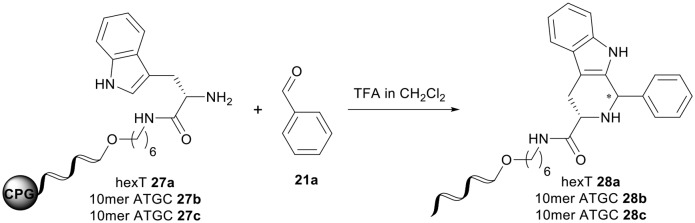
Recently, we established a trifluoroacetic acid-mediated Pictet–Spengler reaction on CPG-bound hexT that was used for the preparation of a DNA-encoded β-carboline library.38 The reaction was successfully reproduced on a CPG-bound oligopyrimidine sequence.43 Encouraged by our results for the Castagnoli–Cushman reaction and the 1,3-dipolar cycloaddition on solid phase-bound and base-protected DNA, we investigated the Pictet–Spengler reaction on both the 10mer ATC- and ATGC-oligonucleotides. Fmoc-protected tryptophan was coupled to the C6-aminolinker of a hexT, 10mer ATC- and 10mer ATGC- sequence followed by on-column Fmoc removal. As expected, the treatment of the CPG-bound hexT-Trp conjugate 27a with 10% TFA in dichloromethane and 1000 equiv. of benzaldehyde for 18 hours resulted in the desired Pictet–Spengler adduct 28a with good conversion (Table 5, entry 1). The amount of TFA could be reduced to 2% without any negative effect on the reaction outcome (Table 5, entry 2). However, the transfer to ATC- and ATGC-sequences was a frustrating experience that clearly showed the limits of the solid phase-based strategy. An incubation of the 10mer ATC-Trp conjugate 27b with 2% or 1% TFA in dichloromethane for 18 hours led to massive degradation of the oligonucleotide by depurination (Table 5, entries 3 and 4). A further reduction of the TFA concentration to 0.5% or 0.2% led still to about 70% degraded DNA material (Table 5, entries 5, 6). In the latter two cases, we could observe formation of minute though isolatable amounts of the target DNA-β-carboline conjugate 28b. (Table 5, entries 5 and 6). Therefore, the reaction time was reduced to 4 hours and 1 hour, respectively (Table 5, entries 7 and 8). Indeed, under these shortened reaction times, it was possible to decrease the degradation of the oligonucleotide to 60% and 40%, respectively. However, the product conversion also dropped to 16% and 8%. In addition, the reaction for 1 hour showed a non identified by-product. Translation of the reaction conditions to a 10mer ATGC-sequence led to only trace amounts of the desired Pictet–Spengler product 28c. DNA degradation was even more pronounced than that in the case of the ATC-sequence (Table 5, entry 9). Reduction of the reaction time to 4 hours and 1 hour led to similar results as that for the ATC-sequence (Table 5, entries 10 and 11), albeit with consistently higher levels of DNA degradation. Thus, acid-mediated reactions with slow reaction kinetics such as the Pictet–Spengler reaction were in our hands not translatable to a DNA code due to competing DNA degradation.
Table 5. Investigations in the synthesis of DNA-β-carboline conjugates by the Pictet–Spengler reaction a .
| |||||
| Entry | DNA Seq. | X [%] | t [h] | Conjugate yield b [%] | DNA degradation b [%] |
| 1 | hexT | 10 | 18 | 52 | <5 |
| 2 | hexT | 2 | 18 | 60 | <5 |
| 3 | 10mer ATC | 2 | 18 | n.d. | >95 |
| 4 | 10mer ATC | 1 | 18 | n.d. | >95 |
| 5 | 10mer ATC | 0.5 | 18 | 17 | 73 |
| 6 | 10mer ATC | 0.2 | 18 | 19 | 73 |
| 7 | 10mer ATC | 0.5 | 4 | 16 | 60 |
| 8 | 10mer ATC | 0.5 | 1 | 8 | 40 |
| 9 | 10mer ATGC | 0.5 | 18 | n.d. | 91 |
| 10 | 10mer ATGC | 0.5 | 4 | 18 | 73 |
| 11 | 10mer ATGC | 0.5 | 1 | <5 | 73 |
aCPG-bound oligonucleotide conjugate 27 (20 nmol), benzaldehyde 2 (1000 equiv., 20 μmol) and trifluoroacetic acid (X%) in 50 μL of CH2Cl2 at ambient temperature. Afterwards, in AMA (30% aqueous ammonia/40% aqueous methylamine, 1 : 1 (vol/vol)) at ambient temperature for 4 h.
bDetermined by analytical RP-HPLC analysis of the crude. 10mer ATC = 5′-TTA CTA CCT A-3′, 10mer ATGC = 5′-GTC ATG ATC T-3′.
Ligation strategy for short DNA codes
The successful exploration of metal ion-mediated imine chemistries with DNA oligonucleotides of different lengths led us to the conclusion that long 31mer DNA oligomers containing both primer and coding sequences might give hard-to-separate mixtures of starting materials and target conjugates, especially when working with split and pool routines, as intended by us in future projects. Small molecule conjugates of short 10–14mer oligonucleotides displayed in our hands a better resolution when purified by ion-pair reverse phase chromatography. Thus, we devised a coding strategy based on a DNA hairpin structure that enabled the use of short, single-stranded DNA sequences as barcodes. The hairpin contained the forward primer, a spacer that prevents read-through by DNA polymerases and an overhang for ligation to the DNA-small molecule conjugate. In order to enable ligation of DNA small molecule conjugates to this hairpin, the connection between the DNA and compound was shifted from the 5′-terminus to a position within the overhang. This DNA-compound conjugation architecture was previously validated for small molecule identification by Vipergen's yoctoreactor technology.15
For the ligation experiments, we synthesized the Castagnoli–Cushman product on a branched 14mer ATGC-oligonucleotide (Scheme S3†). A 14mer oligonucleotide 29 carrying an alkyne modified thymidine in position 4 counted from the 5′ terminus was synthesized by solid phase DNA synthesis. An Fmoc-protected amino-PEG-linker 30 was introduced afterwards by Cu(i)-mediated cycloaddition chemistry. On-column Fmoc-removal and subsequent coupling of 2-(4-formylphenoxy)acetic acid 32 yielded the ATGC-aldehyde conjugate 33. This precursor was used for the Castagnoli–Cushman reaction under optimized conditions which led to the desired product 34 for the ligation experiments with high conversion. The hairpin sequence and the DNA-Castagnoli–Cushman conjugate were 5′-phosphorylated by polynucleotide kinase (PNK). Both the phosphorylated DNA-isoquinolone conjugate and its complementary DNA strand were then ligated to the 5′-phosphorylated hairpin oligonucleotide by T4 ligation. Gel electrophoresis confirmed the successful ligation of the DNA-Castagnoli–Cushman conjugate to the hairpin sequence demonstrating the viability of this encoding strategy (Fig. 3A).
Fig. 3. Encoding strategy for short single-stranded DNA conjugates based on a DNA-hairpin. (A) Gel analysis of the ligation of the DNA-hairpin and DNA-isoquinolone conjugate (sequences of DNAs, hairpin: 3′-CAA ATC CGT TCA S AGG TCG GTG TGA ACG GAT TTG AGT C-5′, code 1: 3′-TCC ATT ACG CT(alkyne)C TC-5′, counter strand 1: 3′-TCA GGA GAA ATG GAT GGA CAT A-5′, S = C6-Spacer). (B) Scheme of a proposed split and pool DNA-encoded library synthesis for the Castagnoli–Cushman reaction based on the hairpin encoding strategy. AMA = 30% aqueous ammonia/40% aqueous methylamine, 1 : 1 (vol/vol).

We propose that this strategy can be used in a split and pool library synthesis starting with a pool of CPG-bound 14mer coding DNA-carboxaldehyde conjugates (Fig. 3B). The 14mer DNA sequence contains a code 1 for the attached aldehyde and stable terminal segments for the recognition of the counter strand. After splitting, for example, the Castagnoli–Cushman reaction is performed using different amines and phthalic anhydrides followed by cleavage of the DNA conjugates from the CPG and isolation of the product pools. The desired DNA-Castagnoli–Cushman conjugates are ligated to the DNA hairpin containing the forward primer as well as to a code 2 identifying the heterocyclic chemistry. This code 2 thus deviates from the classical building block encoding philosophy by encoding a reaction (Fig. 3B). Afterwards, the ligation products are pooled and can be used for further chemical transformations and encoding steps.
Conclusions
Based on previous successful experiments with Au(i)-mediated reactions on solid phase-bound DNA oligonucleotides, we explored the feasibility of tapping into the vast potential of metal-mediated imine chemistry for the synthesis of DNA-tagged heterocycles. Many of these reactions give rise to interesting scaffold architectures under mild reaction conditions. Here, we investigated the ytterbium-promoted Castagnoli–Cushman reaction yielding isoquinolones and a silver-mediated 1,3-azomethine ylide-dipolarophile cycloaddition to densely substituted pyrrolidines. Initial experiments on our hexT adapter, which is a well-suited model oligonucleotide for reaction translation, gave the expected target heterocycles and allowed for the optimization of reaction conditions. To our delight, and in marked contrast to the acid-mediated Pictet–Spengler reaction, both reactions could be translated with a decent reactant scope and little to no DNA degradation to solid support-connected, fully protected 10mer DNA strands that resemble barcodes. Instrumental to successful target heterocycle synthesis was the use of pure organic solvents, even in combination with a dehydrating agent for isoquinolone synthesis. In all cases, the products could be well separated from the DNA-coupled starting material by RP-HPLC. Further, we showed that both reactions have a broad substrate scope regarding diverse amines in the case of the Castagnoli–Cushman reaction or aldehydes in the case of the silver-mediated cycloaddition. Large numbers of amines and aldehydes that are falling into the scope of the successfully tested reactants are commercially available and therefore usable for library synthesis. In both reactions, multiple stereocenters are formed. The chemistry that yields stereoisomers does increase library complexity, and it might place additional demands on the characterization of compounds selected on proteins. On the other hand, this type of chemistry gives access to sp3-rich, three-dimensional molecular architectures, structures that are highly sought in screening decks.41 In the case of the two selected reactions, complexity is reduced as the 1,3-dipolar cycloaddition leads preferentially to the endo-cycloaddition products, and the Yb(OTf)3-mediated Castagnoli–Cushman reaction yields preferentially the cis-isomers.
Finally, we developed a coding strategy for short DNA barcode-small molecule conjugates. To this end, the linker moiety was shifted to a position within the sequence and the barcode was ligated at the 5′-position to a DNA hairpin containing the forward primer and a spacer preventing read-through by polymerases. Currently, we are synthesizing a proof-of-concept encoded library of heterocycles, demonstrating split and pool routines, i.e. using mixtures of solid phase coupled DNA-barcoded building blocks and encoding afterwards heterocycle-forming reactions. As a final remark, solid phase chemistry holds promise for the translation of several metal-mediated heterocycle-forming reactions to DNA-barcoding strategies due to the enhanced stability of protected DNA and the free choice of organic solvents.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by a Boehringer Ingelheim foundation exploration grant and the Mercator Research Center Ruhr (MERCUR) Grant Pr-2016-0010. We thank the group of Prof. Dr. Clever (TU Dortmund University) for access to their DNA synthesizer and technical support.
Footnotes
†Electronic supplementary information (ESI) available: Supplementary data associated with this article can be found in the online version. This includes synthesis and DNA ligation protocols and extended tables for reaction optimization experiments and the reaction scope. Copies of HPLC traces and MALDI-MS spectra are shown for all discussed DNA conjugates. See DOI: 10.1039/c9md00042a
References
- Brenner S., Lerner R. A. Proc. Natl. Acad. Sci. U. S. A. 1992;89:5381. doi: 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. A. Curr. Opin. Chem. Biol. 2010;14:396. doi: 10.1016/j.cbpa.2010.02.017. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E., Dumelin C. E., Liu D. R. Chem. Soc. Rev. 2011;40:5707. doi: 10.1039/c1cs15076f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winssinger N. Chimia. 2013;67:340. doi: 10.2533/chimia.2013.340. [DOI] [PubMed] [Google Scholar]
- Salamon H., Skopic M. K., Jung K., Bugain O., Brunschweiger A. ACS Chem. Biol. 2016;11:296. doi: 10.1021/acschembio.5b00981. [DOI] [PubMed] [Google Scholar]
- Franzini R. M., Randolph C. J. Med. Chem. 2016;59:6629. doi: 10.1021/acs.jmedchem.5b01874. [DOI] [PubMed] [Google Scholar]
- Goodnow Jr R. A., Dumelin C. E., Keefe A. D. Nat. Rev. Drug Discovery. 2017;16:131. doi: 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- High-throughput screening-compatible assay requirements are described in https://www.europeanleadfactory.eu/how-submit/drug-target-assays/requirements.
- Cheng R. K. Y., Fiez-Vandal C., Schlenker O., Edman K., Aggeler B., Brown D. G., Brown G. A., Cooke R. M., Dumelin C. E., Doré A. S., Geschwindner S., Grebner C., Hermansson N., Jazayeri A., Johansson P., Leong L., Prihandoko R., Rappas M., Soutter H., Snijder A., Sundström L., Tehan B., Thornton P., Troast D., Wiggin G., Zhukov A., Marshall F. H., Dekker N. Nature. 2017;545:112. doi: 10.1038/nature22309. [DOI] [PubMed] [Google Scholar]
- Ahn S., Kahsai A. W., Pani B., Wang Q. T., Zhao S., Wall A. L., Stracha R. T., Staus D. P., Wingler L. M., Sun L. D., Sinnaeve J., Choi M., Cho T., Xu T. T., Hansen G. M., Burnett M. B., Lamerdin J. E., Bassoni D. L., Gavino B. J., Husemoen G., Olsen E. K., Franch T., Costanzi S., Chen X., Lefkowitz R. J. Proc. Natl. Acad. Sci. U. S. A. 2017;114:1708. doi: 10.1073/pnas.1620645114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Montalván A. E., Berger M., Kuropka B., Koo S. J., Badock V., Weiske J., Puetter V., Holton S. J., Stöckigt D., ter Laak A., Centrella P. A., Clark M. A., Dumelin C. E., Sigel E. A., Soutter H. H., Troast D. M., Zhang Y., Cuozzo J. W., Keefe A. D., Roche D., Rodeschini V., Chaikuad A., Díaz-Sáez L., Bennet J. M., Fedorov O., Huber K. V. M., Hübner J., Weinmann H., Hartung I. V., Gorjánácz M. ACS Chem. Biol. 2017;12:2730. doi: 10.1021/acschembio.7b00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris P. A., Berger S. B., Jeong J. U., Nagilla R., Bandyopadhyay D., Campobasso N., Capriotti C. A., Cox J. A., Dare L., Dong X., Eidam P. M., Finger J. N., Hoffman S. J., Kang J., Kasparcova V., King B. W., Lehr R., Lan Y., Leister L. K., Lich J. D., MacDonald T. T., Miller N. A., Ouellette M. T., Pao C. S., Rahman A., Reilly M. A., Rendina A. R., Rivera E. J., Schaeffer M. C., Sehon C. A., Singhaus R. R., Sun H. H., Swift B. A., Totoritis R. D., Vossenkämper A., Ward P., Wisnoski D. D., Zhang D., Marquis R. W., Gough P. J., Bertin J. J. Med. Chem. 2017;60:1247. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- Mannocci L., Zhang Y., Scheuermann J., Leimbacher M., De Bellis G., Rizzi E., Dumelin C., Melkko S., Neri D. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17670. doi: 10.1073/pnas.0805130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. A., Acharya R. A., Arico-Muendel C. C., Belyanskaya A. L., Benjamin D. R., Carlson N. R., Centrella P. A., Chiu C. H., Creaser S. P., Cuozzo J. W., Davie C. P., Ding Y., Franklin G. J., Franzen K. D., Gefter M. L., Hale S. P., Hansen N. J., Israel D. I., Jiang J., Kavarana M. J., Kelley M. S., Kollmann C. S., Li F., Lind K., Mataruse S., Medeiros P. F., Messer J. A., Myers P., O'Keefe H., Oliff M. C., Rise C. E., Satz A. L., Skinner S. R., Svendsen J. L., Tang L., van Vloten K., Wagner R. W., Yao G., Zhao B., Morgan B. A. Nat. Chem. Biol. 2009;5:647. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Hansen M. H., Blakskjaer P., Petersen L. K., Hansen T. H., Højfeldt J. W., Gothelf K. V., Hansen N. J. J. Am. Chem. Soc. 2009;131:1322. doi: 10.1021/ja808558a. [DOI] [PubMed] [Google Scholar]
- Halpin D. R., Harbury P. B. PLoS Biol. 2004;2:1015. doi: 10.1371/journal.pbio.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner Z. J., Tse B. N., Grubina R., Doyon J. B., Snyder T. M., Liu D. R. Science. 2004;305:1601. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhao P., Zhang M., Zhao X., Li X. J. Am. Chem. Soc. 2013;135:17727. doi: 10.1021/ja409936r. [DOI] [PubMed] [Google Scholar]
- Melkko S., Scheuermann J., Dumelin C. E., Neri D. Nat. Biotechnol. 2004;22:568. doi: 10.1038/nbt961. [DOI] [PubMed] [Google Scholar]
- Malone M. L., Paegel B. M. ACS Comb. Sci. 2016;18:182. doi: 10.1021/acscombsci.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satz A. L., Cai J., Chen Y., Goodnow R., Gruber F., Kowalczyk A., Petersen A., Naderi-Oboodi G., Orzechowski L., Strebel Q. Bioconjugate Chem. 2015;26:1623. doi: 10.1021/acs.bioconjchem.5b00239. [DOI] [PubMed] [Google Scholar]
- Tian X., Basarab G. S., Selmi N., Kogej T., Zhang Y., Clark M., Goodnow Jr. R. A. Med. Chem. Commun. 2016;7:1316. [Google Scholar]
- Lu X., Roberts S. E., Franklin G. J., Davie C. P. Med. Chem. Commun. 2017;8:1614. doi: 10.1039/c7md00289k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff Y., Berst F. Med. Chem. Commun. 2018;9:1188. doi: 10.1039/c8md00185e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Chai J., Centrella P. A., Gondo C., DeLorey J. L., Clark M. A. ACS Comb. Sci. 2018;20:251. doi: 10.1021/acscombsci.8b00009. [DOI] [PubMed] [Google Scholar]
- de Pedro Beato E., Priego J., Gironda-Martínez A., González F., Benavides J., Blas J., Martín-Ortega M. D., Toledo M. Á., Ezquerra J., Torrado A. ACS Comb. Sci. 2019;21:69–74. doi: 10.1021/acscombsci.8b00142. [DOI] [PubMed] [Google Scholar]
- Ding Y., Clark M. A. ACS Comb. Sci. 2015;17:1. doi: 10.1021/co5001037. [DOI] [PubMed] [Google Scholar]
- Schwope I., Bleczinski C. F., Richert C. J. Org. Chem. 1999;64:4749. doi: 10.1021/jo990036d. [DOI] [PubMed] [Google Scholar]
- Tse B. N., Snyder T. M., Shen Y., Liu D. R. J. Am. Chem. Soc. 2008;130:15611. doi: 10.1021/ja805649f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini R. M., Samain F., Abd Elrahman M., Mikutis G., Nauer A., Zimmermann M., Scheuermann J., Hall J., Neri D. Bioconjugate Chem. 2014;25:1453. doi: 10.1021/bc500212n. [DOI] [PubMed] [Google Scholar]
- Franzini R. M., Ekblad T., Zhong N., Wichert M., Decurtins W., Nauer A., Zimmermann M., Samain F., Scheuermann J., Brown P. J., Hall J., Gräslund S., Schüler H., Neri D. Angew. Chem., Int. Ed. 2015;54:3927. doi: 10.1002/anie.201410736. [DOI] [PubMed] [Google Scholar]
- Klika Škopić M., Bugain O., Jung K., Onstein S., Brandherm S., Kalliokoski T., Brunschweiger A. Med. Chem. Commun. 2016;7:1957. [Google Scholar]
- Usanov D. L., Chan A. I., Maianti J. P., Liu D. R. Nat. Chem. 2018;10:704. doi: 10.1038/s41557-018-0033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstaff M. W., Khan S. I. J. Am. Chem. Soc. 1999;121:4704. [Google Scholar]
- Rist M., Amann N., Wagenknecht H. A. Eur. J. Org. Chem. 2003;13:2498. [Google Scholar]
- Krause A., Hertl A., Muttach F., Jäschke A. Chem. – Eur. J. 2014;20:16613. doi: 10.1002/chem.201404843. [DOI] [PubMed] [Google Scholar]
- Yuen L. H., Franzini R. M. Bioconjugate Chem. 2017;28:1076. doi: 10.1021/acs.bioconjchem.7b00005. [DOI] [PubMed] [Google Scholar]
- Klika Škopić M., Salamon H., Bugain O., Jung K., Gohla A., Doetsch L. J., dos Santos D., Bhat A., Wagner B., Brunschweiger A. Chem. Sci. 2017;8:3356. doi: 10.1039/c7sc00455a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hattum H., Waldmann H. J. Am. Chem. Soc. 2014;136:11853. doi: 10.1021/ja505861d. [DOI] [PubMed] [Google Scholar]
- Welsch M. E., Snyder S. A., Stockwell B. R. Curr. Opin. Chem. Biol. 2010;14:347. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway W. R., Isidro-Llobet A., Spring D. R. Nat. Commun. 2010;1:80. doi: 10.1038/ncomms1081. [DOI] [PubMed] [Google Scholar]
- Vitaku E., Smith D. T., Njardarson J. T. J. Med. Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Klika Škopić M., Willems S., Wagner B., Schieven J., Krause N., Brunschweiger A. Org. Biomol. Chem. 2017;15:8648. doi: 10.1039/c7ob02347b. [DOI] [PubMed] [Google Scholar]
- González-López M., Shaw J. T. Chem. Rev. 2009;109:164. doi: 10.1021/cr8002714. [DOI] [PubMed] [Google Scholar]
- Dar'in D., Bakulina O., Chizhova M., Krasavin M. Org. Lett. 2015;17:3930. doi: 10.1021/acs.orglett.5b02014. [DOI] [PubMed] [Google Scholar]
- Lepikhina A., Bakulina O., Dar'in D., Krasavin M. RSC Adv. 2016;6:83808. [Google Scholar]
- Lepikhina A., Dar'in D., Bakulina O., Chupakhin E., Krasavin M. ACS Comb. Sci. 2017;19:702. doi: 10.1021/acscombsci.7b00118. [DOI] [PubMed] [Google Scholar]
- Chizhova M., Khoroshilova O., Dar'in D., Krasavin M. J. Org. Chem. 2018;83:12722. doi: 10.1021/acs.joc.8b02164. [DOI] [PubMed] [Google Scholar]
- Cushman M., Gentry J., Dekow F. W. J. Org. Chem. 1977;42:1111. doi: 10.1021/jo00427a001. [DOI] [PubMed] [Google Scholar]
- Yu N., Bourel L., Deprez B., Gesquiere J.-C. Tetrahedron Lett. 1998;39:829. [Google Scholar]
- Yadav J. S., Reddy B. V. S., Saritha Raj K., Prasad A. R. Tetrahedron. 2003;59:1805. [Google Scholar]
- Azizian J., Mohammadi A. A., Karimi A. R., Mohammadizadeh M. R. J. Org. Chem. 2005;70:350. doi: 10.1021/jo049138g. [DOI] [PubMed] [Google Scholar]
- Wang L., Liu J., Tian H., Qian C., Sun J. Adv. Synth. Catal. 2005;347:689. [Google Scholar]
- Azizian J., Mohammadi A. A., Soleimani E., Karimi A. R., Mohammadizadeh M. R. J. Heterocyclic Chem. 2006;43:187. [Google Scholar]
- Yadav J. S., Subba Reddy B. V., Ramesh Reddy A., Narsaiah A. V. Synthesis. 2007;20:3191. [Google Scholar]
- Humphries P. S., Balan G., Bechle B. M., Conn E. L., Dirico K. J., Hui Y., Oliver R. M., Southers J. A., Yang X. Tetrahedron Lett. 2009;50:2140. [Google Scholar]
- Karimi A. R., Pashazadeh R. Synthesis. 2010;3:437. [Google Scholar]
- Guranova N., Dar'in D., Krasavin M. Synthesis. 2018;50:2001. [Google Scholar]
- Jiménez-Díaz M. B., Ebert D., Salinas Y., Pradhan A., Lehane A. M., Myrand-Lapierre M.-E., O'Loughlin K. G., Shackleford D. M., de Almeida M. Justino, Carrillo A. K., Clark J. A., Dennis A. S. M., Diep J., Deng X., Duffy S., Endsley A. N., Fedewa G., Guiguemde W. A., Gómez M. G., Holbrook G., Horst J., Kim C. C., Liu J., Lee M. C. S., Matheny A., Martínez M. Santos, Miller G., Rodríguez-Alejandre A., Sanz L., Sigal M., Spillman N. J., Stein P. D., Wang Z., Zhu F., Waterson D., Knapp S., Shelat A., Avery V. M., Fidock D. A., Gamo F.-J., Charman S. A., Mirsalis J. C., Ma H., Ferrer S., Kirk K., Angulo-Barturen I., Kyle D. E., DeRisi J. L., Floyd D. M., Guy R. K. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E5455. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd D. M., Stein P., Wang Z., Liu J., Castro S., Clark J. A., Connelly M., Zhu F., Holbrook G., Matheny A., Sigal M. S., Min J., Dhinakaran R., Krishnan S., Bashyum S., Knapp S., Guy R. K. J. Med. Chem. 2016;59:7950. doi: 10.1021/acs.jmedchem.6b00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothweiler U., Czarna A., Krajewski M., Ciombor J., Kalinski C., Khazak V., Ross G., Skobeleva N., Weber L., Holak T. A. ChemMedChem. 2008;3:1118. doi: 10.1002/cmdc.200800025. [DOI] [PubMed] [Google Scholar]
- Weber L., WO2006/097323A1, 2006.
- Humpries P. S., Benbow J. W., Bonin P. D., Boyer D., Doran S. D., Frisbie R. K., Piotrowski D. W., Balan G., Bechle B. M., Conn E. L., Dirico K. J., Oliver R. M., Soeller W. C., Southers J. A., Yang X. Bioorg. Med. Chem. Lett. 2009;19:2400. doi: 10.1016/j.bmcl.2009.03.082. [DOI] [PubMed] [Google Scholar]
- Hashimoto T., Maruoka K. Chem. Rev. 2015;115:5366. doi: 10.1021/cr5007182. [DOI] [PubMed] [Google Scholar]
- Roscales S., Plumet J. Org. Biomol. Chem. 2019;16:8446. doi: 10.1039/c8ob02072h. [DOI] [PubMed] [Google Scholar]
- Bakulev V. A., Beryozkina T., Thomas J., Dehaen W. Eur. J. Org. Chem. 2018:262. [Google Scholar]
- Arrastia I., Arrieta A., Cossío F. P. Eur. J. Org. Chem. 2018:5889. doi: 10.1002/ejoc.201800911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Wang C. Org. Biomol. Chem. 2018;16:2591. doi: 10.1039/C7OB02686B. [DOI] [PubMed] [Google Scholar]
- Yue G., Wu Y., Dou Z., Chen H., Yin Z., Song X., He C., Wang X., Feng J., Zhang Z., Zoua P., Lu C. New J. Chem. 2018;42:20024. [Google Scholar]
- Maclean D., Schullek J. R., Murphy M. M., Ni Z., Gordon E. M., Gallop M. A. Proc. Natl. Acad. Sci. U. S. A. 1997;94:2805. doi: 10.1073/pnas.94.7.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. C., Lai D., Liu Q. Z., Zhou L., Liu Q., Liu Z. L. Molecules. 2016;21:1665. [Google Scholar]
- Kumar R. S., Almansour A. I., Arumugam N., Mohammad F., Alshahrani W. S., Dupadahalli K., Altaf M., Azama M., Menendez J. C. RSC Adv. 2018;8:41226. doi: 10.1039/c8ra07985d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater M. J., Amphlett E. M., Andrews D. M., Bravi G., Burton G., Cheasty A. G., Corfield J. A., Ellis M. R., Fenwick R. H., Fernandes S., Guidetti R., Haigh D., Hartley C. D., Howes P. D., Jackson D. L., Jarvest R. L., Lovegrove V. L. H., Medhurst K. J., Parry N. R., Price H., Shah P., Singh O. M. P., Stocker R., Thommes P., Wilkinson C., Wonacott A. J. Med. Chem. 2007;50:897. doi: 10.1021/jm061207r. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Liu L., Sun W., Lu J., McEachern D., Li X., Yu S., Bernard D., Ochsenbein P., Ferey V., Carry J., Deschamps J. R., Sun D., Wang S. J. Am. Chem. Soc. 2013;135:7223. doi: 10.1021/ja3125417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.