

Abstract
APOBEC3B (A3B) is a prominent source of mutation in many cancers. To date, it has been difficult to capture the native protein-DNA interactions that confer A3B’s substrate specificity by crystallography due to the highly dynamic nature of wild-type A3B active site. We use computational tools to restore a recent crystal structure of a DNA-bound A3B C-terminal domain mutant construct to its wild type sequence, and run molecular dynamics simulations to study its substrate recognition mechanisms. Analysis of these simulations reveal dynamics of the native A3Bctd-oligonucleotide interactions, including the experimentally inaccessible loop 1-oligonucleotide interactions. A second series of simulations in which the target cytosine nucleotide was computationally mutated from a deoxyribose to a ribose show a change in sugar ring pucker, leading to a rearrangement of the binding site and revealing a potential intermediate in the binding pathway. Finally, apo simulations of A3B, starting from the DNA-bound open state, experience a rapid and consistent closure of the binding site, reaching conformations incompatible with substrate binding. This study reveals a more realistic and dynamic view of the wild type A3B binding site and provides novel insights for structure-guided design efforts for A3B.
Graphical Abstract
Introduction
The APOBEC3 (A3) family of cytidine deaminases is a recently discovered endogenous source of mutation in cancer.1–3 Previously, A3 proteins were studied in the context of their interactions with viruses4, 5 and efforts were undertaken to discover small molecules that modulate the mutational activities of the virally restrictive APOBEC3G enzyme.6–8 Later studies linked A3-driven mutagenesis to cancer progression and recurrence.9–16 A3 proteins have specific substrate sequence preferences, and analyses of some cancer genomes have shown an enrichment of A3 signature mutations, defined as C-to-T and C-to-G in 5’TCW motifs.17–22 The majority of evidence indicates that APOBEC3B (A3B) is the dominant source of these mutations in human cancer.10, 12, 17, 18, 22, 23
A3B is a dual-domain A3 that prefers to deaminate cytosines at TC motifs in single-stranded (ss)DNA, with weaker preference placed on further upstream and downstream bases.17, 24 Each A3 protein consists of either one or two deaminase domains. In A3s with two deaminase domains, like A3B, only the C-terminal domain (ctd) shows significant catalytic activity. In these dual-domain A3s, the role of the N-terminal domain (ntd) is thought to be regulatory and responsible for activities such as subcellular localization.25–27 Despite differentiated divisions of labor, all A3 domains share a minimum sequence identity of 30%, which is associated with a high degree of 3D structure similarity.28, 29
Wild-type A3Bctd has yet to yield to crystallographic approaches. While NMR studies have enabled modeling of solution-state A3Bctd structures30, apo form crystal structures could only be elucidated after introducing numerous stabilizing mutations and truncating loop 3.31 Initial structures with this construct showed loops 1 and 7 in tight contact, resulting in a closed active site.31 Further attempts to capture A3Bctd in an open conformation using different crystal forms resulted in alternative, yet still closed conformations of the active site.32 All atom, explicitly solvated molecular dynamics (MD) simulations also indicated an intrinsic bias for the A3Bctd active site toward a closed conformation via distinct modes of interaction between loop 1 and loop 7.32 Finally, a DNA-bound crystal structure of A3Bctd was achieved but only after an additional A3A loop 1 swap in addition to the previously used loop 3 truncation and solubilizing mutations (Figure 1A).33 Despite its high value, the information in this A3Bctd-ssDNA crystal structure still leaves several questions open about how loop 1 residues interact with ssDNA substrates in wild-type A3Bctd.
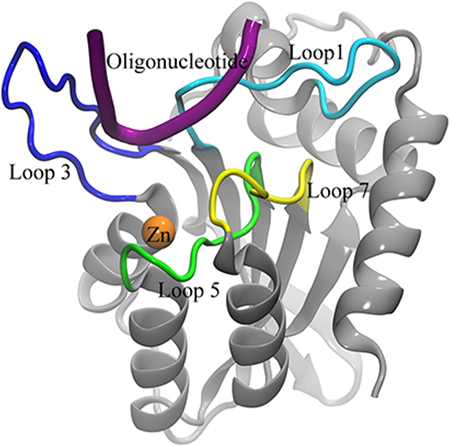
Figure 1:


(A) Cartoon view of the protein, labeling features of interest including loops 1 (light blue), 3 (blue), 5 (green), and 7 (yellow), the substrate oligonucleotide (purple), and the catalytic zinc (orange). (B) Numbering system used to identify atoms in nucleotides. R=DNA (C) Geometry of C2’ endo sugar pucker taken from the starting configuration of the dC simulation, and (D) C3’ endo sugar pucker taken from the second rC simulation. (E) Sugar pucker of the target C, measured in both A3B-dC and (F) A3B-rC simulations. In rC simulation replicate 2, the RNA transitions from a C2’ endo (DNA-preferred) to a C3’ endo (RNA-preferred) sugar pucker. (G) RMSD of target C compared to its initial pose in A3Bctd-DNA (black) and A3Bctd- DNA-rC (red) simulations.
The development of therapies targeting A3 proteins is likely to be assisted by understanding their substrate recognition processes. In general, protein-ssDNA recognition has been found to be mediated by hydrogen bonds and shape complementarity.34–37 Interestingly, the dinucleotide substrate preferences of A3 proteins can be exchanged through the transfer of loop 7 residues.38–40 As expected from these mutagenesis studies, the initial crystal structures of apo A3Bctd showed loop 7 adjacent to the active site.31 Subsequent crystallization of a variant of A3Bctd and A3A in complex with ssDNA substrates confirmed the importance of loop 7 residues and also revealed a role for loop 1 residues in holding ssDNA substrates in the active site cavity.33, 41 Other experimental studies have explored the binding of A3s to chemically modified oligonucleotides, such as those with a ribose-cytidine (rC) base at the target site, as well as other modifications to the oligonucleotide backbone and target base.42–48 Notably, one of these studies found that A3B prefers normal cytosine bases within ssDNA but can bind to and catalyze the deamination of other substrates including 5-methyl-cytosine bases with significantly lower activity.44 DNA and RNA sugars differ in composition by only one hydroxyl group (at the C2’ position), which changes the backbone sugar ring pucker preference from C2’ endo (for DNA) to C3’ endo (for RNA). This difference may contribute to A3B’s selectivity for ssDNA over RNA but the exact mechanism of this selectivity is not known.
One powerful technique to understand the biophysics of proteins and biological interactions is MD computer simulations.49 These simulations model dynamics of all the atoms of a chemical system, starting with an initial geometry and undergoing motion according to the laws of physics at physiologic temperature. MD simulations are capable of modeling not just proteins, but also solvents, ions, and nucleic acids.50–53 With increases in computing power, more questions have fallen within the scope of computational modeling, and MD simulations have begun to find valuable synergies with traditional biochemistry and structural biology. These synergies include showing the atomic-level mechanisms that explain underlying biochemical observations and proposing new routes for further experiments.54–57
In this work, we use explicitly solvated, all atom MD simulations to explore native interactions of wild-type A3Bctd with different oligonucleotide substrates. These simulations reveal the importance of base-specific hydrogen bonds, pocket shape, and backbone sugar conformation in A3Bctd-substrate binding. We also simulate an A3Bctd-substrate complex with a ribose-C as the target nucleotide instead of deoxyribose-C, and observe a ring pucker change to the RNA-preferred C3’ endo conformation. This pucker change leads to a shift away from the crystallized binding pose, revealing a potential intermediate conformation in A3Bctd-oligonucleotide binding that may provide structural insights for further biochemical and therapeutic work. Finally, we simulate apo A3Bctd, and find that, in the absence of a substrate, the DNA binding cleft rapidly and consistently closes, entering a conformation incompatible with substrate binding. Correlations between phenomena in these simulations and prior experimental findings suggest that the atomic-level details of these models can accelerate therapeutic studies of A3B.
Methods
Simulations of A3Bctd and nucleic acid substrates were parameterized using the AMBER FF14SB force field for protein atoms, and FF99BSC0 and FF99BSC0_chi0L3 force field for DNA and RNA atoms, respectively.58–64 The starting coordinates for oligonucleotide-bound simulations were based on PDB entry 5TD5 (Figure 1A). Solvents in the crystal structure other than waters were removed. In each system, amino acid substitutions were reverted to wild-type and missing residues were modeled using the Schrodinger PRIME software suite.65, 66 Crystal waters were left in place, and protonation states and hydrogen coordinates were assigned by VMD PropKa.67, 68 After proton assignment, the protein had a net charge of −7. The catalytic zinc ion and the zinc-coordinating residues in the active site were modeled according to the Cationic Dummy Atom Model.69 The catalytic zinc was bound to a OH− ion, in order to model the pre-catalysis substrate recognition dynamics of A3Bctd. Simulations were embedded in a TIP3P water box generated by LEaP from the AmberTools suite with a buffer distance of 10 A.63 Na and Cl ions were added to neutralize charge and attain a concentration of 0.2 M.61
Three A3Bctd systems were simulated based on the coordinates from PDB ID 5TD5: A DNA-bound system with nucleotide sequence 5’-TTCATG-3’, a hybrid oligonucleotide-bound system with nucleotide sequence 5’-TTrCATG-3’ (where rC indicates a ribonucleotide cytidine), and an apo simulation with all DNA atoms removed. Each system underwent energy minimization in its force field, followed by gradual heating and equilibration with decreasing restraints. AMBER input scripts for each step are provided in the Supplemental Materials. Each system was simulated in triplicate, differing in temperature initialization seed, and each replicate underwent 1 μs of unrestrained MD simulation in an NPT ensemble at 310 K.
The sugar pucker of target nucleotide was monitored using cpptraj.70–72 Analysis of hydrogen bonds was performed using the MDTraj Python package73 and visualized using Matplotlib.74 The existence of hydrogen bonds was defined by Baker-Hubbard criteria.75 The hydrogen bond analysis was performed on snapshots taken at increments of 5 ns in the trajectories. In order to study sequence-specific hydrogen bonding to the oligonucleotide substrates, only hydrogen bonds that involve a nucleobase atom or the target C’s sugar and that appear in at least 15% of any simulation’s snapshots are shown in Table 1.
Table 1:
Frequent protein-oligonucleotide hydrogen bonds made in either the A3Bctd-ssDNA or ssDNA-rC simulations. Colors indicate frequency of hydrogen bonds (% of frames in which bonds occur). Dark red: 0%, light red: 1–33%, yellow: 34–66%, green: 67+%.
| Hydrogen Bond | TTCATG | TTrCATG C2’ endo | TTrCATG C3’ endo |
|---|---|---|---|
| −1 T 3-position NH - Asp314 sidechain O | 94% | 86% | 0% |
| −1 T 3-position NH - Asp316 sidechain O | 0% | 0% | 96% |
| −1 T 2-position O - Tyr 315 backbone NH | 51% | 59% | 0% |
| Target C sugar O2’ - Thr214 sidechain OH | N/A | 3% | 94% |
| Target C sugar O2’H - His253 sidechain Nε | N/A | 18% | 0% |
| Target C sugar O3’ - Asn240 sidechain NH | 22% | 42% | 0% |
| Target C sugar O4’ - Thr214 sidechain OH | 89% | 74% | 0% |
| Target C 2-position O - Ala254 backbone NH | 95% | 76% | 0% |
| Target C 3-position amine - Ser282 backbone C=O | 96% | 85% | 93% |
| Target C 3-position amine - Glu255 sidechain O | 85% | 35% | 0% |
| + 1 A 3-position N - Arg212 sidechain guanidinium | 21% | 17% | 42% |
Pocket volumes were studied using POVME3.076“78 and visualized using Visual Molecular Dynamics.79 The pocket region was defined by a set of inclusion spheres which cover the observed DNA-binding region (Figure S1). This region is defined as running between loops 1 and 7, down into the zinc-containing active site pocket, and out between loops 1 and 3. Because quantitative comparison of the pockets was performed, the POVME convex hull exclusion option was not used, per suggested POVME3.0 best practices.76 All trajectories were aligned by their backbone atoms to the starting structure of the DNA-bound A3Bctd MD simulation (after equilibration).
Results and Discussion
Changing the target nucleotide from DNA to RNA triggers substrate release from A3Bctd active site
The simulations containing the ribose-C (rC) nucleotide at the target position displayed major differences from those containing the deoxyribose-C (dC). In the dC simulations, the −1, +1 and target cytidine nucleotide remained in their original position (RMSD < 2.2 Å). However, in the second rC simulation, the target nucleotide underwent a major shift (RMSD > 3 Å), as shown in Figure 1G. The most characteristic event in this shift is a change in the sugar pucker of the rC nucleotide from the DNA-preferred 2’-endo conformation to the RNA-preferred 3’-endo conformation (Figure 1C–F, Movie S1). This shift in DNA-rC binding pose is simultaneous with a rearrangement of the binding site (Figure 2).
Figure 2:

Binding site-adjacent residues. (A) Whole protein view and (B) catalytic pocket focused view of the simulation with target dC, showing the starting (crystal structure-based) conformation of the substrate oligonucleotide after minimization. (C) Whole protein view and (D) catalytic pocket focused view of the average structure of the C3’ endo portion of the rC simulation, as determined by POVMEpocket shape analysis. Loop 1 is shown in light blue, loop 3 in dark green, loop 5 in light green, and loop 7 in yellow.
The rC pucker change correlates with a change in binding site shape. A comparison of average binding pocket shape between the A3Bctd-ssDNA simulation and C3’ endo snapshots of the A3Bctd-ssDNA-rC is shown in Figure 3. Notably, the change in sugar pucker shifts the DNA-rC substrate toward loop 1, and away from the catalytic glutamate residue and loop 3 (Figure S5).
Figure 3:

Average binding cleft shapes differ between (A) dC simulations (C2’ endo/deoxyribose, red) and (B) the portion of the rC simulation after the sugar pucker change (C3’ endo/ribose, blue). Mesh shows average pocket shape along the DNA binding cleft observed in crystal structures. The atoms in the dC figure structure are the initial coordinates of the simulation, while C3’ endo atomic coordinates are taken from the POVME cluster centroid snapshot. The −1 T in the TTrCATG C3’ endo snapshots leaves the original substrate binding cleft and is no longer in the pocket defined in the 5TD5 crystal. Images are taken from the same angle and structures are RMSD-aligned. Catalytic zinc is shown as a gray sphere.
Interaction footprint of A3Bctd with oligonucleotides
The shifted conformation in the second DNA-rC simulation can also be characterized by movements in the binding pocket residues, as shown in Figure 2, and changes in the network of hydrogen bonds between the protein and target rC. A table of all common hydrogen bonds between base and protein atoms is shown in Table 1, and a more detailed time series plot is provided in Figure S2. As the dC simulations do not have the 2’ O atom, hydrogen bonds involving it are labeled N/A.
After the ring pucker change, the 2’ O atom of the target rC forms a hydrogen bond with Thr214’s sidechain. In non-shifted DNA-rC simulations, the Thr214 sidechain maintains a hydrogen bond to the 4’ O of the target rC. This shift correlates with a breaking of all major protein-base hydrogen bonds in the other nucleotides of the chain, except that between Ser282 and the amine to be hydrolyzed at the 4-position on the rC. In all rC simulations, the initial hydrogen bond between the target C nucleotide’s amine and the catalytic Glu255 residue, is broken partway through each simulation.
Experiments on A3A have shown that Asp131, homologous to A3B’s Asp314, confers the preference for T at the −1 position of the oligonucleotide.33, 41 Both the crystal structures of A3B and our simulations show hydrogen bonds consistently formed between the 3-position NH motif of the T −1 base and the Asp314 sidechain. Interestingly, the 3’-endo portion of the rC simulation has broken this hydrogen bond and replaced it with one to the sidechain of Asp316. Asp316 was shown to be essential for A3B antiviral function and is therefore likely involved in DNA binding.80 Given that this shift only appears in the ssDNA-rC simulation, it is possible that the perturbation caused by the target C’s change to a ribose backbone aided the system in leaving its initial energy well and exploring intermediate binding poses. It is also possible that Asp316 contributes by an indirect electrostatic mechanism when it is not directly forming hydrogen bonds for substrate recognition.
Another less frequent base hydrogen bond with the −1 T intermittently forms with the backbone NH of Tyr315. This hydrogen bond, however, is not specific to the Tyr sidechain, as the contact is to the protein backbone. It is also not specific to thymine, as the other pyrimidine, cytosine, also has an oxygen at the 2-position on its nucleobase. A3G is the only A3 with a strong −1 preference other than T, and it prefers C. The position corresponding to A3B Tyr315 is A3G Asp317, and A3G is the only A3 with a residue other than Tyr or Phe at that position.
While all six nucleotides from the MD simulations were analyzed, only hydrogen bonds involving the −1 T, C, and +1 A nucleotides showed bonding frequencies greater than 15%. The only hydrogen bond involving the +1 A nucleotide was infrequent, which correlates with substrate specificity experiments that found a weaker preference toward the +1 position compared to the −1.17, 24 Thus, while hydrogen bonding can offer an explanation for the target C and −1 T base specificity, the simulations do not reveal specific sidechain-base hydrogen bonding for other nucleotides. The data imply, however, that electrostatic and/or shape-based recognition may take place. In our simulations, the positively charged sidechains of loop 1 residues contact the negatively charged phosphate backbone of the oligonucleotide. These positively charged loop 1 residues are known to be key for activity in A3A and A3Gctd, as A3A H29 and A3G H216 (homologous to A3B R212) could be mutated to an Arg while maintaining residual activity.30, 81 However, when A3G H216 is mutated to Ala, it loses activity.81, 82 While the contacts of these positively charged residues to the phosphate backbone appear to be charge-driven and are not specific to one nucleotide sequence, they may be enabled by the packing of the preferred substrate to the loops, in what may be a conformation- or shape-driven recognition process.
After the change of the rC sugar to the C3’ endo conformation, the target cytosine is too far from the catalytic Glu255 to perform deamination. This new binding mode may be an intermediate conformation in normal DNA binding and/or dissociation, however it was only observed in one of the DNA-rC simulations. It is possible that further simulation of the other DNA-rC systems might eventually show the same shift or further oligonucleotide dissociation.
Notably, the DNA simulations and the C2’ endo portion of the DNA-rC simulations maintain a relatively similar DNA interaction interface (Figure 4). While the C3’ endo portion of the DNA-rC simulation still has the target C buried in the catalytic pocket, the neighboring nucleotides experience an outward shift and define a new interaction surface. This shift moves the −1 T down loop 7, and partially dissociates the −2 T from the protein. The shift of the target rC away from loop 3 brings the +2 T and +3 G in contact with the surface of loop 3 that faces the catalytic pocket, though our hydrogen bond analysis does not indicate that specific interactions drive this association.
Figure 4:

Oligonucleotide interaction surface differences between DNA (left) and DNA-rC (center and right) simulations. Structures from the end of each simulation are shown. Each protein atom is colored by its frequency of contact (<5 Angstrom distance) with the oligonucleotide, on a color scale from blue (no contact) to white (50% contact) to red (100% contact). The oligonucleotides are colored by RMSF, with black corresponding to 0 angstroms and white to 5 Angstroms. The C3’ endo portion of the DNA-rC simulation shows a shift in position of the 0 and −1 nucleotides of the substrate, but low RMSD after the shift. Structures are RMSD-aligned.
Apo A3Bctd simulations show closure of substrate binding cleft
Loops 1, 3, and 7 have been identified as being primarily responsible for substrate recognition, and our data indicate that their interaction patterns are directly affected by the presence of the oligonucleotide (Figure 5). Both the ssDNA-bound and DNA-rC-bound simulations show significantly fewer loop-loop contacts compared to the apo A3Bctd simulations, commensurate with their high number of loop-oligonucleotide interactions.
Figure 5:

Frequency of loop-loop contacts in A3Bctd simulations. White indicates infrequent contacts, and black indicates contacts 50% of the time or more. Contacts are defined as a closest heavy atom distance of < 4 angstroms.
The apo simulations show extensive loop 1 - loop 3 interactions, specifically Arg212 and Gln213 to Asn240, Glu241, Ala242, and Lys243. These contacts are made less frequently in the DNA-bound and DNA-rC-bound simulations. This is to be expected, as the substrate is bound between loops 1 and 3. The almost complete loss of loop 1 - loop 3 contacts after the change to the C3’ endo sugar pucker is due to the substrate adopting a series of non-specific contacts between the +1, +2, and +3 nucleotides and loop 3, thereby preventing loop 3 residues from contacting loop 1.
The apo simulations also show the most loop 1 - loop 7 contacts. This is to be expected, as the substrate oligonucleotide passes directly between these loops. Arg311 in loop 7 makes contact with most residues in the first half of loop 1, from Asn203 to Arg210. The apo simulation is the only simulation in which Tyr313 contacts loop 1, primarily via Arg211, but also less frequently through the flanking Arg210 and Arg212. Both the apo and rC C3’ endo simulations show frequent contacts between Tyr315 on loop 7 and Leu209, Arg210, and Arg211 on loop 1. Generally, the large number and frequency of contacts in the apo and rC C3’ endo snapshots indicate a more closed binding site, again implying that the DNA-rC simulation may have captured an intermediate-bound state of the complex.
The simulations in which ssDNA was removed show a rapid closure of the cleft, leading to a protein conformation incompatible with substrate binding, as seen in Figure 6. This trend was consistent in all three simulation replicates. Interestingly, all three replicates of the apo simulations reach distinct closed states, which can be characterized by different geometries in which arginines 210, 211, and 212 in loop 1 contact tyrosines 313 and 315 in loop 7 (Figure S6). The DNA and DNA-rC simulations maintain an open binding cleft, except for the DNA-rC replicate which experiences a change in sugar pucker and partial unbinding of the substrate. Further, the apo simulation of A3Bctd converges to a much lower pocket volume than was observed in our prior work on A3A (average A3A apo volume shown in magenta).32 This difference is largely due to A3A’s shorter loop 1, and the propensity of the A3A binding cleft to remain open could explain its higher level of deaminase activity relative to A3B.
Figure 6:

Relative pocket volumes of A3Bctd substrate binding cleft duringMD simulations. Each simulation was run in triplicate, with replicates shown as lighter and darker shades of the same color. Average pocket volumes are shown as thick dotted lines. Average volume from the same analysis performed on A3A apo simulations used in prior work shown in magenta.32
Discussion
APOBEC3 enzymes are of high therapeutic interest as anti-viral and anti-cancer strategies.2, 8 Moreover, efforts to use APOBEC3 enzymes as genome engineering tools, for example as A3-Cas9 fusions for targeted base editing, will benefit from a comprehensive understanding of ssDNA binding and deamination mechanisms. ‘ Due to the highly dynamic nature of the A3Bctd active site, it has not been possible to capture its native DNA interaction footprint via x-ray crystallography. In this work, we generate a DNA-bound wild-type A3Bctd model from the recent A3Bctd variant crystal structure in complex with DNA, and investigate the native A3Bctd-DNA interactions and their dynamics using MD simulations. Our simulations offer insight into the mechanisms of substrate recognition and binding. Further, they show significant differences arising from the presence of a non-preferred oligonucleotide. As expected, apo simulations of A3Bctd beginning in the open state experience a rapid and consistent closure of the binding site, reaching a conformation incompatible with substrate binding.
Analyses of MD simulations reveal a set of dynamic hydrogen bonds important for substrate recognition at the −1, 0, and +1 positions of the oligonucleotide. Many of these contacts agree with previous experimental findings, for instance confirming the importance of Asp314 in −1T recognition. Others provide novel routes for further experiments, such as the observation that Asn240 hydrogen bonds more strongly to the 03’ of RNA than DNA, suggesting that an A3B N240V mutant may show reduced overall activity, but stronger selectivity toward DNA over RNA. The interactions of the substrate-recognition loops with each other are also monitored. The loop-loop interactions of the apo protein provide insight into the thermodynamics and kinetics of substrate binding, as many of these contacts must be broken to accommodate a ssDNA substrate.
The perturbation of the DNA substrate to a ribose sugar at only the target nucleotide caused one simulation replicate to undergo a ring pucker change and explore a less tightly-bound conformation of the oligonucleotide substrate. In this novel binding pose, the −1T H-bond with Asp314 is replaced with one to Asp316, the shape of the catalytic pocket is widened, and the target C is shifted away from the catalytic glutamate. This partially unbound pose may represent a binding/dissociation intermediate and could be useful in discovering small molecules capable of A3B inhibition.
Supplementary Material
Acknowledgements
JRW was supported by the NIH Molecular Biophysics Training Grant T32 GM008326. This work was funded in part by the Director’s New Innovator Award Program NIH DP2 0D007237 to REA; R01-GM110129 to DAH, RSH, and REA; R01-GM118000 to RSH, HA, and DAH. Funding and support from the National Biomedical Computation Resource (NBCR) is provided through NIH P41 GM103426.
RSH is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished McKnight University Professor, and an Investigator of the Howard Hughes Medical Institute.
Footnotes
Conflict of Interest Statement:
DAH & RSH are co-founders, consultants, and shareholders of ApoGen Biotechnologies, Inc. REA & HA are consultants of ApoGen Biotechnologies, Inc.
REA is a cofounder of, has equity interest in, and is on the scientific advisory board of Actavalon, Inc.
Supporting Information Available
5 figures, 1 video and MD input files related to this work are prepared as Supporting Information which is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Siriwardena SU; Chen K; Bhagwat AS, Functions and Malfunctions of Mammalian DNA-Cytosine Deaminases. Chem. Rev 2016, 116, 12688–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venkatesan S; Rosenthal R; Kanu N; McGranahan N; Bartek J; Quezada SA; Hare J; Harris RS; Swanton C, Perspective: APOBEC Mutagenesis in Drug Resistance and Immune Escape in HIV and Cancer Evolution. Ann. Oncol 2018, 29, 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helleday T; Eshtad S; Nik-Zainal S, Mechanisms Underlying Mutational Signatures in Human Cancers. Nat. Rev. Genet 2014, 15, 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malim MH; Emerman M, HIV-1 Accessory Proteins--Ensuring Viral Survival in a Hostile Environment. Cell Host Microbe 2008, 3, 388–398. [DOI] [PubMed] [Google Scholar]
- 5.Reuben S Harris JPD, APOBECs and Virus Restriction. Virology 2015, 0, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li M; Shandilya SMD; Carpenter MA; Rathore A; Brown WL; Perkins AL; Harki DA; Solberg J; Hook DJ; Pandey KK; Parniak MA; Johnson JR; Krogan NJ; Somasundaran M; Ali A; Schiffer CA; Harris RS, First-in-Class Small Molecule Inhibitors of the Single-Strand DNA Cytosine Deaminase APOBEC3G. ACS Chem. Biol 2012, 7 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson ME; Li M; Harris RS; Harki DA, Small-Molecule APOBEC3G DNA Cytosine Deaminase Inhibitors Based on a 4-amino-1,2,4-triazole-3-thiol Scaffold. ChemMedChem 2013, 8, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olson ME; Harris RS; Harki DA, APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell Chem Biol 2018, 25, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieuwerts AM; Schrijver WAME; Dalm SU; de Weerd V; Moelans CB; Ter Hoeve N; van Diest PJ; Martens JWM; van Deurzen CHM, Progressive APOBEC3B mRNA Expression in Distant Breast Cancer Metastases. PLoS One 2017, 12, e0171343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sieuwerts AM; Willis S; Burns MB; Look MP; Meijer-Van Gelder ME; Schlicker A; Heideman MR; Jacobs H; Wessels L; Leyland-Jones B; Gray KP; Foekens JA; Harris RS; Martens JWM, Elevated APOBEC3B Correlates with Poor Outcomes for Estrogen-Receptor-Positive Breast Cancers. Horm. Cancer 2014, 5, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cescon DW; Haibe-Kains B; Mak TW, APOBEC3B Expression in Breast Cancer Reflects Cellular Proliferation, while a Deletion Polymorphism is Associated with Immune Activation. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 2841–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law EK; Sieuwerts AM; LaPara K; Leonard B; Starrett GJ; Molan AM; Temiz NA; Vogel RI; Meijer-van Gelder ME; Sweep FCGJ; Span PN; Foekens JA; Martens JWM; Yee D; Harris RS, The DNA Cytosine Deaminase APOBEC3B Promotes Tamoxifen Resistance in ER-Positive Breast Cancer. Sci Adv 2016, 2, e1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker BA; Wardell CP; Murison A; Boyle EM; Begum DB; Dahir NM; Proszek PZ; Melchor L; Pawlyn C; Kaiser MF; Johnson DC; Qiang Y-W; Jones JR; Cairns DA; Gregory WM; Owen RG; Cook G; Drayson MT; Jackson GH; Davies FE; Morgan GJ, APOBEC Family Mutational Signatures are Associated with Poor Prognosis Translocations in Multiple Myeloma. Nat. Commun 2015, 6, 6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagener R; Alexandrov LB; Montesinos-Rongen M; Schlesner M; Haake A; Drexler HG; Richter J; Bignell GR; McDermott U; Siebert R, Analysis of Mutational Signatures in Exomes from B-cell Lymphoma Cell Lines Suggest APOBEC3 Family Members to be Involved in the Pathogenesis of Primary Effusion Lymphoma. Leukemia 2015, 29, 1612–1615. [DOI] [PubMed] [Google Scholar]
- 15.Xu L; Chang Y; An H; Zhu Y; Yang Y; Xu J, High APOBEC3B Expression is a Predictor of Recurrence in Patients with Low-Risk Clear Cell Renal Cell Carcinoma. Urol. Oncol 2015, 33, 340.e1–8. [DOI] [PubMed] [Google Scholar]
- 16.Maura F; Petljak M; Lionetti M; Cifola I; Liang W; Pinatel E; Alexandrov LB; Fullam A; Martincorena I; Dawson KJ; Angelopoulos N; Samur MK; Szalat R; Zamora J; Tarpey P; Davies H; Corradini P; Anderson KC; Minvielle S; Neri A; Avet-Loiseau H; Keats J; Campbell PJ; Munshi NC; Bolli N, Biological and Prognostic Impact of APOBEC-Induced Mutations in the Spectrum of Plasma Cell Dyscrasias and Multiple Myeloma Cell Lines. Leukemia 2018, 32, 1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns MB; Lackey L; Carpenter MA; Rathore A; Land AM; Leonard B; Refsland EW; Kotandeniya D; Tretyakova N; Nikas JB; Yee D; Temiz NA; Donohue DE; McDougle RM; Brown WL; Law EK; Harris RS, APOBEC3B is an Enzymatic Source of Mutation in Breast Cancer. Nature 2013, 494, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns MB; Temiz NA; Harris RS, Evidence for APOBEC3B Mutagenesis in Multiple Human Cancers. Nat. Genet 2013, 45, 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts SA; Lawrence MS; Klimczak LJ; Grimm SA; Fargo D; Stojanov P; Kiezun A; Kryukov GV; Carter SL; Saksena G; Harris S; Shah RR; Resnick MA; Getz G; Gordenin DA, An APOBEC Cytidine Deaminase Mutagenesis Pattern is Widespread in Human Cancers. Nat. Genet 2013, 45, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexandrov LB; Nik-Zainal S; Wedge DC; Aparicio SAJR; Behjati S; Biankin AV; Bignell GR; Bolli N; Borg A; Børresen-Dale A-L; Boyault S; Burkhardt B; Butler AP; Caldas C; Davies HR; Desmedt C; Eils R; Eyfjörd JE; Foekens JA; Greaves M; Hosoda F; Hutter B; Ilicic T; Imbeaud S; Imielinski M; Imielinsk M; Jäger N; Jones DTW; Jones D; Knappskog S; Kool M; Lakhani SR; López-Otín C; Martin S; Munshi NC; Nakamura H; Northcott PA; Pajic M; Papaemmanuil E; Paradiso A; Pearson JV; Puente XS; Raine K; Ramakrishna M; Richardson AL; Richter J; Rosenstiel P; Schlesner M; Schumacher TN; Span PN; Teague JW; Totoki Y; Tutt ANJ; Valdés-Mas R; van Buuren MM; van ‘t Veer L; Vincent-Salomon A; Waddell N; Yates LR; Australian Pancreatic Cancer Genome, I.; Consortium, I. B. C.; Consortium, I. M.-S.; PedBrain I; Zucman-Rossi J; Futreal PA; McDermott U; Lichter P; Meyerson M; Grimmond SM; Siebert R; Campo E; Shibata T; Pfister SM; Campbell PJ; Stratton MR, Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nik-Zainal S; Alexandrov LB; Wedge DC; Van Loo P; Greenman CD; Raine K; Jones D; Hinton J; Marshall J; Stebbings LA; Menzies A; Martin S; Leung K; Chen L; Leroy C; Ramakrishna M; Rance R; Lau KW; Mudie LJ; Varela I; McBride DJ; Bignell GR; Cooke SL; Shlien A; Gamble J; Whitmore I; Maddison M; Tarpey PS; Davies HR; Papaemmanuil E; Stephens PJ; McLaren S; Butler AP; Teague JW; Jönsson G; Garber JE; Silver D; Miron P; Fatima A; Boyault S; Langerød A; Tutt A; Martens JWM; Aparicio SAJR; Borg Å; Salomon AV; Thomas G; Børresen-Dale A-L; Richardson AL; Neuberger MS; Futreal PA; Campbell PJ; Stratton MR; Breast Cancer Working Group of the International Cancer Genome, C., Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bruin EC; McGranahan N; Mitter R; Salm M; Wedge DC; Yates L; Jamal-Hanjani M; Shafi S; Murugaesu N; Rowan AJ; Grönroos E; Muhammad MA; Horswell S; Gerlinger M; Varela I; Jones D; Marshall J; Voet T; Van Loo P; Rassl DM; Rintoul RC; Janes SM; Lee S-M; Forster M; Ahmad T; Lawrence D; Falzon M; Capitanio A; Harkins TT; Lee CC; Tom W; Teefe E; Chen S-C; Begum S; Rabinowitz A; Phillimore B; Spencer-Dene B; Stamp G; Szallasi Z; Matthews N; Stewart A; Campbell P; Swanton C, Spatial and Temporal Diversity in Genomic Instability Processes Defines Lung Cancer Evolution. Science 2014, 346, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanu N; Cerone MA; Goh G; Zalmas L-P; Bartkova J; Dietzen M; McGranahan N; Rogers R; Law EK; Gromova I; Kschischo M; Walton MI; Rossanese OW; Bartek J; Harris RS; Venkatesan S; Swanton C, DNA Replication Stress Mediates APOBEC3 Family Mutagenesis in Breast Cancer. Genome Biol. 2016, 17, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leonard B; Hart SN; Burns MB; Carpenter MA; Temiz NA; Rathore A; Vogel RI; Nikas JB; Law EK; Brown WL; Li Y; Zhang Y; Maurer MJ; Oberg AL; Cunningham JM; Shridhar V; Bell DA; April C; Bentley D; Bibikova M; Cheetham RK; Fan J-B; Grocock R; Humphray S; Kingsbury Z; Peden J; Chien J; Swisher EM; Hartmann LC; Kalli KR; Goode EL; Sicotte H; Kaufmann SH; Harris RS, APOBEC3B Upregulation and Genomic Mutation Patterns in Serous Ovarian Carcinoma. Cancer Res. 2013, 73, 7222–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lackey L; Law EK; Brown WL; Harris RS, Subcellular Localization of the APOBEC3 Proteins during Mitosis and Implications for Genomic DNA Deamination. Cell Cycle 2013, 12, 762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pak V; Heidecker G; Pathak VK; Derse D, The Role of Amino-Terminal Sequences in Cellular Localization and Antiviral Activity of APOBEC3B. J. Virol 2011,85, 8538–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stenglein MD; Matsuo H; Harris RS, Two Regions within the Amino-Terminal Half of APOBEC3G Cooperate To Determine Cytoplasmic Localization. Journal of Virology 2008, 82, 9591–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aydin H; Taylor MW; Lee JE, Structure-Guided Analysis of the Human APOBEC3-HIV Restrictome. Structure 2014, 22, 668–684. [DOI] [PubMed] [Google Scholar]
- 29.Shandilya SMD; Bohn M-F; Schiffer CA, A Computational Analysis of the Structural Determinants of APOBEC3’s Catalytic Activity and Vulnerability to HIV-1 Vif. Virology 2014,471–473, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byeon I-JL; Byeon C-H; Wu T; Mitra M; Singer D; Levin JG; Gronenborn AM, Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry 2016, 55, 2944–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi K; Carpenter MA; Kurahashi K; Harris RS; Aihara H, Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J. Biol. Chem 2015, 290, 28120–28130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi K; Demir O; Carpenter MA; Wagner J; Kurahashi K; Harris RS; Amaro RE; Aihara H, Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci. Rep 2017, 7, 17415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi K; Carpenter MA; Banerjee S; Shaban NM; Kurahashi K; Salamango DJ; McCann JL; Starrett GJ; Duffy JV; Demir O; Amaro RE; Harki DA; Harris RS; Aihara H, Structural Basis for Targeted DNA Cytosine Deamination and Mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol 2016, 24, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luscombe NM; Laskowski RA; Thornton JM, Amino Acid-Base Interactions: a Three-Dimensional Analysis of Protein-DNA Interactions at an Atomic Level. Nucleic Acids Res. 2001, 29, 2860–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siggers T; Gordan R, Protein-DNA Binding: Complexities and Multi-Protein Codes. Nucleic Acids Res. 2014, 42, 2099–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harteis S; Schneider S, Making the Bend: DNA Tertiary Structure and Protein-DNA Interactions. Int. J. Mol. Sci 2014, 15, 12335–12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nadassy K; Wodak SJ; Janin J, Structural Features of Protein-Nucleic Acid Recognition Sites. Biochemistry 1999, 38, 1999–2017. [DOI] [PubMed] [Google Scholar]
- 38.Kohli RM; Abrams SR; Gajula KS; Maul RW; Gearhart PJ; Stivers JT, A Portable Hot Spot Recognition Loop Transfers Sequence Preferences from APOBEC Family Members to Activation-induced Cytidine Deaminase. J. Biol. Chem 2009, 284, 22898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M; Rada C; Neuberger MS, Altering the Spectrum of Immunoglobulin V Gene Somatic Hypermutation by Modifying the Active Site of AID. J. Exp. Med 2010, 207, 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathore A; Carpenter MA; Demir Ö; Ikeda T; Li M; Shaban NM; Law EK; Anokhin D; Brown WL; Amaro RE; Harris RS, The Local Dinucleotide Preference of APOBEC3G Can Be Altered from 5’-CC to 5’-TC by a Single Amino Acid Substitution. J. Mol. Biol 2013, 425, 4442–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kouno T; Silvas TV; Hilbert BJ; Shandilya SMD; Bohn MF; Kelch BA; Royer WE; Somasundaran M; Kurt Yilmaz N; Matsuo H; Schiffer CA, Crystal Structure of APOBEC3A Bound to Single-Stranded DNA Reveals Structural Basis for Cytidine Deamination and Specificity. Nature Communications 2017, 8, 15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nabel CS; Lee JW; Wang LC; Kohli RM, Nucleic Acid Determinants for Selective Deamination of DNA over RNA by Activation-Induced Deaminase. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 14225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carpenter MA; Li M; Rathore A; Lackey L; Law EK; Land AM; Leonard B; Shandilya SMD; Bohn M-F; Schiffer CA; Brown WL; Harris RS, Methylcytosine and Normal Cytosine Deamination by the Foreign DNA Restriction Enzyme APOBEC3A. J. Biol. Chem 2012, 287, 34801–34808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu Y; Ito F; Zhang G; Fernandez B; Yang H; Chen XS, DNA Cytosine and Methylcytosine Deamination by APOBEC3B: Enhancing Methylcytosine Deamination by Engineering APOBEC3B. Biochem. J 2015, 471,25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wijesinghe P; Bhagwat AS, Efficient Deamination of 5-Methylcytosines in DNA by Human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012, 40, 9206–9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suspéne R; Aynaud M-M; Vartanian J-P; Wain-Hobson S, Efficient Deamination of 5-Methylcytidine and 5-Substituted Cytidine Residues in DNA by Human APOBEC3A Cytidine Deaminase. PLoS One 2013, 8, e63461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carpenter MA; Rajagurubandara E; Wijesinghe P; Bhagwat AS, Determinants of Sequence-Specificity within Human AID and APOBEC3G. DNA Repair 2010, 9, 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nabel CS; Jia H; Ye Y; Shen L; Goldschmidt HL; Stivers JT; Zhang Y; Kohli RM, AID/APOBEC Deaminases Disfavor Modified Cytosines Implicated in DNA Demethylation. Nat. Chem. Biol. 2012, 8, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paquet E; Viktor HL, Molecular Dynamics, Monte Carlo Simulations, and Langevin Dynamics: a Computational Review. Biomed Res. Int 2015, 2015, 183918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vangaveti S; Ranganathan SV; Chen AA, Advances in RNA Molecular Dynamics: a Simulator’s Guide to RNA Force Fields. Wiley Interdiscip. Rev. RNA 2017, 8. [DOI] [PubMed] [Google Scholar]
- 51.Galindo-Murillo R; Bergonzo C; Cheatham TE 3rd, Molecular Modeling of Nucleic Acid Structure. Curr. Protoc. Nucleic Acid Chem 2013, 54, Unit 7.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gong Z; Xiao Y; Xiao Y, RNA Stability under Different Combinations of AMBER Force Fields and Solvation Models. J. Biomol. Struct. Dyn 2010, 28, 431–441. [DOI] [PubMed] [Google Scholar]
- 53.Krepl M; Havrila M; Stadlbauer P; Banas P; Otyepka M; Pasulka J; Stefl R; Sponer J, Can We Execute Stable Microsecond-Scale Atomistic Simulations of Protein-RNA Complexes? J. Chem. Theory Comput 2015, 11, 1220–1243. [DOI] [PubMed] [Google Scholar]
- 54.Piana S; Klepeis JL; Shaw DE, Assessing the Accuracy of Physical Models Used in Protein-Folding Simulations: Quantitative Evidence from Long Molecular Dynamics Simulations. Curr. Opin. Struct. Biol 2014, 24, 98–105. [DOI] [PubMed] [Google Scholar]
- 55.Vlachakis D; Bencurova E; Papangelopoulos N; Kossida S, Current State-of-the-Art Molecular Dynamics Methods and Applications. Adv. Protein Chem. Struct. Biol 2014, 94, 269–313. [DOI] [PubMed] [Google Scholar]
- 56.Kalyaanamoorthy S; Chen Y-PP, Modelling and Enhanced Molecular Dynamics to Steer Structure-Based Drug Discovery. Prog. Biophys. Mol. Biol 2014, 114, 123–136. [DOI] [PubMed] [Google Scholar]
- 57.Spyrakis F; Cavasotto CN, Open Challenges in Structure-Based Virtual Screening: Receptor Modeling, Target Flexibility Consideration and Active Site Water Molecules Description. Arch. Biochem. Biophys 2015, 583, 105–119. [DOI] [PubMed] [Google Scholar]
- 58.Pérez A; Marchán I; Svozil D; Sponer J; Cheatham TE 3rd; Laughton CA; Orozco M, Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of Alpha/Gamma Conformers. Biophys. J 2007, 92, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pérez A; Luque FJ; Orozco M, Dynamics of B-DNA on the Microsecond Time Scale. J. Am. Chem. Soc 2007, 129, 14739–14745. [DOI] [PubMed] [Google Scholar]
- 60.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Case DA; Cerutti DS; Cheatham TE III; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW; Greene D; Homeyer N; Izadi S; Kovalenko A; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein D; Merz KM; Monard G; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Simmerling CL; Botello-Smith WM; Swails J; Walker RC; Wang J; Wolf RM; Wu X; Xiao L; York DM; Kollman PA, AMBER 16 and AMBERTools 17. University of California, San Francisco: 2017. [Google Scholar]
- 62.Salomon-Ferrer R; Case DA; Walker RC, An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci 2012, 3, 198–210. [Google Scholar]
- 63.Case DA; Cheatham TE; Darden T; Gohlke H; Luo R; Merz KM; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber Biomolecular Simulation Programs. J. Comput. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banáš P; Hollas D; Zgarbová M; Jurecka P; Orozco M; Cheatham TE; Šponer J. i.; Otyepka M, Performance of Molecular Mechanics Force Fields for RNA Simulations: Stability of UUCG and GNRA Hairpins. J. Chem. Theory Comput 2010, 6, 3836–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacobson MP; Pincus DL; Rapp CS; Day TJF; Honig B; Shaw DE; Friesner RA, A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- 66.Jacobson MP; Friesner RA; Xiang Z; Honig B, On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol 2002, 320, 597–608. [DOI] [PubMed] [Google Scholar]
- 67.Søndergaard CR; Olsson MHM; Rostkowski M; Jensen JH, Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput 2011,7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- 68.Olsson MHM; Søndergaard CR; Rostkowski M; Jensen JH, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput 2011,7, 525–537. [DOI] [PubMed] [Google Scholar]
- 69.Pang Y-P, Novel Zinc Protein Molecular Dynamics Simulations: Steps Toward Antiangiogenesis for Cancer Treatment. J. Mol. Model 1999, 5, 196–202. [Google Scholar]
- 70.Altona C; Sundaralingam M, Conformational Analysis of the Sugar Ring in Nucleosides and Nucleotides. A New Description using the Concept of Pseudorotation. J. Am. Chem. Soc 1972, 94, 8205–8212. [DOI] [PubMed] [Google Scholar]
- 71.Harvey SC; Prabhakaran M, Ribose Puckering: Structure, Dynamics, Energetics, and the Pseudorotation Cycle. J. Am. Chem. Soc 1986, 108, 6128–6136. [Google Scholar]
- 72.Roe DR; Cheatham TE 3rd, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 73.McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernández CX; Schwantes CR; Wang L-P; Lane TJ; Pande VS, MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 2015, 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunter JD, Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng 2007, 9, 90–95. [Google Scholar]
- 75.Baker EN; Hubbard RE, Hydrogen Bonding in Globular Proteins. Prog. Biophys. Mol. Biol 1984, 44, 97–179. [DOI] [PubMed] [Google Scholar]
- 76.Wagner JR; Sørensen J; Hensley N; Wong C; Zhu C; Perison T; Amaro RE, POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput 2017, 13, 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Durrant JD; de Oliveira CAF; McCammon JA, POVME: an Algorithm for Measuring Binding-Pocket Volumes. J. Mol. Graph. Model 2011,29, 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Durrant JD; Votapka L; Sørensen J; Amaro RE, POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics. J. Chem. Theory Comput 2014, 10, 5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Humphrey W; Dalke A; Schulten K, VMD: Visual Molecular Dynamics. J. Mol. Graph 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 80.McDougle RM; Hultquist JF; Stabell AC; Sawyer SL; Harris RS, D316 is Critical for the Enzymatic Activity and HIV-1 Restriction Potential of Human and Rhesus APOBEC3B. Virology 2013, 441, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harjes S; Solomon WC; Li M; Chen K-M; Harjes E; Harris RS; Matsuo H, Impact of H216 on the DNA Binding and Catalytic Activities of the HIV Restriction Factor APOBEC3G. J. Virol 2013, 87, 7008–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen K-M; Harjes E; Gross PJ; Fahmy A; Lu Y; Shindo K; Harris RS; Matsuo H, Structure of the DNA Deaminase Domain of the HIV-1 Restriction Factor APOBEC3G. Nature 2008, 452, 116–119. [DOI] [PubMed] [Google Scholar]
- 83.St Martin A; Salamango D; Serebrenik A; Shaban N; Brown WL; Donati F; Munagala U; Conticello SG; Harris RS, A Fluorescent Reporter for Quantification and Enrichment of DNA Editing by APOBEC-Cas9 or Cleavage by Cas9 in Living Cells. Nucleic Acids Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gerhke JM; Cervantes OR; Kendell Clement M; Pinello L; Keith Joung J, High-Precision CRISPR-Cas9 Base Editors with Minimized Bystander and Off-Target Mutations. bioRxiv 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.