

Abstract
Background:
Alternol is a natural compound isolated from fermentation products of a mutant fungus. Our previous studies demonstrated that Alternol specifically kills cancer cells but spares benign cells.
Methods:
To investigate the mechanism underlying alternol-induced cancer cell-specific killing effect, we took a comprehensive strategy to identify Alternol’s protein targets in prostate cancer cells, including PC-3, C4–2,and 22RV1, plus benign BPH1 cell lines. Major experimental techniques included biotin-streptavidin pulldown assay coupled with mass-spectrometry, in vitro enzyme activity assay for Krebs cycle enzymes and gas chromatography-mass spectrometry (GC-MS) for metabolomic analysis.
Results:
Among 14 verified protein targets, four were Krebs cycle enzymes, fumarate hydratase (FH), malate dehydrogenase-2 (MDH2), dihydrolipoamide acetyltransferase (DLAT) in pyruvate dehydrogenase complex (PDHC) and dihydrolipoamide S-succinyltransferase (DLST) in a-ketoglutarate dehydrogenase complex (KGDHC). Functional assays revealed that PDHC and KGDHC activities at the basal level were significantly higher in prostate cancer cells compared to benign prostate BPH1 cells, while alternol treatment reduced their activities in cancer cells close to the levels in BPH1 cells. Although FH and MDH2 activities were comparable among prostate cancer and benign cell lines at the basal level, Alternol treatment largely increased their activities in cancer cells. Metabolomic analysis revealed that Alternol treatment remarkably reduced the levels of malic acid, fumaric acid, and isocitric acid and mitochondrial respiration in prostate cancer cells. Alternol also drastically reduced mitochondrial respiration and ATP production in PC-3 cells in vitro or in xenograft tissues but not in BPH1 cells or host liver tissues.
Conclusions:
Alternol interacts with multiple Krebs cycle enzymes, resulting in reduced mitochondrial respiration and ATP production in prostate cancer cells and xenograft tissues, providing a novel therapeutic strategy for prostate cancer treatment.
Keywords: alternol, cellular energy, Krebs cycle, metabolic enzymes, mitochondria
1 |. INTRODUCTION
Cancer metabolism is emerging with great interest as a therapeutic target for cancer management.1 Prostate cancer is especially unique in this aspect since benign prostate tissue exhibits truncated Krebs cycle to secret citrate (citrate-producing) due to high level of prostatic zinc concentration, but prostate cancer cells fully oxidize citrate (citrate-oxidizing) for energy production through Krebs cycle.2,3 In addition, most prostate cancer cells maintain functional mitochondria with reprogrammed metabolism4,5 and prostate cancers only present Warburg effect at the late stage of the diseases different from other solid tumors.6 These metabolic features warrant an advantage of targeting Krebs cycle for prostate cancer management.
As the major power house, mitochondria are responsible for about 90% of cellular ATP production by taking up the substrates from the cytoplasm, including pyruvate generated from glycolysis.2 In mitochondrial matrix, pyruvate is converted by the pyruvate dehydrogenase (PDH) complex to acetyl-CoA that enters a series of oxidative phosphorylation (OXPHOS) reactions termed as Krebs cycle.3 This cycle generates two essential electron carriers NADH and FADH2, as well as succinate for the electron transport chain (ETC) on the inner mitochondrial membrane. During this electron transport process, hydrogen ions or protons are pumped into the mitochondrial intermembrane space to establish a proton motive force for ATP production. This process is coupled with the generation of superoxide, the common type of reactive oxygen species (ROS). So far, multiple efforts have focused on targeting specific Krebs cycle enzymes in human cancers, including isocitrate dehydrogenase (IDH) inhibitors for mutated IDH1/2 proteins,4 mitochondrial metabolic disruptor CPI-613,5 and mitochondrial pyruvate carrier (MPC) inhibitor UK5099.6 Since the great flexibility of metabolic reprogramming found in cancer cells,7,8 therefore, novel agents targeting multiple metabolic enzymes or pathways might be able to efficiently constrain aberrant tumor metabolism.
Alternol is a small natural compound purified from fermentation products of a mutant microorganism.9 It was previously shown to inhibit cancer cell proliferation by blocking cell cycle progression and to induce apoptotic cell death in cancer cells.10–13 In our recent work, we demonstrated that Alternol induced apoptosis preferentially in cancer cells through a reactive oxygen species (ROS)-dependent mechanism. Our animal studies also documented Alternol-induced in vivo anti-tumor effect.14 To elucidate the mechanism underlying Alternol-induced anti-tumor effect, we utilized a comprehensive strategy to identify Alternol interacting proteins. Cellular proteins interacted with biotin-labeled Alternol were pulled down by streptavidin beads. After in gel digestion of the eluted proteins, peptides were identified with mass spectrometry technology. Identified protein targets were confirmed by Western blot assay and further verified by cellular thermal stability assay (CETSA).15 Meanwhile, Alternol-induced functional alterations of the target proteins were examined in multiple malignant cells in comparison to benign BPH1 cells. Our data demonstrated that Alternol interacted with multiple Krebs cycle enzymes, leading to functional attenuation of mitochondrial respiration and cellular ATP reduction specifically in malignant cells and xenograft tissues.
2 |. MATERIALS AND METHODS
2.1 |. Cell Culture, antibodies, and other reagents
Prostate cancer PC-3 and 22RV1 cell lines, and benign prostate BPH1 line were obtained from ATCC (Manassas, VA) as described.16,17 C4–2 cell line was obtained from UroCor Inc (Oklahoma City, OK).18 All cell lines were tested for authentication at Genetica DNA Lab (Burlington, NC) and were used within passages of 15–25. Cells were cultured in RPMI 1640 media containing 10% fetal bovine serum and 1% penicillin/streptomycin in a 5% CO2-humidified atmosphere at 37°C. Biotin labeling of Alternol was carried out in house. Succinate assay kit and enzymatic function assay kits for PDHC, KGDHC, FH, and MDH2 were obtained from BioVision (Milpitas, CA). Docetaxel was obtained from Cayman Chemical (Ann Arbor, MI). Alternol was obtained from Sungen Bioscience as reported14 (Shantou, China).
Antibodies for Caspase-3, DLAT, DLST, FH, MDH2, GAPDH, HSPD1, HYOU1, HSP90AB1, HSPA8, ILF2, NPM1, PDIA6, ATP5A1, ANXA2, TUBA1B, and LAMR1 were obtained from Cell Signaling Technology (Danvers, MA). TUNEL immunostaining kit, ETC complex proteins antibodies and Ki-67 antibodies were obtained from Abcam (Cambridge, MA). Beta-actin antibody was obtained from Sigma (St. Louis, MO). Streptavidin-agarose beads and secondary antibodies were obtained from Santa Cruz Biotech (Santa Cruz, CA).
2.2 |. Western blotting, biotin-streptavidin pulldown, and immunohistochemistry
Western blot was performed as previously described.19,20 Total cell lysates were prepared in RIPA buffer supplemented with protease inhibitors and separated by SDS-PAGE and then transferred onto a PVDF membrane. The membrane was incubated with 5% nonfat dry milk for 1 h, followed by primary antibody overnight on a rotating platform at 4°C.
For biotin-streptavidin pulldown assay, cells were treated with biotin-labeled Alternol for 4h and then lysed in cold RIPA buffer as described.21 Protein lysates were incubated with streptavidin-agarose resin overnight at 4C with rotation. After three washes, the elutes were subjected to SDS-PAGE gel or followed by Western blot with antiantibodies as indicated.
PC-3 cells-derived xenograft tissues were fixed in formalin. Paraffin-embedded tissue sections (4 μm) were subjected to dewaxing and rehydration, followed by inactivation of endogenous peroxidase activity. Antigen retrieval was performed in sodium citrate buffer (pH 6.0) with heating. After blocking, tissue sections were incubated with primary antibody overnight at 4°C. Immunosignals were detected with DAKO LSAB System and semi-quantified data were calculated as described in our previous publications.22,23
2.3 |. Mass spectrometry and cellular thermal stability assay
For protein identification, coomassie blue stained SDS-PAGE gel bands were cut out and stored in 0.1% formic acid until analyzed by mass spectrometry as described.24 Briefly, gel slices were reduced, alkylated, digested with trypsin. The peptide extracts were analyzed in triplicate on a LTQ FT mass spectrometer (Thermo Fisher Scientific) coupled with a 2D NanoLC (Eksigent Technologies, CA).
Cellular thermal stability assay (CETSA) with in vitro cell culture models was conducted as described in our publication.25 The in vivo CETSA assay was carried out with PC-3 xenograft tissues treated with Alternol or the solvent using the protocol as described.15
2.4 |. In-silicon docking studies of alternol interaction with protein targets
The crystal structures of FH, DLAT, and MDH2 obtained from Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) were used for docking studies (PDB IDs: FH/3E04, DLAT/3B8K, MDH2/4WLU) with Alternol. The crystal structures containing bound ligands (NAD or L-malate) were removed prior to docking studies. All protein chains present in the PDB structures were retained for docking. Hydrogens were added to both the protein crystal structures and the ligands using Molecular Operating Environment (MOE).26 Docking of Alternol ligand was first performed using AutoDock Vina27 for initial identification of conformations. After initial docking with AutoDock Vina, docking poses were refined further by (1) increasing the exhaustiveness parameter of AutoDock Vina and (2) using MOE docking with both a rigid and flexible receptor protocol.
2.5 |. Mitochondrial respiration assay, ATP measurements, and metabolomic analysis
Oxygen consumption rate (OCR) was measured in real-time using XF24 × 3 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA) as described in our recent publication.28 Briefly, BPH1 and PC-3 cells were seeded in XF24-well plates. Cells were treated with Alternol 10 μM for 4 h. Cells were then subjected to mitochondrial stress test, using the Mito-Stress kit (Seahorse Biosciences Billerica, MA). Protein concentration was used to standardize the OCR among different treatments.
ATP levels in PC-3 cells and xenografts were assessed by the ATPLite™ assay (Promega, Madison). ATP levels in culture cells were also measured with HPLC-based method as described in our recent publication.29 The HPLC system was equipped with a LC-20AD pumps system and SPD-M20A diode array detector (Schimadzu Instruments Inc.) and Supelcosil™ LC-18 150 × 4.6 mm column (5 urn particle size) was used for separation of analytes.
Metabolomic analysis of Krebs cycle intermediates was conducted using a gas chromatography-mass spectrometry (GC-MS) at the University of Utah Metabolomics core facility. Data were collected, metabolites were identified, and their peak area was recorded using MassHunter Quant software (Agilent). Data statistical analysis was performed using Metaboanalsyt 3.0.
2.6 |. Mouse xenograft experiment and alternol treatment
Athymic NCr-nu/nu male mice (Shanghai Laboratory Animal Center, China) were maintained in accordance with the procedure guidelines and the experimental protocol approved by the Institutional Animal Care and Use Committee of Xi’An Jiaotong University. Xenograft tumors were generated in 20 mice with PC-3 cells. Alternol was dissolved in 20% DMSO/PBS solution and the dose was set for 50 mg/kg body weight. Once xenografts reached 3–5 mm in size (diameter), mice (n = 10per group) were treated with Alternol orthesolventthree timesa weekintraperitoneally as described.14 At the end of experiments, major organs and xenograft tissue specimens were harvested for analysis.
2.7 |. Statistical analysis
Quantitative data shown are presented as mean ±SEM from at least three independent experiments. Images of Western blots and immunostaining experiments were shown from a representative result. Statistically significant was analyzed using the statistical software SPSS 20.0 software (SPSS, Inc., Chicago, IL).
3. |. RESULTS
3.1 |. Alternol interacts with multiple cellular proteins including Krebs cycle enzymes
To look for the cellular protein targets that are interacted with Alternol, we used a biotin-streptavidin pulldown assay coupled with mass-spectrometry protein identification approach. PC-3 cells were treated with biotin-labeled Alternol for 4 h and protein elutes from streptavidin beads pulldown were separated on SDS-PAGE gel (Figure 1A). After staining, protein bands were subjected for in-gel digestion followed by mass-spectrometry approach. A total of 26 cellular proteins were identified including four physiologically biotin-interacting proteins, indicating the success of biotin-based pulldown experiment (Table 1).
FIGURE 1.
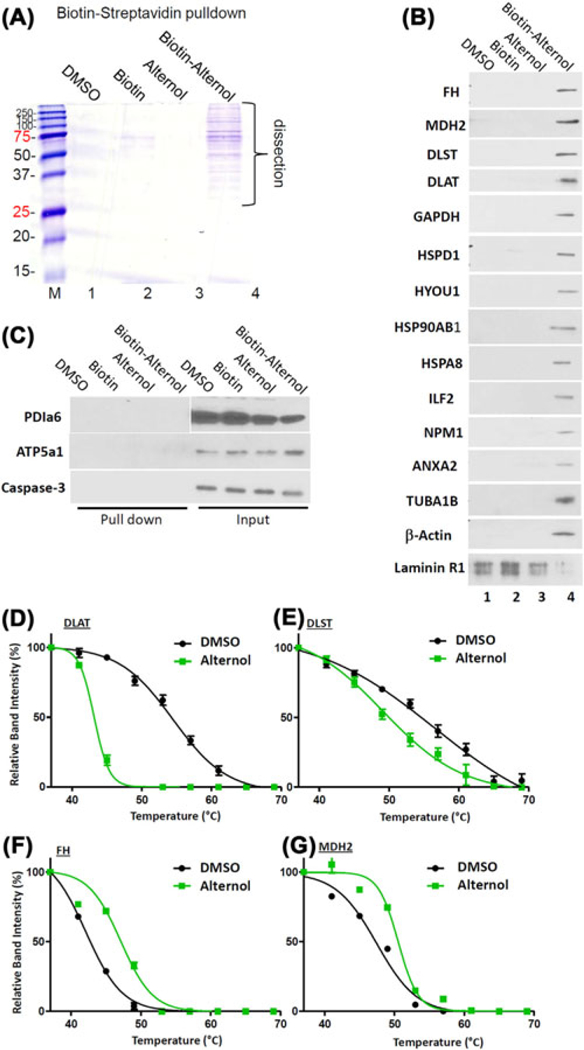
Alternol interacts with multiple cellular proteins. A, PC-3 cells were teated with the solvent DMSO, biotin, Alternol, biotin-labeled Alternol (10 μM) for 4h. Cell lysates were incubated with Streptavidin agarose resin overnight at 4°C. After three washes, the elutes were run on SDS-PAGE followed by comassine blue staining. Protein bands were dissected for Mass-Spectrometry. B, The Biotin-Streptavidin pulldown elutes were used for Western blots with antibodies as indicated. C, Both pulldown elutes and whole cell protein lysates were used for the Western blots. Caspase-3 blot was included as a negative target control. D-G, PC-3 cells were treated with the solvent DMSO or Alternol for 4 h and cellular proteins were extracted for CETSA assay as described in the text. Protein band densities were acquired with ImageJ software and the curve plots were generated with Graphpad Prism 5.0 software
TABLE 1.
Biotin-alteronol pull-down proteins identified by mass-spectrometry
| Protein category | Alternol-interacting proteins | MW (KD) |
|---|---|---|
| 4 biotin-binding proteins | Methylcrotonoyl-CoA carboxylase beta, MCCC2 | 62 |
| Propionyl-CoA carboxylase alpha, PCCA | 80 | |
| Propionyl-CoA carboxylase beta, PCCB | 59 | |
| Pyruvate carboxylase, PC | 130 | |
| 6 metabolic proteins | Malate dehydrogenase 2, MDH2 | 37 |
| Fumarate hydratase, FH | 56 | |
| Dihydrolipoyllysine-residue succinyltransferase, DLST | 50 | |
| Dihydrolipoyllysine-residue acetyltransferase, DLAT | 71 | |
| Glyceraldehyde-3-phosphate dehydrogenase, GAPDH | 32 | |
| ATP synthase subunit alpha, ATP5A1 | 61 | |
| 4 heat-shock proteins | Heat shock protein 60kD, HSPd1 | 63 |
| Heat shock cognate 71 kDa, HSPa8 | 71 | |
| Heat shock protein HSP 90-beta, HSP90ab1 | 80 | |
| Heat shock 70kDa protein 1-like isoform 5, HSPa1L | 71 | |
| 3 ER proteins | Heat shock protein 90kDa beta family member 1, HSP90B1 | 90 |
| Protein disulfide-isomerase A6, PDIa6 | 55 | |
| Hypoxia up-regulated protein 1, HYOU1 | 110 | |
| 4 cytoskeleton proteins | Actin b, ACTB | 41 |
| Tubulin beta 4b, TUBB4B | 49 | |
| Tubulin alpha 1B, TUBA1B | 50 | |
| Filamin a, FLNa | 290 | |
| 3 membrane proteins | Integrin alpha-2, ITGa2 | 130 |
| Laminin receptor 1, LAMR1 | 33 | |
| Annexin A2 isoform 2, ANXa2 | 37 | |
| 2 cell signaling proteins | Nucleophosmin isoform X1, NPM1 | 32 |
| Interleukin enhancer-binding factor 2, ILF2 | 43 |
We then used the co-immunoprecipitation coupled with Western blot assays toverify these targets using the protein elutes from the biotin-streptavidin pulldown assay. As shown in Figure 1B, 14 protein targets were verified except that Laminin receptor 1 (LAMR1) was defined as a non-specific target. In addition, PDIa6 and ATP5a1 were not detectable in the pulldown elutes but detectable in the whole cellular lysate, indicating as false-positive targets (Figure 1C). In addition, HSPD1, HSPA1L, Tubulin-β4B (TUBB4B), Filamin-α (FLNA), and integrin-α2 (ITGA2) were not successfully verified due to antibody failure.
Then, we focused on four Krebs cycle enzymes; FH, MDH2, DLAT, and DLST. CETSA assays were conducted to further verify their interaction with Alternol. As shown in Figure 1D–G, Alternol treatment in PC-3 cells largely shifted the thermo-stability curve of these protein targets, in which the curves for DLAT and DLST were left-shifted while the curves for FH and MDH2 were right-shifted. Detailed parameters for CETSAdata were summarized in Table 2. These data clearly demonstrated a strong interaction of Alternol with these metabolic enzyme proteins.
TABLE 2.
In vitro CETSA data summary
| Treatment | Tm 50 | ΔTm50 | R2 value | |
|---|---|---|---|---|
| DLAT | DMSO | 54.80 | 0.9931 | |
| Alteronol | 43.28 | −11.52 | 0.9988 | |
| DLST | DMSO | 56.12 | 0.9804 | |
| Alteronol | 48.81 | −7.28 | 0.9871 | |
| FH | DMSO | 42.18 | 0.9983 | |
| Alteronol | 47.56 | 5.38 | 0.9817 | |
| MDH2 | DMSO | 47.91 | 0.9866 | |
| Alteronol | 50.48 | 2.57 | 0.9876 |
To get insight for the mechanism of Alternol interaction with the target proteins, we performed an in silicon docking study with published crystal structure data for FH, DLAT, and MDH2, except for DLST that is not available. There was a strong binding between Alternol and FH at a single site situated near residues 348, 350, and 354 of chain C and the binding affinity is −8.7kcal/mol (Figure 2A). Alternol also strongly bound to MDH2 at a single site corresponding to NAD and L-malate binding sites (residues 86,182, 216) with a binding affinity of −9.0kcal/mol (Figure 2B). Interestingly, Alternol strongly bound to DLAT at two sites on chain A, the first site near residues 476 and 542 (binding affinity −8.1 kcal/mol) (Figure 2C) and the second site near residues 353 and 473 (binding affinity −8.0 kcal/mol) (Figure 2D).
FIGURE 2.
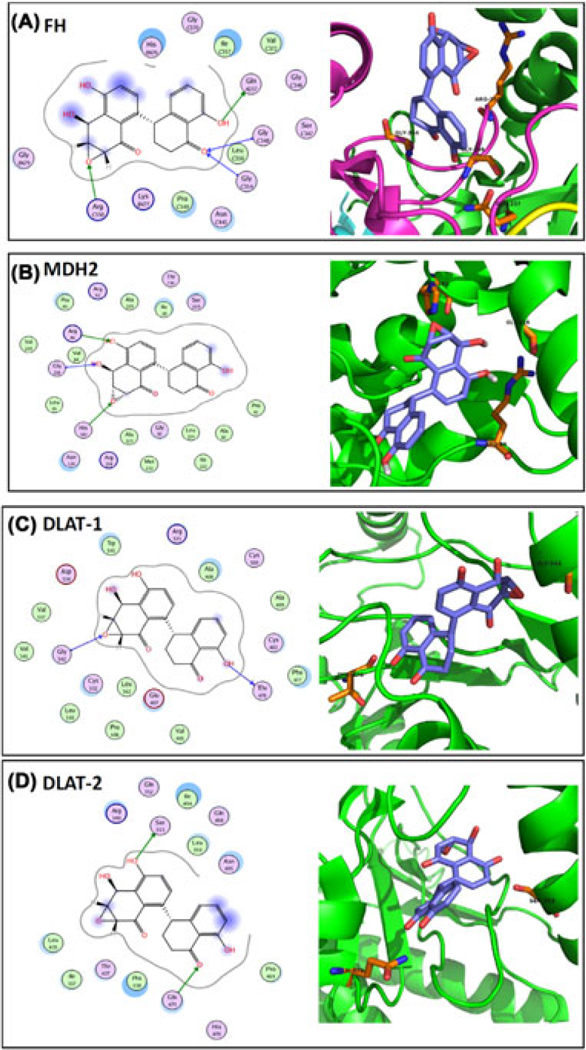
In-silicon docking analysis. The crystal structure of the target proteins were extracted from the RCSB PDB database and the docking analysis were conducted as described in the text. The image on the left side in each panel shows the interaction diagram of Alternol molecule with the amino acid residues on the target protein. The green arrows indicate sidechain donor or acceptor; The blue arrows indicate backbone donor or acceptor. The images on the right side shows the cartoon view of the interaction site. Purple sticks indicate Alternol molecule and orange sticks indicate amino acide residues that interact with Alternol
3.2 |. Alternol attenuates Krebs cycle enzyme activities
The functional consequences of Alternol interaction with these Krebs cycle enzyme proteins were assessed in enzymatic assays in vitro using multiple prostate cancer cell lines plus a benign BPH1 cell line that served as a benign control since BPH1 cells were less susceptible to Alternol-induced cell death.14 Since DLAT is a component of PDH complex (PDHC) and DLST is a component of KGDH complex (KGDHC), their activities were assessed as an enzymatic complex, respectively. As shown in Figure 3A–D, the basal activities of PDHC and KGDHC complexes were significantly higher in malignant cells compared to BPH1 cells, indicating an elevated Krebs cycle flux in cancer cells. After Alternol treatment, PDHC and KGDHC activities were significantly reduced only in malignant cells, reaching to the level close or lower than that in BPH1 cells. In contrast, the basal activities of FH and MDH enzymes were comparable among the malignant and benign cells. Alternol treatment significantly increased FH and MDH activities in malignant cells but only slightly in BPH1 cells. These data demonstrated that Alternol attenuated the activities of these four Krebs cycle enzymes in malignant but not in benign cells.
FIGURE 3.
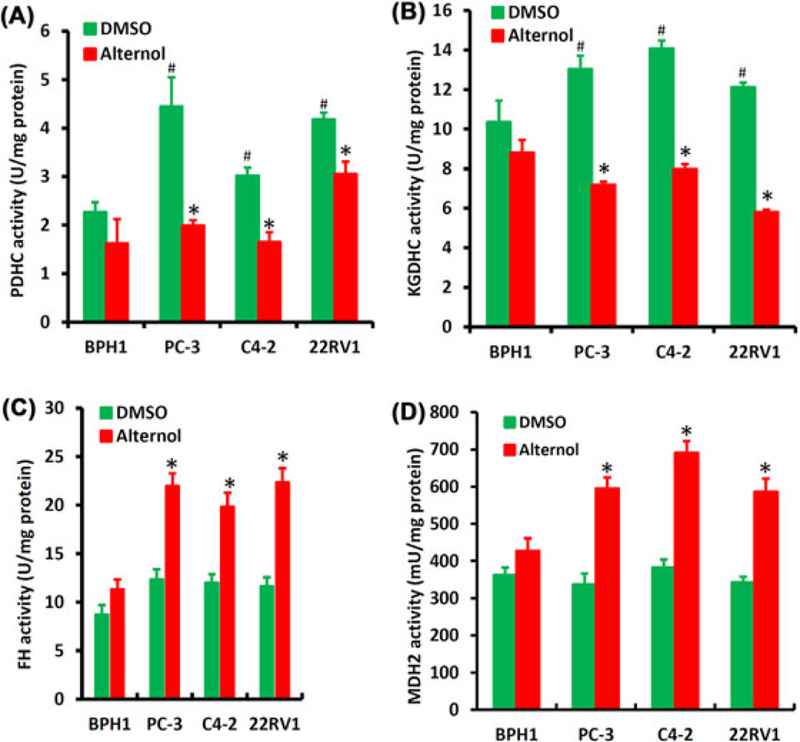
Alternol attenuates Krebs cycle enzymatic activities. A-D, Prostate cancer cells and BPH1 cells were treated with the solvent DMSO or Alternol (10 μM) for 4 h and whole cellular proteins were extracted for the enzymatic assays as indicated with the pre-assembled assay kits from BioVision. The asterisks indicate significant differences (Student’s t-test, P < 0.05) between Alternol treatment and DMSO control. The # signs indicate significant differences (Student’s t-test, P < 0.05) between malignant cells and BPH1 cells in DMSO control group
3.3 |. Alternol reduces mitochondrial respiration and cellular ATP production
Since Krebs cycle flux is to facilitate mitochondrial respiration and ATP synthesis, we then determined if Alternol treatment disturbs mitochondrial respiration (oxygen consumption) and cellular ATP production. The Seahorse system coupled with a mitochondrial stress test kit was used to examine the effect of Alternol treatment on mitochondrial oxygen respiration. As shown in Figures 4A and 4B, Alternol treatment significantly reduced basal oxygen consumption rate (OCR) and sharply suppressed the maximal OCR and ATP-coupled OCR but largely increased the proton leak (a sign of uncoupled respiration) in PC-3 cells. Conversely, Alternol only caused a slight reduction of the maximal OCR in BPH1 cells. These data indicate a severe attenuation of mitochondrial respiration in malignant cells.
FIGURE 4.
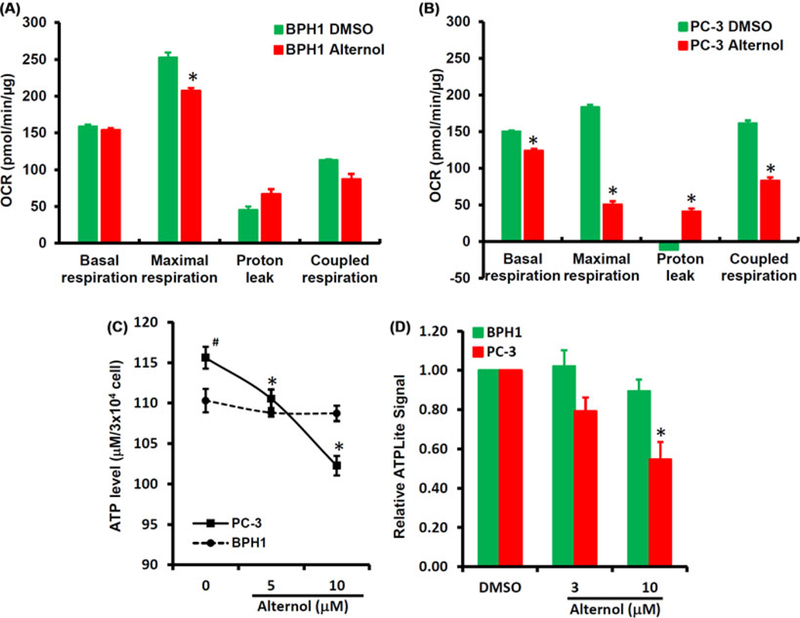
Alternol reduces mitochondrial respiration and ATP production. A-B, BPH1 or PC-3 cells were seeded in XF24-well plates and the OCR was determined with XF24 × 3 Extracellular Flux Analyzer as described in the text.28 The asterisk indicates a significant difference compared to DMSO control (Student’s t-test, P < 0.05). C, PC-3 and BPH1 cells were left untretaed or treated with Alternol (5–10 μM) for 4 h. ATP contents were analyzed using a HPLC-based approach as described.29 The asterisk indicates a significant difference compared to the untreated cells (Student’s t-test, P < 0.05) and the # sign indicates a significant difference compared to BPH1 cells (Student’s t-test, P < 0.05). D, PC-3 and BPH1 cells were treated with DMSO, Alternol (3–10 μM) for 4 h and cellular ATP contents were determined using ATPLite™ assay kit. The relative ATP levels were calculated when the reading for DMSO-treated cells was set at 1 after normalized with total protein levels in each samples. The asterisk indicates a significant difference compared to DMSO control (Student’s t-test, P < 0.05).
Next, HPLC-based direct measurement and luciferase assay-based indirect measurement were utilized to examine cellular ATP levels. As shown in Figure 4C, the basal ATP levels in PC-3 cells were significantly higher than that in BPH1 cells, indicating an active ATP synthesis in malignant cells. Alternol treatment dramatically reduced ATP levels in a dose-dependent manner in PC-3 cells but not in BPH1 cells. Similarly, the luciferase-based assay also revealed a significant reduction of ATP levels after Alternol treatment in PC-3 cells but not in BPH1 cells (Figure 4D). These data suggest that Alternol treatment suppressed mitochondrial oxygen respiration and ATP synthesis, a severe consequence of Krebs cycle disturbance.
To investigate Alternol-reduced ATP production in vivo, we generated subcutaneous xenograft tumors with PC-3 cells in nude mice. Alternol was administered by intraperitoneal injection at 50 mg/kg bodyweight three days a week for 4 weeks. As shown in Figure 5A, Alternol therapy significantly reduced xenograft tumor growth as measured by tumor volume compared to the solvent control. At the end of the experiment, xenograft tumor specimens were dissected for ATP content assessment. Consistent with the tumor suppressing effect, Alternol therapy largely reduced ATP levels in xenografts compared to the solvent control (Figure 5B). However, ATP levels in mouse liver tissues did not show significant reduction in Alternol-treated animals compared to the solvent control (Figure 5B). In parallel, immunosignals for Ki-67, a cellular marker of cell proliferation, was significantly reduced in Alternol-treated xenograft tumors compared to the solvent control, indicating a reduced tumor cell proliferation (Figures 5C and 5D). Consistent with our previous report,14 immunosignals for cleaved caspase-3 and TUNEL were significantly increased in Alternol-treated xenografts compared to the control. These data strongly suggest that Alternol therapy reduces ATP synthesis preferentially in malignant but not in normal tissues, leading to reduced tumor growth and increased apoptotic cell death.
FIGURE 5.
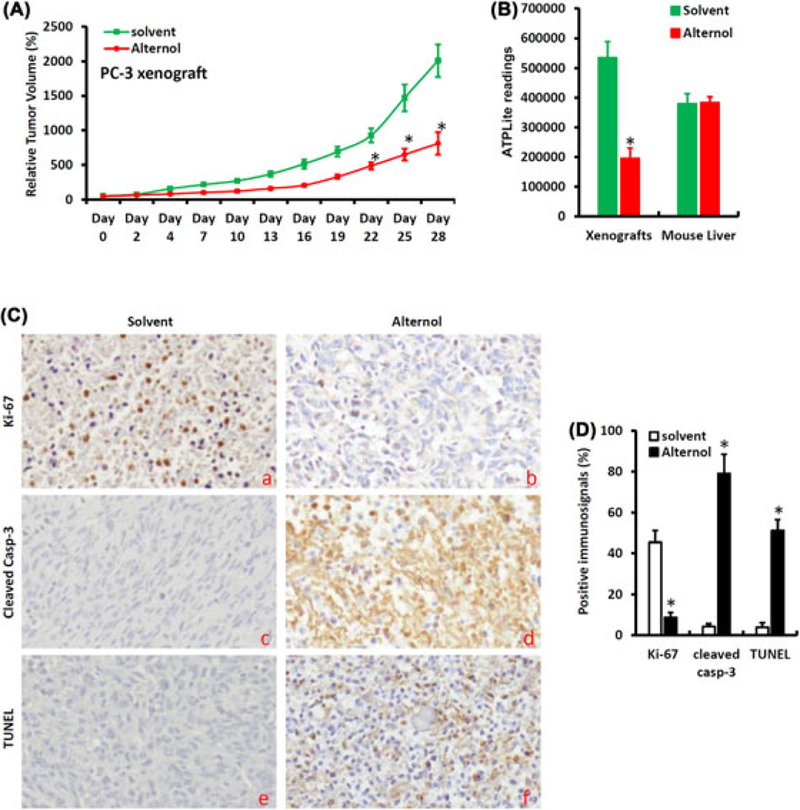
Alternol suppresses xenograft tumor growth and reduces ATP production in malignant tissues. A, Subcutaneous xenografts were established with PC-3 cells in male nude mice and animals were treated with the solvent (as control, n = 10) or Alternol (10 mg/kg body weight, n = 10) by intraperitoneal injection three-time a week. Tumor size was measured at each treatment. Asterisks indicate significant difference compared to the control group (Student’s t-test, P < 0.05). B, Xenograft tumors and host liver tissues were harvested from animals at the end of experiments. Protein lysates were used for the ATPLite™ measurement as described earlier. The asterisk indicates a significant difference compared to the solvent control (Student’s t-test, P < 0.05). C-D, Xenograft tumors were fixed and paraffin-embeded for immunostaining with the antibodies as indicated. Semi-quantified data for the immnunosignals were summarized in panel D. The asterisk indicates a significant difference compared to the solvent control (Student’s t-test, P < 0.01)
3.4 |. Alternol interacts with the enzyme proteins in vivo
An in vivo CETSA assay with the xenograft tumor specimens was conducted to investigate Alternol interaction with the target proteins. As shown in Figure 6A–D and Table 3, the thermo-stability curves for DLAT, DLST, and FH proteins were largely shifted in Alternol-treated xenografts compared to the control, which were in an agreement with the data obtained from in vitro cell culture models (Figure 1D–G and Table 2), clearly indicating Alternol interaction with these metabolic enzymes. A weak engagement for Alternol with MDH2 protein was observed in this in vivo model that a further verification is needed.
FIGURE 6.
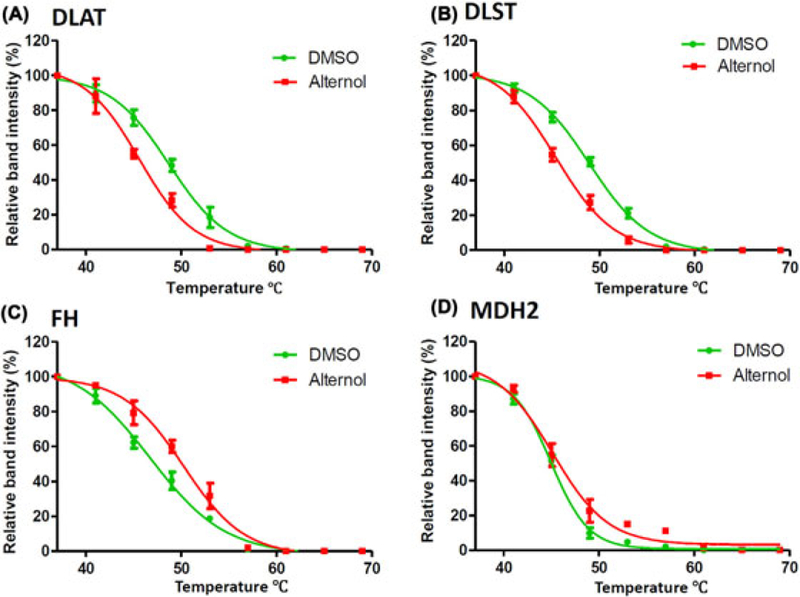
Alternol interacts with Krebs cycle enzyme proteins in vivo. A-D, Protein lysates from xenograft tumors treated with the solvent or Alternol were used for the CETSA assay as described above. Protein band density data were acquired with ImageJ software and analyzed with Graphpad Prizm 5.0 software. Detailed CETSA values were summarized in Table 3
TABLE 3.
In vivo CETSA data summary
| Treatment | Tm 50 | ΔTm50 | R2 value | |
|---|---|---|---|---|
| DLAT | Solvent | 48.69 | 0.9917 | |
| Alteronol | 45.61 | −3.08 | 0.9865 | |
| DLST | Solvent | 48.92 | 0.9963 | |
| Alteronol | 45.42 | −3.58 | 0.9949 | |
| FH | Solvent | 46.73 | 0.9923 | |
| Alteronol | 50.27 | 3.54 | 0.9866 | |
| MDH2 | Solvent | 44.96 | 0.9963 | |
| Alteronol | 45.12 | 0.16 | 0.9820 |
3.5 |. Alternol induces significant metabolic disturbances in malignant cells
We next conducted a metabolomic analysis of Krebs cycle intermediates using a gas chromatography-mass spectrometry (GC-MS) approach. PC-3 and BPH1 cells were treated with the solvent or Alternol for 4h and cell pellets were subjected to metabolites extraction. Fifty-eight metabolites including Krebs cycle intermediates, carbohydrates, amino acids, and lipids were successfully recovered for analysis using the online tool at www.metaboanalyst.ca (Table S1). At the basal condition, four Krebs cycle metabolites (citric acid, succinic acid, fumaric acid, and malic acid) were significantly higher in PC-3 cells compared to that in BPH1 cells (Figure 7A–D and S1), indicating a much more active Krebs cycle flux in malignant cells.
FIGURE 7.
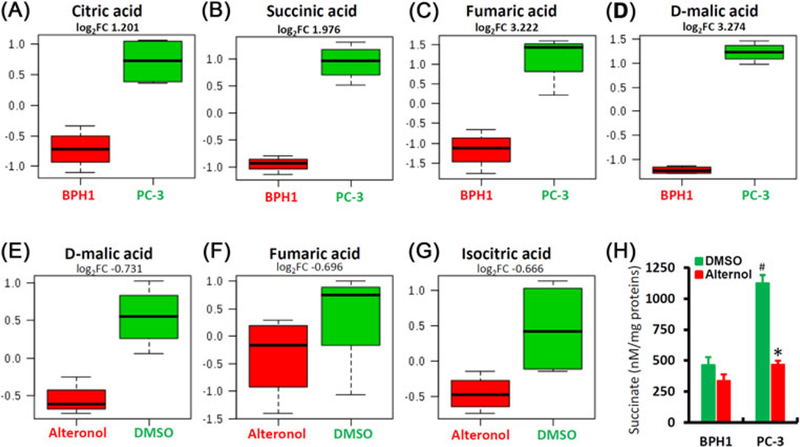
Alternol attenuates mitochondrial metabolism. A-D, Comparison of four major Krebs cycle intermediates between PC-3 and BPH1 cells. PC-3 and BPH1 cells were harvested for metabolomic analysis using the gas chromatography-mass spectrometry (GC-MS) as described in the text. Individual comparison for each mitabolites was shown with fold change values (log2FC). E-G, PC-3 cells treated with DMSO or Alternol (10 μM) for 4 h were harvested for metabolomic analysis as described above and the The individual comparison was shown with fold change values (log2FC). H, PC-3 and BPH1 cells were treated with DMSO or Alternol (10 μM) for 4 h. Whole cell lysates were used for the measurement of succinate levels using an assay kit from BioVision. Succinate levels were normalized with protein concentrations. The asterisk indicates a significant difference compared to DMSO control (Student’s t-test, P < 0.05) and the # sign indicates a significant difference compared to BPH1 cells (Student’s t-test, P < 0.05)
After Alternol treatment, three Krebs cycle intermediates (isocitric acid, fumaric acid, and malic acid) were significantly reduced in PC-3 cells (Figures 7E–G and S2). However, only two metabolites (methionine and hypotaurine) were significantly reduced in BPH1 cells after Alternol treatment (Figure S3). Since succinate, a major connecting factor between Krebs cycle and mitochondrial respiration, was not recovered from this metabolomic assay, we analyzed its levels in a separate assay. As shown in Figure 6H, the basal levels of succinate were extremely higher in PC-3 cells than that in BPH1 cells, indicating higher metabolic activities in malignant cells. Alternol treatment largely reduced succinate levels in PC-3 cells to the basal level in BPH1 cells. These data clearly indicated that Alternol treatment eliminated the excessive Krebs cycle flux in malignant cells to the levels in benign cells.
4 |. DISCUSSION
In this study, we identified and confirmed 14 Alternol-interacting proteins in prostate cancer cells. Four of them are Krebs cycle enzymes (FH, MDH2, DLAT, and DLST). Functional analysis revealed that Alternol treatment attenuated their enzymatic activities, led to reduced mitochondrial respiration and ATP production in cancer cells. Sparklingly, Alternol treatment eliminated excessive ATP production in cancer cells or its derived xenograft tumors but had not significant reduction in benign cells or the host normal liver tissue. These data indicate a superior specificity in cancer cell/tissue and a huge safety advantage of Alternol as an anti-cancer agent. As the authors’ awareness, Alternol is the first natural compound reported targeting multiple Krebs cycle enzymes in cancer cells.
We confirmed the Alternol interaction with the Krebs cycle enzyme targets utilizing the CETSA assay that was recently developed to conveniently analyze protein-drug interaction.15 Our data demonstrated a very consistent profile of Alternol-induced thermo-stability shifts for DLAT, DLST, and FH in both in vitro cell culture and in vivo xenograft models. Although MDH2’s melting curve was largely shifted in cell culture model, there was only a minor shift when xenograft tissues were assessed. This discordance might be due to lower drug concentration in xenograft tissues in vivo compared to cell culture model in vitro. On the other hand, the in silicon protein docking analysis indicated that Alternol strongly interacts with MDH2, thus, further analysis with additional assays are needed to verify Alternol-interaction with MDH2 in vivo.
Metabolic abnormalities or reprogramming at the genetic, epigenetic, and proteomic levels have emerged as common events in human cancers including prostate cancers.30,31 Specifically, at the protein levels, eight Krebs cycle enzymes including Alternol-targeted protein MDH2 and FH were highly expressed in prostate cancers compared to BPH tissues,30 indicating them as potential therapeutic targets. However, there is no report about their activities in prostate cancers. In this study, we analyzed their enzymatic activities in multiple prostate cancer cells. In comparison to BPH1 cells, prostate cancer cells consistently showed higher activities of PDHCand KGDHC complexes. Metabolomic analysis revealed that the metabolic intermediates downstream of these two complexes were higher in prostate cancer cells than that in benign cells. Interestingly, Alternol treatment reduced PDHCand KGDHC activities in prostate cancer cells to the levels close to benign cells. Meanwhile, metabolic intermediates such as malic acid, fumaric acid, and isocitric acid were also reduced by Alternol treatment in prostate cancer cells. These data strongly suggest that Alternol represents a brand-new category of agents that eliminates excessive PDHC and KGDHC activities, representing an excellent safety profile.
In Krebs cycle, PDH/DLAT complex converts the glycolysis metabolite pyruvate to acetyl-CoA at the entering step of the Krebs cycle and KGDH/DLST complex catalyzes the critical step to produce metabolic intermediate succinate used by mitochondrial ETC.32 Our data revealed that Alternol treatment reduced PDHC and KGDHC activities but enhanced FH and MDH2 activities, which might be the reason for succinate reduction, resulting in attenuation of oxygen respiration and ATP production (illustrated in Figure 8). Our data also demonstrated Alternol as a multiple protein disturber, especially interacting with multiple enzymes in the Krebs cycle to attenuate cancer cell metabolism. In this aspect, Alternol has much better property than other synthetic inhibitors of mitochondrial enzymes such as the inhibitors for mutant IDH1/2 enzymes33,34 or ETC inhibitor Metformin.8
FIGURE 8.
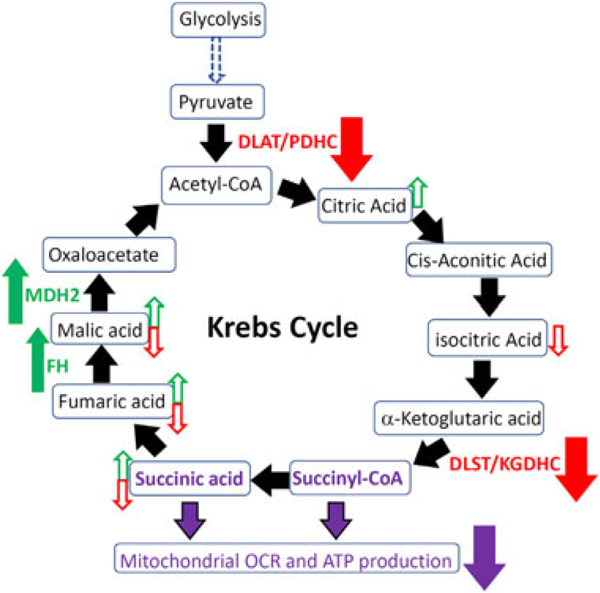
A schematic model for Alternol-induced attenuation of Krebs cycle metabolism. Alternol interacts with DLAT/PDH and DLST/KGDH complexes and reduces their enzymatic activities (solid red arrow). Alternol also interacts with FH and MDH2 to increase their activities (solid green arrow). Compared to BPH1 cells, the basal levels of citric acid, malic aicd, fumaric acid and succnic acid were higher in PC-3 cells (open green arrow). Alternol treatment in PC-3 cells reduces the levels of malic acid, fumaric acid, isocitric acid and succinic acid (open red arrow). Alternol had no effect on Krebs cycle metabolites in BPH1 cells. All events lead to reduced succinate levels, attenuated mitochondrial respiration and less ATP production in PC-3 cells (purple arrow)
Taken together, we identified and verified that Alternol interacts with 14 cellular proteins in prostate cancer cells including four Krebs cycle enzymes. Alternol treatment attenuated their enzymatic activities, reduced mitochondrial respiration, and eliminated excessive ATP production in prostate cancer cells. Our data indicate Alternol as a novel metabolism orientated anti-cancer agent for prostate cancer.
5 |. CONCLUSION
Ideally, a therapeutic drug for curing cancer should have a cancer preference with a minimum side effect on benign tissues. We demonstrated that Alternol exerts a cancer-specific killing effect with very limited effect on benign prostate cells. Through a comprehensive approach, we identified Alternol-interacting protein targets including four Krebs cycle enzymes. Alternol disturbs their functions and reduces mitochondrial respiration and ATP production only in malignant cells or tissues while sparing benign cells. Therefore, it possesses a great potential to be developed as a successful clinical therapy. As the authors aware, Alternol is the first natural compound reported targeting multiple Krebs cycle enzymes in cancer cells.
Supplementary Material
ACKNOWLEDGEMENTS
We are very grateful for the generous gift of Alternol reagent from Dr Jiepeng Chen at Sungen Biosciences (Shantou, China). This work was supported in part by the Natural Science Foundation of Shandong Province, China (ZR2016HL25). The in silicon docking work was conducted by Dr Aaron Smalter, the former Director for the Molecular Graphics & Modeling Lab in the University of Kansas Molecular Structures Group.
Funding information
Natural Science Foundation of Shandong Province,China, Grant number: ZR2016HL25
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigor of preclinical research recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Footnotes
CONFLICT OF INTEREST
All authors declare no conflicts of interest.
AVAILABILITY OF DATA AND MATERIAL
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Finley LW, Zhang J, Ye J, Ward PS, Thompson CB. SnapShot: cancer metabolism pathways. Cell Metab. 2013;17:466–466 e462. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Karakhanova S, Hartwig W, et al. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016;231:2570–2581. [DOI] [PubMed] [Google Scholar]
- 3.Krebs HA. The citric acid cycle and the Szent-Gyorgyi cycle in pigeon breast muscle. Biochem J. 1940;34:775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stuart SD, Schauble A, Gupta S, et al. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hildyard JC, Ammala C, Dukes ID, Thomson SA, Halestrap AP. Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim Biophys Acta. 2005;1707:221–230. [DOI] [PubMed] [Google Scholar]
- 7.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amoedo ND, Obre E, Rossignol R. Drug discovery strategies in the field of tumor energy metabolism: limitations by metabolic flexibility and metabolic resistance to chemotherapy. Biochim Biophys Acta. 2017;1858:674–685. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Wang J, Sun B, Zhang Y, Zhu J, Li C. Cell growth inhibition, G2M cell cycle arrest, and apoptosis induced by the novel compound Alternol in human gastric carcinoma cell line MGC803. Invest New Drugs. 2007;25:505–517. [DOI] [PubMed] [Google Scholar]
- 10.Yao Y, Zhang B, Chen H, et al. Alteronol inhibits proliferation in HeLa cells through inducing a G1-phase arrest. J Pharm Pharmacol. 2012;64: 101–107. [DOI] [PubMed] [Google Scholar]
- 11.Yeung ED, Morrison A, Plumeri D, et al. Alternol exerts prostate- selective antitumor effects through modulations of the AMPK signaling pathway. Prostate. 2012;72:165–172. [DOI] [PubMed] [Google Scholar]
- 12.Zhu XL, Wang YL, Chen JP, et al. Alternol inhibits migration and invasion of human hepatocellular carcinoma cells by targeting epithelial-to-mesenchymal transition. Tumour Biol. 2014;35: 1627–1635. [DOI] [PubMed] [Google Scholar]
- 13.Zuo D, Zhou Z, Wang H, et al. Alternol, a natural compound, exerts an anti-tumour effect on osteosarcoma by modulating of STAT3 and ROS/MAPK signalling pathways. J Cell Mol Med. 2017;21:208–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Y, Chen R, Huang Y, et al. Natural compound alternol induces oxidative stress-dependent apoptotic cell death preferentially in prostate cancer cells. Mol Cancer Ther. 2014;13:1526–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez Molina D, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. [DOI] [PubMed] [Google Scholar]
- 16.Chen R, Zhao Y, Huang Y, et al. Nanomicellar TGX221 blocks xenograft tumor growth of prostate cancer in nude mice. Prostate. 2015;75:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J, Xie SX, Huang Y, et al. Prostate-targeted biodegradable nanoparticles loaded with androgen receptor silencing constructs eradicate xenograft tumors in mice. Nanomedicine (Lond). 2012;7: 1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pang J, Yang YW, Huang Y, et al. P110beta inhibition reduces histone H3K4 di-methylation in prostate cancer. Prostate. 2017;77:299–308. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Q, Youn H, Tang J, et al. Phosphoinositide 3-OH kinase p85alpha and p110beta are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene. 2008;27:4569–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun A, Li C, Chen R, et al. GSK-3beta controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate. 2016;76:172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Havel LS, Kline ER, Salgueiro AM, Marcus AI. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene. 2015;34:1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun A, Tawfik O, Gayed B, et al. Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. Prostate. 2007;67:203–213. [DOI] [PubMed] [Google Scholar]
- 23.Sun A, Tang J, Terranova PF, Zhang X, Thrasher JB, Li B. Adeno-associated virus-delivered short hairpin-structured RNA for androgen receptor gene silencing induces tumor eradication of prostate cancer xenografts in nude mice: a preclinical study. Int J Cancer. 2010;126:764–774. [DOI] [PubMed] [Google Scholar]
- 24.Prasannan CB, Villar MT, Artigues A, Fenton AW. Identification of regions of rabbit muscle pyruvate kinase important for allosteric regulation by phenylalanine, detected by H/D exchange mass spectrometry. Biochemistry. 2013;52:1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He C, Duan S, Dong L, et al. Characterization of a novel p110beta- specific inhibitor BL140 that overcomes MDV3100-resistance in castration-resistant prostate cancer cells. Prostate. 2017;77: 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maier JK, Labute P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins. 2014;82:1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavan H, Christudoss P, Mickey K, et al. Arsenite effects on mitochondrial bioenergetics in human and mouse primary hepatocytes follow a nonlinear dose response. Oxid Med Cell Longev. 2017;2017: 9251303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen P, Yu J, Chalmers B, et al. Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anticancer Drugs. 2012;23:437–444. [DOI] [PubMed] [Google Scholar]
- 30.Latonen L, Afyounian E, Jylha A, et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat Commun. 2018;9:1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sajnani K, Islam F, Smith RA, Gopalan V, Lam AK. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie. 2017;135:164–172. [DOI] [PubMed] [Google Scholar]
- 32.Chinopoulos C Which way does the citric acid cycle turn during hypoxia? The critical role of alpha-ketoglutarate dehydrogenase complex. J NeurosciRes. 2013;91:1030–1043. [DOI] [PubMed] [Google Scholar]
- 33.Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. [DOI] [PubMed] [Google Scholar]
- 34.Deng G, Shen J, Yin M, et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J Biol Chem. 2015;290:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.