

Abstract
The tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), is a potent lung carcinogen and exerts its carcinogen effects upon metabolic activation. The identification and quantitation of NNK metabolites could identify potential biomarkers of bio-activation and detoxification of this potent carcinogen and may be used to predict lung cancer susceptibility among smokers. Here, we have used in vivo isotope labeling and high-resolution mass spectrometry-based methods for the comprehensive profiling of all known and unknown NNK metabolites. The sample enrichment, LC/MS, and data analysis workflow, including the custom script for automated d0-d4 m/z pair peak detection, enabled unbiased identification of numerous NNK metabolites. The structures of the metabolites were confirmed using targeted LC-MS2 with retention time (tR) and MS2 fragmentation comparison to standards when possible. Eleven known metabolites and the unchanged NNK were identified simultaneously. More importantly, our workflow revealed new and novel NNK metabolites including: the 1,3-Diol (13); α-OH-methyl-NNAL-Gluc (14); nitro-NK-N-oxide (15); nitro-NAL-N-oxide (16); γ-OH NNAL (17); and three N-acetylcysteine (NAC) metabolites (18a-c). We measured the differences in the relative distribution of a panel of nitroso-containing NNK-specific metabolites in rats before and after phenobarbital (PB) treatment, and this serves as a demonstration of a general strategy for the detection of metabolic differences in animal and cell systems. Lastly, we have generated a d4-labeled NNK metabolite mixture to be used as internal standards (d4-rat urine) for relative quantitation of NNK metabolites in humans, and this new strategy will be used to assess carcinogen exposure and ultimately to evaluate lung cancer risk and susceptibility in smokers.
Keywords: NNK, tobacco carcinogens, metabolites profiling, mass spectrometry, stable isotope labeling
INTRODUCTION
Lung cancer is the most common and preventable form of cancer worldwide. In the United States, it is the leading cause of cancer mortality among men and women, and in 2017 it was estimated that 1 out of 4 cancer deaths were due to lung cancer.1 The strong correlation between lung cancer and tobacco smoking is well documented, however, despite the 90% lung cancer incidence caused by tobacco smoking, only a small fraction (11–24%) of smokers will develop the disease over their lifetime.2,3 It is hypothesized that this disparity may be due to inter-individual genetics differences, which result in variations in the uptake and metabolism of tobacco carcinogens leading to differing levels of carcinogen-derived metabolites.4–6 The quantitation of these metabolites could provide insights into the relative contributions of the different pathways involved in the bio-activation and detoxification of carcinogens, and could enable rational strategies for cancer prevention based on targeted surveillance of high-risk and susceptible populations.
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is a potent lung carcinogen present in all tobacco products and one of the most powerful carcinogens in tobacco smoke. It is classified as a Group 1 human carcinogen by the International Agency for Research on Cancer. In order to exert its carcinogenic effects, NNK must be metabolically activated into highly reactive species capable of modifying DNA. These modifications may result in DNA damage that, if not repaired, can lead to mutations causing cell growth dysregulation and cancer.
Previous studies have extensively characterized the metabolic pathways of bio-activation and detoxification of NNK in laboratory animals.7–12 Figure 1 summarizes the structures of known NNK metabolites as determined in animal models. NNK is primarily metabolized by N-oxidation, α-hydroxylation, and carbonyl reduction to 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), also a potent carcinogen, which is detoxified by conversion to its glucuronide conjugates. Profiling of NNK metabolites has been previously reported using tritium labeled [5-3H]-NNK and radio flow high-performance liquid chromatography (HPLC).13–19 Using this approach, the retention times of metabolites were compared with the retention times of synthetic standards for qualitative confirmation. While useful in identifying major NNK metabolites, this approach lacks the selectivity for the identification of co-eluting metabolites and does not provide structural information for the characterization of unknown metabolites. The quantitation of some of the identified metabolites have been used to measure carcinogen exposure, uptake, activation, and detoxification in humans. For instance, NNAL and its glucuronides (NNAL-O-Gluc and NNAL-N-Gluc) have been extensively used to measure NNK exposure and detoxification, respectively.20–22 However, these methods rely on the quantitation of a few predetermined NNK metabolites with known MS fragmentation spectra, and the resulting measurements do not allow for the determination of the relative contributions of the different metabolic pathways responsible for NNK-induced carcinogenesis. A comprehensive method that is able to identify potentially all metabolites and understand the contribution of the various pathways of NNK metabolism is needed.
Figure 1.
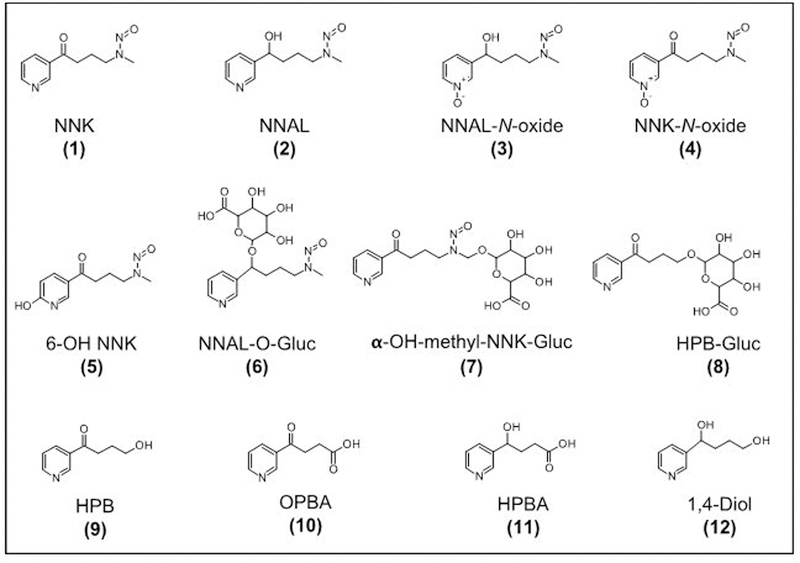
Structures of NNK and its known metabolites as determined in laboratory animals.
Mass spectrometry-based metabolomics have been extensively used for profiling metabolites resulting from various perturbations in biological systems. Isotope labeling approaches have also been incorporated into these assays, allowing for confident identification and improved quantitation of metabolites. As a result, a number of stable isotope labeling (SIL)-assisted software tools have been developed for untargeted metabolomics, however, most of these tools have been designed for specific applications, which use various labeling and measurement strategies.23–28 Here, we have developed a workflow to complement these existing tools for processing high-resolution LC/MS mass spectral data obtained from stable isotope labeling experiments. The sample enrichment, LC/MS, and data analysis workflow presented in this work, including the custom script for the automated d0-d4 m/z pair peak detection, enabled unbiased identification of numerous metabolites, including the identification of new and novel NNK metabolites in rats. Chemical structures for the previously unidentified NNK metabolites are proposed, several of which were expected but had not been detected yet in either animal or human model systems. This new panel of metabolites could serve as potential biomarkers to assess NNK uptake, metabolic activation and detoxification, and ultimately contribute to our understanding lung cancer risk and susceptibility among smokers.
MATERIALS AND METHODS
Caution:
d0-NNK, d4-NNK, 13C6-NNAL, and NNAL are carcinogens and must be handled carefully and with adequate precautions.
Materials.
F344 rats (Male, 250–300 g) were obtained from Charles River (Kingston, NY). NNAL, [13C6]-NNAL (1,2’,3’,4’,5’,6’−13C6), NNAL-O-Gluc, d0-NNK, d4-NNK (pyridine-d4; isotopic purity > 98%), and 1,4-Diol were obtained from Toronto Research Chemicals (TRC, Ontario, Canada). NNK-N-oxide, NNAL-N-oxide, β-OH NNAL, β-OH NNK-N-oxide, 1,3-Diol, HPBA, and OPBA were synthesized as previously described.11 β-OH NNK was a kind gift from Dr. Stephen Hecht. Oasis HLB and Oasis MCX solid-phase extraction cartridges were purchased from Waters (Milford, MA). Strata-X polymeric reverse-phase cartridge was purchased from Phenomenex (Torrance, CA). All acids and organic solvents were HPLC or MS grade.
In vivo stable isotope labeling of NNK metabolites.
d0-NNK and d4-NNK were used to treat F344 rats following a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota. Figure 2a illustrates the experimental design for the animal experiment and the four urine samples collected corresponding to the different urine collections (BU, U1, U2, and U3). Baseline urine samples (BU) were collected for 24 hours before the NNK treatment. After which, rats were dosed with an intraperitoneal (i.p.) injection of d0-NNK (0.1 mg/kg) to Rat A and d4-NNK to Rat B. Then 24-h urine samples (U1) were collected. To account for individual variability, a cross-over experiment was performed on the next day where d0-NNK was administered to Rat B and d4-NNK to Rat A. The 24-h urine samples (U2) were again collected. To enhance the formation and identification of NNK metabolites in rat urine, phenobarbital (PB) (40 mg/kg per day) was administered for three days. PB induces α-hydroxylation of NNK and various UDP-glucuronosyl transferases (UGTs).15 Following the three-day induction, another i.p. injection of d0-NNK to Rat A and d4-NNK to Rat B was administered and urine samples (U3) were collected for 24 hours. All urine samples were collected on cooling blocks to prevent degradation of metabolites and to prevent bacterial growth. A 1:1 (v/v) of each urine sample collection were combined to yield BU*, U1*, U2*, U3*, which were used for the NNK metabolites profiling (Figure 2a).
Figure 2.
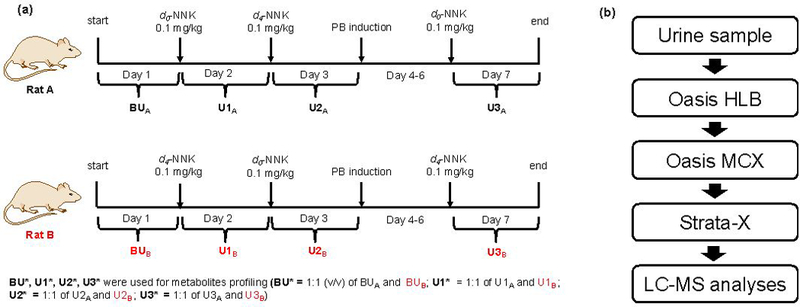
(a) Experimental scheme for characterizing NNK metabolites in rats. (b) Optimized sample enrichment strategy wherein most of the known NNK metabolites in PB-induced rat urine were detected.
Rat urine sample preparation.
The sample cleanup and enrichment protocols were optimized using a mixture of synthetic standards consisting of NNAL, [13C6]-NNAL, NNAL-O-Gluc, NNK-N-oxide, NNAL-N-oxide, 1,4-Diol, HPBA, and OPBA. The methods were further refined using a 1:1 ratio (v/v) of d0-labeled PB-induced rat urine (U3A) and d4-labeled PB-induced rat urine (U3B) and monitoring for known NNK metabolites. During the refinement, a protein precipitation step was introduced to eliminate potential interference of high abundant proteins during LC/MS analysis. One hundred (100 μL) microliters of PB-induced d0-NNK rat urine (U3A) and 100 μL PB-induced d4-NNK rat urine (U3B) samples were combined. The urine sample was spiked with labeled NNAL (13C6-NNAL, 0.5 pmol) and deproteinized by adding 4 volumes of 1:1:1 acetonitrile (ACN): methanol (MeOH): acetone. The mixture was vortexed and incubated for 15 min on ice. After which, the sample was centrifuged, the supernatant collected and dried under vacuum. The sample was subjected to a series of solid phase extraction systems, with different modes of separation (see Figure S9b for detailed protocol). Figure 2b shows the flow chart of the optimized sample preparation and cleanup strategy used for the LC/MS analyses of rat urine samples.
LC/MS parameters.
All LC/MS experiments were performed on a Thermo Orbitrap Fusion mass spectrometer (Thermo Scientific, Waltham, MA) coupled to a Dionex RSLC UPLC (Thermo Scientific, Sunnyvale, CA). Reverse phase chromatography was performed on a home-packed Luna C18 column (5 μm, 120 Å, 200 mm x 75 μm ID, Phenomenex, Torrance, CA) at room temperature and flow rate of 0.3 μL min−1 using 5 mM ammonium acetate as mobile phase A and methanol as mobile phase B. The LC gradient started with 5% B for the first 5 min with a flow rate of 1.0 μL min−1, followed by switching of the injection valve to remove the 5 μL loop from the flow path, and reducing the flow rate to 0.3 μL min−1 over 1 minute. A linear gradient from 5% to 98% B over 19 min was used followed by a constant 98% B for 3 min. Finally, re-equilibration was performed by changing the mobile phase composition from 95% to 5% B over 2 min and increasing the flow rate to 1.0 μL min−1 over 2 min with a 5% B hold for 6 min. The total run time was 38 min. An optimal S-Lens RF setting of 60% was used for analysis. Instrument sensitivity and mass accuracy was checked using a mixture of synthetic NNK metabolites standard before all LC/MS analyses. Mass spectra were recorded in positive ion mode with nano-electrospray voltage set to 2.2 kV and an ion transfer tube temperature of 350 °C. All LC/MS analyses were operated with at least 30,000 mass resolving power (FWHM) with internal mass calibration using the EASY-IC feature of the Orbitrap Fusion.
Untargeted MS-based method for screening known and unknown NNK metabolites in rats.
An untargeted MS-based approach was used for the comprehensive profiling of known and unknown NNK metabolites in the four rat urine samples corresponding to the four urine collections (BU, U1, U2, and U3) as shown in Figure 2a. For each collection, urine from Rat A and Rat B was combined in a 1:1 (v/v) ratio and processed using the sample cleanup protocol shown in Figure 2b. This was done to have both d0-and d4-metabolites in each sample, except for the baseline urine (BU). Pairs of d0-and d4-NNK metabolites in each of the four samples were then measured during LC/MS analysis using high-resolution full scan data acquisition at a mass range of m/z 150–900. The LC/MS and data-analysis workflow for the untargeted profiling for all known and unknown NNK metabolites is illustrated in Figure 3a. The following MS parameters were used for the high-resolution accurate mass full scan data acquisition: mass range, m/z 150–900; resolution setting, 240,000; quadrupole isolation; AGC target setting, 4e5; maximum injection time, 100 ms; and RF lens, 60%. The full scan mass spectral data were acquired in centroid mode and analyzed with Compound Discoverer (CD) 2.0 (Thermo Scientific, Sunnyvale CA) to provide feature detection, which further processed following the data analysis workflow shown in Figure 3a. Feature detection in CD was performed using the following parameters: retention time alignment using the adaptive curve alignment model; 5 ppm mass tolerance; 0.2 min retention time tolerance; intensity tolerance of 30% (relative intensity tolerance used for isotope search); S/N threshold of 3; minimum peak intensity of 500; ions detected as [M+H]+, [M+NH4]+, [M+Na]+, [M+K]+, [M+H+MeOH]+, [2M+H]+, [M+2H]2+; minimum element counts (C H); maximum element counts (C90, H190, Br3, Cl4, K2, N10, Na2, O15, P2, S5). The resulting features were exported to Excel (Microsoft) and analyzed for automatic d0-d4 m/z pair identification using a custom JAVA script implemented on the Google spreadsheet platform, and available at https://docs.google.com/spreadsheets/d/1YN8xwjhylBRloBxMKqau_eAqVHX1FHvpCC7cadzzn3I/edit?ts=5962dd1f#gid=0. The script identifies pairs of features with m/z values separated by exactly 4.0251 Da (±0.001 Da). Further analysis is performed to identify those m/z pairs that have retention times within 0.2 min, and pairs in samples U1*, U2*, and U3* combined into a single list, which is further scrutinized by: (1) elimination of m/z pairs detected in the baseline urine (BU*) sample, (2) manual evaluation of candidate d0-d4 pairs using Xcalibur 3.0 (Thermo Scientific, Sunnyvale, CA) to ensure similar peak shape and intensity, and (3) elimination of redundant features by removing multiple instances of m/z pairs. The resulting candidate d0-d4 m/z pairs are assigned to known metabolites based on their accurate masses as determined from the high-resolution full scan data. For those m/z pairs which do not correspond to a known metabolite, the measured high-resolution accurate d0 m/z mass is searched against the METLIN database (Scripps, CA) for putative metabolite identification.29 Both the tentatively assigned NNK metabolites and unknown ones were further confirmed and characterized using targeted LC-MS2 as described below.
Figure 3.
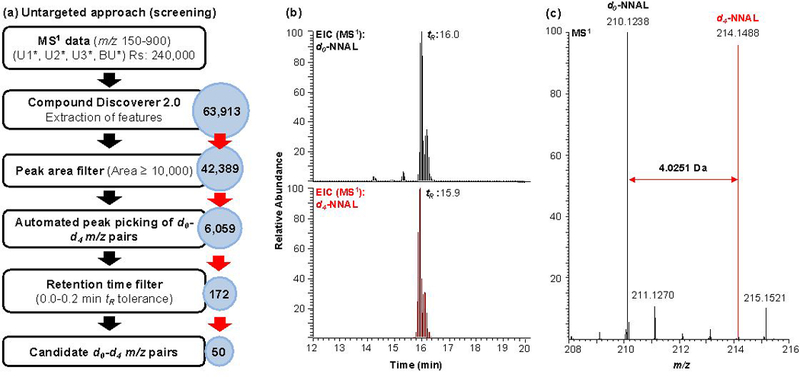
LC/MS and data analysis workflow for the comprehensive profiling of all known and unknown NNK metabolites in rats. (a) Untargeted MS-based profiling approach. Features extracted using Compound Discoverer from high-resolution full scan data (Rs=240,000) were subjected to a series of filters. The peak area filter was used to exclude low abundant features (Peak area <10,000) before the automatic peak picking of m/z pairs using a custom script. The resulting list of m/z pairs were then filtered using a specified retention window (0–0.2 min) resulting to a list of candidate m/z pairs/metabolites. (b) Representative extracted ion chromatogram (EIC) of NNAL, a major NNK metabolite, from the full scan mass spectral data. (c) MS1 spectrum of d0-NNAL and d4-NNAL.
Targeted MS-based approach (structural confirmation).
To gain structural confirmation, the list of m/z pairs detected with the untargeted MS-based profiling approach were used to create a targeted mass inclusion list for LC-MS2 analysis (Figure 4), with reinjection of the samples using the following parameters: quadrupole isolation window; 1.5 m/z; HCD (High-energy collisional dissociation); 25%; Orbitrap resolution setting, 30,000; scan range, m/z 80–500; RF lens, 60%; AGC target, 5e4; maximum injection time, 54 ms; all parallelizable injection time feature, enabled. The MS2 data were acquired in profile mode with the EASY-IC internal mass calibration feature enabled. The high-resolution accurate masses of fragment ions of expected and known NNK metabolites were extracted from the MS2 mass spectral data within 5 ppm mass tolerance. For accurate and confident identification of candidate m/z pair, manual evaluation of candidate d0-d4 pairs using Xcalibur 3.0 (Thermo Scientific, Sunnyvale, CA) was performed to ensure that the m/z pairs have similar peak shape, intensity ratio (d0/d4), and have at least 2–3 MS2 fragment ions for both m/z values. Where possible, the retention time and MS2 fragmentation pattern of synthetic standards were used to confirm the identity of NNK metabolites.
Figure 4.
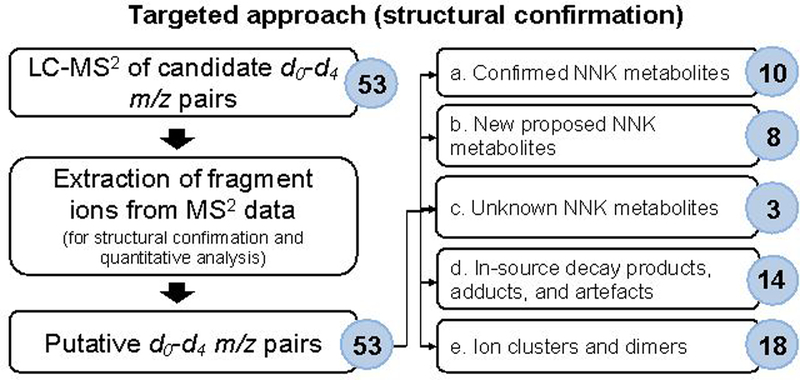
Targeted MS-based approach using LC-MS2 for structural confirmation of the candidate m/z pairs/metabolites identified in the untargeted profiling approach. The identities of several analytes were confirmed by comparing their tR and MS2 fragmentation pattern to that of synthetic standards. While unknown metabolites were identified using their MS2 mass spectral fragmentation data and database search in METLIN when possible.
RESULTS AND DISCUSSION
Profiling of NNK metabolites in biological fluids from animals has been traditionally accomplished using tritium labeling and radio flow high-performance liquid chromatography (HPLC). 13–19 In addition, methods based on mass spectrometry, often with selected reaction monitoring (SRM) have been used to quantify selected NNK metabolites in both animal models and humans.20,21,30–32 While useful in measuring a few NNK metabolites at a time, these methods do not provide structural information for identification and characterization of unknown compounds. Since, the traditional SRM approach measures only a few metabolites per analysis, limited insights are gained on the relative contributions of the individual metabolic pathways to NNK bioactivation and detoxification. Here, we have developed a sample enrichment strategy, HR-AM LC/MS method, and data analysis workflow for the untargeted profiling and simultaneous characterization of known and unknown NNK metabolites. To obtain a comprehensive profile of all NNK metabolites in rats, we analyzed the urine samples of rats treated with d0-NNK and d4-NNK.
Untargeted MS-based method for screening known and unknown NNK metabolites in rats.
A sample processing and data analysis workflow was developed to screen for previously characterized NNK metabolites as well as to simultaneously identify new metabolites in rat urine from a d0- d4-NNK exposure experiment. We have tested different combinations of SPE columns to provide orthogonal modes of cleanup and enrichment of the different NNK metabolites in the urine sample, and found that the combination of the three SPEs as presented in the manuscript gave the best detection and recovery of most of the known NNK metabolites. For example, using 13C6-NNAL spiked into baseline rat urine (1:1 ratio of urine from Rat A and Rat B), the recovery obtained was 73%. The optimized sample preparation method was further refined and applied to the analysis of urine from NNK treated rats (Figure 2a,b). PB treatment was used to induce cytochrome P450 2B (the catalyst of NNK alpha-hydroxylation), and UDP-glucuronosyltransferases.13,15 To generate a comprehensive NNK metabolism profile of known and unknown NNK metabolites, LC/MS analysis was carried out on 1:1 mixtures of urine samples from two rats (A and B). Four urine samples corresponding to different urine collections were collected from each rat (Figure 2a). Baseline urine (BU) samples were collected on Day 1 before NNK treatment. U1 urine was collected on Day 2 wherein Rat A was treated with d0-NNK and Rat B with d4-NNK. On Day 3, the exposures were switched, and Rat A received d4-NNK and Rat B received d0-NNK and urine was again collected for 24 h (U2). This cross-over experiment was performed to account for the biological variability in the metabolism of NNK in the two rats. Lastly, urine samples (U3) were collected on Day 7 after the 3-day PB-treatment and final NNK treatment, where Rat A received d0-NNK and Rat B d4-NNK.
The data analysis workflow used for the untargeted profiling of NNK metabolites in rats is illustrated in Figure 3a. The urine from Rat A and B was combined for each urine collection (Figure 2a) to obtain three samples containing both d0 and d4-NNK: U1*, U2*, and U3*. The analysis of the three treated samples generated a combined total number of 63,913 features or m/z values after feature extraction using Compound Discoverer (~21,000 features detected in each sample) (Table S1). The 63,913 features include features that are common in all three samples as well as features that are unique to each sample. Peak area filtering of features with ion abundance greater than 10,000 yielded 42,389 m/z values, which were then uploaded, along with corresponding retention times and peak areas, and analyzed using the automated d0-d4 m/z pair identification script implemented on a Google spreadsheet platform (Table S2). The custom script was used to select pairs of m/z values separated by 4.0251 Da (±0.001 Da), the mass difference expected for the d0- and d4-NNK metabolite pairs. This resulted in 6,059 m/z pairs (~2,000 in each sample, Table S3). The data were further filtered by an initial retention time (tR) window of 12 s or 0.2 min to yield a total of 172 d0-d4 m/z pairs for U1*, U2*, and U3* (Table S4). Fifty (50) non-redundant d0-d4 m/z pairs were obtained upon subtraction of m/z pairs detected in the baseline urine (BU) sample (Table S5), manual evaluation of each m/z pair using Xcalibur to ensure that the peak shape and intensity ratio of the d0-d4 signals are similar, and exclusion of redundant metabolites or features with similar m/z and tR that are found in at least two urine sample dataset. Redundant metabolites or features are those detected in more than one data set. For instance, NNAL (2), was detected in all samples, U1*, U2*, and U3* but was counted only as one of the metabolites in the final 50 d0-d4 m/z pairs (Table S6).
From the 50 d0-d4 m/z pairs, 10 metabolites were tentatively identified based on the agreement of the measured accurate masses with those of NNK metabolites previously reported in literature (Table S7). A representative EIC of NNAL (2), a major NNK metabolite, from the full scan mass spectral data is presented in Figure 3b. Both d0-NNAL (m/z 210.1238) and d4-NNAL (m/z 214.1488) are present with similar retention times (d4-NNAL, 15.9 min; d0-NNAL, 16.0 min). The two peaks observed for both masses correspond to the (E)- and (Z)- isomers of NNAL. The MS spectrum of these peaks contained pairs of m/z values separated by 4.0251 Da (±0.001 Da), m/z 210.1238 and m/z 214.1488 corresponding to the d0-NNAL and d4-NNAL, respectively (Figure 3c). Although full scan data acquisition is not as sensitive as the targeted approach, we were able to simultaneously identify 9 of 11 known metabolites of NNK and the unchanged NNK (Figure 1, Table S7).
Targeted MS-based method for structural confirmation of known metabolites and characterization of new and novel NNK metabolites.
In order to confirm the identity of the 10 tentatively assigned d0-d4 m/z pairs detected in the untargeted approach (Table S7) and to characterize the remaining 40 pairs, the samples were re-injected for targeted LC-MS2 structural confirmation (Figure 4). LC-MS2 analysis of 53 total target pairs, including three additional pairs of NNK metabolites, was performed and fragment ions were extracted. The two undetected NNK metabolites were 6-OH NNK (5) and HPB (9). One additional mass pair, m/z 242.1135 (16) and m/z 246.1386, was included since a relatively abundant tentative d0-NNK metabolite, m/z 240.0978 (Table 1, 15) was observed in the untargeted approach. We hypothesized that a reduced version, which would be a metabolite of NNAL (m/z 242.1135) may also be formed. The complete list of the 53 m/z pairs used for the LC-MS2 assay are listed in Table S8.
Table 1.
Identification of confirmed NNK metabolites, new proposed NNK metabolites, and unknown NNK metabolites using untargeted (UT) and targeted (T) mass spectrometry-based methods. Structural characterization by mass spectrometry (MS) was performed using the measured high-resolution accurate precursor masses in the full scan (MS1) and MS2 mass spectral data and when possible, comparing with standards. For NMR, 1H-NMR and 1H-1H COSY were performed.
| Confirmed metabolites | ||||||
|---|---|---|---|---|---|---|
| d0 m/z | d4 m/z | d0 tR (min) | d4 tR (min) | Description | MS method (Untargeted, UT; Targeted, T) |
Structural Confirmation |
| 182.0811 | 186.1061 | 8.2 | 8.1 | HPBA (11) | UT, T | Synthetic standard (tR, MS) |
| 386.1556 | 390.1806 | 12.6 | 12.6 | NNAL-O-Gluc (6) | UT, T | Synthetic standard (tR, MS) |
| 226.1184* | 230.1434 | 12.8 | 12.7 | NNAL-N-oxide (3) | UT, T | Synthetic standard (tR, MS) |
| 342.1180 | 346.1430 | 12.9 | 12.9 | HPB-Gluc (8) | UT, T | Literature (MS) |
| 180.0656 | 184.0906 | 13.0 | 12.9 | OPBA (10) | UT, T | Synthetic standard (tR, MS) |
| 224.1032 | 227.1221 (d3) | 14.1 | 14.0 | 6-OH NNK (5) | T | Literature (MS) |
| 224.1032 | 228.1280 | 14.1 | 14.0 | NNK-N-oxlde (4) | UT, T | Synthetic standard (tR, MS) |
| 400.1350 | 404.1601 | 14.5 | 14.5 | α-OH-methyl-NNK-Gluc (7) | UT, T | Literature (MS) |
| 168.1019 | 172.1269 | 14.3 | 14.2 | 1,4-Diol (Diol 1 ) (12) | UT, T | Synthetic standard (tR, MS) |
| 210.1243 | 214.1486 | 16.0 | 15.9 | NNAL (2) | UT, T | Synthetic standard (tR, MS) |
| 166.0863 | 170.1113 | 16.7 | 16.7 | HPB (9) | T | Literature (MS) |
| 208.1081 | 212.1331 | 17.5 | 17.4 | NNK (1) | UT, T | Synthetic standard (tR, MS) |
| New proposed metabolites | ||||||
| 168.1019 | 172.1269 | 15.6 | 15.5 | 1,3-Diol (Diol 2) (13) | UT, T | Synthetic standard (tR, MS) |
| 402.1508 | 406.1759 | 11.3 | 11.2 | α-OH-methyl-NNAL-Gluc (14) | UT, T | MS |
| 240.0977 | 244.1229 | 14.7 | 14.7 | nitro-NK-N-oxide (15) | UT, T | MS, NMR |
| 242.1135 | 246.1386 | 13.6 | 13.5 | nitro-NAL-N-oxide (16) | T | MS |
| 226.1185 | 230.1437 | 13.9 | 13.9 | γ-OH NNAL (OH-NNAL 2) (17) | UT, T | MS |
| 313.1214 | 317.1462 | 15.8 | 15.8 | NAC-PHB 1 (18a) | UT, T | MS |
| 313.1213 | 317.1465 | 16.4 | 16.3 | NAC-PHB 2 (18b) | UT, T | MS |
| 313.1214 | 317.1465 | 17.2 | 17.2 | NAC-PHB 3 (18c) | UT, T | MS |
| Unknown metabolites | ||||||
| 168.1019 | 172.1270 | 16.1 | 16.0 | Unknown 1 (Diol 3) (19) | UT, T | Unknown |
| 226.1185 | 230.1437 | 13.6 | 13.5 | Unknown 2 (OH-NNAL 1) (20) | T | Unknown |
| 254.1135 | 258.1385 | 14.1 | 14.0 | Unknown 3 (21) | UT, T | Unknown |
m/z 226.1185, tR12.8 min (Table 1, confirmed metabolites) vs. tR:13.4 min (Figure S5a, Panel III)
For accurate and confident identification of NNK metabolites in the targeted LC-MS2, high-resolution accurate masses of the fragment ions were extracted from the MS2 data. The following criteria were used to characterize the metabolites, (1) the identified metabolites should have both d0 and d4 separated by 4.0251 (±0.001 Da) with a tR difference of less than 0.2 min, (2 ) the m/z error of the precursor and fragment ions should be less than 5 ppm for both labeled and unlabeled versions, (3) at least 2 or 3 fragment ions separated by 4.0251 (±0.001 Da) should be present in the MS2 of the d0-d4 m/z pairs, (4) both labeled and unlabeled analytes should form similar adducts (H, Na, NH4), and (5) should not be present in the baseline urine, BU* (without NNK treatment). Using the LC-MS2 analysis for structural confirmation, the 53 d0-d4-m/z pairs were classified as (a) confirmed metabolites, (b) new proposed metabolites, (c) unknown metabolites (d) in-source decay products (ISD), adducts, and artefacts, and (e) ion clusters and dimers (Table 1 and Table S11).
The identities of the 9 tentatively assigned metabolites and the unchanged NNK (10 total, Table S7) from the untargeted screening approach were confirmed based on their MS2 fragmentation, tR comparison with synthetic standards, and from available data in the literature. Two known NNK metabolites, 6-OH NNK (6) and HPB (8), that were not observed in the untargeted screening were identified using the targeted approach (Table 1). One potential limitation of the current approach is that, due to the position of the deuterium atoms on the NNK, metabolites having the pyridine ring substitution could not be identified. For instance, 6-OH NNK (5) was not identified in the screening approach since the metabolite contains only three deuteriums. Because it is a known NNK metabolite, we have added this compound in the targeted LC-MS2 analysis to confirm its presence in the rat urine. Our approach could be improved by the use of 13C-labeled precursors, but the availability of these labeled compounds is limited and very expensive.
In summary, we have definitively confirmed the presence of NNK and 11 of its metabolites in the urine of F344 rats treated with NNK. In addition to the confirmed metabolites, our workflow revealed new and novel NNK metabolites not previously reported (Table 1). The identities and structures for eight of the unknown m/z pairs were proposed based on their high-resolution accurate masses and MS2 mass spectra, NMR data when possible, and data in the literature. These metabolites included the 1,3-Diol (13); α-OH-NNAL-Gluc (14); nitro-NK-N-oxide (15); nitro-NAL-N-oxide (16); γ-OH NNAL (17); and three N-acetylcysteine (NAC) metabolites (18a, 18b, 18c), which were identified, for the first time as NNK metabolites in the rat. The characterization of these metabolites is described in detail below. Three additional potential metabolites were detected, but data supporting their identification is limited (Table 1).
Characterization of new and novel NNK metabolites.
Using the in vivo d0-d4-NNK labeling strategy for the comprehensive characterization of NNK metabolites in rats, we were able to detect and identify new and novel NNK metabolites. The structures and identities of eight of these metabolites were proposed based on high-resolution accurate masses of both precursor ions and MS2 fragment ions, and comparison to related compounds (Table 1). The 1,3-Diol (13) was confirmed as a metabolite of NNK in the rat for the first time here (Figure 5 and Table 1). This compound has previously been identified as a solvolysis product of model compounds that generate the pyridylhydroxybutylating diazohydroxide.33 The data supporting this is as follows: three chromatographic peaks with m/z of 168.1019 and m/z 172.1270 pairs were detected (Figure S1a, b). The first, tR: 14.3 min co-elutes with and has the identical MS2 spectra as the standard of the 1,4-Diol (Figure 1, 12 and Figure S1 a,b,g,h). Whereas, the peak at tR: 15.6 min has an identical tR and MS2 spectra to the 1,3-Diol (Figure 5, 13; Figure S1a,b,d,i–k). The third peak with tR: 16.5 min (Figure S1a,b,e,f), which appears to be another diol (Table 1, 19) has an MS2 spectra consistent with a structure where the hydroxyl groups are attach on the same carbon (Figure S1e, f). Although it is typical for geminal diols to lose water and form more stable ketones, in this particular case, this feature seems to lose •OH radical forming a carbene (m/z 108.0445), which can further rearrange to the isobaric aldehyde by intramolecular H-shift.34 Further structural analyses are needed to confirm the exact identity of the third diol.
Figure 5.
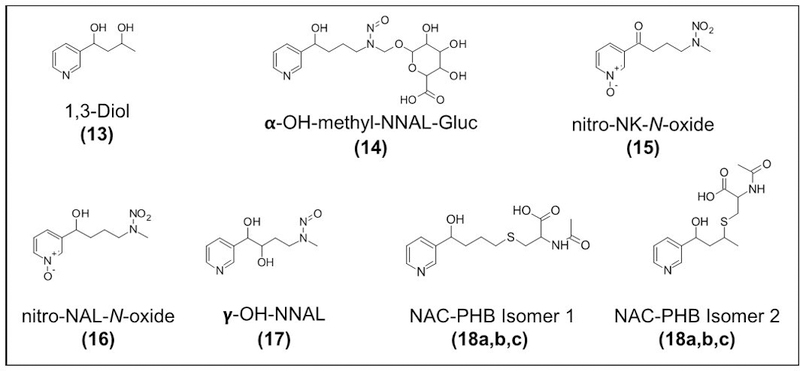
Proposed structures of new and novel NNK metabolites identified in rats including the 1,3-Diol (13); α-OH-methyl-NNAL-Gluc (14); nitro-NK-N-oxide (15); nitro-NAL-N-oxide (16); γ-OH NNAL (17); and three N-acetylcysteine (NAC) metabolites (18a, 18b, 18c).
The NNK metabolite, α-OH-methyl-NNAL-Gluc (14) is related to the glucuronide of α-OH-methyl-NNK (Figure 1, 7), which was characterized as a metabolite of NNK in the rat a number of years ago.14 Therefore, we expected that the analogous glucuronide of α-OH-methyl-NNAL (Figure 5, 14) would be formed and d0 and d4-m/z metabolite pair with the correct masses of d0-α-OH-methyl-NNAL-Gluc, m/z 402.1508 and d4-α-OH-methyl-NNAL-Gluc, m/z 406.1759 was detected eluting 3.2 min earlier than the α-OH-methyl-NNK-Gluc (Figure S2a, b). The fragmentation pattern of this new metabolite was analogous to that of the well-characterized fragmentation pattern of α-OH-methyl-NNK-Gluc (Figure S2c, d). Pairs of fragment ions separated by 4 Da due to the 4 deuterium were observed in the MS2 of the two compounds. The proposed structures of the fragment ions in the MS2 spectra of these compounds are presented in Figure S2c, d. The relative ion abundance of α-OH-methyl-NNAL-Gluc was at least 5 times lower than the α-OH-methyl-NNK-Gluc.
The NNK metabolite, nitro-NK-N-oxide (15) identified in the rat is characterized by d0- m/z 240.0977 and d4-m/z 244.1229 and tR: 14.7 min. The MS2 spectra showed fragment ions consistent with the structures shown in Figure S3b. The fragmentation data of this compound is similar to that of NNK-N-oxide. However, the loss of 62.9956 Da (HNO3) from the molecular ion peak (m/z 240.0977) is observed for this compound, whereas the loss of 47.0007 Da (HNO2) is observed in the MS2 of NNK-N-oxide. We hypothesized that this compound could be a hydroxylated NNK-N-oxide or a nitroamine, wherein the nitroso group is further oxidized to NO2. This compound was found to be a minor contaminant of the NNK-N-oxide standard. Therefore, we isolated it from a solution of the synthetic NNK-N-oxide and obtained NMR spectra (Figure S3c). The NMR (1H-NMR, 1H-1H COSY) data were consistent with the nitroamine of NNK-N-oxide (Figure 5, 15; Figure S3d, e).
A second, related novel metabolite, nitro-NAL-N-oxide was observed with m/z 242.1135) (16). The MS2 fragmentation data for this compound is consistent with a nitroamine of NNAL-N-oxide (Figure 5, 16). The MS2 fragmentation of m/z 242.1135 has many features common to the fragmentation of NNAL-N-oxide, except that a loss of 62.9956 Da (HNO3) from the molecular ion peak, m/z 242.1135 (Figure S4b) was observed, not the loss of 47.0007 Da (HNO2) that was observed for the MS2 of NNAL-N-oxide.
Three metabolites with m/z 226.1186 were observed. This mass is that of either a hydroxylated NNAL metabolites (OH-NNAL) or the most abundant NNK metabolite present in rat urine, NNAL-N-oxide (Figure 1, 3). The NNAL-N-oxide (tR= 12.8 min) was detected at relatively high levels and the two additional compounds were present in lower abundance (Table 1). We hypothesized these compounds are hydroxylated NNAL metabolites, OH-NNAL 2 (17) and OH-NNAL 1 (20). The major MS2 fragment of OH-NNAL 2 (17) is m/z 135.0678 (Figure 5Sb, Panel III). This fragment is not present in the fragmentation spectrum of NNAL-N-oxide standard (Figure S5d, Panel I), which has two characteristic fragments, m/z 161.10735 and m/z 179.1179. An EIC of m/z 135.0678 and m/z 179.1184 clearly illustrated the presence of two additional metabolites (tR: 13.6 and 14.3) that are isobaric with NNAL-N-oxide. Hydroxylation of NNAL can occur in α, β, or γ positions from the N-nitroso group and on the pyridine ring. We ruled out hydroxylation on the pyridine ring since the d4-NNK is deuterated on the pyridine ring and no loss of deuterium was observed. α-hydroxylation is the main pathway involved in the metabolic activation of NNK, however α-OH NNK and α-OH NNAL are unstable and typically detected as their glucuronides or as metabolites of non-enzymatic degradation products. To confirm the identity of the hydroxylated NNAL, a synthetic standard of β-OH NNAL was prepared. LC/MS and co-injection experiments confirmed that no β-OH NNAL was present in the rat urine. The hydroxylated NNAL metabolites (17) and (20) did not co-elute with β-OH NNAL (Figure S5e). We hypothesized that OH-NNAL 2 (17) could be γ-OH NNAL as the MS2 fragmentation data are consistent with a structure with the hydroxyl group in the γ position (Figure 5, 17). Unfortunately, OH-NNAL 1 (20) partially co-elutes with another metabolite, NNAL-N-oxide, therefore a clean MS2 spectra was not obtained for this compound limiting our ability to hypothesize its structure. Further confirmation of the structures of the two newly identified hydroxylated NNAL metabolites is underway.
In addition to the metabolites mentioned above, our method allowed the detection of N-acetylcysteine-PHB (NAC-PHB) (18a, 18b, 18c) in the urine of treated rats. Figure S6a shows the EICs of m/z 313.1214 (d0) and m/z 317.1464 (d4) showing three distinct peaks at retention times of: 16.6 min, 17.2 min, and 17.9 min, which could correspond to three possible isomers. The MS2 spectra of the chromatographic peaks at 17.2 min and 17.9 min showed similar fragment ions and are consistent with isomers of N-acetylcysteine-PHB (Figure S6b–c). Because the ion abundance of the chromatographic peak at 16.6 min is too low, we were not able to get a good MS2 mass spectral data for this compound. The two isomers of PHB-NAC metabolites shown in Figure S6d have been previously identified in rat hemoglobin,35 while the identity of the third isomer needs to be elucidated. An unknown metabolite (m/z 214.0893), which we hypothesized to contain a thiol group based on its measured high-resolution accurate mass was also identified. This metabolite could be a breakdown product of in-source decay (ISD), and the exact identity of the precursor compound still needs to be elucidated (Table S11; Figure S7a,b). Lastly, another unknown compound (21) was also detected and characterized by the initial loss of HNO2 (Table 1, Figure S8a, b). Based on the measured high-resolution accurate mass of its precursor [M+H]+ and MS2 fragment ions, we proposed that this compound could be a methoxylated NNK metabolite, however, the exact identity of this unknown compound needs further investigation.
Identification of in-source decay ions (ISD), artifacts, metabolite adducts, ion clusters (C) and dimers (D).
A number of m/z pairs corresponding to ISDs, adducts, and artefacts from sample preparation conditions were also identified (Table S11). The detection of ISD ions, with exact retention times of the precursor ions for several known metabolites (e.g. NNAL-N-oxide (3), NNK-N-oxide (4), NNAL (2), and NNK (1); Table S11), further confirmed their identities. Artefactual formation of NNK metabolites generated during sample cleanup was also identified and is a critical finding for the design of suitable sample cleanup procedures for the quantitation of these metabolites. Previous studies have quantified intact NNAL glucuronides in human urine and have used the SRM transition m/z 386→m/z 210.21 However, here we have identified a methyl ester of NNAL-O-Gluc (6, m/z 400.1713). We have confirmed that the artefactual formation of the methyl ester occurs with a synthetic standard of NNAL-O-Gluc, which has been processed by the sample enrichment strategy illustrated in Figure 2b (Figure S9b). If some of the glucuronides are esterified as a result of sample preparation conditions, quantitation of these compounds would be affected, and amounts underestimated. This artefactual derivatization was also observed with other glucuronidated NNK metabolites such as α-OH-methyl-NNK-Gluc (7).
Finally, our workflow revealed extensive dimerization of NNK metabolites and ion clustering with solvent molecules such as methanol, and other high-abundant endogenous compounds present in the sample during ESI-MS analysis (Table S11). For example, N-oxides in general can form extensive dimerization during ESI-MS.36,37 Clusters of NNAL-N-oxide and methyl guanosine was observed at m/z 523.2259 (d0) and m/z 527.2513 (d4) (Figure S10, Panel I and II). Upon HCD, the intact precursor and fragment ions from both NNAL-N-oxide (m/z 226.11858) and methyl guanosine (m/z, 298.11444) were observed in the MS2 spectra (Figure S10c). This cluster disappeared when the sample was diluted 1000 times, which indicates that this clustering is due to the high-concentration of NNAL-N-oxide and methyl guanosine in the urine samples (Figure S10d, Panel I and II).
Relative distribution of NNK metabolites after phenobarbital (PB) induction.
One of the future goals of this work is to be able to use this workflow to characterize all NNK metabolites in humans to predict lung cancer risk and susceptibility among smokers. We hypothesized that genetic differences would result in variations in the capacity to activate and detoxify carcinogens, and therefore will result in differing levels of NNK metabolites in biological fluids. To mimic probable dysregulation and disparities in the metabolic enzyme activities, we induced the enzymes responsible for NNK metabolism using PB. Because our focus is on the identification and quantitation of NNK-specific metabolites in humans, we used those metabolites containing the nitroso group (Figure 1), as other metabolites of NNK (non-nitroso containing) may be derived from nicotine. Figure 6 shows the NNK metabolic profiles of Rat A before and after PB induction. The levels of several metabolites changed with PB induction. For example, the level of NNK (1) after induction decreased only slightly (+PB/-PB = 0.8), but dramatic differences were observed in the case of NNAL (2), NNAL-O-Gluc (3), and α-OH-methyl-NNK-Gluc (7). The NNAL levels decreased by two-fold, while the levels of NNAL-O-Gluc and α-OH-methyl-NNK-Gluc increased after PB induction, which is consistent with increased enzyme activity of UGTs responsible for the glucuronidation of various xenobiotics. These results are consistent with previous studies on rats upon induction of PB or 3-methylcholantrene (MC).14,15 The ratio of NNAL (+PB/-PB = 0.5) and α-OH-methyl-NNK-Gluc (+PB/-PB = 2.7) using MS-based absolute quantitation (data not shown) is in agreement with the ratio of NNAL (+PB/-PB = 0.5) and only slightly with α-OH-methyl-NNK-Gluc (+PB/-PB = 4.1) obtained using our LC-MS2 approach.
Figure 6.
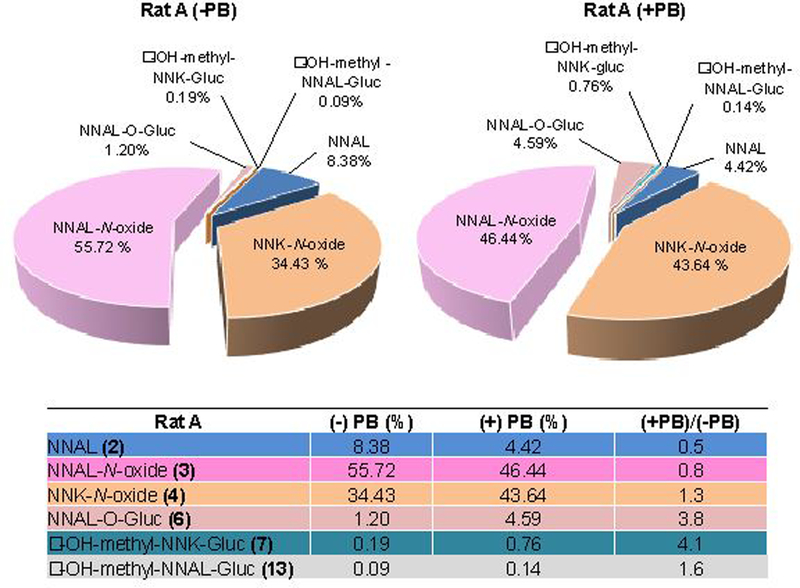
Relative distribution of selected nitroso-containing NNK metabolites in rats before and after induction by PB.
CONCLUSIONS
We have used an in vivo isotope labeling approach using d0-NNK and d4-NNK for the comprehensive profiling and simultaneous identification of known and unknown NNK metabolites in rats. The combined sample preparation, HR-AM LC/MS, and data analysis workflow presented here revealed and confirmed known NNK metabolites as well as m/z pairs corresponding to new and novel NNK metabolites. Several proposed NNK metabolites including the 1,3-Diol (13); α-OH-methyl-NNAL-Gluc (14); nitro-NK-N-oxide (15); nitro-NAL-N-oxide (16); γ-OH NNAL (17); and three N-acetylcysteine (NAC) metabolites (18a-c) were characterized for the first time in rat urine. Our current workflow detected metabolites that could have been missed using only targeted MS-based approaches. In addition, we detected in-source decay products (ISD), salt adducts, artefacts, and extensive formation of ion clusters and dimers. The identification of the source of these compounds is necessary to develop an accurate method for NNK metabolite detection. This strategy will be used to monitor and quantify NNK metabolites in human urine using a panel of metabolite biomarkers to assess carcinogen uptake, metabolic activation and detoxification, and ultimately for the evaluation of lung cancer risk and identification of susceptible smokers. The characterized rat urine from [pyridine-d4]NNK treated rats in this study may be used as internal standards for the relative quantitation of NNK metabolism in humans. Finally, the custom script reported here for the automated d0-d4 m/z pair peak detection could be integrated into the Compound Discoverer software package to allow users to interrogate high-resolution orbitrap-acquired mass spectral data using stable isotope labeling approaches.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant CA-138338 from the National Cancer Institute. R.D. was partially supported by the 2017 American Society for Mass Spectrometry Postdoctoral Career Development Award. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support Grant CA-077598. Salary support for P.W.V. was provided by the U.S. National Institutes of Health and National Cancer Institute [Grant R50-CA211256]. We thank Dr. Stephen Hecht (University of Minnesota), Dhimant Desai, Ph.D. and Shantu Amin, Ph.D. (Penn State Milton S. Hershey Medical Center, Hershey, PA) for providing the synthesized β-OH NNK, Drs. Lisa Peterson and Irina Stepanov for their helpful comments on this work, and Robert Carlson for editorial assistance.
Abbreviations:
- NNK
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNAL
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNK-N-oxide
4-[Methyl(nitroso)amino]-1-(1-oxido-3-pyridinyl)-1-butanone
- NNAL-N-oxide
4-[Methyl(nitroso)amino]-1-(1-oxido-3-pyridinyl)-1-butanol
- α-OH-methyl-NNK-Gluc
3,4,5-trihydroxy-6-((nitroso(4-oxo-4-(pyridin-3-yl)butyl)amino)methoxy)tetrahydro-2H-pyran-2-carboxylic acid
- α-OH-methyl-NNAL-Gluc
3,4,5-Trihydroxy-6-(((4-hydroxy-4-(pyridin-3-yl)butyl)(nitroso)amino)methoxy)tetrahydro-2H-pyran-2-carboxylic acid
- NNAL-O-Gluc
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol-O-glucuronide
- nitro-NK-N-oxide
3-(4-(Methyl(nitro)amino)butanoyl)pyridine 1-oxide; nitro-NAL-N-oxide, 3-(1-Hydroxy-4-(methyl(nitro)amino)butyl)pyridine 1-oxide
- γ-OH NNAL
N-(3,4-Dihydroxy-4-(pyridin-3-yl)butyl)-N-methylnitrous amide
- β-OH NNK
3-Hydroxy-4-[methyl(nitroso)amino]-1-(pyridine-3-yl)butan-1-one
- β-OH NNAL
4-[Methyl(nitroso)amino]-1-(pyridine-3-yl)butane-1,3-diol
- β-OH NNK-N-oxide
3-Hydroxy-4-[methyl(nitroso)amino]-1-(1-oxidopyridin-3-yl)butan-1-one
- 6-OH NNK
4-(Methylnitrosamino)-1-[3-(6-hydroxypyridyl)-1-butanone
- HPB
4-Hydroxy-1-(3-pyridyl)-1-butanone
- HPB-Gluc
4-Hydroxy-1-(3-pyridyl)-1-butanone glucuronide
- HPBA
4-Hydroxy-4-(3-pyridyl)-butyric acid
- OPBA
4-Oxo-4-(3-pyridyl)butyric acid
- 1,4-Diol
4-Hydroxy-1-(3-pyridyl)-1-butanol
- 1,3-Diol
3-Hydroxy-1-(3-pyridyl)-1-butanol
- NAC
N-acetylcysteine
- PB
Phenobarbital
- UDPGT
UDP-glucuronosyl transferase
Footnotes
Supporting Information Available
Supporting Information. Excel spreadsheets containing datasets from U1*, U2*, U3*, and BU* (Table S1–Table S5), custom script for the automated peak pair detection, and PDF file with Figure S1–S10 and Table S6–Table S11. The information is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A CA Cancer J Clin 2017, 67, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Hecht SS Nat Rev Cancer 2003, 3, 733–744. [DOI] [PubMed] [Google Scholar]
- (3).Hecht SS Chem Res Toxicol 2008, 21, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rodriguez-Antona C; Gomez A; Karlgren M; Sim SC; Ingelman-Sundberg M Hum Genet 2010, 127, 1–17. [DOI] [PubMed] [Google Scholar]
- (5).Schwartz AG; Prysak GM; Bock CH; Cote ML Carcinogenesis 2007, 28, 507–518. [DOI] [PubMed] [Google Scholar]
- (6).Taioli E Carcinogenesis 2008, 29, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Balbo S; Johnson CS; Kovi RC; James-Yi SA; O’Sullivan MG; Wang M; Le CT; Khariwala SS; Upadhyaya P; Hecht SS Carcinogenesis 2014, 35, 2798–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hecht SS; Carmella SG; Foiles PG; Murphy SE; Peterson LA Environ Health Perspect 1993, 99, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hecht SS; Peterson LA; Spratt TE IARC Sci Publ 1994, 91–106. [PubMed] [Google Scholar]
- (10).Hecht SS; Carmella SG; Murphy SE; Akerkar S; Brunnemann KD; Hoffmann D N Engl J Med 1993, 329, 1543–1546. [DOI] [PubMed] [Google Scholar]
- (11).Hecht SS; Young R; Chen CB Cancer Res 1980, 40, 4144–4150. [PubMed] [Google Scholar]
- (12).Hecht SS Proc Soc Exp Biol Med 1997, 216, 181–191. [DOI] [PubMed] [Google Scholar]
- (13).Peterson LA; Ng DK; Stearns RA; Hecht SS Chem Res Toxicol 1994, 7, 599–608. [DOI] [PubMed] [Google Scholar]
- (14).Murphy SE; Spina DA; Nunes MG; Pullo DA Chem Res Toxicol 1995, 8, 772–779. [DOI] [PubMed] [Google Scholar]
- (15).Murphy SE; Nunes MG; Hatala MA Chem Biol Interact 1997, 103, 153–166. [DOI] [PubMed] [Google Scholar]
- (16).Maertens LA; Upadhyaya P; Hecht SS; Zimmerman CL Drug Metab Dispos 2010, 38, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jalas JR; Ding X; Murphy SE Drug Metab Dispos 2003, 31, 1199–1202. [DOI] [PubMed] [Google Scholar]
- (18).Jalas JR; McIntee EJ; Kenney PM; Upadhyaya P; Peterson LA; Hecht SS Chem Res Toxicol 2003, 16, 794–806. [DOI] [PubMed] [Google Scholar]
- (19).Schrader E; Hirsch-Ernst KI; Richter E; Foth H Naunyn Schmiedebergs Arch Pharmacol 1998, 357, 336–343. [DOI] [PubMed] [Google Scholar]
- (20).Carmella SG; Le Ka KA; Upadhyaya P; Hecht SS Chem Res Toxicol 2002, 15, 545–550. [DOI] [PubMed] [Google Scholar]
- (21).Yao L; Zheng S; Guan Y; Yang J; Liu B; Wang W; Zhu X Anal Chim Acta 2013, 788, 61–67. [DOI] [PubMed] [Google Scholar]
- (22).Park SL; Carmella SG; Ming X; Vielguth E; Stram DO; Le Marchand L; Hecht SS Cancer Epidemiol Biomarkers Prev 2015, 24, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Leeming MG; Isaac AP; Pope BJ; Cranswick N; Wright CE; Ziogas J; O’Hair RA; Donald WA Anal Chem 2015, 87, 4104–4109. [DOI] [PubMed] [Google Scholar]
- (24).Bueschl C; Kluger B; Neumann NKN; Doppler M; Maschietto V; Thallinger GG; Meng-Reiterer J; Krska R; Schuhmacher R Anal Chem 2017, 89, 9518–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Capellades J; Navarro M; Samino S; Garcia-Ramirez M; Hernandez C; Simo R; Vinaixa M; Yanes O Anal Chem 2016, 88, 621–628. [DOI] [PubMed] [Google Scholar]
- (26).Huang X; Chen YJ; Cho K; Nikolskiy I; Crawford PA; Patti GJ Anal Chem 2014, 86, 1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kessler N; Walter F; Persicke M; Albaum SP; Kalinowski J; Goesmann A; Niehaus K; Nattkemper TW PLoS One 2014, 9, e113909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chokkathukalam A; Jankevics A; Creek DJ; Achcar F; Barrett MP; Breitling R Bioinformatics 2013, 29, 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Smith CA; O’Maille G; Want EJ; Qin C; Trauger SA; Brandon TR; Custodio DE; Abagyan R; Siuzdak G Ther Drug Monit 2005, 27, 747–751. [DOI] [PubMed] [Google Scholar]
- (30).Kotandeniya D; Carmella SG; Pillsbury ME; Hecht SS J Chromatogr B Analyt Technol Biomed Life Sci 2015, 1007, 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cao B; Zhang Q; Ji H; Liu J; Lang H; Feng M; Zhang J Anal Bioanal Chem 2014, 406, 4465–4471. [DOI] [PubMed] [Google Scholar]
- (32).Hu CW; Hsu YW; Chen JL; Tam LM; Chao MR Arch Toxicol 2014, 88, 291–299. [DOI] [PubMed] [Google Scholar]
- (33).Spratt TE; Peterson LA; Confer WL; Hecht SS Chem Res Toxicol 1990, 3, 350–356. [DOI] [PubMed] [Google Scholar]
- (34).Buck H International Journal of Quantum Chemistry 2012, 112, 3711–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Carmella SG; Kagan SS; Spratt TE; Hecht SS Cancer Res 1990, 50, 5453–5459. [PubMed] [Google Scholar]
- (36).Ibrahim H; Couderc F; Perio P; Collin F; Nepveu F Rapid Commun Mass Spectrom 2013, 27, 621–628. [DOI] [PubMed] [Google Scholar]
- (37).Butler M; Arroyo Mañez P; Cabrera GM J Mass Spectrom 2010, 45, 536–544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.