

Abstract

The electronic properties of amide linkers, which are intricate components of biomolecules, offer a wealth of unexplored possibilities. Herein, we demonstrate how the different modes of attaching an amide to a pyrene chromophore affect the electrochemical and optical properties of the chromophore. Thus, although they cause minimal spectral shifts, amide substituents can improve either the electron-accepting or electron-donating capabilities of pyrene. Specifically, inversion of the amide orientation shifts the reduction potentials by 200 mV. These trends indicate that, although amides affect to a similar extent the energies of the ground and singlet excited states of pyrene, the effects on the doublet states of its radical ions are distinctly different. This behavior reflects the unusually strong orientation dependence of the resonance effects of amide substituents, which should extend to amide substituents on other types of chromophores in general. These results represent an example where the Hammett sigma constants fail to predict substituent effects on electrochemical properties. On the other hand, Swain–Lupton parameters are found to be in good agreement with the observed trends. Examination of the frontier orbitals of the pyrene derivatives and their components reveals the underlying reason for the observed amide effects on the electronic properties of this polycyclic aromatic hydrocarbon and points to key molecular-design strategies for electronic and energy-conversion systems.
1. Introduction
The importance of amides as building blocks in living systems cannot be overstated. Peptide bonds hold proteins together, and the amide propensity for hydrogen bonding defines the secondary structures and the functionality of these biopolymers.1−3 Since the invention of nylon,4 synthetic polyamides have been at the forefront of materials science and engineering.5−9 Due to their extended π-conjugation, amide bonds are rigid and assume planer conformations.10−14
In addition to functioning as bonding elements and hydrogen-bonding cross-linkers, amides also have interesting electronic properties. The amide dipoles, ranging between 3 and 5 D,15 render many vital enzymatic and cellular processes possible.16−18 Ordered amide and hydrogen bonds give rise to the enormous macrodipoles of protein helices,18−21 providing guidelines for bioinspired designs.22−32 In addition to making ion channels functional,17,33 such dipolar amide conjugates prove invaluable for rectifying the directionality of electron transfer and transport.21,34−39 Aliphatic amides oxidize at about 1.5 V versus saturated calomel electrode (SCE)40 and have optical gaps of 5.6 eV between their highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs), corresponding to an n−π* transition around 222 nm,11 making them electron-rich π-conjugated UV absorbers.
Except for a few reports on how the orientation of amide bonds affects molecular properties, such as enantioselectivity, catalytic activity, and the biological uptake of nanoconstructs,41−43 the understanding of the effects of this important substituent on the characteristics of organic chromophores remains in the realm of empirical deductions. Even the classical Hammett constants predict that, regardless of their orientation, amides should be electron-withdrawing groups.44 However, the Swain–Lupton resonance parameters, accounting for the mesomeric effects of functional groups,45 do suggest that there should be a difference between the effects of amides attached via their nitrogens or via their carbonyl carbons.44,45
Substituent effects on electronically excited states correlate roughly with ground-state descriptors such as Hammett constants. However, because it is the difference between the ground and excited states that defines the optical properties of a molecule, if a substituent affects the energies of both the ground and excited states in the same manner, the shifts in the optical spectra will be negligible, regardless of how strong these effects are. Furthermore, despite all advances in physical organic chemistry during the 20th century, the search for reliable descriptors of substituent effects on excited-state properties is still in its infancy.46 Thus, systematic studies on how amides and other substituents affect transitions between different electronic states are important for filling key voids in physical organic chemistry and other pertinent fields.
The question of interest here is, aside from serving as linkers, hydrogen-bonding sites, and dipole sources, how do amide functionalities impact the electronic properties of π-conjugated moieties to which they are attached?
To test this capability of amides, we focus on pyrene as the π-conjugated moiety. Pyrene and its derivatives are among the most widely used chromophores due to their good fluorescence quantum yields (φf) and the unusually long lifetime (τ) of the singlet excited states.47−50 The potentials at which they oxidize and reduce make pyrenes a popular choice as electron donors and acceptors.51−57 Along with their attractive optical and electrochemical properties, the strong propensity for self-assembly via π-stacking, which pyrene and its derivatives exhibit,58−60 allows them to serve as building blocks for electronic and photonic materials and interfaces.61−71
To examine how amides affect the electronic properties of pyrene, we attach them in opposite orientations directly to the aromatic ring (PyN-C and PyC-N, Chart 1). Insertion of a methylene linker (PymN-C and PymC-N, Chart 1) breaks the π-conjugation between the amide and the pyrene moiety, permitting an evaluation of the amide effects in the absence of strong electronic coupling with the polycyclic aromatic hydrocarbons (PAHs). Unlike other π-conjugating substituents, such as nitro, nitroso, and alkanoyl groups,54,72 amides, even when directly attached to pyrene, cause small shifts in the UV/visible absorption and fluorescence spectra. The most profound effect of the amides is on the reduction potentials of oxidation and reduction of pyrene. Thus, although amides affect to a similar extent the energies of the ground and singlet excited states of pyrene, their effects on the doublet states of their radical ions are quite different.
Chart 1. Structures of Pyrene Derivatives with Differently Oriented Secondary Amides Attached Directly (PyN-C and PyC-N) and via a Methylene Linker (PymN-C and PymC-N).

2. Results
2.1. Molecular-Design Considerations
Amide coupling between the corresponding amine and carboxylic-acid derivatives yields the pyrene–amide conjugates in good yields after chromatographic purification and recrystallization steps (Chart 1). The positions 1, 3, 6, and 8 (PyN-C on Chart 1) are the most susceptible to electrophilic and radical substitutions.72−74 Therefore, this study focuses on pyren-1-yl amides (Chart 1).
2.2. Amide Effects on Optical Properties of Pyrene
The optical spectra of the four pyrene–amide
derivatives
(Chart 1) investigated
here show features characteristic of pyrene. In addition to the transitions
to upper excited states, the substituents perturb the symmetry of
pyrene, enhancing the intensity of the symmetry-forbidden S0 → S1 electronic absorption at 375 nm (Figure 1a). Although the
perturbation of the symmetry is stronger for PyN-C and PyC-N than for the methylene-linked derivatives, the amide substituents
do not cause large spectral shifts (Figure 1a). The spectra of PymN-C and PymC-N resemble those of alkyl-substituted pyrenes,58,75 indicating that methylene linkers do indeed effectively reduce the
interaction between the amide and the pyrene π-conjugated system.
Direct attachment of the amide via its carbon, PyC-N, broadens the absorption and emission
bands (Figure 1a) and
decreases the zero-to-zero transition energy 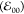 by less than kBT (Table 1). Attaching the amide moiety via its nitrogen, PyN-C, enhances the spectral broadening
and decrease
by less than kBT (Table 1). Attaching the amide moiety via its nitrogen, PyN-C, enhances the spectral broadening
and decrease 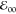 a bit further.
a bit further.
Figure 1.

Optical properties of the pyrene–amide derivatives in acetonitrile (MeCN), purged with argon. (a) Absorption and fluorescence spectra (λex = 335 nm) with the various transitions designated. The dotted lines represent the absorption spectra amplified by a factor of 20 for visualization of the S0 → S1 forbidden transitions. (b) Time-correlated single-photon counting (TCSPC) emission-decay curves recorded at the fluorescence maxima (λex = 278 nm; excitation-pulse width = 0.9 ns).
Table 1. Photophysical Properties of the Pyrene–Amide Derivatives for Various Solvents.
|
hΔν (eV)b |
||||||
|---|---|---|---|---|---|---|
| solvent | φf | τ (ns) |
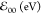 a a
|
S2c | S1d | |
| PymN-C | MeCN | 0.41 | 217 | 3.30 | 0.34 | 0.013 |
| DCM | 0.38 | 150 | 3.30 | 0.31 | 0.011 | |
| CHCl3 | 0.38 | 99.6 | 3.31 | 0.31 | 0.009 | |
| PymC-N | MeCN | 0.44 | 216 | 3.30 | 0.34 | 0.018 |
| DCM | 0.39 | 139 | 3.29 | 0.31 | 0.020 | |
| CHCl3 | 0.29 | 111 | 3.29 | 0.31 | 0.015 | |
| PyN-C | MeCN | 0.23 | 13.7 | 3.23 | 0.42 | 0.029 |
| DCM | 0.26 | 13.7 | 3.22 | 0.40 | 0.025 | |
| CHCl3 | 0.19 | 12.2 | 3.23 | 0.39 | 0.026 | |
| PyC-N | MeCN | 0.44 | 69.3 | 3.28 | 0.37 | 0.039 |
| DCM | 0.42 | 36.6 | 3.27 | 0.37 | 0.041 | |
| CHCl3 | 0.42 | 29.2 | 3.26 | 0.37 | 0.050 | |
From the crossover point of the normalized S0 → S1 absorption and the fluorescence bands.
Stoke’s shifts: Δν is the difference between the frequencies of the absorption and fluorescence maxima, and h is the Planck constant.
Using the S0 → S2 absorption maximum for Δν.
Using the S0 → S1 absorption maximum.
The emission-decay kinetics shows the same trends (Figure 1b). PymN-C and PymC-N exhibit large values of τ (Table 1), which are comparable to those of alkyl-substituted pyrenes.58,60,75 Attaching the amide directly via its carbonyl carbon, PyC-N, shortens τ, reflecting the enhancement of the intensity of the symmetry-forbidden S1 → S0 transition. Attaching the amide group via its nitrogen, PyN-C, shortens τ even further (Figure 1b and Table 1).
Stoke’s shifts (Δν), estimated from the absorption and emission maxima at about 340 and 380 nm, respectively, encompass S2 → S1 internal conversion as well as relaxation of S1. The latter is small, since the Δν values using the S0 → S1 absorption are minute and are comparable to those of quantum dots and solid-state materials (Table 1).76−78 This finding indicates that solvation effects following photoexcitation do not appreciably perturb the geometry of these pyrene derivatives.
The photophysical trends (Figure 1 and Table 1) reflect the consequences of π-conjugation of the amide
with the PAH. The effects are, indeed, more pronounced when the amide
is directly attached via its nitrogen than its carbonyl carbon. Also,
the close resemblance between the properties of PymN-C and PymC-N indicates that the amide electric dipoles, which generate
large fields even across methylene linkers,39 are not responsible for the observed effects. The lack of a significant
solvent dependence of 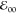 and Δν (Table 1) further confirms that amide
dipoles do
not contribute to the differences between the photophysics of the
four derivatives. The decrease in φf and τ
in chlorinated solvents (Table 1), which was also reported for pyrene itself,50 may be a polarity effect. Photoinduced charge-transfer
interactions with the solvent, however, might account for it
as well.79
and Δν (Table 1) further confirms that amide
dipoles do
not contribute to the differences between the photophysics of the
four derivatives. The decrease in φf and τ
in chlorinated solvents (Table 1), which was also reported for pyrene itself,50 may be a polarity effect. Photoinduced charge-transfer
interactions with the solvent, however, might account for it
as well.79
2.3. Amide Effects on Reduction and Oxidation Properties of Pyrene
Cyclic voltammetry of the pyreneamides, dissolved
in MeCN, shows that all derivatives except PyC-N have reduction potentials (EPy|Py•–) around −2 V versus
SCE (Figure 2 and Table 2). For PyC-N, EPy|Py•– = −1.8 V versus SCE. The potentials
at which pyrenes oxidize, expressed as reduction potentials of the
radical cations (EPy•+|Py), range from 1.2 V versus SCE for PyN-C to 1.4 V versus SCE for PyC-N (Table 2).
The electrochemical HOMO–LUMO gaps, i.e., EPy•+|Py – EPy|Py•–, match the optical 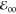 quite well (Tables 1 and 2). This finding
suggests a negligible difference between the energies of the solvated
ions and the photoexcited species.80
quite well (Tables 1 and 2). This finding
suggests a negligible difference between the energies of the solvated
ions and the photoexcited species.80
Figure 2.

Electrochemical properties of the pyrene–amide derivatives in acetonitrile purged with argon in the presence of different concentrations, Cel, of supporting electrolyte, (n-C4H9)4NPF6. (a) Cyclic voltammograms for Cel = 25 mM (scan rate = 100 mV s–1). For reversible reduction, the half-wave potentials, E(1/2), are determined from the average between the anodic and cathodic peak potentials. For irreversible oxidation, E(1/2) is estimated from the inflection-point potentials of the anodic waves.81 (b) Dependence of E(1/2) on Cel. The solid lines represent the data fits, and the dotted lines show extrapolations of the potentials to Cel = 0, to yield E(1/2) for neat solvent.82,83
Table 2. Reduction and Oxidation Properties of the Four Pyrene–Amide Derivatives.
|
E(1/2) (Cel = 0) (V vs SCE)a |
|||||
|---|---|---|---|---|---|
| solvent | EPy•+|Pyc | EPy|Py•–d | IE (eV)b | EA (eV)b | |
| PymN-C | MeCN | 1.30 ± 0.17 | –2.01 ± 0.01 | 5.58 | 2.35 |
| DCM | 1.44 ± 0.05 | 5.74 | 2.17 | ||
| CHCl3 | 5.82 | 2.00 | |||
| PymC-N | MeCN | 1.29 ± 0.01 | –2.04 ± 0.08 | 5.55 | 2.32 |
| DCM | 1.44 ± 0.02 | 5.65 | 2.18 | ||
| CHCl3 | 5.81 | 2.00 | |||
| PyN-C | MeCN | 1.21 ± 0.38 | –2.04 ± 0.01 | 5.36 | 2.33 |
| DCM | 1.33 ± 0.13 | 5.48 | 2.17 | ||
| CHCl3 | 5.62 | 1.98 | |||
| PyC-N | MeCN | 1.38 ± 0.01 | –1.81 ± 0.01 | 5.63 | 2.49 |
| DCM | 1.60 ± 0.03 | 5.75 | 2.34 | ||
| CHCl3 | 5.89 | 2.17 | |||
Half-wave reduction potentials for neat solvents obtained from extrapolation to Cel = 0 (Figure 2b).82
Ionization energy (IE) from the difference between the density functional theory (DFT)-calculated energies of the ground state and the radical cation and electron affinity (EA) from the difference between the energies of the ground state and the radical anion, as implemented by Koopman’s theorem. For the calculations, the B3LYP density functional and the 6-311+G(d,p) basis set were used together with implementation of the polarizable continuum model (PCM).
Potentials at which oxidation occurs, expressed as the reduction potentials of the radical cations, Py•+ + e– → Py.
Reduction potential of the pyrene derivatives, i.e., Py + e– → Py•–.
Overall, attaching the
amide group to pyrene via the carbonyl carbon
lowers the energy of the HOMO and LUMO, enhancing the propensity of
the pyrene chromophore to act as an electron acceptor. Conversely,
attaching the amide group via the nitrogen elevates the energy of
the pyrene HOMO without affecting the energy of the LUMO. This narrowing
of the HOMO–LUMO gap, reflected in the value of 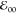 , makes PyN-C a better electron donor than the other three
derivatives.
, makes PyN-C a better electron donor than the other three
derivatives.
For dichloromethane (DCM), the potentials at which the pyrenes oxidize, expressed as reduction potentials of the radical cations, show the same trends but are shifted to more positive values by about 100–200 mV in comparison with those for MeCN (Table 2). Thus, a decrease in the polarity of the medium destabilizes the radical cations that are formed upon oxidation and increases the corresponding reduction potentials of the oxidized pyreneamides, EPy•+|Py. Unfortunately, the reduction potentials of the pyrene derivatives, EPy|Py•–, are outside the electrochemical window of DCM and the potentials at which both the oxidation and reduction occur are both outside the electrochemical window of CHCl3.
To address the lack
of electrochemical information for these cases,
we resort to density functional theory (DFT) calculations of the ionization
energies (IE) and electron affinities (EA) of the pyrene derivatives.
The values of IE and EA show the same trends as those of the reduction
potentials and provide information on the reduction and oxidation
properties that are electrochemically inaccessible for some of the
solvents. For each of the pyrene derivatives, IE increases and EA
decreases with a decrease in the solvent polarity (Table 2), which is consistent with
destabilization of both doublet states, i.e., of the radical cation
and the radical anion. Thus, a decrease in medium polarity increases
the electrochemical HOMO–LUMO gap, i.e., EPy•+|Py – EPy|Py•– ≈ IE – EA, while 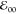 is invariant with the solvent.
For low-polarity
solvents, therefore, the stabilization due to Coulombic interactions
between the photogenerated hole and the unpaired electron in the singlet
excited state becomes particularly important.
is invariant with the solvent.
For low-polarity
solvents, therefore, the stabilization due to Coulombic interactions
between the photogenerated hole and the unpaired electron in the singlet
excited state becomes particularly important.
In contrast to the amide effects on the optical properties of the pyrene derivatives, which appear to be subtle, the effects on their electrochemical behavior are substantial when the amide is directly attached to the chromophore. Inverting the orientation of the amide causes a 200 mV shift in the reduction potentials, which can represent a huge difference in the design of molecules for electronics and optoelectronics.
Attaching an amide via its carbonyl carbon causes positive shifts
in both reduction potentials, EPy•+|Py and EPy|Py•– (Table 2),
and some broadening of the absorption and emission bands (Figure 1a) but does not lead
to dramatic bathochromic shifts or to a decrease in 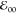 (Figure 1a and Table 1). In contrast, attaching the amide via its nitrogen
results
in much more evident changes in the optical properties of pyrene,
broadening the absorption and emission spectra and decreasing
(Figure 1a and Table 1). In contrast, attaching the amide via its nitrogen
results
in much more evident changes in the optical properties of pyrene,
broadening the absorption and emission spectra and decreasing  , φf, and τ
(Figure 1 and Table 1). In addition, PyN-C exhibits a negative shift
of EPy•+|Py and no shift
of EPy|Py•– (Table 2), affecting the energy
of the
HOMO but not of the LUMO of the pyrene chromophore. This narrowing
of the HOMO–LUMO gap, which is related to π-conjugation
between the pyrene chromophore and the amide substituent, is characteristic
of PyN-C as compared with
the other three derivatives.
, φf, and τ
(Figure 1 and Table 1). In addition, PyN-C exhibits a negative shift
of EPy•+|Py and no shift
of EPy|Py•– (Table 2), affecting the energy
of the
HOMO but not of the LUMO of the pyrene chromophore. This narrowing
of the HOMO–LUMO gap, which is related to π-conjugation
between the pyrene chromophore and the amide substituent, is characteristic
of PyN-C as compared with
the other three derivatives.
3. Discussion
The evidence suggests that π-conjugation of the amides with the pyrene rings is the underlying reason for observed trends. Amides have substantial permanent electric dipoles15 that can readily affect not only the electrochemical behavior of these derivatives by stabilizing or destabilizing the formed radical ions but also may affect the optical properties of the polarizable pyrene via an intramolecular Stark effect.84 The amide dipole, however, points from the oxygen to the nitrogen of the bond,15 which is tangential to the pyrene rings for the four derivatives under investigation. The two methylene-linked derivatives, PymN-C and PymC-N, provide the most important evidence that the amide dipoles do not play a significant role in the observed trends. When the amide substituent cannot participate in direct π-conjugation with the pyrene chromophore, the inversion of the orientation of the amide dipole does not alter the reduction or oxidation propensities of these pyrenes (PymC-N vs PymN-C, Table 2). Furthermore, if the amide dipoles had exerted significant effects, a lowering of the solvent polarity should induce differences between the reduction potentials or the IE and EA of PymN-C and PymC-N. Therefore, it is safe to assume that direct electronic interactions between the amides and the pyrene chromophore govern the observed trends in this study. Thus, field effects, prevalent for PyC-N, represent induction through σ bonds, rather than through-space interactions provoked by the orientation of the amide dipole-generated fields.
The Hammett substituent constants, σp and σm, suggest that amide groups should be preferentially electron-withdrawing, regardless of the orientation in which they are bound to an aromatic ring. When attached via carbon, e.g., −CONHCH3, the Hammett sigma constants are σp = 0.34 and σm = 0.35,44 characterizing the amide as a moderately good electron-withdrawing group in ground-state reactions. Likewise, when the amide is attached via its nitrogen, e.g., −NHCOCH3,44 the values are σp = 0.00 and σm = 0.21, which makes it electron-withdrawing in the meta and neither electron withdrawing nor electron donating in the para position on a benzene ring, contradicting our spectroscopic and electrochemical findings.
Amide bonds oxidize at about 1.3–1.8 V versus SCE, which is relatively close to the EPy•+|Py of pyrene, ensuring effective mixing between the HOMOs of the pyrene and the amide substituent. The large HOMO–LUMO gap of amides, however, indicate that the reduction of an amide group should occur at potentials more negative than about −4 V versus SCE. Hence, this precludes strong mixing between the high-energy LUMO of the amide and the much lower energy LUMO of pyrene.
Considering the HOMOs of pyrene and the amide reveals a drastic difference between the propensities for π-conjugation of the carbon-bonded and nitrogen-bonded orientations of the substituent. Driven by protein science, semiclassical studies of the electronic structures of amides clearly reveal that the HOMO of the amide group is a nonbonding orbital on the oxygen and HOMO – 1 is a π-orbital, also with a nonbonding character, localized on nitrogen and oxygen with a node on the carbon.11 Although HOMO – 2 of the amide is delocalized over the carbon, nitrogen, and oxygen, it is much lower in energy than the HOMO – 1.11
DFT calculations of the simplest carboxamide, i.e., formamide, illustrate these trends (Figure 3). Many of the frontier orbitals have nonbonding or σ-character distribution over the plane of the amide bond and cannot mix with the π-orbitals of coplanar aromatic rings. Even though the amide LUMO has a π character, its high energy level makes such π-conjugation unfavorable, which is consistent with the observed trends in the reduction potentials and the EA (Table 2).
Figure 3.

Molecular-orbital diagram
of CHONH2 showing the frontier
orbitals obtained from DFT calculation at the B3LYP/6-311G(3df,3dp)
level of theory with implementation of MeCN as a solvation medium
(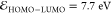 , IE – EA = 7.2 eV,
, IE – EA = 7.2 eV, 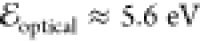 , i.e., 222
nm, EA•+|A ≈
1.8 V vs SCE40).
, i.e., 222
nm, EA•+|A ≈
1.8 V vs SCE40).
The HOMO – 1 of the amide reveals the underlying reason for the orientation dependence of the observed effects. The HOMO – 1 has a π-character with a node on the carbon (Figure 3) that would suppress π-conjugation with coplanar aromatic rings connected directly to the carbonyl carbon. The HOMOs of pyrene connected to the amide nitrogen, however, could interact with the HOMO – 1 of the amide. The HOMO – 2 of the amide is its only π-orbital with no orthogonal nodal planes but its much lower energy (than the HOMO) makes mixing with the frontier orbitals of pyrene energetically unfavorable. Overall, attaching the amide group directly to the pyrene chromophore via the nitrogen favors mixing between the HOMO – 1 of the amide and the HOMOs of pyrene. Conversely, mixing with the HOMOs of pyrene is much less favorable when the amide is attached via its carbon.
Unlike conventional Hammett sigma constants, the Swain–Lupton
field  and resonance
and resonance 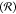 parameters
for substituent effects45 of an amide group
capture this dependence on
the bonding orientation and thus provide a much better rationalization
of the experimentally observed trends than the Hammett sigma values.
For −CONHCH3, the Swain–Lupton parameters
are
parameters
for substituent effects45 of an amide group
capture this dependence on
the bonding orientation and thus provide a much better rationalization
of the experimentally observed trends than the Hammett sigma values.
For −CONHCH3, the Swain–Lupton parameters
are  and
and 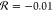 and for −NHCOCH3,
and for −NHCOCH3, 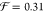 and
and  .44 Hence, the
inductive or field effects are indicative of electron withdrawal and
would be expected to be similar for both bonding orientations. In
contrast, the electron-donating mesomeric effect of the electron pair
on nitrogen should dominate in the latter orientation, consistent
with the observed difference between the properties of PyN-C and PyC-N.
.44 Hence, the
inductive or field effects are indicative of electron withdrawal and
would be expected to be similar for both bonding orientations. In
contrast, the electron-donating mesomeric effect of the electron pair
on nitrogen should dominate in the latter orientation, consistent
with the observed difference between the properties of PyN-C and PyC-N.
The Swain–Lupton empirical parameters can also rationalize
the differences between the effects observed here for amides and those
of other carbonyl substituents. As an example, for an acetyl group,
−COCH3,  and
and 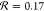 . These values indicate that the propensity
of −COCH3 for mesomeric electron withdrawal should
be stronger than that of −CONHCH3. Indeed, alkanoyl
pyrene derivatives not only undergo reduction at potentials that are
less negative than those of PyC-N(54) but also exhibit a bathochromic
shift of the first π → π* absorption band to 400
nm.85
. These values indicate that the propensity
of −COCH3 for mesomeric electron withdrawal should
be stronger than that of −CONHCH3. Indeed, alkanoyl
pyrene derivatives not only undergo reduction at potentials that are
less negative than those of PyC-N(54) but also exhibit a bathochromic
shift of the first π → π* absorption band to 400
nm.85
This example emphasizes another important point. Bathochromic spectral shifts indicate that the substituent lowers the energies of the excited states more than that of the ground state. In our compounds, such shifts do not occur even when the amide is directly attached to the pyrene moiety. Such direct attachment, however, does affect the reduction potentials (Table 2), implying that the amide substituents have much stronger effects on the radical ions than on the singlet excited states. In classical terms, this trend is consistent with through-resonance, which is pronounced for electron-donating substituents bound to aromatic rings with positively charged groups and for electron-withdrawing substituents bound to aromatics with negatively charged groups. The pyrene derivatives under investigation have only one substituent. Nevertheless, upon electrochemical oxidation or reduction, the doublet states of the radical ions are considerably more susceptible to through-resonance interactions with the amides than is the case for the electroneutral singlet states.
Although the Swain–Lupton parameters for methylene-linked
amides suggest the possibility of a hyperconjugation effect across
the methylene group (i.e., for −CH2NHCOCH3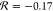 and for −CH2CONHCH3
and for −CH2CONHCH3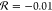 ),44 DFT calculations
reveal that the frontier orbitals of PymN-C and PymC-N are
practically the same: localized on the pyrene chromophore with a negligible
delocalization over the amide group. The HOMO of PyC-N shows similar patterns. In contrast,
the HOMO and the LUMO of PyN-C are heavily delocalized over the amide and pyrene rings (Figure 4). These DFT results
agree with the spectroscopic and electrochemical trends and confirm
that the amide effects arise from differences in the propensity for
π-conjugation between the different bonding orientations.
),44 DFT calculations
reveal that the frontier orbitals of PymN-C and PymC-N are
practically the same: localized on the pyrene chromophore with a negligible
delocalization over the amide group. The HOMO of PyC-N shows similar patterns. In contrast,
the HOMO and the LUMO of PyN-C are heavily delocalized over the amide and pyrene rings (Figure 4). These DFT results
agree with the spectroscopic and electrochemical trends and confirm
that the amide effects arise from differences in the propensity for
π-conjugation between the different bonding orientations.
Figure 4.

Frontier orbitals of the pyrene derivatives obtained from DFT calculations at the B3LYP/6-311+G(d,p) level of theory: HOMO and LUMO, showing the different extents of delocalization over the amide substituents, which is most pronounced for PyN-C and least pronounced for PymN-C and PymC-N. In fact, the delocalization in the HOMO of PyC-N is quite similar to that in the HOMOs of PymN-C and PymC-N.
4. Conclusions
This study demonstrates that amides are not benign linking groups and manifest additional complexity in the interaction of their electronic structures with π-conjugated chromophores. Along with their enormous electric dipoles and ability to form ordered hydrogen-bonded networks, the judicious inclusion and orientation of amide substituents provides a powerful tool for molecular, supramolecular, and materials designs. Elucidation of the origins of the observed effects in the present set of duly chosen pyrene conjugates suggests that the present findings can be safely extrapolated to other amide-substituted polycyclic aromatic hydrocarbon derivatives, as well as to other π-conjugated chromophores in general.
5. Experimental Section
5.1. Synthetic Procedures
All reagents and solvents were purchased from TCI America, Sigma-Aldrich or Alfa Aesar and used as received. The reaction progress was monitored by means of thin-layer chromatography, which was performed with aluminum foil plates, covered with silica gel 60 F254 (Merck). Product purification was done by the means of column chromatography on Kieselgel 60 (Merck). All reported 1H NMR and 13C NMR spectra were recorded on 300 or 400 MHz spectrometers. Chemical shifts (δ ppm) were determined using the solvent peaks as internal references. High-resolution mass spectra (HRMS) were obtained via electrospray ionization mass spectrometry.
5.1.1. N-(Heptan-4-yl)-2-(pyren-1-yl)acetamide (PymC-N)
1-Pyreneacetic acid (200 mg, 0.77 mmol), chloro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate (TCFH, 384 mg, 1.15 mmol), and 4-dimethylamino pyridine (10 mg, 0.082 mmol) were placed in a 50 mL round bottom flask equipped with a magnetic stir bar. While purging with argon, 5 mL of dry DCM was added and the reaction vessel was immersed in a dry ice/acetone bath. 4-Heptylamine (92 μL, 0.615 mmol) was slowly added, followed by the slow addition of 4-methylmorpholine (0.5 mL, 4.5 mmol). The reaction mixture was allowed to warm up to room temperature and was stirred overnight. The solution was diluted with 25 mL of DCM and washed with water (3 × 100 mL). The organic layer was collected, dried over Na2SO4, and concentrated in vacuo. Purification using flash chromatography (stationary phase: silica gel: eluent gradient: from 100% hexanes to 100% ethyl acetate) afforded 118 mg (0.330 mmol, 43% yield) of PymC-N as a yellow powder; 55 mg (0.15 mmol) of the sample was recrystallized from ethanol to afford 37 mg (0.10 mmol, 67% recrystallization yield and 29% overall yield) of PymC-N. 1H NMR (400 MHz, CDCl3) δ/ppm: 8.07 (8H, m), 7.86 (1H, d, J = 7.6 Hz), 4.91 (1H, d, J = 8.8 Hz), 4.26 (2H, s), 3.91 (1H, td, J1 = 8.3, J2 = 4.2 Hz), 1.2 (2H, m), 1.02 (6H, m), 0.72 (6H, t, J = 7.1 Hz). 13C NMR (400 MHz, CDCl3) δ/ppm: 170.58, 131.41, 131.15, 130.98, 129.70, 128.79, 128.65, 128.35, 127.71, 127.47, 126.36, 125.62, 125.55, 125.25, 124.77, 123.19, 49.00, 42.43, 37.21, 19.03, 14.01. HRMS (m/z): [M + H]+ calculated for C25H28NO+, 358.2171; found 358.2174.
5.1.2. 2-Propyl-N-(pyren-1-ylmethyl)pentanamide (PymN-C)
1-Pyrenemethylamine hydrochloride (200 mg, 0.75 mmol) was placed in a 50 mL round bottom flask equipped with a magnetic stir bar. While purging with argon, 5 mL of dry DCM was added and the reaction vessel was immersed in a dry ice/acetone bath. 2-Propylpentanoyl chloride (193 μL, 1.125 mmol) was slowly added, followed by the slow addition of 4-methylmorpholine (0.6 mL, 5.45 mmol). The reaction mixture was allowed to warm up to room temperature and was stirred overnight. The solution was diluted with 25 mL of DCM and washed with water (3 × 100 mL). The organic layer was collected, dried over Na2SO4, and concentrated in vacuo. The crude solid was dissolved in 5 mL of DCM, and the clear solution was slowly diluted with 25 mL of hexane. The precipitate formed was collected and dried to produce 213 mg (0.6 mmol, 80%) of PymN-C; 45 mg (0.13 mmol) of the sample was recrystallized from ethanol to afford 25 mg (0.07 mmol, 57% recrystallization yield and 46% overall yield) of PymN-C. 1H NMR (300 MHz, CDCl3) δ/ppm: 8.15 (3H, m), 8.08 (1H, s), 8.05 (1H, d, J = 2.3 Hz), 7.97 (3H, m), 7.88 (1H, d, J = 7.9 Hz), 5.83 (1H, s), 5.07 (2H, d, J = 5.3 Hz), 2.00 (1H, dt, J1 = 9.2 Hz, J2 = 4.7 Hz), 1.65 (2H, m), 1.28 (6H, m), 0.85 (6H, m). 13C NMR (300 MHz, CDCl3) δ/ppm: 176.21, 131.84, 131.78, 131.34, 129.59, 128.60, 128.09, 127.89, 126.65, 125.92, 125.56, 125.26, 123.61, 48.37, 42.47, 35.89, 21.50, 14.76. HRMS (m/z): [M + H]+ calculated for C25H28NO+, 358.2171; found 358.2187.
5.1.3. N-(Heptan-4-yl)pyrene-1-carboxamide (PyC-N)
1-Pyrenecarboxylic acid (205 mg, 0.83 mmol), chloro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate (TCFH, 404 mg, 1.22 mmol), and 4-dimethylamino pyridine (10 mg, 0.082 mmol) were placed in a 50 mL round bottom flask equipped with a magnetic stir bar. While purging with argon, 5 mL of dry DCM was added and the reaction vessel was immersed in a dry ice/acetone bath. 4-Heptylamine (101 μL, 0.675 mmol) was slowly added, followed by the slow addition of 4-methylmorpholine (0.5 mL, 4.5 mmol). The reaction mixture was allowed to warm up to room temperature and stirred overnight. The solution was diluted with 25 mL of DCM and washed with water (3 × 100 mL). The organic layer was collected, dried over Na2SO4, and concentrated in vacuo. Purification using flash chromatography (stationary phase: silica gel; eluent gradient: from 100% hexanes to 100% ethyl acetate) afforded 100 mg (0.29 mmol, 36% column yield) of PyC-N as a white powder; 49 mg (0.14 mmol) of the sample were recrystallized from ethanol to afford 38 mg (0.11 mmol, 78% recrystallization yield and 28% overall yield) of PyC-N. 1H NMR (400 MHz, CDCl3) δ/ppm: 8.51 (1H, d, J = 9.2 Hz), 8.18 (2H, d, J = 7.6 Hz), 8.08 (3H, m), 8 (3H, m), 5.84 (1H, d, J = 9.2 Hz), 4.34 (1H, m), 1.56 (9H, m), 1.01 (6H, m). 13C NMR (400 MHz, CDCl3) δ/ppm: 169.84, 132.49, 131.12, 131.39, 130.94, 128.79, 128.67, 128.62, 127.30, 126.48, 125.90, 125.83, 124.94, 124.66, 124.62, 124.46, 124.43, 49.85, 37.93, 19.58, 14.37. HRMS (m/z): [M + H]+ calculated for C24H26NO+, 344.2014; found 344.2001.
5.1.4. 2-Propyl-N-(pyren-1-yl)pentanamide (PyN-C)
1-Aminopyrene (104 mg, 0.48 mmol) was placed in a 50 mL round bottom flask equipped with a magnetic stir bar. While purging with argon, dry DCM (5 mL) was added and the reaction vessel was immersed in a dry ice/acetone bath. 2-Propylpentanoyl chloride (118 μL, 0.69 mmol) was slowly added, followed by the slow addition of pyridine (116 μL, 1.4 mmol). The reaction mixture was allowed to warm up to room temperature and was stirred overnight. The solution was diluted with 25 mL of DCM and washed with water (3 × 100 mL). The organic layer was collected, dried over Na2SO4, and concentrated in vacuo. Purification using flash chromatography (stationary phase: silica gel; eluent gradient: from 100% hexanes to 100% ethyl acetate) afforded 115 mg (0.336 mmol, 70% column yield) of PyN-C as a white powder; 55 mg (0.16 mmol) of the column-purified sample was recrystallized from ethanol to afford 49 mg (0.14 mmol, 88% recrystallization yield and 62% overall yield) of PyN-C. 1H NMR (400 MHz, CDCl3) δ/ppm: 8.36 (1H, d, J = 7.8 Hz), 8.1 (4H, m), 7.97 (1H, m), 7.79 (1H, s), 2.47 (1H, m), 1.83 (2H, m), 1.54 (8H, m), 1.01 (6H, m). 13C NMR (400 MHz, CDCl3) δ/ppm: 175.33, 131.52, 130.97, 130.47, 129.28, 128.16, 127.54, 127.00, 126.31, 125.67, 125.34, 125.20, 124.94, 123.79, 122.84, 120.29, 48.98, 35.83, 21.32, 14.48. HRMS (m/z): [M + H]+ calculated for C24H26NO+, 344.2014; found 344.2140.
5.2. Optical Spectroscopy
Steady-state absorption spectra are recorded in the transmission mode using a JASCO V-670 spectrophotometer (Tokyo, Japan). The steady-state emission spectra and the time-correlated single-photon counting (TCSPC) fluorescence decays are measured using a FluoroLog-3 spectrofluorometer (Horiba Jobin Yvon, Edison, NJ), equipped with a pulsed diode laser (λ = 278 nm, 0.9 ns pulse width), as previously reported.86−88
The wavelengths of the maxima of the absorption and emission
spectra were obtained from fitting the spectral peaks with Gaussian
functions. For estimating zero-to-zero energy, 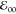 , of a
conjugate, we plot its absorption
and fluorescence spectra on the same graph where the fluorescence
maximum is adjusted to be equal to the maximum of the band at the
red edge of the absorption spectrum.
, of a
conjugate, we plot its absorption
and fluorescence spectra on the same graph where the fluorescence
maximum is adjusted to be equal to the maximum of the band at the
red edge of the absorption spectrum. 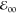 is estimated
from the wavelength at which
the thus normalized spectra cross (Table 1a). The fluorescence quantum yields, φf (Table 1),
were determined by comparing the integrated emission intensities of
the samples with those of the reference, an ethanol solution of Coumarin
151 (φf0 = 0.49)89−93
is estimated
from the wavelength at which
the thus normalized spectra cross (Table 1a). The fluorescence quantum yields, φf (Table 1),
were determined by comparing the integrated emission intensities of
the samples with those of the reference, an ethanol solution of Coumarin
151 (φf0 = 0.49)89−93
| 1 |
where F(λ) is the fluorescence intensity at wavelength λ, A(λex) is the absorbance at the excitation wavelength, n is the refractive index of the medium, and the subscript “0” indicates the quantities for the reference solution.
5.3. Electrochemistry
Cyclic voltammetry was conducted using a Reference 600TM potentiostat/galvanostat/ZRA (Gamry Instruments, PA), connected to a three-electrode cell, as previously described.82,83 Glassy carbon electrode and platinum wire were used for the working and counter electrodes, respectively. A saturated calomel electrode (Gamry Instruments) was used for a reference electrode. To prevent contamination, the reference electrode was brought in contact with the sample solution via a salt bridge. When not in use, the reference electrode is stored submersed in saturated potassium chloride solution.
Anhydrous aprotic solvents with different polarity, dichloromethane (DCM) and acetonitrile (MeCN), were employed with different concentrations of tetrabutylammonium hexafluorophosphate ((n-C4H9)4NPF6) as the supporting electrolyte. Prior to recording each voltammogram, the sample was extensively purged with argon while maintaining its volume constant by adding more anhydrous solvent. For each solvent, a set of voltammograms was recorded from 25 to 200 mM, increasing the electrolyte concentration in increments of 25 mM. The half-wave potentials, E(1/2), were determined from the midpoints between the cathodic and anodic peak potentials for reversible or quasireversible voltammograms and from the inflection points of the waves for irreversible oxidation and reduction. The anodic and cathodic peak potentials, Ea and Ec, respectively, were determined from the zeros of the first derivatives of the voltammograms, i.e., as the potentials at which ∂I/∂E = 0 when ∂E/∂t = constant. The inflection points were determined from the zeros of the second derivatives of the voltammograms, ∂2I/∂E2 = 0 at ∂E/∂t = constant.81,94 The second derivatives of the reversible and quasireversible voltammograms indicated that the inflection-point potentials were quite close to the midpoints between Ea and Ec, ensuring the reliability for the estimates of E(1/2) from the inflection points of the irreversible voltammograms. The voltammograms were recorded at a scan rate of 100 mV s–1. To correct for potential drifts in the reference electrode (SCE, connected to the cell via a salt bridge), ferrocene was used as a standard (E(1/2) = 0.45 ± 0.01 V vs SCE for MeCN, 100 mM NBu4BF4).82 Voltammograms of the standard were recorded before and after each set of measurements. To correct for the dependence of E(1/2) on the electrolyte concentration, the potentials in neat electrolyte-free solvent were estimated from extrapolations to zero supporting electrolyte concentration (Figure 2b).82,83
5.4. Computational Analysis
The pyrene derivatives (Chart 1) were modeled using density functional theory (DFT). For simplicity, the aliphatic chains were truncated to two carbons, i.e., to ethyl groups (Figure 3). The DFT calculations were performed at the at the B3LYP/6-311G(3df,3dp) level of theory for the formamide and at the B3LYP/6-311+G(d,p) level of theory for the pyrene derivatives95−97 for the gas phase using Gaussian 09.98 Although the calculations for formamide are sensitive to the selection of basis set, the results for the pyrene derivatives were practically the same for the different levels of theory tested (see Supporting Information). On the basis of Koopman’s theorem, we estimate IE and EA from the energies of the radical-ion doublet states and the ground states. Spin-unrestricted calculations are used for the calculations of the radical-cation and radical-anion doublet states. Solvent effects were estimated by comparing the results from gas-phase calculations to those in an integral equation formalism polarizable continuum model (PCM).99
Acknowledgments
We thank the U.S. National Science Foundation (grants CHE 1465284 and CHE 1800602) for funding this work. V.I.V. extends his gratitude to the Fulbright Commission Brazil for supporting his collaboration with the University of São Paulo. F.H.Q. thanks the CNPq (Universal grant 408181/2016-3), INCT-Catálise (CNPq 465454/2014-3), and NAP-PhotoTech for support and the CNPq for a research productivity fellowship. We also extend our gratitude to Dr. Maksim Y. Livshits and Prof. Elena Jakubikova for their assistance with the DFT calculations.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01581.
1H and 13C NMR spectra of the synthesized pyrene–amide derivatives, along with their ionization energies and the electron affinities calculated at different levels of theory (PDF)
Author Present Address
# PrimusLabs, Santa Maria, California 93455, United States (D.B.).
Author Present Address
∇ Swampscott High School, 200 Essex Street, Swampscott, Massachusetts 01907, United States (B.G.).
The authors declare no competing financial interest.
Supplementary Material
References
- Brylinski M.; Gao M.; Skolnick J. Why not consider a spherical protein? Implications of backbone hydrogen bonding for protein structure and function. Phys. Chem. Chem. Phys. 2011, 13, 17044–17055. 10.1039/c1cp21140d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor W. R.; Thornton J. M. Prediction of super-secondary structure in proteins. Nature 1983, 301, 540–542. 10.1038/301540a0. [DOI] [PubMed] [Google Scholar]
- Williams R. W.; Dunker A. K. Determination of the secondary structure of proteins from the amide I band of the laser Raman spectrum. J. Mol. Biol. 1981, 152, 783–813. 10.1016/0022-2836(81)90127-3. [DOI] [PubMed] [Google Scholar]
- Carothers W. H.Linear Polyamides Suitable for Spinning into Strong Pliable Fibers. US2130523A1938.
- Jang Y.; Champion J. A. Self-assembled materials made from functional recombinant proteins. Acc. Chem. Res. 2016, 49, 2188–2198. 10.1021/acs.accounts.6b00337. [DOI] [PubMed] [Google Scholar]
- Song Z.; Han Z.; Lv S.; Chen C.; Chen L.; Yin L.; Cheng J. Synthetic polypeptides: From polymer design to supramolecular assembly and biomedical application. Chem. Soc. Rev. 2017, 46, 6570–6599. 10.1039/C7CS00460E. [DOI] [PubMed] [Google Scholar]
- Rueda F.; Cespedes M. V.; Conchillo-Sole O.; Sanchez-Chardi A.; Seras-Franzoso J.; Cubarsi R.; Gallardo A.; Pesarrodona M.; Ferrer-Miralles N.; Daura X.; Vazquez E.; Garcia-Fruitos E.; Mangues R.; Unzueta U.; Villaverde A. Bottom-up instructive quality control in the biofabrication of smart protein materials. Adv. Mater. 2015, 27, 7816–7822. 10.1002/adma.201503676. [DOI] [PubMed] [Google Scholar]
- Deming T. J. Polypeptide materials. New synthetic methods and applications. Adv. Mater. 1997, 9, 299–311. 10.1002/adma.19970090404. [DOI] [Google Scholar]
- Crespo L.; Sanclimens G.; Pons M.; Giralt E.; Royo M.; Albericio F. Peptide and amide bond-containing dendrimers. Chem. Rev. 2005, 105, 1663–1681. 10.1021/cr030449l. [DOI] [PubMed] [Google Scholar]
- Fischer G. Chemical aspects of peptide bond isomerisation. Chem. Soc. Rev. 2000, 29, 119–127. 10.1039/a803742f. [DOI] [Google Scholar]
- Bulheller B. M.; Rodger A.; Hirst J. D. Circular and linear dichroism of proteins. Phys. Chem. Chem. Phys. 2007, 9, 2020–2035. 10.1039/b615870f. [DOI] [PubMed] [Google Scholar]
- Temussi P. A.; Tancredi T.; Quadrifoglio F. Conformational rigidity of the amide bond. Variable-temperature nuclear magnetic resonance study of the system Ag+-n,n-dimethylacetamide. J. Phys. Chem. 1969, 73, 4227–4232. 10.1021/j100846a032. [DOI] [Google Scholar]
- Tonelli A. E. On the stability of cis and trans amide bond conformations in polypeptides. J. Am. Chem. Soc. 1971, 93, 7153–7155. 10.1021/ja00755a007. [DOI] [PubMed] [Google Scholar]
- Vasudev P. G.; Chatterjee S.; Shamala N.; Balaram P. Structural chemistry of peptides containing backbone expanded amino acid residues: Conformational features of β, γ, and hybrid peptides. Chem. Rev. 2011, 111, 657–687. 10.1021/cr100100x. [DOI] [PubMed] [Google Scholar]
- Upadhyayula S.; Bao D.; Millare B.; Sylvia S. S.; Habib K. M. M.; Ashraf K.; Ferreira A.; Bishop S.; Bonderer R.; Baqai S.; Jing X.; Penchev M.; Ozkan M.; Ozkan C. S.; Lake R. K.; Vullev V. I. Permanent electric dipole moments of carboxyamides in condensed media: What are the limitations of theory and experiment?. J. Phys. Chem. B 2011, 115, 9473–9490. 10.1021/jp2045383. [DOI] [PubMed] [Google Scholar]
- Suydam I. T.; Snow C. D.; Pande V. S.; Boxer S. G. Electric fields at the active site of an enzyme: Direct comparison of experiment with theory. Science 2006, 313, 200–204. 10.1126/science.1127159. [DOI] [PubMed] [Google Scholar]
- Doyle D. A.; Cabral J. M.; Pfuetzner R. A.; Kuo A.; Gulbis J. M.; Cohen S. L.; Chait B. T.; MacKinnon R. The structure of the potassium channel: Molecular basis of k+ conduction and selectivity. Science 1998, 280, 69–77. 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Hol W. G. J.; van Duijnen P. T.; Berendsen H. J. C. The α-helix dipole and the properties of proteins. Nature 1978, 273, 443–446. 10.1038/273443a0. [DOI] [PubMed] [Google Scholar]
- Wada A. Dielectric properties of polypeptide solutions. I. The electric dipole moment of the α helix in dioxane. J. Chem. Phys. 1958, 29, 674–675. 10.1063/1.1744559. [DOI] [Google Scholar]
- Shin Y.-g. K.; Newton M. D.; Isied S. S. Distance dependence of electron transfer across peptides with different secondary structures: The role of peptide energetics and electronic coupling. J. Am. Chem. Soc. 2003, 125, 3722–3732. 10.1021/ja020358q. [DOI] [PubMed] [Google Scholar]
- Vullev V. I. From biomimesis to bioinspiration: What’s the benefit for solar energy conversion applications?. J. Phys. Chem. Lett. 2011, 2, 503–508. 10.1021/jz1016069. [DOI] [Google Scholar]
- Ferrand Y.; Huc I. Designing helical molecular capsules based on folded aromatic amide oligomers. Acc. Chem. Res. 2018, 51, 970–977. 10.1021/acs.accounts.8b00075. [DOI] [PubMed] [Google Scholar]
- Zhang D.-W.; Zhao X.; Hou J.-L.; Li Z.-T. Aromatic amide foldamers: Structures, properties, and functions. Chem. Rev. 2012, 112, 5271–5316. 10.1021/cr300116k. [DOI] [PubMed] [Google Scholar]
- Ziach K.; Chollet C.; Parissi V.; Prabhakaran P.; Marchivie M.; Corvaglia V.; Bose P. P.; Laxmi-Reddy K.; Godde F.; Schmitter J.-M.; Chaignepain S.; Pourquier P.; Huc I. Single helically folded aromatic oligoamides that mimic the charge surface of double-stranded B-DNA. Nat. Chem. 2018, 10, 511–518. 10.1038/s41557-018-0018-7. [DOI] [PubMed] [Google Scholar]
- Ashraf M. K.; Pandey R. R.; Lake R. K.; Millare B.; Gerasimenko A. A.; Bao D.; Vullev V. I. Theoretical design of bioinspired macromolecular electrets based on anthranilamide derivatives. Biotechnol. Prog. 2009, 25, 915–922. 10.1002/btpr.189. [DOI] [PubMed] [Google Scholar]
- Xia B.; Bao D.; Upadhyayula S.; Jones G.; Vullev V. I. Anthranilamides as bioinspired molecular electrets: Experimental evidence for a permanent ground-state electric dipole moment. J. Org. Chem. 2013, 78, 1994–2004. 10.1021/jo301942g. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Geib S. J.; Hamilton A. D. Oligoanthranilamides. Non-peptide subunits that show formation of specific secondary structure. J. Am. Chem. Soc. 1996, 118, 7529–7541. 10.1021/ja9539857. [DOI] [Google Scholar]
- Li X.; Markandeya N.; Jonusauskas G.; McClenaghan N. D.; Maurizot V.; Denisov S. A.; Huc I. Photoinduced electron transfer and hole migration in nanosized helical aromatic oligoamide foldamers. J. Am. Chem. Soc. 2016, 138, 13568–13578. 10.1021/jacs.6b05668. [DOI] [PubMed] [Google Scholar]
- Méndez-Ardoy A.; Markandeya N.; Li X.; Tsai Y.-T.; Pecastaings G.; Buffeteau T.; Maurizot V.; Muccioli L.; Castet F.; Huc I.; Bassani D. M. Multi-dimensional charge transport in supramolecular helical foldamer assemblies. Chem. Sci. 2017, 8, 7251–7257. 10.1039/C7SC03341A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffs M.; Delsuc N.; Veldman D.; Vân Anh N.; Williams R. M.; Meskers S. C. J.; Janssen R. A. J.; Huc I.; Schenning A. P. H. J. Helical aromatic oligoamide foldamers as organizational scaffolds for photoinduced charge transfer. J. Am. Chem. Soc. 2009, 131, 4819–4829. 10.1021/ja809367u. [DOI] [PubMed] [Google Scholar]
- Larsen-Clinton J. M.; Espinoza E. M.; Mayther M. F.; Clark J.; Tao C.; Bao D.; Larino C. M.; Wurch M.; Lara S.; Vullev V. I. Fluorinated aminoanthranilamides: Non-native amino acids for bringing proteomic approaches to charge-transfer systems. Phys. Chem. Chem. Phys. 2017, 19, 7871–7876. 10.1039/C7CP00432J. [DOI] [PubMed] [Google Scholar]
- Dutzler R.; Campbell E. B.; Cadene M.; Chait B. T.; MacKinnon R. X-ray structure of a clc chloride channel at 3.0 å reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294. 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Bao D.; Upadhyayula S.; Larsen J. M.; Xia B.; Georgieva B.; Nunez V.; Espinoza E. M.; Hartman J. D.; Wurch M.; Chang A.; Lin C.-K.; Larkin J.; Vasquez K.; Beran G. J. O.; Vullev V. I. Dipole-mediated rectification of intramolecular photoinduced charge separation and charge recombination. J. Am. Chem. Soc. 2014, 136, 12966–12973. 10.1021/ja505618n. [DOI] [PubMed] [Google Scholar]
- Galoppini E.; Fox M. A. Effect of the electric field generated by the helix dipole on photoinduced intramolecular electron transfer in dichromophoric α-helical peptides. J. Am. Chem. Soc. 1996, 118, 2299–2300. 10.1021/ja951555a. [DOI] [Google Scholar]
- Yasutomi S.; Morita T.; Imanishi Y.; Kimura S. A molecular photodiode system that can switch photocurrent direction. Science 2004, 304, 1944–1947. 10.1126/science.1098489. [DOI] [PubMed] [Google Scholar]
- Shlizerman C.; Atanassov A.; Berkovich I.; Ashkenasy G.; Ashkenasy N. De novo designed coiled-coil proteins with variable conformations as components of molecular electronic devices. J. Am. Chem. Soc. 2010, 132, 5070–5076. 10.1021/ja907902h. [DOI] [PubMed] [Google Scholar]
- Garbuio L.; Antonello S.; Guryanov I.; Li Y.; Ruzzi M.; Turro N. J.; Maran F. Effect of orientation of the peptide-bridge dipole moment on the properties of fullerene-peptide-radical systems. J. Am. Chem. Soc. 2012, 134, 10628–10637. 10.1021/ja303696s. [DOI] [PubMed] [Google Scholar]
- Krzeszewski M.; Espinoza E. M.; Červinka C.; Derr J. B.; Clark J. A.; Borchardt D.; Beran G. J. O.; Gryko D. T.; Vullev V. I. Dipole effects on electron transfer are enormous. Angew. Chem., Int. Ed. 2018, 57, 12365–12369. 10.1002/anie.201802637. [DOI] [PubMed] [Google Scholar]
- Odonnell J. F.; Mann C. K. Controlled-potential oxidation of aliphatic amides. J. Electroanal. Chem. Interfacial Electrochem. 1967, 13, 157–162. 10.1016/0022-0728(67)80108-6. [DOI] [Google Scholar]
- Nichols E. M.; Derrick J. S.; Nistanaki S. K.; Smith P. T.; Chang C. J. Positional effects of second-sphere amide pendants on electrochemical CO2 reduction catalyzed by iron porphyrins. Chem. Sci. 2018, 9, 2952–2960. 10.1039/C7SC04682K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srujan M.; Chandrashekhar V.; Reddy R. C.; Prabhakar R.; Sreedhar B.; Chaudhuri A. The influence of the structural orientation of amide linkers on the serum compatibility and lung transfection properties of cationic amphiphiles. Biomaterials 2011, 32, 5231–5240. 10.1016/j.biomaterials.2011.03.059. [DOI] [PubMed] [Google Scholar]
- Han X.; Remsburg J. W.; He L.; Beesly T. E.; Armstrong D. W. Effect of the orientation of amide linkage groups on the enantioselectivity of two related synthetic polymeric chiral stationary phases. Chromatographia 2008, 67, 199–210. 10.1365/s10337-007-0490-5. [DOI] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Swain C. G.; Lupton E. C. Jr. Field and resonance components of substituent effects. J. Am. Chem. Soc. 1968, 90, 4328–4337. 10.1021/ja01018a024. [DOI] [Google Scholar]
- Dobrowolski J. C.; Lipiński P. F. J.; Karpińska G. Substituent effect in the first excited singlet state of monosubstituted benzenes. J. Phys. Chem. A 2018, 122, 4609–4621. 10.1021/acs.jpca.8b02209. [DOI] [PubMed] [Google Scholar]
- Østergaard M. E.; Hrdlicka P. J. Pyrene-functionalized oligonucleotides and locked nucleic acids (lnas): Tools for fundamental research, diagnostics, and nanotechnology. Chem. Soc. Rev. 2011, 40, 5771–5788. 10.1039/c1cs15014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vullev V. I.; Jiang H.; Jones G. II. Excimer Sensing. In Advanced Concepts in Fluorescence Sensing; Geddes C. D., Lakowicz J. R., Eds.; Topics in Fluorescence Spectroscopy; Springer: Boston, MA, 2005; vol. 10, pp 211–239. [Google Scholar]
- Winnik F. M. Photophysics of preassociated pyrenes in aqueous polymer solutions and in other organized media. Chem. Rev. 1993, 93, 587–614. 10.1021/cr00018a001. [DOI] [Google Scholar]
- Karpovich D. S.; Blanchard G. J. Relating the polarity-dependent fluorescence response of pyrene to vibronic coupling. Achieving a fundamental understanding of the py polarity scale. J. Phys. Chem. 1995, 99, 3951–3958. 10.1021/j100012a014. [DOI] [Google Scholar]
- Kathiravan A.; Srinivasan V.; Khamrang T.; Velusamy M.; Jaccob M.; Pavithra N.; Anandan S.; Velappan K. Pyrene based d-π-a architectures: Synthesis, density functional theory, photophysics and electron transfer dynamics. Phys. Chem. Chem. Phys. 2017, 19, 3125–3135. 10.1039/C6CP08180K. [DOI] [PubMed] [Google Scholar]
- Hofmann O. T.; Glowatzki H.; Buerker C.; Rangger G. M.; Broeker B.; Niederhausen J.; Hosokai T.; Salzmann I.; Blum R. P.; Rieger R.; Vollmer A.; Rajput P.; Gerlach A.; Muellen K.; Schreiber F.; Zojer E.; Koch N.; Duhm S. Orientation-dependent work-function modification using substituted pyrene-based acceptors. J. Phys. Chem. C 2017, 121, 24657–24668. 10.1021/acs.jpcc.7b08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. M.; Vân Anh N.; van Stokkum I. H. M. Triplet formation by charge recombination in thin film blends of perylene red and pyrene: Developing a target model for the photophysics of organic photovoltaic materials. J. Phys. Chem. B 2013, 117, 11239–11248. 10.1021/jp402086p. [DOI] [PubMed] [Google Scholar]
- Jones G. II; Vullev V. I. Photoinduced electron transfer between non-native donor-acceptor moieties incorporated in synthetic polypeptide aggregates. Org. Lett. 2002, 4, 4001–4004. 10.1021/ol026656+. [DOI] [PubMed] [Google Scholar]
- Vullev V. I.; Jones G. Photoinduced electron transfer in alkanoylpyrene aggregates in conjugated polypeptides. Tetrahedron Lett. 2002, 43, 8611–8615. 10.1016/S0040-4039(02)01895-6. [DOI] [Google Scholar]
- Jones G. II; Vullev V.; Braswell E. H.; Zhu D. Multistep photoinduced electron transfer in a de novo helix bundle: Multimer self-assembly of peptide chains including a chromophore special pair. J. Am. Chem. Soc. 2000, 122, 388–389. 10.1021/ja981936z. [DOI] [Google Scholar]
- Jones G. II; Lu L. N.; Vullev V.; Gosztola D.; Greenfield S.; Wasielewski M. Photoactive peptides. 6. Photoinduced electron transfer for pyrenesulfonamide conjugates of tryptophan-containing peptides. Mitigation of fluoroprobe behavior in n-terminal labeling experiments. Bioorg. Med. Chem. Lett. 1995, 5, 2385–2390. 10.1016/0960-894X(95)00416-Q. [DOI] [Google Scholar]
- Jones G. II; Vullev V. I. Ground- and excited-state aggregation properties of a pyrene derivative in aqueous media. J. Phys. Chem. A 2001, 105, 6402–6406. 10.1021/jp010087q. [DOI] [Google Scholar]
- Daugherty D. L.; Gellman S. H. A fluorescence assay for leucine zipper dimerization: Avoiding unintended consequences of fluorophore attachment. J. Am. Chem. Soc. 1999, 121, 4325–4333. 10.1021/ja990178p. [DOI] [Google Scholar]
- Jones G. II; Vullev V. I. Contribution of a pyrene fluorescence probe to the aggregation propensity of polypeptides. Org. Lett. 2001, 3, 2457–2460. 10.1021/ol016123l. [DOI] [PubMed] [Google Scholar]
- Figueira-Duarte T. M.; Muellen K. Pyrene-based materials for organic electronics. Chem. Rev. 2011, 111, 7260–7314. 10.1021/cr100428a. [DOI] [PubMed] [Google Scholar]
- Mateo-Alonso A. Pyrene-fused pyrazaacenes: From small molecules to nanoribbons. Chem. Soc. Rev. 2014, 43, 6311–6324. 10.1039/C4CS00119B. [DOI] [PubMed] [Google Scholar]
- Guo L.; Wang M.; Cao D. A novel Zr-MOF as fluorescence turn-on probe for real-time detecting H2S gas and fingerprint identification. Small 2018, 14, 1703822 10.1002/smll.201703822. [DOI] [PubMed] [Google Scholar]
- Noh H.; Kung C.-W.; Islamoglu T.; Peters A. W.; Liao Y.; Li P.; Garibay S. J.; Zhang X.; DeStefano M. R.; Hupp J. T.; Farha O. K. Room temperature synthesis of an 8-connected zr-based metal-organic framework for top-down nanoparticle encapsulation. Chem. Mater. 2018, 30, 2193–2197. 10.1021/acs.chemmater.8b00449. [DOI] [Google Scholar]
- Thomas S. S.; Tang H.; Gaudes A.; Baggesen S. B.; Gibb C. L. D.; Gibb B. C.; Bohne C. Tuning the binding dynamics of a guest-octaacid capsule through noncovalent anchoring. J. Phys. Chem. Lett. 2017, 8, 2573–2578. 10.1021/acs.jpclett.7b00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore J. D.; Gupta A.; Warren J. J.; Brunschwig B. S.; Gray H. B. Noncovalent immobilization of electrocatalysts on carbon electrodes for fuel production. J. Am. Chem. Soc. 2013, 135, 18288–18291. 10.1021/ja4099609. [DOI] [PubMed] [Google Scholar]
- Tang H.; de Oliveira C. S.; Sonntag G.; Gibb C. L. D.; Gibb B. C.; Bohne C. Dynamics of a supramolecular capsule assembly with pyrene. J. Am. Chem. Soc. 2012, 134, 5544–5547. 10.1021/ja301278p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malig J.; Romero-Nieto C.; Jux N.; Guldi D. M. Integrating water-soluble graphene into porphyrin nanohybrids. Adv. Mater. 2012, 24, 800–805. 10.1002/adma.201103697. [DOI] [PubMed] [Google Scholar]
- Bartelmess J.; Ballesteros B.; de la Torre G.; Kiessling D.; Campidelli S.; Prato M.; Torres T.; Guldi D. M. Phthalocyanine-pyrene conjugates: A powerful approach toward carbon nanotube solar cells. J. Am. Chem. Soc. 2010, 132, 16202–16211. 10.1021/ja107131r. [DOI] [PubMed] [Google Scholar]
- Schulz-Drost C.; Sgobba V.; Gerhards C.; Leubner S.; Calderon R. M. K.; Ruland A.; Guldi D. M. Innovative inorganic-organic nanohybrid materials: Coupling quantum dots to carbon nanotubes. Angew. Chem., Int. Ed. 2010, 49, 6425–6429. 10.1002/anie.200906891. [DOI] [PubMed] [Google Scholar]
- Udit A. K.; Hill M. G.; Bittner V. G.; Arnold F. H.; Gray H. B. Reduction of dioxygen catalyzed by pyrene-wired heme domain cytochrome p450 bm3 electrodes. J. Am. Chem. Soc. 2004, 126, 10218–10219. 10.1021/ja0466560. [DOI] [PubMed] [Google Scholar]
- Espinoza E. M.; Xia B.; Darabedian N.; Larsen J. M.; Nunez V.; Bao D.; Mac J. T.; Botero F.; Wurch M.; Zhou F.; Vullev V. I. Nitropyrene photoprobes: Making them, and what are they good for?. Eur. J. Org. Chem. 2016, 2016, 343–356. 10.1002/ejoc.201501339. [DOI] [Google Scholar]
- Casas-Solvas J. M.; Howgego J. D.; Davis A. P. Synthesis of substituted pyrenes by indirect methods. Org. Biomol. Chem. 2014, 12, 212–232. 10.1039/C3OB41993B. [DOI] [PubMed] [Google Scholar]
- Feng X.; Hu J.-Y.; Redshaw C.; Yamato T. Functionalization of pyrene to prepare luminescent materials-typical examples of synthetic methodology. Chem. – Eur. J. 2016, 22, 11898–11916. 10.1002/chem.201600465. [DOI] [PubMed] [Google Scholar]
- Zimerman O. E.; Weiss R. G. Static and dynamic fluorescence from α,ω-di(1-pyrenyl)alkanes in polyethylene films. Control of probe conformations and information about microstructure of the media. J. Phys. Chem. A 1998, 102, 5364–5374. 10.1021/jp972758j. [DOI] [Google Scholar]
- Saliba M.; Correa-Baena J.-P.; Grätzel M.; Hagfeldt A.; Abate A. Perovskite solar cells: From the atomic level to film quality and device performance. Angew. Chem., Int. Ed. 2018, 57, 2554–2569. 10.1002/anie.201703226. [DOI] [PubMed] [Google Scholar]
- Guo S.; Bao D.; Upadhyayula S.; Wang W.; Guvenc A. B.; Kyle J. R.; Hosseinibay H.; Bozhilov K. N.; Vullev V. I.; Ozkan C. S.; Ozkan M. Photoinduced electron transfer between pyridine coated cadmium selenide quantum dots and single sheet graphene. Adv. Funct. Mater. 2013, 23, 5199–5211. 10.1002/adfm.201203652. [DOI] [Google Scholar]
- Klimov V. I. Optical nonlinearities and ultrafast carrier dynamics in semiconductor nanocrystals. J. Phys. Chem. B 2000, 104, 6112–6123. 10.1021/jp9944132. [DOI] [Google Scholar]
- Namiki A.; Nakashima N.; Yoshihara K. Fluorescence quenching due to the electron-transfer - indole-chloromethanes in rigid ethanol glass. J. Chem. Phys. 1979, 71, 925–930. 10.1063/1.438383. [DOI] [Google Scholar]
- Uno B.; Okumura N.. Molecular Scientific Approach in Electroorganic Chemistry. Recent Research Developments in Pure & Applied Analytical Chemistry; Gifu Pharmaceutical University: Gifu, 1998; Vol. 2, pp 83–99. [Google Scholar]
- Bao D.; Millare B.; Xia W.; Steyer B. G.; Gerasimenko A. A.; Ferreira A.; Contreras A.; Vullev V. I. Electrochemical oxidation of ferrocene: A strong dependence on the concentration of the supporting electrolyte for nonpolar solvents. J. Phys. Chem. A 2009, 113, 1259–1267. 10.1021/jp809105f. [DOI] [PubMed] [Google Scholar]
- Espinoza E. M.; Larsen J. M.; Vullev V. I. What makes oxidized n-acylanthranilamides stable?. J. Phys. Chem. Lett. 2016, 7, 758–764. 10.1021/acs.jpclett.5b02881. [DOI] [PubMed] [Google Scholar]
- Bao D.; Ramu S.; Contreras A.; Upadhyayula S.; Vasquez J. M.; Beran G.; Vullev V. I. Electrochemical reduction of quinones: Interfacing experiment and theory for defining effective radii of redox moieties. J. Phys. Chem. B 2010, 114, 14467–14479. 10.1021/jp101730e. [DOI] [PubMed] [Google Scholar]
- Lockhart D. J.; Kim P. S. Internal stark-effect measurement of the electric-field at the amino terminus of an alpha-helix. Science 1992, 257, 947–951. 10.1126/science.1502559. [DOI] [PubMed] [Google Scholar]
- Armbruster C.; Knapp M.; Rechthaler K.; Schamschule R.; Parusel A. B. J.; Kohler G.; Wehrmann W. Fluorescence properties of 1-heptanoylpyrene: A probe for hydrogen bonding in microaggregates and biological membranes. J. Photochem. Photobiol., A 1999, 125, 29–38. 10.1016/S1010-6030(99)00099-4. [DOI] [Google Scholar]
- Lu H.; Bao D.; Penchev M.; Ghazinejad M.; Vullev V. I.; Ozkan C. S.; Ozkan M. Pyridine-coated lead sulfide quantum dots for polymer hybrid photovoltaic devices. Adv. Sci. Lett. 2010, 3, 101–109. 10.1166/asl.2010.1110. [DOI] [Google Scholar]
- Ghazinejad M.; Kyle J. R.; Guo S.; Pleskot D.; Bao D.; Vullev V. I.; Ozkan M.; Ozkan C. S. Non-invasive high-throughput metrology of functionalized graphene sheets. Adv. Funct. Mater. 2012, 22, 4519–4525. 10.1002/adfm.201200434. [DOI] [Google Scholar]
- Upadhyayula S.; Nunez V.; Espinoza E. M.; Larsen J. M.; Bao D.; Shi D.; Mac J. T.; Anvari B.; Vullev V. I. Photoinduced dynamics of a cyanine dye: Parallel pathways of non-radiative deactivation involving multiple excited-state twisted transients. Chem. Sci. 2015, 6, 2237–2251. 10.1039/C4SC02881C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby G. A.; Demas J. N. The measurement of photoluminescence quantum yields. A review. J. Phys. Chem. 1971, 75, 991–1024. 10.1021/j100678a001. [DOI] [Google Scholar]
- Jung B.; Vullev V. I.; Anvari B. Revisiting indocyanine green: Effects of serum and physiological temperature on absorption and fluorescence characteristics. IEEE J. Sel. Top. Quantum Electron. 2014, 20, 7000409 10.1109/jstqe.2013.2278674. [DOI] [Google Scholar]
- Vasquez J. M.; Vu A.; Schultz J. S.; Vullev V. I. Fluorescence enhancement of warfarin induced by interaction with β-cyclodextrin. Biotechnol. Prog. 2009, 25, 906–914. 10.1002/btpr.188. [DOI] [PubMed] [Google Scholar]
- Sjöback R.; Nygren J.; Kubista M. Absorption and fluorescence properties of fluorescein. Spectrochim. Acta, Part A 1995, 51, L7–L21. 10.1016/0584-8539(95)01421-P. [DOI] [Google Scholar]
- Zhang X. F.; Zhang J. L.; Liu L. M. Fluorescence properties of twenty fluorescein derivatives: Lifetime, quantum yield, absorption and emission spectra. J. Fluoresc. 2014, 24, 819–826. 10.1007/s10895-014-1356-5. [DOI] [PubMed] [Google Scholar]
- Roth H. G.; Romero N. A.; Nicewicz D. A. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 2016, 27, 714–723. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Krishnan R.; Binkley J. S.; Seeger R.; Pople J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.et al. Gaussian 09, revision a.1; Gaussian Inc.: Wallingford, CT, 2009.
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.