

Abstract
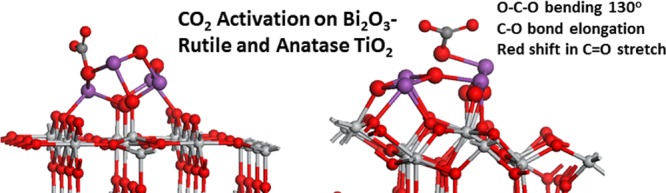
The capture and conversion of CO2 are of significant importance in enabling the production of sustainable fuels, contributing to alleviating greenhouse gas emissions. While there are a number of key steps required to convert CO2, the initial step of adsorption and activation by the catalyst is critical. Well-known metal oxides such as oxidized TiO2 or CeO2 are unable to promote this step. In addressing this difficult problem, a recent experimental work shows the potential for bismuth-containing materials to adsorb and convert CO2, the origin of which is attributed to the role of the bismuth lone pair. In this paper, we present density functional theory (DFT) simulations of enhanced CO2 adsorption on heterostructures composed of extended TiO2 rutile (110) and anatase (101) surfaces modified with Bi2O3 nanoclusters, highlighting in particular the role of heterostructure reduction in activating CO2. These heterostructures show low coordinated Bi sites in the nanoclusters and a valence band edge that is dominated by Bi–O states, typical of the Bi3+ lone pair. The reduction of Bi2O3–TiO2 heterostructures can be facile and produces reduced Bi2+ and Ti3+ species. The interaction of CO2 with this electron-rich, reduced system can produce CO directly, reoxidizing the heterostructure, or form an activated carboxyl species (CO2–) through electron transfer from the reduced heterostructure to CO2. The oxidized Bi2O3–TiO2 heterostructures can adsorb CO2 in carbonate-like adsorption modes, with moderately strong adsorption energies. The hydrogenation of the nanocluster and migration to adsorbed CO2 is feasible with H-migration barriers less than 0.7 eV, but this forms a stable COOH intermediate rather than breaking C–O bonds or producing formate. These results highlight that a reducible metal oxide heterostructure composed of a semiconducting metal oxide modified with suitable metal oxide nanoclusters can activate CO2, potentially overcoming the difficulties associated with the difficult first step in CO2 conversion.
1. Introduction
Increasing energy demand is leading to fossil fuel supply issues, and the use of fossil fuels has pushed the atmospheric CO2 concentration to 410 ppm. Continuing CO2 emissions are projected to result in a higher than 2 °C rise in average global temperatures. Given these severe societal problems, there is an urgent need to find materials that can convert the CO2 produced by the combustion of fossil fuels back to the fuels or to the precursors for the production of more useful chemicals. This will reduce our reliance on nonsustainable fossil fuels and contribute to curbing anthropogenic CO2 emissions. The solar-driven photoreduction or thermal reduction of CO2 to CO or directly to liquid fuels will enable a sustainable approach to producing fuels and storing solar energy in high-energy chemical bonds.1−11
Using solar energy, for example, through solar concentrators, coupled with catalysts that can promote activation of CO2 to drive the CO2 reduction process, is an attractive strategy to meet this ambitious aim. However, to date, there are no practical catalysts that can exploit solar energy to efficiently reduce CO2 to useful chemical species. A range of different catalysts for CO2 conversion using electrocatalysis, thermochemical catalysis, and photocatalysis has been investigated in recent years, and an example of such a photocatalyst is Pt-modified TiO2 nanotubes.8,12,13 However, the efficiencies for methane production are extremely low, and Pt will never be an economically viable catalyst for large-scale CO2 conversion.
Irrespective of how electrons are provided to reduce CO2, the key step in any CO2 capture and use/conversion process is the initial adsorption of CO2, which should promote the “activation” of the molecule. This activation can be a charge transfer to CO2, distortions to the molecular structure such as bending of O–C–O away from the linear elongations in the molecular C–O distances, or hydrogenation. CO2 should either dissociate to a CO molecule or undergo hydrogenation to produce methane or methanol, via formate or carboxyl pathways. The activation should also avoid the production of overstabilized inactive surface-bound carbonates which would poison the catalyst.
In recent years, catalysts based on metallic Cu, oxide-derived Cu, or Cu with mixed oxidation states have gained great interest and have been demonstrated to reduce CO2 to useful molecules such as methane, methanol, or ethanol.14−22 Recent experimental work indicates the ability of nanocatalysts containing a mix of Cu+ and Cu2+ oxidation states and in oxide-like structures to promote CO2 reduction1,2,10,14−16,18,19,21−29 and this includes Cu metal, oxide-derived Cu, and mixed oxidation state Cu. Modeling the interaction of CO2 with copper oxides using density functional theory (DFT) has provided useful results for further development of catalysts for CO2 conversion. Wu et al. studied the adsorption of CO2 at the Cu2O(111) surface in which excess electrons were produced through the formation of oxygen vacancies18 and found that dissociative adsorption of CO2 was thermodynamically unfeasible, and although a CO2δ− anion could be formed, this was not favorable, and hence excess electrons are not necessarily sufficient to promote CO2 capture and conversion. Wu et al. also studied the adsorption of CO2 and its derivatives at Cu2O(111) using hybrid DFT21 and found that only the adsorption of linear CO2 was favorable. This is supported by the work of Benavid and Carter who showed that physisorption of linear CO2 is favored over adsorption of bent anionic CO2.19 Mishra and colleagues reported strong CO2 chemisorption at the high-energy Cu–O-terminated (110) surface of Cu2O15 and exothermic adsorption of CO2 at the high-energy (011) surface of CuO.2 Uzunova et al. studied the conversion of CO2 to methanol on Cu2O nanolayers and clusters16 using hybrid DFT. The authors considered water as the source of H atoms for the hydrogenation of CO2 and described a reaction pathway from a carboxyl group (*COOH) to formic acid, which was the rate-limiting step, and then onto formaldehyde and finally methanol. This work would suggest other pathways for CO2 conversion beyond the dissociation into CO. In the work of Favaro et al.,24 a model of Cu with subsurface oxygen was required for copper to adsorb CO2 in the first place and understand the experimental findings. However, despite these advances, there still remains an urgent need from both a fundamental and a technoeconomic perspective to discover new materials that can activate and reduce CO2, in particular to produce sustainable fuels, particularly if renewable energy can be used, either for electroreduction or to produce renewable hydrogen from water.
Another popular metal oxide is titanium dioxide, and the adsorption of CO2 at different titania surfaces and nanostructures has been widely studied. The roles of excess electrons introduced by oxygen vacancies, low coordinated atomic sites, and the surface structure in CO2 adsorption and activation have been discussed.30−32 The presence of excess electrons and holes was shown to influence the adsorption and activation of CO2 at rutile (110), and both bent CO2– anion and CO2+ cation configurations were identified from the DFT calculations.33 Lee and Kanai32 used DFT to explore the difference between anatase (101) and a nanostructured TiO2 quantum dot for CO2 activation and hydrogenation. They found that the TiO2 quantum dot promoted CO2 activation and hydrogenation when compared to the extended anatase (101) surface. Yang and colleagues showed that subnanometer Pt clusters supported on the anatase (101) surface enhanced CO2 activation through the provision of additional adsorption sites and the transfer of electron density to the TiO2 substrate.34
Various other metal oxides have been investigated for CO2 conversion, and several theoretical studies have been conducted into the reaction pathways involving the hydrogenation of CO and CO2 at a variety of catalytic surfaces, including Cu/CeO2 and Cu/CeO2/TiO217 or Cu/ZnO/Al2O3.35 An enhanced photoreduction of CO2 with H2O vapor has been reported for dispersed CeO2/TiO2.36 Here, the ceria is modeled as a small cluster supported on rutile TiO2, similar to the work on water gas shift,37−39 and the role of Ce3+ in visible light absorption, photogenerated charge separation, and strengthening of the CO2–surface bonding was highlighted.
Recent experimental works from Rosenthal and co-workers40−43 and Walker et al.44 strongly suggest that non-transition-metal based catalysts, in particular those containing bismuth or tin, are able to activate CO2 or convert it to other molecules with good efficiency. The catalysts in refs40,43 are composed of Bi–glassy carbon and use ionic liquids or Bi nanoparticles, which have large amounts of Bi3+. The former materials can convert CO2 to alcohols with high efficiency, and even though ionic liquids are used, the role of Bi in the catalyst remains crucial to promote the conversion of CO2.
The bismuth pyrochlore oxide Bi2Ti2O7 was studied in ref (44) and was shown to have high CO2 chemisorption capacity. In this system, low-energy ion scattering (LEIS) shows clearly that the surface layer is terminated by a bismuth oxide layer. This surface structure arises from the well-known stereochemical lone pair in Bi3+.45,46 By contrast, in the pyrochlore Y2Ti2O7, there is no surface enrichment with Y3+ (lacking the lone pair). This Bi-oxide-terminated pyrochlore was examined for its capacity to adsorb and activate CO2 using infrared spectroscopy. The features in the infrared spectrum corresponding to the adsorbed CO2 were clearly present upon heating to 308 K. Y2Ti2O7, by contrast, does not strongly adsorb CO2. This suggests that the adsorption of CO2 arises from the presence of Bi3+ and a bismuth oxide surface layer in the pyrochlore surface.
From the studies in refs,40,44 it is reasonable to propose that the presence of the Bi3+ cation, with its stereochemically active lone pair, in a Bi-oxide structure will be active in promoting the interaction of Bi-containing oxides with CO2. However, the mechanism of CO2 adsorption and conversion on Bi3+-containing materials, as a good exemplar of the p-block metal oxides, needs to be further investigated, and this is one of the aims of this study.
Previously, we have used first-principles DFT simulations to design heterostructured materials which are composed of TiO2 (rutile or anatase) surfaces modified with metal oxide nanoclusters. In our earlier work, the emphasis has been on new heterostructured materials with predicted visible light absorption47−59 and reduced charge recombination.48−52,60−62 We have recently studied the interaction of molecules, such as CO2, with tin(II) oxide,47 zirconia,47 manganese oxide,63 alkaline earth oxide,64 and ceria65 nanocluster-modified TiO2 heterostructures. We have found that such nanocluster-modified TiO2 heterostructures can adsorb CO2,47 but that some heterostructures, such as reduced MnOx–TiO2, do not show favorable CO2 adsorption, even though reduced cations are present.63 There is still much work to be done to understand the factors that drive CO2 activation on metal oxides.
In the present paper, we take the ideas from the experimental work on CO2 activation on Bi-containing materials, the ability of nanostructures to activate CO2, and the activity of nanocluster-modified TiO2 and use first-principles DFT to examine in detail the interaction and adsorption of CO2 at Bi2O3 nanocluster-modified rutile and anatase TiO2. We explore the role of the Bi-oxide, the low coordinated sites in nanoclusters and reduction of the Bi2O3–TiO2 heterostructures in CO2 adsorption and activation. In some examples, we examine further interaction with hydrogen for the first steps in hydrogenation and formation of intermediates such as COOH or HCOO. The role of heterostructure reduction in CO2 activation is crucial, wherein the charge transfer from the reduced oxide heterostructure can result in the formation of a carboxylate or direct formation of CO.
2. Methods
In the DFT computations, we follow our approach from previous work61 and prepare heterostructures of Bi2O3 nanoclusters supported on the extended low-energy rutile (110) and anatase (101) surfaces; although other rutile and anatase surfaces can be present in, for example, typical TiO2 nanoparticles, these are the dominant surface facets and are well-studied.
All DFT computations use a three-dimensional periodic surface slab within the VASP code66−69 and a plane-wave basis set to describe the valence electrons. Projector-augmented wave potentials,70,71 with 4, 5, 6, 4, and 1 valence electrons for Ti, Bi, O, C, and H, respectively, describe the core–valence electron interactions, and this setup was extensively tested in our earlier work on Bi2O3-modified TiO2.61 The cutoff for the kinetic energy is 396 eV, and the exchange–correlation functional is the Perdew–Wang 9172 approximation. A Monkhorst–Pack (2 × 1 × 1) k-point sampling grid is used. For consistency with our previous work, we apply the DFT + U approach73,74 to describe the Ti 3d states, with a value of U = 4.5 eV. The convergence criteria for the electronic and ionic relaxations are 0.0001 eV and 0.02 eV/Å. Methfessel–Paxton smearing is used with σ = 0.1 eV (also for the broadening of the peaks in the projected density of states). We use the Newton–Raphson relaxation algorithm, and all calculations are spin-polarized throughout, with no constraints on the spin.
The rutile (110) surface is terminated by twofold coordinated bridging O atoms, with threefold coordinated in-plane oxygen atoms in the surface layer. In the same layer, the Ti atoms take fivefold and sixfold coordinations. The anatase (101) surface is characterized by twofold coordinated oxygen atoms terminating the surface layer, and the outermost Ti atoms are fivefold coordinated. Surface supercell expansions of (2 × 4) and (4 × 2) are employed for rutile and anatase, respectively, and the vacuum gap in all cases is 12 Å.
In forming these composite structures, we first relax the gas-phase metal oxide nanoclusters and the unmodified rutile and anatase surfaces using the same computational setup described above. The oxide nanocluster modifier is adsorbed at the TiO2 surfaces in different configurations, and each of these are relaxed, as described in refs.48,57,61,75 We selected the most stable Bi2O3–TiO2 composites for the study of CO2 activation in the present paper. We will use the term Bi2O3–TiO2 throughout this paper to indicate the general Bi2O3 nanocluster-modified TiO2 heterostructure, using the precise nanocluster composition and TiO2 surface when necessary.
The stability of the heterostructure is characterized by the computed nanocluster adsorption energy, Eads
| 1 |
where E[(Bi2O3)n–TiO2] is the computed total energy of the Bi2O3 nanocluster-modified TiO2 surface (in which n is the number of Bi2O3 units in the nanocluster, so that in this paper n = 2 or 3), E[(Bi2O3)n] is the computed total energy of the free (Bi2O3)n nanocluster, and E[(TiO2)] is the computed total energy of the unmodified TiO2 (rutile/anatase) surface.
The reduction of the Bi2O3–TiO2 heterostructures is studied by removing oxygen from the nanocluster modifier (which is always more stable than removing oxygen from the TiO2 support) and computing the formation energy of the oxygen vacancy as follows
| 2 |
where E{((Bi2O3–x)–TiO2) is the total energy of the Bi2O3–TiO2 composite with one oxygen removed and 1/2E(O2) is the reference energy for oxygen, namely half the total energy of the O2 molecule. We correct the oxygen vacancy formation energy for entropy, which, at 298 K, is 0.60 eV. Despite the known errors in the DFT energy of the reference O2 molecule, the trends in oxygen vacancy formation are independent of this error. We also use ab initio thermodynamics to determine the stability of the reduced Bi2O3–TiO2 heterostructures formed by removing oxygen over a range of temperatures.
To analyze the stability of the reduced heterostructured models when exposed to an environment of oxygen, we use the DFT-derived total energies as an input into an atomistic thermodynamics framework, which considers the effect of the surrounding gas phase as a reservoir that is in thermodynamic equilibrium with the heterostructure. If we assume that the oxygen reservoir exchanges particles with the system without affecting its chemical potential, the oxygen vacancy formation energy is calculated as follows
| 3 |
where E(Bi2O3−δ–TiO2) and E(Bi2O3–TiO2) are the total DFT energies of the Bi2O3–TiO2 heterostructures with and without the oxygen vacancy, NO is the number of oxygen atoms removed, and μO(p,T) is the chemical potential of oxygen. Approximating oxygen as an ideal gas, we explicitly introduce pressure and temperature in our model through the analytic relation between the chemical potentials and the temperature and pressure of the two gas reservoirs as follows
| 4 |
Here, T and p represent the temperature and partial pressure of oxygen, p0 denotes atmospheric pressure, and kB is the Boltzmann constant. μ′ is the energetic term that includes contributions from the rotations and vibrations of the molecule, as well as the ideal-gas entropy at 1 atm, which can be calculated or taken from the experimental values listed in the thermodynamic tables. Thus, for a given pressure, the stability of oxygen vacancies can be determined over a range of temperatures.
The CO2 adsorption energy at Bi2O3-modified TiO2 heterostructures is defined in eq 5
| 5 |
where E(CO2@(Bi2O3)n–TiO2) is the computed total energy of the relaxed adsorption structure of CO2 at Bi2O3-modified TiO2 composites, whether stoichiometric or reduced. We examined many CO2 adsorption structures, and those shown in the following text are the most stable that we have found. Although van der Waals corrections can be added to these computations, we have tested and found that the change in the adsorption energies is ca. 0.15 eV, which is not significant compared to the magnitude of the computed CO2 adsorption energies, and furthermore, there is no effect of the inclusion of vdW corrections on the relative stability of different CO2 adsorption structures.
3. Results and Discussion
3.1. Bi2O3-Modified Rutile and Anatase TiO2 Heterostructures
Figure 1 shows the atomic structures of the nanocluster-modified anatase (101) and rutile (110) surfaces of Bi4O6 and Bi6O9. We have described these heterostructured systems in detail in previous work,61 and we briefly summarize the key features required for this paper. The adsorption energies can be computed relative to the corresponding gas-phase stoichiometric Bi2O3 nanoclusters, and these are −5.18, −5.18, and −5.72 eV for Bi4O6-rutile, Bi6O9-rutile, and Bi4O6-anatase, respectively. If we consider these energies in terms of the deposition of the nanoclusters at very low loading/coverage through atomic layer deposition, chemisorption–calcination, or incipient wetness impregnation, then the above energies can be related to the desorption of the Bi2O3 nanoclusters from the TiO2 supports. The magnitude of the computed energies indicates a strong binding of the nanocluster modifiers at both TiO2 surfaces and suggests they would only desorb at very high temperatures.
Figure 1.

Relaxed atomic structure for Bi2O3 nanocluster-modified rutile (110) and anatase (101). (a) Bi4O6-rutile (110), (b) Bi6O9-rutile (110), and (c) Bi4O6-anatase (101). The color coding in this and subsequent figures is Ti = light gray sphere, O = red sphere, and Bi = purple sphere. Bi and O atoms in the adsorbed nanoclusters are depicted by larger radius spheres.
We can also consider the stability against sintering into larger nanoclusters. In the gas phase, the gain in energy when two nanoclusters of composition Bi2O3 coalesce into a larger Bi4O6 nanocluster is −6.1 eV, and the coalescence of a gas-phase Bi2O3 and a gas-phase Bi4O6 nanocluster results in a gain of 3.1 eV. On the surface, we use the total energies of the bismuth oxide nanocluster-modified rutile heterostructures to assess the nanocluster stability against coalescence into larger nanoclusters.75 For the example of Bi2O3 and Bi4O6 coalescing to a Bi6O9 nanocluster
The computed energy is +2.69 eV against coalescence of the two smaller nanoclusters supported on rutile (110). Thus, when the Bi2O3 nanoclusters are supported on rutile or anatase, they will be stable against desorption or aggregation. The high stability of the supported Bi2O3 nanoclusters is primarily driven by the formation of new interfacial bonds between the nanocluster and the surface. We also note that the extensive work of Graciani et al. on ceria–TiO2 used small Ce2O3 species supported on rutile (110),38,39 which were also shown to be highly stable.
On Bi4O6-rutile, two Bi cations bind to the bridging surface oxygen, with typical Bi–O distances of 2.17 Å. These Bi cations are threefold coordinated, whereas the remaining Bi cations are twofold coordinated. Three oxygen atoms from the nanocluster bind to the fivefold coordinated Ti surface atoms, with the Ti–O distances in the range of 1.83–2.05 Å. In the Bi6O9-rutile nanocluster, one Bi cation binds to a bridging oxygen, with a Bi–O distance of 2.37 Å. Two oxygen atoms from the nanocluster bind to the surface Ti atoms, with the Ti–O distances of 1.94 and 1.86 Å. Here, the terminal Bi cations, furthest away from the cluster–surface interface, are threefold coordinated, and there are twofold coordinated oxygen sites in the nanocluster.
In Bi4O6-anatase, three Bi cations bind to the surface oxygen, with the Bi–O distances of 2.14, 2.14, 2.24, and 2.17 Å. The two Bi cations furthest away from the cluster–surface interface are threefold coordinated and the remaining Bi cations are fourfold coordinated. Four oxygen atoms in the nanocluster bind to the surface Ti, with the distances Ti–O in the range of 1.87–2.06 Å. There are three twofold coordinated oxygen sites: one is the terminal oxygen in the nanocluster and the remaining two sites bridge the nanocluster and the anatase (101) surface.
The other important aspect of the Bi2O3–TiO2 composites for the interaction with CO2 is their electronic properties, and in Figure 2 we show the projected electronic density of states (PEDOS) for the Bi2O3-rutile and Bi2O3-anatase systems. Bi3+ is an interesting species as it has a stereochemically active lone pair, which results in the presence of Bi-derived electronic states at the top of the valence band. This gives rise to the distorted structure of bulk Bi2O346 and the Bi-rich surface region in the pyrochlore Bi2Ti2O7.44 The other metal oxides with +3 cations do not show these structural distortions.
Figure 2.

PEDOS projected onto the Bi 6s, Ti 3d, and O 2p states (nanocluster and surface) for (a) Bi4O6 rutile (110), (b) Bi6O9-rutile (110), and (c) Bi4O6-anatase (101). The zero of energy in all cases is the Fermi level. The left panels show the cation PEDOS and the right panels show the oxygen 2p PEDOS.
In the PEDOS of the Bi2O3–TiO2 heterostructures, the Bi electronic states are present at the valence band edge of all the composite systems studied. For modified rutile (110), we see Bi (6s + 6p) and O 2p states originating from the nanoclusters lying at a higher energy than the corresponding TiO2 valence and conduction band edges, which should result in a small red shift in light absorption. For anatase, the Bi2O3-derived states lie just below the anatase valence band edge, which results in no predicted red shift. The key finding is that the Bi states lie at the top of the valence band edge, and this can be important in the reduction or CO2 adsorption on the Bi2O3–TiO2 heterostructures.
Finally, we present the results for the reduction of the Bi2O3–TiO2 heterostructures. The reduced heterostructures are prepared by removing oxygen atoms from the Bi2O3 nanoclusters and computing the oxygen vacancy formation energy. From a practical perspective, reduction can be facilitated by a thermochemical process using concentrated solar radiation or by the introduction of hydrogen in a redox process. Relaxing the structure after the removal of oxygen, the most stable reduced heterostructures are shown in Figure 3. The computed formation energies for the most stable oxygen vacancy sites are 1.75 eV (1.12 eV) for Bi4O6-rutile (110), 1.64 eV (1.01 eV) for Bi6O9-rutile (110), and 3.0 eV (2.37 eV) for Bi4O6-anatase (101), where the formation energies corrected for TΔS at 298 K are given in parentheses. These formation energies that show a moderate cost for oxygen vacancy formation in the supported Bi2O3 nanoclusters are notably smaller than that on the corresponding bare TiO2 surfaces (3.5 eV for rutile (110) and 3.6–4.1 eV for anatase (101)76), so that reduction should be possible under moderate conditions. The other oxygen sites have formation energies larger than 2 eV on Bi4O6-rutile and between 3.4 and 3.7 eV on Bi4O6-anatase. Figure 4 shows the stability of the most stable oxygen vacancy sites in each heterostructure as a function of temperature at a pressure of 1 atm. From this plot, we can see that reduction should take place at 500–600 K, which is certainly feasible in the context of CO2 reduction/conversion processes. For the rutile-supported Bi6O9 nanocluster, the larger formation energy of the vacancy clearly results in a higher temperature for vacancy formation, although this is still notably lower than that on bare rutile (110).
Figure 3.

Atomic structure and spin density isosurfaces of the most stable reduced Bi2O3–TiO2 heterostructures: (a) Bi4O6-rutile (110), (b) Bi6O9-rutile (110), and (c) Bi4O6-anatase (101). Spin density isosurfaces enclose spin densities up to 0.02 electrons/Å3. The color coding of the atoms is the same as in Figure 1.
Figure 4.
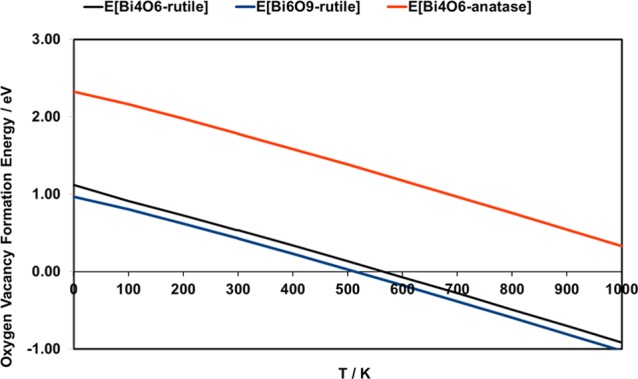
Computed oxygen vacancy formation energy for Bi2O3-modified TiO2 as a function of temperature at a pressure of 1 atm.
After the reduction of the Bi2O3 nanoclusters, the local atomic structure shows some changes. On Bi4O6-rutile (110), the removal of oxygen results in the formation of a structure that shows symmetry, with two twofold coordinated oxygen atoms terminating the nanocluster. The Bi cations are threefold coordinated, and each Bi atom binds to a bridging oxygen atom from the rutile (110) surface, with the Bi–O distances in the range of 2.17–2.18 Å. Furthermore, each Bi atom also coordinates to one oxygen from the nanocluster, which itself binds to a surface Ti atom, with the Bi–O distances of 2.07–2.15 Å and Ti–O distances of 2.03 Å.
On Bi6O9-rutile, the formation of the most stable oxygen vacancy results in small changes to the nanocluster structure. Two Bi cations are coordinated to the surface-bridging oxygen sites, with the Bi–O distances of 2.20–2.23 Å. Two nanocluster oxygen bind to the surface fivefold coordinated Ti, with the Ti–O distances of 1.89 and 2.02 Å, whereas after relaxation, four oxygen atoms take a twofold coordination environment by breaking the bonds with Bi cations (giving long Bi–O distances of 2.51 and 2.56 Å).
On Bi4O6-anatase, two Bi cations are threefold coordinated and the remaining cations are fourfold coordinated. The typical Bi–O distances range from 1.86 to 2.15 Å, and the Bi–O distances to the surface range from 2.12 to 2.16 Å. One oxygen atom from the surface migrates outward to bind to a Bi cation in the nanocluster, with three oxygen atoms in the nanocluster binding to the surface Ti sites; the Bi–O distances are in the range 1.86–1.90 Å.
The localization of the two electrons released upon removal of the neutral oxygen atom is shown by the spin density isosurfaces in Figure 3. For both Bi2O3 nanocluster-modified rutile (110) composites, the electrons preferentially localize on the Ti sites in the rutile (110) surface, with the computed Ti Bader charges of +1.65/1.71 electrons in Bi4O6-rutile and +1.65/1.69 electrons in Bi6O9-rutile and spin magnetization in the range 0.92–0.94 μB. For reference, Ti4+ cations have computed Bader charges of +1.30 electrons and zero spin magnetization. The first electron localizes on a subsurface Ti site and the second electron localizes onto a surface fivefold coordinated Ti site that has no interaction with the supported nanocluster. These values are typical of the reduced Ti3+ species in rutile (110).
In contrast, on Bi4O6-anatase (101), the two electrons localize on a surface fivefold coordinated Ti3+ site and on a Bi site in the nanocluster, which results in the formation of a reduced Bi2+ species. The computed Bader charges are +1.74 electrons on Ti and +2.99 electrons on Bi; the latter is comparable to a computed Bi Bader charge of +2.1 electrons for Bi3+ cations, indicating the reduction of the Bi species. The computed spin magnetizations on Ti and Bi are 0.96 and 0.7 μB. This Bi site binds to the oxygen that migrated out of the surface, and this particular oxygen bridges the two reduced Ti3+ and Bi2+ sites.
3.2. Adsorption of CO2 at Reduced Bi2O3–TiO2 Heterostructures
We now discuss the adsorption of CO2 at the Bi2O3 nanocluster-modified TiO2 heterostructures, and in this section we focus on the reduced heterostructures. We examined the interaction of the reduced Bi2O3–TiO2 heterostructures with CO2 in a number of adsorption configurations. Figure 5 shows the most stable relaxed structures after the relaxation of adsorbed CO2 on each reduced Bi2O3–TiO2 heterostructure. The computed interaction energies for CO2 are −0.45 eV on reduced Bi4O6-rutile (110), −0.53 eV on reduced Bi6O9-rutile (110), and −0.93 eV on reduced Bi4O6-anatase (101). These energies indicate a moderately strong interaction between the initially reduced metal oxide heterostructure and the CO2 molecule.
Figure 5.

Atomic structure of the most stable CO2 adsorption structures on reduced Bi2O3–TiO2 composites: (a) Bi4O6-rutile (110), (b) Bi6O9-rutile (110), and (c) Bi4O6-anatase (101). The color coding is the same as in Figure 1, with an additional inclusion of a gray sphere for carbon. The Bi and O atoms in the supported Bi2O3 nanoclusters are depicted with larger diameter spheres.
CO2 binds to reduced Bi4O6-rutile (110) with the formation of two Bi–O bonds, with the Bi–O distances of 2.45 and 2.52 Å. The carbon of CO2 binds to the oxygen in the nanocluster, with a C–O distance of 1.38 Å. The C–O distances in the adsorbed CO2 are elongated to 1.27 Å, and the O–C–O angle is 130°. Thus, the CO2 molecule adsorbs moderately exothermically on the reduced supported Bi4O6 nanocluster in a manner that can be considered carbonate-like with a tridentate adsorption configuration, although one of the C–O bonds is significantly longer than the typical C–O carbonate distance.
Examining the electronic structure in the CO2–Bi4O6-rutile system, the computed Bader charges on the previously reduced Ti atoms are now +1.3 electrons, signifying the presence of oxidized Ti4+ species. Interestingly, we find that the charge on two Bi cations in the nanocluster changes to 2.8 and 3.2 electrons, indicating that these Bi cations are partially reduced. One of these Bi cations binds to the oxygen of CO2, suggesting a rearrangement of charge upon CO2 adsorption.
On Bi6O9-rutile (110), the relaxation of initially adsorbed CO2 leads to spontaneous breaking of a C–O bond in the molecule, which results in the release of a free CO molecule, with no energy barrier to this process. Although one might expect a significant energy gain in breaking a C–O bond, we have to consider that this process heals the vacancy site on the Bi2O3 nanocluster, which has a high stability, and there would also be an energy cost to break the C–O bond in the molecule. In any case, the key point is the exothermic release of CO.
When CO is released, the previous supported Bi6O8 nanocluster is reoxidized, with the Bi6O9 composition. Examining the atomic structure, we see that the free CO has a C–O distance of 1.14 Å, which is typical of the gas-phase CO, and the Bi–O distances are similar to those in the stoichiometric Bi6O9-rutile system. In the Bi6O9-rutile system, the computed Bader charges of Ti and Bi are +1.3 and +2.2 electrons, consistent with the presence of only oxidized Ti4+ and Bi3+ species. The electrons on the TiO2 surface after reduction are therefore transferred to CO2 to allow the formation of new Bi–O bonds and a free CO molecule.
To examine this in more detail and complete a redox cycle, we adsorb H2 at the site of the oxygen species that results from CO2 dissociation, that is, oxygen that reoxidized reduced Bi6O8-rutile. Upon relaxation, a water molecule spontaneously forms, with an exothermic energy gain of 1.13 eV which reduces the Bi6O9 nanocluster; removal of water from the supercell requires 0.43 eV, and the reduced supported nanocluster is available for reaction with another CO2. Thus, the Bi6O9 nanocluster supported on rutile TiO2 appears to show a pathway for CO2 dissociation through a redox process involving water formation in a reverse water gas shift-like process.
Finally, on Bi4O6-anatase (101), the computed Bader charges on the CO2 molecule indicate a charge transfer of ca. 1.5 electrons to the molecule from the reduced composite. The computed Bader charges of 1.3 and 2.1 electrons on previously reduced Ti and Bi further support this electron transfer, which reoxidises Ti and Bi. This process then results in the formation and stabilization of a carboxyl, CO2– species. This is one of the activated CO2 species that is important in CO2 conversion. In the gas phase, the potential required for the formation of CO2– is prohibitively high, but in the reduced Bi2O3-modified anatase system, the presence of both low coordinated active sites and excess electrons after reduction appears to facilitate electron transfer to the adsorbed CO2 molecule. In adsorbed carboxyl, the C–O distances are 1.25 and 1.27 Å, again showing a significant elongation over the free molecule. The bending of CO2 upon interaction with the nanocluster is also apparent, with an O–C–O angle of 130°. CO2 binds to the nanocluster through a Bi–O bond, which has a distance of 2.42 Å and a Bi–C bond of 2.37 Å involving a second Bi atom in the nanocluster.
The computed vibrational frequencies for CO2 adsorbed on reduced heterostructures are shown in Table 1. The computed gas-phase vibrational modes of CO2 are 2354, 1325, and 632 cm–1, with the latter being degenerate. These correspond to the asymmetric C=O stretch, the symmetric C=O stretch, and the O–C–O bending mode. The large red shift in the C=O stretching mode of 760 and 767 cm–1 on Bi4O6-rutile and Bi4O6-anatase is consistent with the strong adsorption of CO2, as is the breaking of the degeneracy in the CO2 bending mode upon adsorption. We note that on Bi4O6-anatase the splitting of the degeneracy in the CO2 bending mode is larger, the origin of which is likely due to one oxygen of CO2 not binding with the Bi2O3 nanocluster. By contrast, the two oxygen atoms in CO2 bind with the nanocluster in Bi4O6-rutile, so that the split in the degeneracy of the two modes is smaller.
Table 1. Computed CO2 Vibrational Frequencies for Activated CO2 at Reduced Bi2O3–TiO2a.
| reduced Bi2O3–TiO2 heterostructure | CO2 vibrational frequencies/cm–1 |
|---|---|
| Rutile (110) | |
| Bi4O6-rutile (110) | 1594, 1254, 941, 774 |
| Bi6O9-rutile (110) | gas-phase CO |
| Anatase (101) | |
| Bi4O6-anatase (101) | 1587, 1203, 700, 531 |
Our computed vibrational frequencies of free CO2 are 2354, 1325, and 632 cm–1, which can be compared with the experimental data: 2349 cm–1 (C=O asymmetric stretch), 1388 cm–1 (C=O symmetric stretch), and 667 cm–1 (C=O bonding, degenerate).
In addition to the excess electrons introduced by reduction and the presence of Bi–O electronic states at the top of the valence band, the atomic structure of the supported Bi2O3 nanoclusters can facilitate CO2 adsorption because of the presence of low coordinated atomic sites and the flexibility in these supported nanoclusters that allows strong relaxations after the adsorption of molecules. We can also relate these findings to the work of Walker et al.44 on the Bi-containing pyrochlore. In this system, the surface is terminated by a Bi–O layer, similar to our Bi2O3 nanocluster-modified TiO2, and the Bi 6s/6p electronic states are mixed with O 2p states at the top of the valence band. Thus, the pyrochlore has a suitable electronic structure to permit interaction with CO2 and an atomic structure that also facilitates the interaction with CO2.
Given that the adsorption of CO2 is the crucial first step in the catalytic conversion of CO2, which is generally not favorable or could form highly stable carbonates on metal oxides, our finding that CO2 interacts moderately strongly with the reducible Bi2O3-modified TiO2 and can form CO or a carboxyl intermediate is a key result of this work.
3.3. CO2 Adsorption at Oxidized Bi2O3–TiO2 Heterostructures
Table 2 presents the computed adsorption energies of the two most stable adsorption configurations of CO2 at each oxidized Bi2O3–TiO2 heterostructure, and Figure 6 shows the relaxed atomic structures for these CO2 adsorption configurations. From Table 2, we can see that there is at least one moderately strong interacting CO2 adsorption configuration at each Bi2O3– TiO2 heterostructure, with the computed adsorption energies of −0.54, −1.01, and −0.82 eV on Bi4O6-rutile, Bi6O9-rutile, and Bi4O6-anatase, respectively. Compared to the computed adsorption energies of CO2 on other oxidized metal oxide-modified TiO2 systems, for example, in ref (47) or on polymorphs of TiO2 itself,10,30,31 the adsorption energies of CO2 on Bi2O3-modified TiO2 are notably stronger, indicating that these heterostructures are more favorable toward CO2 adsorption, although we note that there is no direct formation of CO or carboxylate, suggesting that the oxidized Bi2O3–TiO2 heterostructures may facilitate CO2 capture.
Table 2. Computed Adsorption Energies, in eV, of CO2 at Bi2O3-Modified Rutile (110) and Anatase (101) Heterostructures.
| adsorption configuration | Eads/eV |
|---|---|
| On Rutile (110) | |
| Bi4O6-rutile (110) + CO2 | –0.37 |
| Bi4O6-rutile (110) + CO2 | –0.54 |
| Bi6O9-rutile (110) + CO2 (linear) | –0.28 |
| Bi6O9-rutile (110) + CO2 | –1.01 |
| On Anatase (101) | |
| Bi4O6-anatase (101) + CO2 (linear) | –0.51 |
| Bi4O6-anatase (101) + CO2 | –0.82 |
Figure 6.

Relaxed adsorption structures of CO2 at Bi2O3-modified rutile (110) and anatase (101) composites; the composition is indicated in each case. (a,b) CO2 adsorption at Bi4O6-rutile (110) (c,d) CO2 adsorption at Bi6O9-rutile (110), and (e,f) CO2 adsorption at Bi4O6-anatase (101). The color coding is the same as in Figure 3.
Examining the atomic structures for the adsorbed CO2, we see that there are adsorption configurations on Bi6O9-rutile and Bi4O6-anatase, namely the configurations in Figure 6c,e, which have the weakest interactions and show a linear CO2 that is unchanged from the gas phase. This is a CO2 physisorption mode, which we do not expect to be important.
The other adsorption modes in Figure 6 are of more interest, and these show new C–O interactions with the supported nanoclusters, and the adsorption structure is consistent with a carbonate-like adsorption mode, albeit without the strong stabilization usually found for carbonate adsorption,64,65 and consistent with the CO2 adsorption on the Bi-oxide-terminated pyrochlore.44
Table 3 presents the important C–O distances, O–C–O bending angles, and the computed CO2 vibrational modes. The common feature of these adsorption modes of CO2 is that the molecule is clearly bent (gas-phase CO2 is linear, with an O–C–O angle of 180°). The O–C–O angles in the adsorbed CO2 are 131°, 127°, and 130° on Bi4O6-rutile, Bi6O9-rutile, and Bi4O6-anatase. This is consistent with the bending of activated CO2 observed on other materials, including transition-metal carbides, or on ceria-modified rutile (110).4,77,78
Table 3. Computed C–O and Bi–O Distances and CO2 Vibrational Frequencies for CO2 Adsorbed at Bi2O3-Modified Rutile (110) and Anatase (101)a.
| structure | C–O/Å | O–C–O angle/° | CO2 vibrational frequencies/cm–1 |
|---|---|---|---|
| Rutile (110) | |||
| Bi4O6-rutile (110) + CO2 | 1.27, 1.26, 1.39 | 131 | 1617, 1250, 924, 774 |
| Bi6O9-rutile (110) + CO2 | 1.26, 1.29, 1.39 | 127 | 1531, 1304, 1028, 774 |
| Anatase (101) | |||
| Bi4O6-anatase (101) + CO2 | 1.26, 1.27, 1.39 | 130 | 1593, 1243, 917, 790 |
The first two C–O distances in column 2 are in the adsorbed molecule and the third C–O distance is to oxygen in the Bi2O3 nanocluster. Our computed vibrational frequencies of free CO2 are 2354, 1325, and 632 cm–1, with the latter being degenerate.
On Bi4O6-rutile, the C–O distances in the adsorbed CO2 elongate to 1.26 and 1.27 Å, whereas the C–O distance to nanocluster oxygen is 1.39 Å. This is a notable elongation in the C–O distances over those in the gas-phase molecule (which is 1.16 Å). Similarly, on Bi6O9-rutile, the resulting bidentate adsorption configuration has elongated C–O distances of 1.26 and 1.29 Å in the molecule, whereas the C–O distance to the nanocluster is 1.39 Å. On Bi4O6-anatase, the carbon atom binds in a monodentate fashion to one oxygen in the nanocluster, with a C–O distance of 1.39 Å, whereas the C–O distances in the molecule are 1.26 and 1.27 Å.
Clearly, on Bi2O3-modified TiO2, although the CO2 molecule does not adsorb in a typical carbonate adsorption mode in which the three C–O distances are equal (∼1.29 Å) and the O–C–O angles are 120°, we can nonetheless describe the adsorption as carbonate-like. Although a new C–O interaction with a C–O distance of 1.39 Å is present, we can propose that this interaction participates in helping to stabilize the adsorbed CO2, so that it can further interact with hydrogen (see Section 3.3).
We have also computed the vibrational modes of adsorbed CO2 on Bi2O3-modified TiO2. Upon the adsorption of CO2 at the oxidized Bi2O3–TiO2 heterostructures, there are significant red shifts in the asymmetric C=O stretch of 737, 823, and 761 cm–1 on Bi4O6-rutile, Bi6O9-rutile, and Bi4O6-anatase, respectively. The degeneracy of the O–C–O bending mode is lifted upon adsorption at Bi2O3–TiO2. CO2 adsorbed on Bi6O9-rutile shows the largest red shift of the C=O stretch, which is consistent with its larger adsorption energy and distortions away from the gas-phase CO2 geometry. This shift in the C=O stretching mode correlates with the strength of the CO2–Bi2O3–TiO2 interaction, and it has been observed in the experimental studies of CO2 adsorption on a range of materials.79−81
Finally, we briefly examine the electronic structure. Figure 7 shows the PEDOS for C and O atoms in CO2, and Ti and Bi atoms, as the examples of CO2 adsorbed on Bi6O9-rutile and Bi4O6-anatase. In both cases, the CO2-derived C 2p and O 2p electronic states lie at the top of the valence band. Furthermore, there is a strong interaction between the Bi 6s/6p states and the carbon and oxygen 2p states of CO2. This is consistent with the strong interaction and distortion of CO2 when adsorbed at the supported Bi2O3 nanoclusters. We have also computed the Bader atomic charges upon CO2 adsorption. These do not show a significant charge transfer between Bi2O3–TiO2 and CO2, indicating that there is no charge transfer upon the adsorption of CO2.
Figure 7.
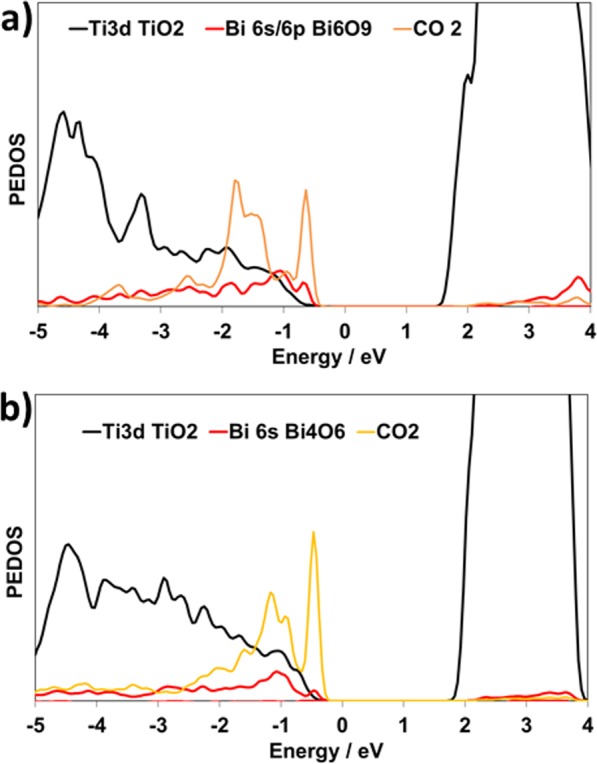
PEDOS projected onto the Ti 3d, Bi 6s/6p, and CO2 2p states after the adsorption of CO2 onto (a) Bi6O9-rutile and (b) Bi4O6-anatase.
In understanding the strong CO2 adsorption without any significant charge transfer, we consider the atomic and electronic structures of Bi2O3 nanocluster-modified TiO2. The nature of the valence band of Bi2O3–TiO2, with high-lying Bi2O3-derived electronic states, permits the interaction of these Bi–O electronic states with the C and O atoms of CO2. This can be considered similar to the case of the alkaline earth oxides, which show increased basicity and higher energy valence band states on going from Mg to Ca to Ba, and the interaction with CO2 is stronger with the increased basic character along this series.
3.4. Hydrogenation of Adsorbed CO2
Starting from the CO2 adsorption structures in Section 3.3, which show moderate adsorption energies and distortions to CO2 but no direct CO formation, we first examined the dissociation and formation of CO and adsorbed oxygen. However, such starting structures relax back to those already described, and if we remove CO, this is endothermic by up to 3 eV.
We have therefore examined the first steps in hydrogenation to explore if the adsorbed CO2 species can be hydrogenated. We focus on Bi4O6-rutile and Bi4O6-anatase, with the most favorable CO2 adsorption modes discussed above. We adsorb a hydrogen atom at the Bi2O3–TiO2 heterostructure to simulate the dissociation of H2 and the formation of a hydroxyl on the nanocluster. We then use the climbing image NEB method with three images to determine the activation barrier for H migration, from which either a formate or carboxyl species can be produced.
Figure 8a,b shows the structure of the H atom adsorbed as a hydroxyl on Bi4O6-rutile and Bi4O6-anatase. The energy gain relative to 1/2 H2 (in gas phase) is 1.42 eV on Bi4O6-rutile and 0.63 eV on Bi4O6-anatase. These are moderate energy gains for hydrogen adsorption. Upon H adsorption at Bi4O6-rutile, the O–H distance is 0.98 Å, and the Bi–O distances involving this oxygen are 2.25 and 2.44 Å. On Bi4O6-anatase, the O–H distance is 1.01 Å, and the Bi–O distances for this oxygen are 2.31 and 2.18 Å.
Figure 8.
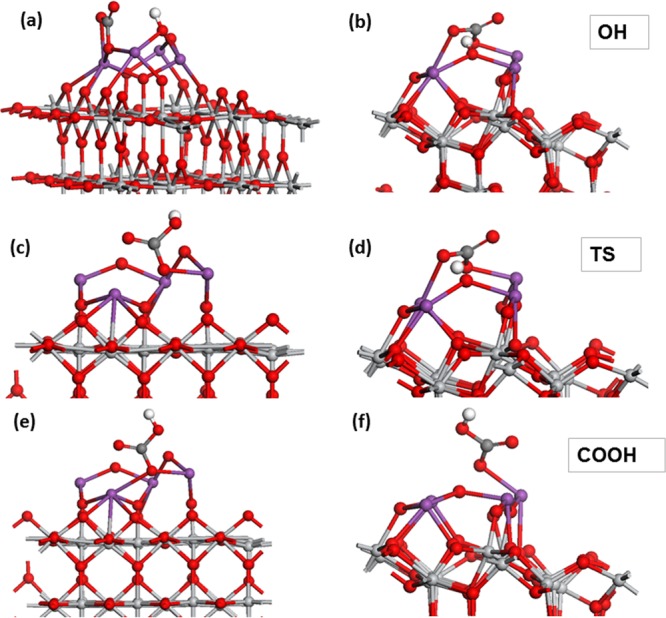
(a) Atomic structure of a hydroxyl species on Bi4O6-rutile with adsorbed CO2. Hydrogen is denoted by the white sphere. (b) Atomic structure of a hydroxyl species on Bi46O6-anatase with adsorbed CO2. (c) Atomic structure of the transition state for H migration to adsorbed CO2 on Bi4O6-rutile. (d) Atomic structure of the transition state for H migration to adsorbed CO2 on Bi4O6-anatase. (e) Atomic structure of a COOH intermediate on Bi4O6-rutile. (f) Atomic structure of a COOH intermediate on Bi4O6-anatase.
The computed barrier for H migration from the hydroxyl to oxygen in CO2 to form a carboxyl intermediate is 0.77 eV on Bi4O6-rutile (110) and 0.68 eV on Bi4O6-anatase (101). These are moderate barriers for the H transfer to the adsorbed CO2. The atomic structure of the transition state is shown in Figure 8c,d. In the transition state on Bi4O6-rutile, the H atom has migrated away from the nanocluster toward the oxygen of the adsorbed CO2. On Bi4O6-anatase, the transition state involves the H atom migrating away from the oxygen of the nanocluster by 0.25 Å toward CO2.
The atomic structure of the carboxyl intermediate on Bi4O6-rutile and Bi4O6-anatase is shown in Figure 8e,f. For the COOH intermediates, the C=O, C–O(H), OH, and C–O(Bi) distances are presented in Table 4. We also examined the formation of a formate intermediate. However, formate either lies higher by ca. 3 eV, on Bi4O6-rutile, or is not stable, relaxing to COOH on Bi4O6-anatase, and thus the COOH intermediate is preferred. If we introduce a second hydrogen atom to make COOH2, this always relaxes to water and CO2, removing oxygen from the Bi2O3 nanocluster, and thus we conclude that although CO2 can adsorb at the oxidized Bi2O3–TiO2 nanoclusters, it is difficult to hydrogenate to useful products.
Table 4. Computed C=O, C–O(H), OH, and C–O(Bi) Distances in COOH Intermediates on Bi4O6-Rutile and Bi4O6-Anatase in Å.
| Bi4O6-rutile (110) | Bi4O6-anatase (101) | ||
|---|---|---|---|
| C=O | 1.24 | C=O | 1.24 |
| C–O(H) | 1.35 | C–O(H) | 1.37 |
| OH | 0.98 | OH | 0.98 |
| C–O(Bi) | 1.31 | C–O(Bi) | 1.31 |
4. Conclusions
We have studied the interaction and activation of CO2 at the novel heterostructure of Bi2O3 nanoclusters supported on rutile and anatase TiO2 surfaces. Previous experimental work has demonstrated that Bi-containing materials can activate and convert CO2 to more useful molecules, but the origin of this is not yet clear.
The Bi2O3–TiO2 heterostructures can be reduced, with moderate energy costs, which should be possible through a thermochemical process or by the addition of hydrogen. This results in the reduction of Ti sites, and Bi sites in the nanocluster can also be reduced. The interaction of the reduced heterostructures with CO2 can result in direct exothermic CO formation, or charge transfer to CO2 (reoxidizing Bi and Ti cations), to give an activated carboxylate species. The CO can be removed, and the addition of hydrogen is an exothermic process that forms water and reduces the nanocluster. This suggests a promising approach for CO2 conversion via CO.
We find that CO2 adsorbs moderately strongly at the oxidized Bi2O3–TiO2 heterostructures, with the computed adsorption energies ranging from −0.54 to 1.01 eV; van der Waals interactions simply shift these energies by ca. 0.15 eV, but do not change the overall trends. CO2 adsorbs in a carbonate-like adsorption mode (but is not overstabilized), with no charge transfer to the molecule. Finally, the computed vibrational modes of the adsorbed CO2 show a significant red shift of over 750 cm–1 in the C=O stretching mode and a breaking of the degeneracy in the O–C–O bending mode. The magnitude of the red shift in the C=O stretching mode correlates with the strength of the adsorption of CO2. The position of the high-lying valence band states derived from Bi3+–O interactions appear to drive the activation of CO2.
We find that the CO2 adsorbed in this fashion can be hydrogenated to a COOH intermediate, with the migration barriers from the nanocluster to the adsorbed CO2 of less than 0.8 eV, whereas CO2 dissociation is not favorable. However, further hydrogenation of COOH does not result in any useful products, and we conclude that reduction is the key to the activation and subsequent conversion of CO2 on Bi2O3-modified TiO2 heterostructures.
These results show that the unique properties of bismuth-containing oxides can be exploited to adsorb and activate carbon dioxide, which is the key first step in the reductive conversion of CO2 to useful molecules. The combination of a non-bulk-like nanocluster-containing Bi3+ species, which is reducible, provides an interesting material that can activate CO2, whether that be by strong adsorption and distortion, electron transfer to form a carboxylate, or by direct breaking of a C–O bond, and we propose that these heterostructures can be synthesized and tested for CO2 activation.
Acknowledgments
Financial Support from Science Foundation Ireland through the Starting Investigator Research Grant Project EMOIN SFI/09/SRIG/I2160 and the Science Foundation Ireland US-Ireland R&D Partnership Program Project SusChem SFI/US/14/E2915 is gratefully acknowledged. Access to the computational resources at the Science Foundation Ireland/Higher Education Authority funded Irish Center for High End Computing is acknowledged. We are grateful for support from the COST Action CM1104 “Reducible Metal Oxides, Structure and Function”. We acknowledge important discussions with Profs. K. A. Gray, E. Weitz, and J. A. Byrne.
The author declares no competing financial interest.
References
- Chang X.; Wang T.; Gong J. CO2 photo-reduction: insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. 10.1039/c6ee00383d. [DOI] [Google Scholar]
- Mishra A. K.; Roldan A.; de Leeuw N. H. CuO Surfaces and CO2 Activation: A Dispersion-Corrected DFT+U Study. J. Phys. Chem. C 2016, 120, 2198–2214. 10.1021/acs.jpcc.5b10431. [DOI] [Google Scholar]
- Kwon S.; Liao P.; Stair P. C.; Snurr R. Q. Alkaline-earth metal-oxide overlayers on TiO2: application toward CO2 photoreduction. Catal. Sci. Technol. 2016, 6, 7885–7895. 10.1039/c6cy01661h. [DOI] [Google Scholar]
- Kunkel C.; Viñes F.; Illas F. Transition metal carbides as novel materials for CO2 capture, storage, and activation. Energy Environ. Sci. 2016, 9, 141–144. 10.1039/c5ee03649f. [DOI] [Google Scholar]
- Peng C.; Reid G.; Wang H.; Hu P. Perspective: Photocatalytic reduction of CO2 to solar fuels over semiconductors. J. Chem. Phys. 2017, 147, 030901. 10.1063/1.4985624. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Pan F.; Li Y. A review on the effects of TiO 2 surface point defects on CO 2 photoreduction with H 2 O. J. Materiomics 2017, 3, 17–32. 10.1016/j.jmat.2016.12.001. [DOI] [Google Scholar]
- Song C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing, Adsorption and Chemical Processing. Catal. Today 2006, 115, 2–32. 10.1016/j.cattod.2006.02.029. [DOI] [Google Scholar]
- Roy S. C.; Varghese O. K.; Paulose M.; Grimes C. A. Toward solar fuels: photocatalytic conversion of carbon dioxide to hydrocarbons. ACS Nano 2010, 4, 1259–1278. 10.1021/nn9015423. [DOI] [PubMed] [Google Scholar]
- Mikkelsen M.; Jørgensen M.; Krebs F. C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. 10.1039/b912904a. [DOI] [Google Scholar]
- Habisreutinger S. N.; Schmidt-Mende L.; Stolarczyk J. K. Photocatalytic Reduction of CO2on TiO2and Other Semiconductors. Angew. Chem., Int. Ed. 2013, 52, 7372–7408. 10.1002/anie.201207199. [DOI] [PubMed] [Google Scholar]
- Goeppert A.; Czaun M.; Jones J.-P.; Surya Prakash G. K.; Olah G. A. Recycling of carbon dioxide to methanol and derived products - closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. 10.1039/c4cs00122b. [DOI] [PubMed] [Google Scholar]
- Varghese O. K.; Paulose M.; LaTempa T. J.; Grimes C. A. High-Rate Solar Photocatalytic Conversion of CO2and Water Vapor to Hydrocarbon Fuels. Nano Lett. 2009, 9, 731–737. 10.1021/nl803258p. [DOI] [PubMed] [Google Scholar]
- Varghese O. K.; Paulose M.; LaTempa T. J.; Grimes C. A. High-Rate Solar Photocatalytic Conversion of CO2and Water Vapor to Hydrocarbon Fuels. Nano Lett. 2010, 10, 750. 10.1021/nl903742x. [DOI] [PubMed] [Google Scholar]
- Durand W. J.; Peterson A. A.; Studt F.; Abild-Pedersen F.; Nørskov J. K. Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces. Surf. Sci. 2011, 605, 1354–1359. 10.1016/j.susc.2011.04.028. [DOI] [Google Scholar]
- Mishra A. K.; Roldan A.; de Leeuw N. H. A density functional theory study of the adsorption behaviour of CO2 on Cu2O surfaces. J. Chem. Phys. 2016, 145, 044709. 10.1063/1.4958804. [DOI] [PubMed] [Google Scholar]
- Uzunova E. L.; Seriani N.; Mikosch H. CO2 conversion to methanol on Cu(i) oxide nanolayers and clusters: an electronic structure insight into the reaction mechanism. Phys. Chem. Chem. Phys. 2015, 17, 11088–11094. 10.1039/c5cp01267h. [DOI] [PubMed] [Google Scholar]
- Graciani J.; Mudiyanselage K.; Xu F.; Baber A. E.; Evans J.; Senanayake S. D.; Stacchiola D. J.; Liu P.; Hrbek J.; Sanz J. F.; Rodriguez J. A. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550. 10.1126/science.1253057. [DOI] [PubMed] [Google Scholar]
- Wu H.; Zhang N.; Wang H.; Hong S. Adsorption of CO2 on Cu2O (111) oxygen-vacancy surface: First-principles study. Chem. Phys. Lett. 2013, 568–569, 84–89. 10.1016/j.cplett.2013.03.032. [DOI] [Google Scholar]
- Bendavid L. I.; Carter E. A. CO2 Adsorption on Cu2O(111): A DFT+U and DFT-D Study. J. Phys. Chem. C 2013, 117, 26048–26059. 10.1021/jp407468t. [DOI] [Google Scholar]
- Liu L.; Zhao C.; Li Y. Spontaneous Dissociation of CO2 to CO on Defective Surface of Cu(I)/TiO2-x Nanoparticles at Room Temperature. J. Phys. Chem. C 2012, 116, 7904–7912. 10.1021/jp300932b. [DOI] [Google Scholar]
- Wu H.; Zhang N.; Cao Z.; Wang H.; Hong S. The adsorption of CO2, H2CO3, HCO3– and CO32– on Cu2O (111) surface: First-principles study. Int. J. Quantum Chem. 2012, 112, 2532–2540. 10.1002/qua.23250. [DOI] [Google Scholar]
- Grabow L. C.; Mavrikakis M. Mechanism of Methanol Synthesis on Cu through CO2and CO Hydrogenation. ACS Catal. 2011, 1, 365–384. 10.1021/cs200055d. [DOI] [Google Scholar]
- Wang L.; Gupta K.; Goodall J. B. M.; Darr J. A.; Holt K. B. In situ spectroscopic monitoring of CO2 reduction at copper oxide electrode. Faraday Discuss. 2017, 197, 517–532. 10.1039/c6fd00183a. [DOI] [PubMed] [Google Scholar]
- Favaro M.; Xiao H.; Cheng T.; Goddard W. A.; Yano J.; Crumlin E. J. Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 6706–6711. 10.1073/pnas.1701405114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A. A.; Abild-Pedersen F.; Studt F.; Rossmeisl J.; Nørskov J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. 10.1039/c0ee00071j. [DOI] [Google Scholar]
- Wu H.; Zhang N.; Wang H.; Hong S. Adsorption of CO2 on Cu2O (111) oxygen-vacancy surface: First-principles study. Chem. Phys. Lett. 2013, 568–569, 84–89. 10.1016/j.cplett.2013.03.032. [DOI] [Google Scholar]
- Gao D.; Zegkinoglou I.; Divins N. J.; Scholten F.; Sinev I.; Grosse P.; Roldan Cuenya B. Plasma-activated copper nanocube catalysts for efficient carbon dioxide electroreduction to hydrocarbons and alcohols. ACS Nano 2017, 11, 4825–4831. 10.1021/acsnano.7b01257. [DOI] [PubMed] [Google Scholar]
- Mistry H.; Varela A. S.; Bonifacio C. S.; Zegkinoglou I.; Sinev I.; Choi Y.-W.; Kisslinger K.; Stach E. A.; Yang J. C.; Strasser P.; Cuenya B. R. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 2016, 7, 12123. 10.1038/ncomms12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhade S. A.; Luo W.; Nie X.; Asthagiri A.; Janik M. J. Theoretical insight on reactivity trends in CO2 electroreduction across transition metals. Catal. Sci. Technol. 2016, 6, 1042–1053. 10.1039/c5cy01339a. [DOI] [Google Scholar]
- Indrakanti V. P.; Kubicki J. D.; Schobert H. H. Photoinduced activation of CO2 on TiO2 surfaces; Quantum chemical modeling of CO2 adsorption on oxygen vacancies. Fuel Process. Technol. 2011, 92, 805–811. 10.1016/j.fuproc.2010.09.007. [DOI] [Google Scholar]
- Pipornpong W.; Wanbayor R.; Ruangpornvisuti V. Adsorption of CO2 on the perfect and oxygen vacancy defect surfaces of anatase TiO2 and its photocatalytic mechanism of conversion to CO. Appl. Surf. Sci. 2011, 257, 10322–10328. 10.1016/j.apsusc.2011.06.013. [DOI] [Google Scholar]
- Lee D.; Kanai Y. Role of four-fold coordinated titanium and quantum confinement in CO2 reduction at titania surface. J. Am. Chem. Soc. 2012, 134, 20266–20269. 10.1021/ja309871m. [DOI] [PubMed] [Google Scholar]
- Yin W.-J.; Wen B.; Bandaru S.; Krack M.; Lau M. W.; Liu L.-M. The effect of excess electron and hole on CO2 adsorption and activation on rutile (110) surface. Sci. Rep. 2016, 6, 23298. 10.1038/srep23298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-T.; Wood B. C.; Bhethanabotla V. R.; Joseph B. CO2 Adsorption on Anatase TiO2 (101) Surfaces in the Presence of Subnanometer Ag/Pt Clusters: Implications for CO2 Photoreduction. J. Phys. Chem. C 2014, 118, 26236–26248. 10.1021/jp509219n. [DOI] [Google Scholar]
- Behrens M.; Studt F.; Kasatkin I.; Kuhl S.; Havecker M.; Abild-Pedersen F.; Zander S.; Girgsdies F.; Kurr P.; Kniep B.-L.; Tovar M.; Fischer R. W.; Norskov J. K.; Schlogl R. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. 10.1126/science.1219831. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhao J.; Wang T.; Li Y.; Li X.; Yin J.; Wang C. CO2 photoreduction with H2O vapor on highly dispersed CeO2/TiO2 catalysts: Surface species and their reactivity. J. Catal. 2016, 337, 293–302. 10.1016/j.jcat.2015.12.030. [DOI] [Google Scholar]
- Graciani J.; Sanz J. F. Designing a new generation of catalysts: Water gas shift reaction example. Catal. Today 2015, 240, 214–219. 10.1016/j.cattod.2014.03.071. [DOI] [Google Scholar]
- Park J. B.; Graciani J.; Evans J.; Stacchiola D.; Senanayake S. D.; Barrio L.; Liu P.; Sanz J. F.; Hrbek J.; Rodriguez J. A. Gold, Copper, and Platinum Nanoparticles Dispersed on CeOx/TiO2(110) Surfaces: High Water-Gas Shift Activity and the Nature of the Mixed-Metal Oxide at the Nanometer Level. J. Am. Chem. Soc. 2009, 132, 356–363. 10.1021/ja9087677. [DOI] [PubMed] [Google Scholar]
- Park J. B.; Graciani J.; Evans J.; Stacchiola D.; Ma S.; Liu P.; Nambu A.; Sanz J. F.; Hrbek J.; Rodriguez J. A. High catalytic activity of Au/CeOx/TiO2(110) controlled by the nature of the mixed-metal oxide at the nanometer level. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4975–4980. 10.1073/pnas.0812604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMeglio J. L.; Rosenthal J. Selective Conversion of CO2 to CO with High Efficiency Using an Inexpensive Bismuth-Based Electrocatalyst. J. Am. Chem. Soc. 2013, 135, 8798–8801. 10.1021/ja4033549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Ramos J.; DiMeglio J. L.; Rosenthal J. Efficient Reduction of CO2 to CO with High Current Density Using in Situ or ex Situ Prepared Bi-Based Materials. J. Am. Chem. Soc. 2014, 136, 8361–8367. 10.1021/ja501923g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Ramos J.; Pupillo R. C.; Keane T. P.; DiMeglio J. L.; Rosenthal J. Efficient Conversion of CO2 to CO Using Tin and Other Inexpensive and Easily Prepared Post-Transition Metal Catalysts. J. Am. Chem. Soc. 2015, 137, 5021–5027. 10.1021/ja5121088. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Chi M.; Veith G. M.; Zhang P.; Lutterman D. A.; Rosenthal J.; Overbury S. H.; Dai S.; Zhu H. Rational Design of Bi Nanoparticles for Efficient Electrochemical CO2 Reduction: The Elucidation of Size and Surface Condition Effects. ACS Catal. 2016, 6, 6255–6264. 10.1021/acscatal.6b01297. [DOI] [Google Scholar]
- Walker R. J.; Pougin A.; Oropeza F. E.; Villar-Garcia I. J.; Ryan M. P.; Strunk J.; Payne D. J. Surface Termination and CO2 Adsorption onto Bismuth Pyrochlore Oxides. Chem. Mater. 2016, 28, 90–96. 10.1021/acs.chemmater.5b03232. [DOI] [Google Scholar]
- Walsh A.; Payne D. J.; Egdell R. G.; Watson G. W. Stereochemistry of post-transition metal oxides: revision of the classical lone pair model. Chem. Soc. Rev. 2011, 40, 4455–4463. 10.1039/c1cs15098g. [DOI] [PubMed] [Google Scholar]
- Walsh A.; Watson G. W. Polymorphism in Bismuth Stannate: A First-Principles Study. Chem. Mater. 2007, 19, 5158–5164. 10.1021/cm0714279. [DOI] [Google Scholar]
- Fronzi M.; Daly W.; Nolan M. Reactivity of metal oxide nanocluster modified rutile and anatase TiO 2 : Oxygen vacancy formation and CO 2 interaction. Appl. Catal., A 2016, 521, 240–249. 10.1016/j.apcata.2015.11.038. [DOI] [Google Scholar]
- Nolan M.; Iwaszuk A.; Lucid A. K.; Carey J. J.; Fronzi M. Design of Novel Visible Light Active Photocatalyst Materials: Surface Modified TiO2. Adv. Mater. 2016, 28, 5425–5446. 10.1002/adma.201504894. [DOI] [PubMed] [Google Scholar]
- Fronzi M.; Iwaszuk A.; Lucid A.; Nolan M. Metal oxide nanocluster-modified TiO2 as solar activated photocatalyst materials. J. Phys.: Condens. Matter 2016, 28, 074006. 10.1088/0953-8984/28/7/074006. [DOI] [PubMed] [Google Scholar]
- Tada H.; Jin Q.; Iwaszuk A.; Nolan M. Molecular-scale transition metal oxide nanocluster surface-modified titanium dioxide as solar-activated environmental catalysts. J. Phys. Chem. C 2014, 118, 12077–12086. 10.1021/jp412312m. [DOI] [Google Scholar]
- Iwaszuk A.; Lucid A. K.; Razeeb K. M.; Nolan M. First principles investigation of anion-controlled red shift in light absorption in ZnX (X = O, S, Se) nanocluster modified rutile TiO2. J. Mater. Chem. A 2014, 2, 18796–18805. 10.1039/c4ta03334e. [DOI] [Google Scholar]
- Iwaszuk A.; Nolan M. SnO-nanocluster modified anatase TiO2 photocatalyst: exploiting the Sn(ii) lone pair for a new photocatalyst material with visible light absorption and charge carrier separation. J. Mater. Chem. A 2013, 1, 6670–6677. 10.1039/c3ta10647k. [DOI] [Google Scholar]
- Iwaszuk A.; Nolan M.; Jin Q.; Fujishima M.; Tada H. Origin of the visible-light response of nickel(II) oxide cluster surface modified titanium(IV) dioxide. J. Phys. Chem. C 2013, 117, 2709–2718. 10.1021/jp306793r. [DOI] [Google Scholar]
- Jin Q.; Fujishima M.; Iwaszuk A.; Nolan M.; Tada H. Loading effect in copper(II) oxide cluster-surface-modified titanium(IV) oxide on visible- and UV-light activities. J. Phys. Chem. C 2013, 117, 23848–23857. 10.1021/jp4085525. [DOI] [Google Scholar]
- Jin Q.; Fujishima M.; Nolan M.; Iwaszuk A.; Tada H. Photocatalytic Activities of Tin(IV) Oxide Surface-Modified Titanium(IV) Dioxide Show a Strong Sensitivity to the TiO2 Crystal Form. J. Phys. Chem. C 2012, 116, 12621–12626. 10.1021/jp302493f. [DOI] [Google Scholar]
- Nolan M. First-Principles Prediction of New Photocatalyst Materials with Visible-Light Absorption and Improved Charge Separation: Surface Modification of Rutile TiO2 with Nanoclusters of MgO and Ga2O3. ACS Appl. Mater. Interfaces 2012, 4, 5863–5871. 10.1021/am301516c. [DOI] [PubMed] [Google Scholar]
- Iwaszuk A.; Nolan M. Reactivity of sub 1 nm supported clusters: (TiO2)n clusters supported on rutile TiO2 (110). Phys. Chem. Chem. Phys. 2011, 13, 4963–4973. 10.1039/c0cp02030c. [DOI] [PubMed] [Google Scholar]
- Nolan M. Surface modification of TiO2 with metal oxide nanoclusters: a route to composite photocatalytic materials. Chem. Commun. 2011, 47, 8617–8619. 10.1039/c1cc13243a. [DOI] [PubMed] [Google Scholar]
- Nolan M. Electronic coupling in iron oxide-modified TiO2 leads to a reduced band gap and charge separation for visible light active photocatalysis. Phys. Chem. Chem. Phys. 2011, 13, 18194–18199. 10.1039/c1cp21418g. [DOI] [PubMed] [Google Scholar]
- Rhatigan S.; Nolan M. Impact of surface hydroxylation in MgO-/SnO-nanocluster modified TiO2 anatase (101) composites on visible light absorption, charge separation and reducibility. Chin. Chem. Lett. 2017, 29, 757–764. 10.26434/chemrxiv.5447101. [DOI] [Google Scholar]
- Lucid A.; Iwaszuk A.; Nolan M. A first principles investigation of Bi2O3-modified TiO2 for visible light Activated photocatalysis: The role of TiO2 crystal form and the Bi3+ stereochemical lone pair. Mater. Sci. Semicond. Process. 2014, 25, 59–67. 10.1016/j.mssp.2014.01.005. [DOI] [Google Scholar]
- Iwaszuk A.; Nolan M. Lead oxide-modified TiO2 photocatalyst: tuning light absorption and charge carrier separation by lead oxidation state. Catal. Sci. Technol. 2013, 3, 2000–2008. 10.1039/c3cy00194f. [DOI] [Google Scholar]
- Schwartzenberg K. C.; Hamilton J. W. J.; Lucid A. K.; Weitz E.; Notestein J.; Nolan M.; Byrne J. A.; Gray K. A. Multifunctional photo/thermal catalysts for the reduction of carbon dioxide. Catal. Today 2017, 280, 65–73. 10.1016/j.cattod.2016.06.002. [DOI] [Google Scholar]
- Nolan M. Alkaline earth metal oxide nanocluster modification of rutile TiO2 (110) promotes water activation and CO2 chemisorption. J. Mater. Chem. A 2018, 6, 9451–9466. 10.1039/c8ta01789a. [DOI] [Google Scholar]
- Rhatigan S.; Nolan M. CO2 and water activation on ceria nanocluster modified TiO2 rutile (110). J. Mater. Chem. A 2018, 6, 9139–9152. 10.1039/c8ta01270a. [DOI] [Google Scholar]
- Kresse G.; Hafner J. Ab initiomolecular dynamics for liquid metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47, 558–561. 10.1103/physrevb.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab initiomolecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 49, 14251–14269. 10.1103/physrevb.49.14251. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of Ab-initio Total Energy Calculations for Metals and Semiconductors using a Plane-wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758–1775. 10.1103/physrevb.59.1758. [DOI] [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953–17979. 10.1103/physrevb.50.17953. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Chevary J. A.; Vosko S. H.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B: Condens. Matter Mater. Phys. 1992, 46, 6671–6687. 10.1103/physrevb.46.6671. [DOI] [PubMed] [Google Scholar]
- Anisimov V. I.; Zaanen J.; Andersen O. K. Band theory and Mott insulators: HubbardUinstead of StonerI. Phys. Rev. B: Condens. Matter Mater. Phys. 1991, 44, 943–954. 10.1103/physrevb.44.943. [DOI] [PubMed] [Google Scholar]
- Dudarev S. L.; Botton G. A.; Savrasov S. Y.; Humphreys C. J.; Sutton A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B: Condens. Matter Mater. Phys. 1998, 57, 1505–1509. 10.1103/physrevb.57.1505. [DOI] [Google Scholar]
- Iwaszuk A.; Mulheran P. A.; Nolan M. TiO2 nanocluster modified-rutile TiO2 photocatalyst: a first principles investigation. J. Mater. Chem. A 2013, 1, 2515–2525. 10.1039/c2ta01582j. [DOI] [Google Scholar]
- Ha M.-A.; Alexandrova A. N. Oxygen vacancies of anatase(101): extreme sensitivity to the density functional theory method. J. Chem. Theory Comput. 2016, 12, 2889–2895. 10.1021/acs.jctc.6b00095. [DOI] [PubMed] [Google Scholar]
- Vidal A. B.; Feria L.; Evans J.; Takahashi Y.; Liu P.; Nakamura K.; Illas F.; Rodriguez J. A. CO2 Activation and Methanol Synthesis on Novel Au/TiC and Cu/TiC Catalysts. J. Phys. Chem. Lett. 2012, 3, 2275–2280. 10.1021/jz300989e. [DOI] [PubMed] [Google Scholar]
- Posada-Pérez S.; Viñes F.; Ramirez P. J.; Vidal A. B.; Rodriguez J. A.; Illas F. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 2014, 16, 14912–14921. 10.1039/c4cp01943a. [DOI] [PubMed] [Google Scholar]
- Schneider W. F. Qualitative Differences in the Adsorption Chemistry of Acidic (CO2, SOx) and Amphiphilic (NOx) Species on the Alkaline Earth Oxides. J. Phys. Chem. B 2004, 108, 273–282. 10.1021/jp036323+. [DOI] [Google Scholar]
- Jensen M. B.; Pettersson L. G. M.; Swang O.; Olsbye U. CO2Sorption on MgO and CaO Surfaces: A Comparative Quantum Chemical Cluster Study. J. Phys. Chem. B 2005, 109, 16774–16781. 10.1021/jp052037h. [DOI] [PubMed] [Google Scholar]
- Duan Y.; Sorescu D. C. CO2 capture properties of alkaline earth metal oxides and hydroxides: A combined density functional theory and lattice phonon dynamics study. J. Chem. Phys. 2010, 133, 074508. 10.1063/1.3473043. [DOI] [PubMed] [Google Scholar]