

Abstract

Promoting the production of industrially important aromatic chloroamines over transition-metal nitrides catalysts has emerged as a prominent theme in catalysis. This contribution provides an insight into the reduction mechanism of p-chloronitrobenzene (p-CNB) to p-chloroaniline (p-CAN) over the γ-Mo2N(111) surface by means of density functional theory calculations. The adsorption energies of various molecularly adsorbed modes of p-CNB were computed. Our findings display that, p-CNB prefers to be adsorbed over two distinct adsorption sites, namely, Mo-hollow face-centered cubic (fcc) and N-hollow hexagonal close-packed (hcp) sites with adsorption energies of −32.1 and −38.5 kcal/mol, respectively. We establish that the activation of nitro group proceeds through direct pathway along with formation of several reaction intermediates. Most of these intermediaries reside in a significant well-depth in reference to the entrance channel. Central to the constructed mechanism is H-transfer steps from fcc and hcp hollow sites to the NO/–NH groups through modest reaction barriers. Our computed rate constant for the conversion of p-CNB correlates very well with the experimental finding (0.018 versus 0.033 s–1 at ∼500 K). Plotted species profiles via a simplified kinetics model confirms the experimentally reported high selectivity toward the formation of p-CAN at relatively low temperatures. It is hoped that thermokinetics parameters and mechanistic pathways provided herein will afford a molecular level understanding for γ-Mo2N-mediated conversion of halogenated nitrobenzenes into their corresponding nitroanilines; a process that entails significant industrial applications.
1. Introduction
Aromatic haloamine compounds are important intermediates that are extensively deployed in the manufacturing of various prominent chemicals, most notably, drugs, pesticides, and pigments.1 The main preparation routes of these amines utilize metal–acid systems or selective hydrogenation to the analogous haloanilines (HANs) over noble-metal catalysts such as platinum and palladium.2−4 However, several parameters have hindered a mass-scale utilization of these approaches in the production of HANs. These factors encompass high operating cost and low selectivity toward the formation of HANs. The latter is mainly attributed to hydrodehalogenation reactions that also result in the formation of aniline (AN) and nitrobenzene (NB).5 Various factors may contribute to the catalytic hydrogenation of Mo2N-based materials. The selectivity underpinning reaction kinetics toward the synthesis of the desired haloamine is greatly influenced by the synthesis conditions, most notably, the Mo and N-precursors and other operational conditions (deployed solvents, temperature, and pressure).1,6,7 The catalyst-assisted hydrogenation reduction of aromatic molecules, in which molecular hydrogen plays a vital role as a reducing agent, has attained considerable interest. This is owing to the generation of less quantities of acid wastes when compared with homogeneous chemical reduction reactions (i.e., the traditional Bechamp reaction) or with the metal–acid system.8 In general, surface-mediated reduction of haloamines outperforms homogeneous gas phase and acid-based reduction based on generating less acidic wastes.9 Additionally, the production of hydrogen chloride (HCl) from the hydrodechlorination reaction affects the catalytic activity of the reactor.10 Thus, it is essential to improve heterogeneous catalysts that enforce the occurrence of selective hydrogenation of the nitro groups to the corresponding amino groups.
Recent related developments have decreased the rate of inhibiting the hydrodehalogenation during the reduction of halonitrobenzene via the use of a variety of metal catalysts. For instance, an activated carbon decorated with Pd nanoparticles of a size larger than 25 nm in a 500 mL reactor was found to be highly selective toward the hydrogenation of halogenated nitrobenzene of about 99.90%.11 Ma et al. investigated the influence of using Ir nanoparticles as a support in activated carbon catalyst (Ir/C) for the hydrogenation of halogenated nitrobenzene. They reported that high selectivity (>99%) to p-chloroaniline (p-CAN) was accomplished with a particle size of (<3 nm) Ir nanoparticles.12 Furthermore, a study of gas-phase hydrogenation of ortho-chloronitrobenzene (o-CNB) to ortho-chloroaniline over Pd/Al2O3 catalyst promoted by several alkali metals was reported by Vishwanathan et al.13 It was revealed that the selectivity of the reduction of o-CNB depends on the polarity of the N=O bond in the −NO2 group.
However, the use of noble metals such as Pt and Pd is undesirable due to the high cost and low catalytic stability, in which active sites could be readily blocked under high pressure of deployed hydrogen.14 It is well established that transition-metal nitride catalysts exhibit high catalytic performance in an array of hydrogenation reactions. The partial hydrogenation of acetylene over two unsupported phases of β-Mo2N and γ-Mo2N has recently been investigated.15
These synthesized catalysts were prepared by the temperature-programmed reaction of MoO3 with N2 + H2. It was found that high selectivity (77–90%) toward ethylene formation could be attained over both β-Mo2N and γ-Mo2N crystallite phases. However, trivial amount of green oil formation has been detected by X-ray photoelectron spectroscopy over γ-Mo2N in particular.
Cárdenas-Lizana et al.16 stated that a 100% selectivity of haloanilines was obtained during liquid-phase hydrogenation of p-chloronitrobenzene (p-CNB), over the crystal-phase Mo nitride catalysts. Furthermore, they performed a reaction over alumina-supported palladium as a benchmark catalyst. Nonselective behavior toward the production of nitrobenzene (NB) and aniline (AN) from a combined hydrodechlorination/hydrogenation has been detected. In another experimental work carried out under atmospheric pressure at 493 K by the same group, p-CAN was the only intermediate from the −NO2 group’s reduction via a reaction rate constant of k = 2.0 min–1.17 From theoretical point of view, a computational study reported adsorption energies and reaction barriers for the elementary steps involved in the p-CNB reduction paths over two sites of Pd catalysts, namely, terrace and step.18 Their findings suggested that the reaction barriers of the involved reactions are about the same over Pd(111) and Pd(211) surfaces. However, to the best of our knowledge, this contribution is the first to explore the reaction pathways encountered during the p-CNB reduction over the transition-metal nitride surface γ-Mo2N(111) via the state-of-the-art density functional theory (DFT) calculations.
Routes for the formation of several reaction intermediates such as chloronitrosobenzene (CNSB), chlorophenylhydroxylamine (CPHA), dichloroazoxybenzene (CAOB), dichloroazobenzene (CAB), and dichlorohydrazobenzene (CHAB) have comprehensively been reported. These intermediates are highly toxic and resist oxidative decomposition, and, thus, their formation should be minimized. The accumulation of these compounds on the surface of the catalyst generally hinders the formation of p-CAN.5 This study has a 2-fold aim: (i) to report reaction pathways for the reduction mechanism and (ii) to predict kinetics parameters for all scanned steps. Thermokinetic parameters reported herein should be instrumental in the pursuit to formulate novel γ-Mo2N-based catalyst tailored for specific industrial applications pertinent to sustainable chemical treatment of halonitrobenzenes.
2. Computational Details
All geometrical optimizations and energy and vibrations calculations were carried out by the DMol3 code19 using the Perdew–Burke–Ernzerhof generalized gradient approximation as the exchange–correlation functional.20 Localized double-numerical basis sets along with polarization functions21 afford the atomic environments. A Monkhorst–Pack k-points mesh of (1 × 3 × 1) was used in the integration of the irreducible part of the Brillion zone. In all calculations, we set the thermal smearing at 0.01 Hartree. The convergence thresholds for energy, forces, and displacements were 2 × 10–5 Ha, 4 × 10–3 Ha/Å, and 5 × 10–4 Å, respectively, along with a global cutoff of 4.2 Å. Dispersion correction term (DFT-D2) has also been implemented.22 The γ-Mo2N(111) surface as reported in our previous work23 was found to be the most energetically stable plane based on the ab initio atomistic thermodynamic approach. This plane was constructed by a periodic four-layer slab with a (2 × 2) supercell and a vacuum layer of 12 Å to avoid interactions between periodic configurations along the z-direction. During the calculations, all the layers and the adsorbates were allowed to fully relax. A test on one reaction (i.e., two structures) using a global cutoff at 5.0 Å and 1 × 5 × 1 k-points mesh changed its reaction energy only marginally by ∼4%.
The transition states (TSs) were derived by the linear synchronous and quadratic synchronous transit method.24 Vibrational frequencies yield activation enthalpies and entropies as a function of selected temperatures based on the Arrhenius formula: k(T) = A exp(−Ea/RT). The two kinetic parameters (the A-factor and activation energy, Ea) were estimated by fitting reaction rate constants, k(T), with the inverse of temperature (1/T) according to the conventional transition-state theory (TST).25
We calculated the adsorption energies of γ-Mo2N/species systems according to
where Esurface+species refers to the energy of the studied system and Esurface+fixed species denotes the energy of a noninteracting system in which the chemical species is fixed in the middle of the slab separated from the surface by at least 6 Å. Finally, the activation barriers represent the difference in energy between optimized transition structures and reactants in each investigated step.
3. Results and Discussion
3.1. Molecular Adsorption of p-Chloronitrobenzene Over the γ-Mo2N(111) Surface
While searching for the most energy preferable adsorption site of p-CNB over the γ-Mo2N(111) surface, various orientations have been examined. The nature of the interaction of the p-CNB molecule is primarily based on the position in which the phenylene and nitro groups contribute in the adsorption process. Consequently, the role of orientations in the molecular adsorbates via the phenylene (flat) and nitro (upright) groups were often regarded to be of prominence.4 Attempts to optimize a physisorbed state along multiple rotations always converge to either a vertical or a horizontal structure. The physisorbed energies of p-CNB over the γ-Mo2N(111) surface are listed in Table 1, whereas the side and top views of all the feasible geometries are shown in Figure 1. In the A1 configuration, the physisorption of p-CNB occurs when both O atoms of the nitro group are directly adsorbed at the top of two Mo atoms via double Mo–O bonding with an average distance of 2.15 Å, yielding an adsorption energy of −27.5 kcal/mol. The structure of A2 configuration depicted in Figure 1 infers that when the phenylene group is positioned flat to the 3-fold face-centered cubic (fcc) (i.e., molybdenum vacant site), the adsorption occurs only through a single Mo–O bond with a distance of 2.3 Å with an associated adsorption energy of −32.1 kcal/mol.
Table 1. Energies and Adsorption Sites for the p-CNB Molecule Over the γ-Mo2N(111) Surface.
| adsorption configuration | adsorption description | Eads (kcal/mol) |
|---|---|---|
| A1 | molecular p-CNB adsorbs in an upright position at bridge site | –27.5 |
| A2 | molecular p-CNB adsorbs in a flat posture at 3-fold fcc site (underneath Mo atom) | –32.1 |
| A3 | molecular p-CNB adsorbs in a flat position at 3-fold hcp site (underneath N atom) | –22.0 |
| A4 | phenylene group p-CNB adsorbs in a flat position on-top of surface N atom | –28.8 |
| A5 | nitro group of p-CNB adsorbs vertically on 3-fold hcp site | –38.5 |
| A6 | phenylene group of p-CNB adsorbs parallel on N-top site | –29.1 |
| A7 | nitro p-CNB adsorbs vertically on Mo-bridge site | –4.2 |
Figure 1.

Top and side views of the optimized configurations of adsorbed p-CNB molecule over the γ-Mo2N(111) surface. Light blue: Mo; dark blue: N; red: O; gray: C; white: H; and green: Cl. (The same color code applies throughout the article.)
When the p-CNB molecule is positioned horizontally and above the 3-fold hexagonal close-packed (hcp) nitrogen vacant site, another adsorption structure can be obtained, namely, the A3 configuration. In this configuration, all of the C atoms of the phenylene group are bounded to the surface Mo atoms through atomic distances of ∼2.26 Å with an adsorption energy of −22.0 kcal/mol. A4 and A6 structures entail very similar adsorption energies of −28.8 and −29.1 kcal/mol, respectively, in which the p-CNB is placed at the top of a surface N atom. In the A5 structure, the nitro group is linked to the surface via two O atoms at the top of two neighboring Mo atoms, i.e., the phenylene group is perpendicularly positioned above the γ-Mo2N(111) plane. The formed two Mo–O bonds amount to 2.27 Å, with an estimated adsorption energy of −38.5 kcal/mol, indicating genuine chemical bonds. However, when the nitro group was initially situated upright at the top of a surface nitrogen atom (A7), the p-CNB molecule directly desorbed with a height of ∼4.01 Å following full optimization. This could be attributed to the repulsion between the two negatively charged N and O entities. This phenomenon incurs the highest adsorption energy of only −4.2 kcal/mol. In a comparison with other systems, DFT calculations were utilized to estimate the adsorption energies of p-CAN over several Pd surfaces (including steeped, flat) and clusters. Adsorption energies were found to reside in the range of −39.2 to −61.11 kcal/mol.26
The overall aim of the hydrohalogenation process is to convert the nitro group (−NO2) of p-CNB into an amine group (−NH2) while leaving the chlorine atom at the para site of the phenyl ring. Careful inspection of the geometries in Figure 1 reveals that the horizontal-like adsorbed A2 structure is a structurally suitable to undergo hydrogenation of the nitro group. This adsorption mode is most likely to happen on the catalyst surface because it affords the lowest adsorption energy. Furthermore, the flat-like configuration of A2 makes it more geometrically oriented toward surface-mediated hydrogenation of its nitro group. For these reasons, we elect to consider the A2 configuration as the initial structure in the hydrohalogenation mechanism of the p-CNB molecule.
3.2. Mechanisms of Chloronitrobenzene Hydrogenation to Chloroaniline
Literature presents various reaction routes for the hydrogenation of p-CNB molecule into p-CAN over a wide array of potential catalysts. Herein, guided by the experimental results16,27 and in view of the previously suggested analogous mechanisms,12,18 we investigate the reduction mechanism of p-CNB into p-CAN mediated by the γ-Mo2N(111) surface. To survey plausible formation of side products, various routes were inspected, depending on which bond is easier to break, the C–Cl bond or the N=O bond.28Scheme 1 depicts the proposed pathways for the surface-assisted conversion of p-CNB into p-CAN. The direct reduction mechanism proceeds via the formation of nitroso and hydroxylamine compounds (via mechanism A). However, throughout condensation–reduction, the formation of azoxybenzene and azobenzene intermediates (mechanism C) takes place. Furthermore, the formation of the undesired toxic intermediates (i.e., azoxybenzene and azobenzene) must be suppressed during the hydrogenation mechanism. Further hydrogenolysis of the substituted position in the parent p-CNB molecule or intermediates leads to the formation of aniline (AN) and nitrobenzene (NB),29 although not to a large extent.
Scheme 1. Proposed Reaction Pathways for the Hydrogenation of p-CNB to p-CAN.

Bold arrows refer to the direct reduction (mechanism A), dashed arrows depict (mechanism B), whereas plain arrows represent the condensation reduction routes (mechanism C).
3.2.1. Direct Dissociation Pathways
Reduction of p-CNB to p-CAN involves subsequent fission of N–O bond by which chloronitrosobenzene (CNSB) and chlorophenylhydroxylamine (CPHA) compounds are formed as byproducts. Two possible pathways feature hydrogen-induced detachment reactions (i.e., mechanisms A and B), whereas a third potential mechanism involves a self-condensation route (mechanism C). Acquiring detailed potential energy surfaces for these three alternate routes determines the most kinetically preferred route for the experimentally observed products of the hydrogenation process.
3.2.1.1. Mechanism A
It is very experimentally challenging to determine to what extent the prehydrogenation of p-CNB contributes to the N–O bond rupture in reference to thermal fission process (i.e., noncatalytic process). The calculated adsorption energy of the p-CNB molecule over prehydrogenated surface was −7.3 kcal/mol. This value is substantially lower than the corresponding adsorption energy of the p-CNB molecule over the neat surface at −22.0 kcal/mol (for the A3 structure later). However, based on the facile process dictating the dissociative adsorption of hydrogen molecules,30 it is expected that active sites of the Mo2N(111) surface are prehydrogenated at the temperature of the experiment.
This has motivated us to investigate the dissociation pathway in which the N–O bond is cleaved directly without the involvement of adsorbed hydrogen atoms. Figure 2 displays the potential energy profile for the direct N–O bond fission. The calculated reaction energy for the reaction ClC6H4NO2* (D1) → ClC6H4NO* + O* is slightly exothermic by 2.8 kcal/mol; nevertheless, it associated with a sizable energy barrier of 47.7 kcal/mol via the transition state structure (TSD). Clearly, such a high barrier hinders the occurrence of the direct surface-assisted N–O bond scission at the experimental temperature of 493 K.16 This finding correlates with the high barrier computed (37.1 kcal/mol) for the corresponding direct fission of the N–O bonds in the case of nitrobenzene (C6H5NO2) reduction to aniline (C6H5NH2) over Pt catalyst.31
Figure 2.
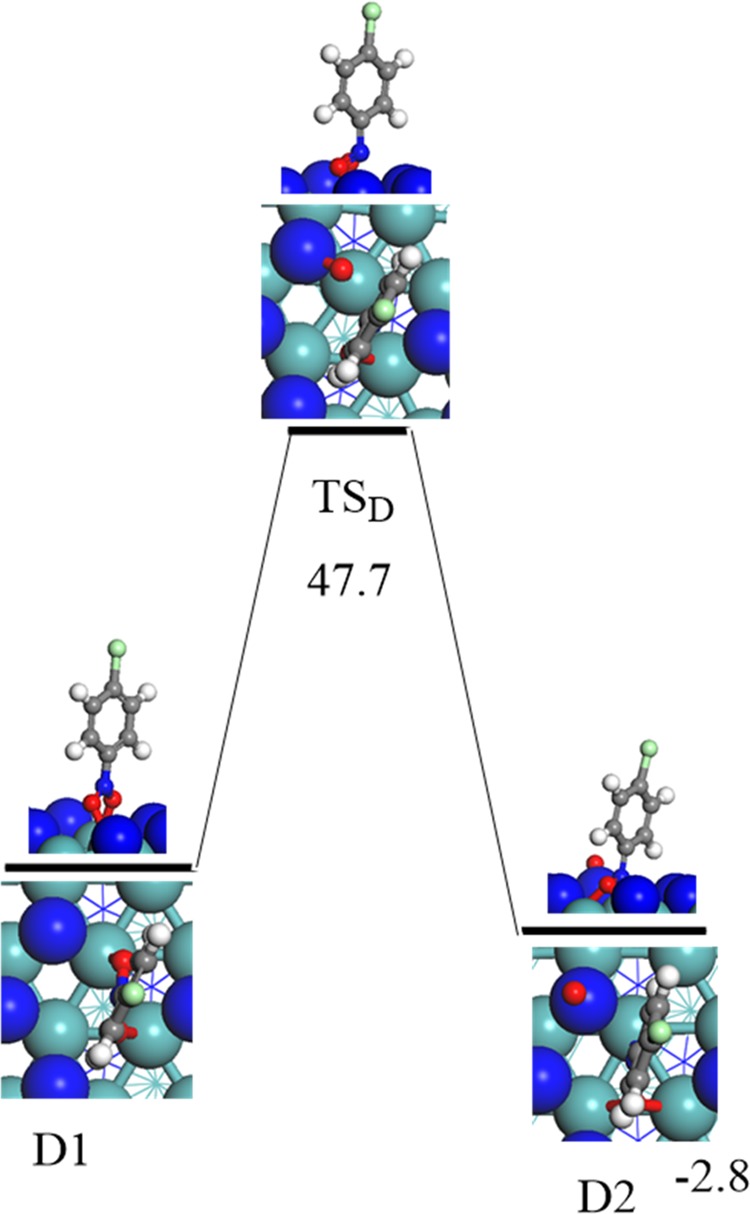
Reaction energy and activation barrier (in kcal/mol) for the direct fission of the N–O bond.
Figure 3 shows the structures, reaction, and activation energies for all the steps involved in Mechanism A, whereas Figure 4 depicts the corresponding potential energy surface in reference to the initial reactant. The direct dissociation pathway via hydrogen transfer from the surface proceeds by several elementary steps initiated by hydrogenating the first O atom of the nitro group (−NO2). This process weakens the relatively strong N–O bond (70.7 kcal/mol)32 and thus facilitates the subsequent N–O bond dissociations. Hydrogen-transfer reactions from different adsorption sites, i.e., top of N atom and 3-fold hcp vacant site have been investigated. We found that hydrogen transfer from the N top site requires a significantly higher barrier in reference to the analogous transfer from the 3-fold hcp N-hollow site, 13.3 versus 25.3 kcal/mol. The dependency of the catalytic hydrogenation capacity of Mo2N largely depends on the existence of nitrogen vacant sites, as we demonstrated in our previous work on the selective hydrogenation of acetylene to ethene over Mo2N surfaces.33 Starting with the first atomic H* transfer to the O atom in p-CNB via the reaction (1) ClC6H4NO2* (M1) + H* → ClC6H4NO2H*, the computed reaction energy was found to be 8.1 kcal/mol above the entrance channel. The corresponding activation barrier amounts to 13.3 kcal/mol characterized by the transition structure TS1 as shown in Figure 3.
Figure 3.

Structures of the intermediates and transition states for the steps involved in the conversion of p-CNB to p-CAN on γ-Mo2N(111) (mechanisms A (black) and B (red) arrows). Activation barriers and reaction energies are computed in reference to reactants in each step. All values are in kcal/mol.
Figure 4.

Potential energy surface for the conversion of p-CNB to p-CAN on γ-Mo2N(111) (mechanisms A (black) and B (red) dotted lines). All energies are in reference to M1.
To demonstrate the catalytic capacity of the γ-Mo2N(111), we have considered the homogeneous uncatalyzed first step hydrogenation step of the NO site in the parent p-CBN molecule. H transfer from a hydrogen molecule to the NO site was found to incur a sizable barrier of 59.2 kcal/mol. This value significantly overshoots the analogous catalyzed H transfer step at 13.3 kcal/mol.
Upon the hydrogenation of the first N–O bond into N–OH, its length stretches from 1.25 to 1.36 Å, marking its activation. Subsequent to this step, the second reaction (2A) ClC6H4NO2H* (M2) + H* → ClC6H4NO* + H2O* proceeds with an energy barrier of 23.1 kcal/mol (TS2A) forming adsorbed chloronitrosobenzene (CNSB) ClC6H4NO* as an intermediate and water molecule. This reaction is exothermic with energy of 42.0 kcal/mol (M3A). Desorption of H2O* from surface into a gas-phase water molecule requires only 10.0 kcal/mol. Although this path would expedite the deoxygenation in comparison with the direct N–O fission route (23.1 versus 47.7 kcal/mol), further hydrogenation is necessary to complete the catalytic cycle into p-CAN. We accordingly presumed that, after water desorption, the initial configuration of ClC6H4NO* and the adsorbed atomic H remain in the same initial state as in reaction (2A).
In the transition structure TS3A (reaction (3A) ClC6H4NO* (M3A) + H* → ClC6H4NOH*), an adsorbed H* migrates into the oxygen atom in the ClC6H4NO* radical. This step results in the formation of the adsorbed ClC6H4NOH (M4A), in which the N–O bond elongates by 19% in reference to the analogous distance in M3A. This reaction is exothermic by 8.9 kcal/mol, with a modest hydrogenation barrier of 14.2 kcal/mol via TS3A. Subsequent hydrogen transfer into the adsorbed ClC6H4NOH* moiety via reaction (R4A), ClC6H4NOH* (M4A) + H* → ClC6H4NHOH* (M5) affords the adduct moiety of ClC6H4NHOH (M5). This step ensues in an exothermic reaction by 12.0 kcal/mol and occurs via an activation barrier of 25.3 kcal/mol characterized by TS4A. In the next step (R5), a water molecule is eliminated through the movement of a surface H* atom into the hydroxyl group of the M5 adduct. The activation barrier for this step amounts to 16.2 kcal/mol via TS5.
Finally, an adsorbed p-CAN molecule is produced through the attachment of an H* atom to the NH group in ClC6H4NH* (M6) as revealed in reaction (6). The reaction ClC6H4NH* (M6) + H* → ClC6H4NH2* (M7) completes the reduction mechanism of p-CNB into p-CAN. This final H* transfer is accomplished through the transition-state structure that signifies a reaction barrier of 12.5 kcal/mol (TS6). The adsorbed p-CAN in the M7 configuration resides in a well-depth of 28.3 kcal/mol in reference to M6. Consequently, it is inferred that the hydrogenation step of chlorophenylhydroxylamine (CPHA, M5) in reaction (R4A) incurs the highest activation barrier and is most likely to resemble the rate-limiting step. The overall reaction in mechanism A, as given in Figure 4, is exothermic by 20.2 kcal/mol.
3.2.1.2. Mechanism B
This mechanism differs from mechanism A with regard to the nature of reactions (2)–(4). Figure 3 represents the structure of elementary reactions in the hydrogenation of p-CNB (mechanism B, in red arrows). Figure 4 depicts the corresponding potential energy surface. In reaction (2B) ClC6H4NO2H* (M2) + H* → ClC6H4NO2H2*, the second O’s atom of the nitro group is hydrogenated via another hydrogen transfer step termed as the double H-induced dissociation, in which the nitro group is doubly hydrogenated to N(OH)2 group before the dissociation of the first N–OH bond. This step requires an energy barrier of 19.0 kcal/mol (TS2B). The reaction energy of the new synthesized intermediate ClC6H4NO2H2* (M3B) is found to be slightly endothermic by 3.3 kcal/mol. After double hydrogenation of the nitro group, the hydrogen transfer from the 3-fold hcp vacant site to the hydroxyl’s group in M3B liberates a water molecule, in analogous reactions to those illustrated in mechanism A, i.e., via the reaction (3B) ClC6H4NO2H2* (M3B) + H* → ClC6H4NOH* + H2O*. One of the OH group in the ClC6H4NO2H2* intermediate departs the M3B moiety simultaneously with the movement of an adsorbed H* atom forming adsorbed ClC6H4NOH* (M4B) adduct, as characterized by the transition structure TS3B via a very sizable activation barrier of 55.8 kcal/mol. This elementary step appears to be exothermic by 23.0 kcal/mol. The desorption energy of released water molecule is predicted to be 18.9 kcal/mol, leaving behind an adsorbed ClC6H4NOH* adduct. The next step (ClC6H4NOH* (M4B) + H* → ClC6H4NHOH* (M5)) features a hydrogen transfer into the OH group via TS4B. Barrier for this step stands at 38.7 kcal/mol. Comparing the activation energies required for the corresponding steps in both mechanisms A and B, it is obvious that the activation energy of the mechanism A is generally lower than that in the analogous steps in mechanism B (23.1/14.2/25.3 versus 19.0/55.8/38.7 kcal/mol); nonetheless, the barrier for the opening step in mechanism B is slightly lower than that of mechanism A (i.e., 23.1 versus 19.0 kcal/mol). By reaching the M5 intermediate, the rest of the steps are the same as in mechanism A.
3.2.2. Aniline Formation via Indirect Reaction Pathway (i.e., Condensation Pathway, Mechanism C)
Another expected reduction route proceeds through the formation of dichloroazoxybenzene (CAOB) as an important intermediate in the surface-assisted coupling of ClC6H4NO* (the structure M3A in mechanisms A). The structure of dichloroazobenzene (Cl2C12H8N2*) (CAB) is generated by another hydrogen transfer reaction of dichloroazoxybenzene (Cl2C12H8N2O*), (CAOB). Furthermore, dichlorohydrazobenzene (Cl2C12H8N2H2*) (CHAB) is the final moiety before the production of p-CAN by breaking the azo bonds. The latter term refers to the (−N=N−) bond in which two halophenylene rings are linked. Figure 5 displays the configurations involved in the condensation pathways that constitute unimolecular and bimolecular reactions of mechanism C. Figure 6 depicts the potential energy surface for the condensation pathway. Table 2 lists the activation barriers and the reactions energies for all the elementary steps involved in the condensation pathway.
Figure 5.

Potential energy surface for the condensation pathway (mechanism C). Activation barriers and reaction energies are computed in reference to reactants in each step. All values are in kcal/mol.
Figure 6.

Potential energy surface for the condensation pathway (mechanism C). Values are in reference to the initial reactant M1C.
Table 2. Calculated Activation Energies (Ea) and Reaction Energies (ΔE) of Steps Involved in the Condensation Pathway.
| reaction | reaction scheme | Ea (kcal/mol) | ΔE (kcal/mol) |
|---|---|---|---|
| (1C) | ClC6H4NO* + ClC6H4NHOH* → Cl2C12H8N2O* + H2O* dichloroazoxybenzene (CAOB) | 53.3 | –29.0 |
| (2C) | Cl2C12H8N2O* + H* → Cl2C12H8N2OH* | 69.6 | 58.4 |
| (3C) | Cl2C12H8N2OH* + H* → Cl2C12H8N2* + H2O* dichloroazobenzene (CAB) | 54.6 | –20.7 |
| (4C) | Cl2C12H8N2* + H* → Cl2C12H8N2H* | 60.2 | –32.6 |
| (5C) | Cl2C12H8N2H* + H* → Cl2C12H8N2H2* dichlorohydrazobenzene (CHAB) | 23.0 | –19.4 |
| (6C) | Cl2C12H10N2* + H* → ClC6H4NH2* + ClC6H4NH* | 58.7 | –47.1 |
| (7C) | ClC6H4NH2* + ClC6H4NH* + H* → 2ClC6H4NH2* | 34.6 | 24.5 |
The first four reactions are exactly the same as in mechanisms A and B. Thus, we commence tracking mechanism C by the step that leads to the generation of chlorophenylhydroxylamine (structure M2C Figure 5) via the presence of adjacent preadsorbed ClC6H4NO* and ClC6H4NHOH* adducts (M1C). In the structure of M2C formed via the reaction (ClC6H4NO* + ClC6H4NHOH* (M1C) → ClC12H8N2O* + H2O*), an azo bond accompanies the elimination of a water molecule. The formation of the dichloroazoxybenzene (CAOB) (ClC12H8N2O*) intermediate is exothermic by 29.0 kcal/mol. Most intermediates in the condensation pathway are linked with the surface via bonding O atoms with a Mo surface site. Desorption of H2O into a gas-phase water molecule requires a desorption energy of 29.2 kcal/mol. Although this coupling reaction is noticeably exothermic, it demands a sizable barrier of 53.3 kcal/mol (TS1C). Clearly, such a high barrier presents a bottleneck for the condensation pathway and indicates that the condensation route is highly unlikely to proceed kinetically via flat placed molecules. The barrier of TS1C is in well accord with the analogous barrier for the corresponding step encountered in the condensation pathway over the Pd3/Pt(111) surface (i.e., 52.12 kcal/mol).34 Considering the near-flat initial ClC6H4NO* + ClC6H4NHOH* structures, the activation barrier of TS1C is increased by ∼8.0 kcal/mol compared with the near-vertical structures depicted in Figure 5.
The reduction reaction of dichloroazoxybenzene (M2C) in the reaction (R2C) Cl2C12H8N2O* (M2C) + H* → Cl2C12H8N2OH* was found to be endothermic by 58.4 kcal/mol (M3C), with a very sizable activation energy 69.6 kcal/mol via (TS2C). This high barrier is attributed to the energy required for breaking the surface Mo–O bond in the M2C configuration. Upon hydrogen transfer, the N–O bond is stretched by 0.10 Å in reference to the N–O bond in the M2C structure. A third hydrogen transfer reaction generates dichloroazobenzene (CAB) (M4C) via the reaction (R3C): Cl2C12H8N2OH* (M3C) + H* → Cl2C12H8N2* + H2O*. This step proceeds via a considerable activation barrier of 54.6 kcal/mol through (TS3C) in reference to the M3C intermediate. The formed dichloroazobenzene (M4C) consequently reacts with an adsorbed H* atom, resulting in the formation of the Cl2C12H8N2H* intermediate (M5C) through reaction (R4C). This step requires an energy barrier of 60.2 kcal/mol via transition state structure (TS4C) and releases an excess energy of about −32.6 kcal/mol. The next step in the proposed mechanism is a reaction (R5C) in which dichlorohydrazobenzene moiety can be generated as a result of hydrogen transfer from 3-fold fcc vacant site. This reaction was found to be exothermic reaction by 19.4 kcal/mol (M6C), with an estimated barrier of 23.0 kcal/mol as represented in the transition structure (TS5C). Fragmentation of dichlorohydrazobenzene into adsorbed p-CAN molecule and ClC6H4NH* radical proceeds by the attachment of hydrogen atom to the N atom in dichlorohydrazobenzene reaction (R6C). This reaction is highly exothermic by 47.1 kcal/mol and necessitates a high activation energy of about 58.7 kcal/mol over M7C through the transition structure TS6C, correspondingly. A p-CAN molecule forms via attachment of a surface H* with the NH group in the ClC6H4NH* moiety (M7C) along the reaction (R7C) with an activation barrier of 34.6 kcal/mol (TS7C). All in all, the high activation barriers for these reactions could be attributed to the strong N–O and N–N bonds that are broken during the course of the mechanism.
Finally, it is of interest to shed some light on the barrier for a plausible chlorination of p-CNB to nitrobenzene (NB). We therefore investigate the dechlorination reactions of p-CNB into nitrobenzene via the fission of the C–Cl bond (ClC6H4NO2* → C6H4NO2* + Cl*). Figure 7 portrays the optimized structures involved in this reaction. Our findings indicate that the direct fission of the Cl–C bond is associated with a high energy barrier of 58.0 kcal/mol. This high barrier supports the experimental observation of Perret et al.,27 who tested two crystallographic phases of molybdenum nitride, namely, β-Mo2N and γ-Mo2N. They concluded that both the studied crystals promoted the selective nitro group reduction into amino group and suppressed the dehalogenation step.
Figure 7.
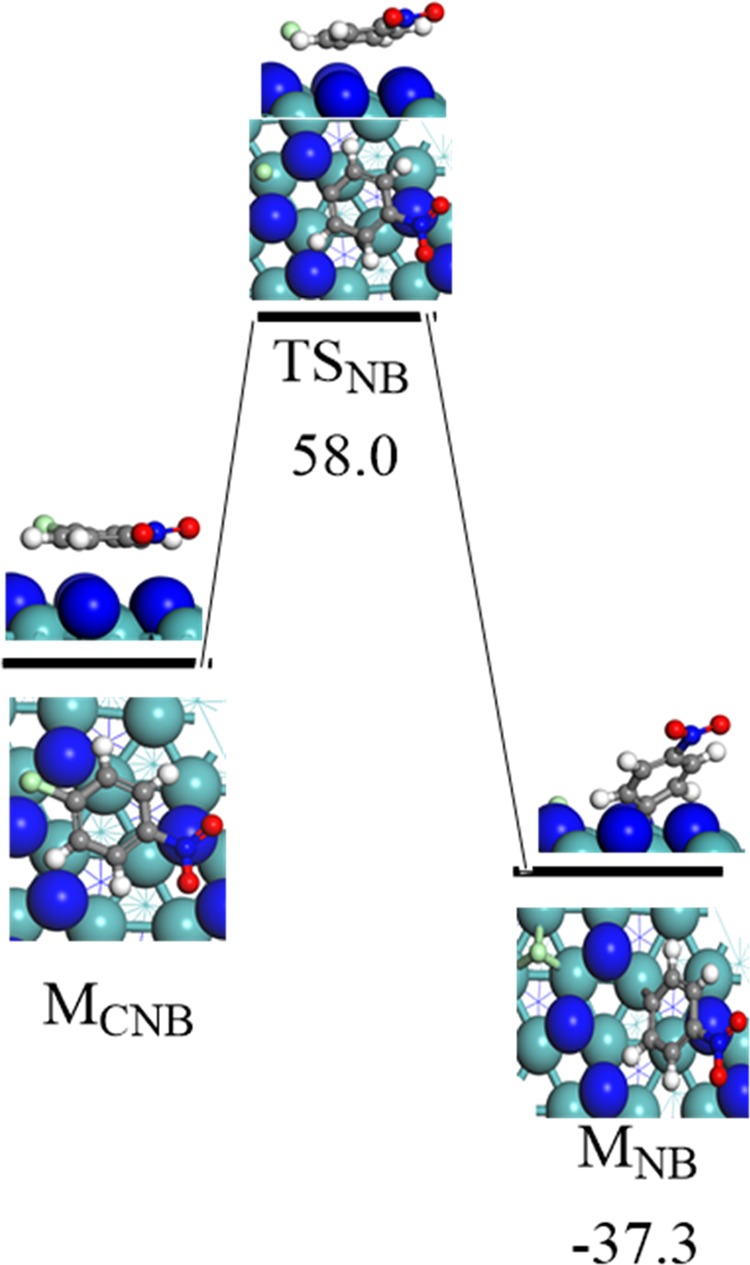
Reaction energy and activation barrier for the optimized geometries involved in the surface-mediated cleavage of the C–Cl bond in p-CNB. Values are in kcal/mol.
3.3. Reaction Rates and Kinetics
Both β and γ phases of Mo2N display similar adsorption sites. The unit cells for both surfaces only differ in their c-lattice, and, hence, our calculated kinetics for the γ phase should resemble the experimental values for the γ phase.35 We predict the thermokinetic parameters for the hydrogenation reaction of the p-CNB molecule in the molybdenum nitride system. Table 3 reports the Arrhenius parameters and the activation barriers for the reactions in mechanisms A and B, whereas Figure 8 demonstrates the Arrhenius plots for reactions in the forward directions involved in direct dissociation routes. A useful feature of the reaction rate parameters provided in Table 3 is to assess the relative importance of the potentially competing reactions. For instance, based on the reaction rate constants reported in Table 3, reaction (R2A) largely predominates the competing reaction (R2B).
Table 3. Fitted Kinetic Parameters for Forward and Reverse Surface Reactions Involved in the Reduction Mechanism of Chloronitrobenzene Over the γ-Mo2N(111) Surface.
| reaction | reaction scheme | A (s–1) | Ea (kcal/mol) | |
|---|---|---|---|---|
| M1 → M2 | R1f | ClC6H4NO2* + H* → ClC6H4NO2H* | 8.6 × 1012 | 12.7 |
| M2 → M1 | R1r | ClC6H4NO2H* → ClC6H4NO2* + H* | 3.3 × 1016 | 3.6 |
| M2 → M3A | R2f | ClC6H4NO2H* + H* → ClC6H4NO* + H2O* | 2.4 × 1015 | 24.0 |
| M3A → M2 | R2r | ClC6H4NO* + H2O* → ClC6H4NO2H* + H* | 1.7 ×1011 | 64.7 |
| M3A → M4A | R3f | ClC6H4NO* + H* → ClC6H4NOH* | 7.9 × 108 | 15.0 |
| M4A → M3A | R3r | ClC6H4NOH* → ClC6H4NO* + H* | 1.0 × 1011 | 22.6 |
| M2 → M3B | R2f | ClC6H4NO2H* + H* → ClC6H5NO2H2* | 7.7 × 1015 | 15.6 |
| M3B → M2 | R2r | ClC6H5NO2H2* → ClC6H4NO2H* + H* | 8.14 × 1015 | 7.6 |
| M3B → M4B | R3f | ClC6H4NO2H2* + H* → ClC6H4NOH* + H2O* | 1.0 × 1013 | 54.9 |
| M4B → M3B | R3r | ClC6H4NOH* + H2O* → ClC6H4NO2H2* + H* | 9.9 × 1011 | 71.2 |
| M4 → M5 | R4f | ClC6H4NOH* + H* → ClC6H4NHOH* | 3.6 × 1012 | 37.0 |
| M5 → M4 | R4r | ClC6H4NHOH* → ClC6H4NOH* + H* | 3.7 × 1014 | 45.0 |
| M5 → M6 | R5f | ClC6H4NHOH* + H* → ClC6H4NH + H2O* | 4.3 × 1012 | 15.8 |
| M6 → M5 | R5r | ClC6H4NH* + H2O* → ClC6H4NHOH* + H* | 1.5 × 1013 | 63.5 |
| M6 → M7 | R6f | ClC6H4NH* + H* → ClC6H4NH2* | 2.8 × 1016 | 19.2 |
| M7 → M6 | R6r | ClC6H4NH2* → ClC6H4NH* + H* | 1.8 × 107 | 45.6 |
Figure 8.
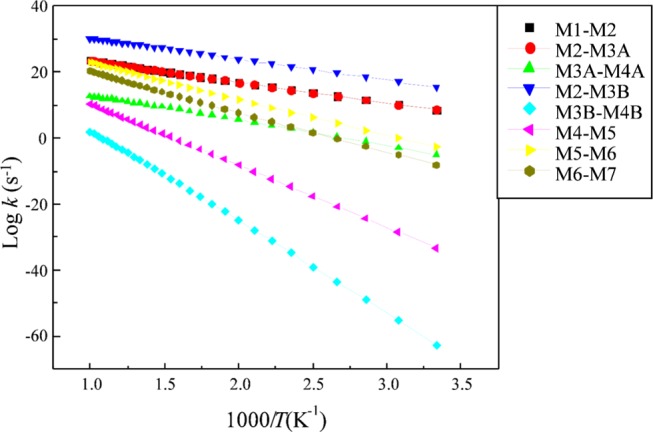
Arrhenius plots of forward hydrogenation reactions that operates in the conversion of p-CNB into p-CAN over the γ-Mo2N(111) surface, fitted in the range of 300–1000 K.
By plotting the residence time (τ) in the plug flow reactor (PFR) against ln (1/1 – X), where X is the conversion of p-CNB, Cárdenas-Lizana et al.16 attained the overall consumption rate of ∼2.0 min–1 (i.e., 0.033 s–1) for p-CNB at 493 K and τ between 1800 and 7800 s. Now, we are in a position to compare our kinetic analysis with the experimentally obtained overall rate constant.16 Inspection of the potential energy surface in Figure 4 reveals that the overall activation barrier for the reduction of p-CNB into p-CAN corresponds to the R2A reaction with a net activation barrier of 31.2 kcal/mol (i.e., the height of TS2A in reference to M1). By considering a typical value of the A factor at 1.0 × 1013 s–1, we obtain an overall consumption rate for p-CNB at 0.018 s–1 (at 493 K). Clearly, our computed rate constant for the conversion of p-CNB correlates very well with the experimental finding of Cárdenas-Lizana et al. (0.018 versus 0.033 s–1).
Following the formation
of the M3A structure, all the intermediates
and transition states reside below the entrance channel. Thus, the
formation of p-CAN from p-CNB could
be modeled based on the two opening reactions (R1) and (R2A). Herein,
we consider a simplified plug flow reactor (PFR) model to compute
the conversion (X) of p-CAN from p-CNB. Central to this simplified treatment is the assumption
that all the reactions are treated as unimolecular reactions in view
of the great excess of p-CNB in reference to inlet
H2 in the experimental conditions (i.e., 0.99 versus 0.01).
The simplified PFR model utilizes the specified experimental conditions
by Cárdenas-Lizana et al. These conditions comprise a reactor
volume of 8.0 × 10–5 m3 (with an
internal diameter of 0.015 m) and an inlet molar flow rate of p-CNB at 8.0 × 10–8 mol/s. The initial
concentration of p-CNB was fixed at 0.24 mol/m3. The model consists of design PFR differential equations 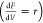 that
relate to the change in the molar
flow rate (f) of the three considered species (M1,
M2, and M3A), with the PFR’s volume (V) and
the rate of the reaction (r) along the reaction sequence
that
relate to the change in the molar
flow rate (f) of the three considered species (M1,
M2, and M3A), with the PFR’s volume (V) and
the rate of the reaction (r) along the reaction sequence
The reactions are considered to be elementary with respect to the concentration (C)
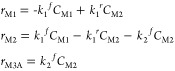 |
Rate constant values at 493 K for reactions (R1) and (R2) are taken from Table 2. The reverse reaction (R2A) was added to account for the fast reaction in the backward direction (i.e., activation energy is only 5.5 kcal/mol). Figure 9a contrasts our modeled X values with their analogous experimental values for τ between 1800 and 7800 s. The latter can be varied by adjusting the reactor’s volume in reference to the volumetric flow rate. Our computed X values slightly overshoot the corresponding experimental estimates. Nonetheless, our simplified PFR model reasonably reproduces the experimental profile pertinent to the consumption of p-CNB. Figure 9b,c portrays the computed concentrations of species and conversion/selectivity (S) down the length of the PFR reactor. The latter signifies the ratio for the molar flow rate of species M3A to the inlet molar flow rate of M1. It serves to provide an estimate for the fraction of M1 that is converted into M3A. As elaborated earlier and based on the facile nature of all the subsequent reactions (R3A–R6), we envisage that the calculated S value for the intermediate M3A largely represents the analogous value for p-CAN.
Figure 9.

(a) Computed and experimental conversion values. (b) Calculated concentration values for M1, M2, and M3A as a function of the reactor’s length. (c) Conversion of p-CNB and selectivity (S) for the formation of M3A. aRef (16).
4. Conclusions
In this study, via density functional theory (DFT-D2) calculations, we map out mechanisms underlying the selective hydrogenation of p-CNB to p-CAN over the γ-Mo2N(111) surface catalyst. The two most energetically favorable adsorption sites are found when chloronitrobenzene is flat and upright positioned to the Mo-hollow fcc and the N-hollow hcp of γ-Mo2N(111) surface, respectively. We establish that the reduction of p-CNB to p-CAN proceeds through the direct and indirect (condensation) pathways; however, the direct pathway systematically requires lower activation barriers. Based on the TST theory, reaction rate constants are evaluated for the reactions involved in direct pathways (mechanisms A and B). The reduction process involves several steps of H* transfer from the surface (mainly at fcc and hcp sites) into NO moieties. The high barrier for direct fission of the C–Cl bond clearly hinders the formation of an aniline molecule. The hydrogenation of chloronitrosobenzene is considered to be the rate-limiting step with an energetic barrier of 55.8 kcal/mol. Desorption of water molecules entails lower desorption energies. Mechanisms and kinetic analyses (via a simplified PFR’s model) presented herein illustrate the pathways for the experimentally observed hydrogenation of the nitro group at relatively low temperatures. Our findings systematically provide a comprehensive understanding into the selective catalysis of such large aromatic molecules and reveal the promising role of γ-Mo2N surface in producing commercially important organic amino compounds.
Acknowledgments
This study has been funded by the Australian Research Council (ARC). Calculations in this study were carried out at the Pawsey Supercomputing Centre in Perth, as well as at the National Computational Infrastructure (NCI) in Canberra, Australia. Z.J. and H.M. gratefully thank the Ministry of Higher Education and Scientific Research of Iraq for the award of postgraduate scholarships and financial support.
The authors declare no competing financial interest.
References
- Wang X.; Liang M.; Zhang J.; Wang Y. Selective hydrogenation of aromatic chloronitro compounds. Curr. Org. Chem. 2007, 11, 299–314. 10.2174/138527207779940856. [DOI] [Google Scholar]
- Králik M.; Vallušová Z.; Major P.; Takáčová A.; Hronec M.; Gašparovičová D. Hydrogenation of chloronitrobenzenes over Pd and Pt catalysts supported on cationic resins. Chem. Pap. 2014, 68, 1690–1700. 10.2478/s11696-014-0565-3. [DOI] [Google Scholar]
- Kratky V.; Kralik M.; Mecarova M.; Stolcova M.; Zalibera L.; Hronec M. Effect of catalyst and substituents on the hydrogenation of chloronitrobenzenes. Appl. Catal., A 2002, 235, 225–231. 10.1016/S0926-860X(02)00274-0. [DOI] [Google Scholar]
- Mahata A.; Rai R. K.; Choudhuri I.; Singh S. K.; Pathak B. Direct vs. indirect pathway for nitrobenzene reduction reaction on a Ni catalyst surface: a density functional study. Phys. Chem. Chem. Phys. 2014, 16, 26365–26374. 10.1039/C4CP04355C. [DOI] [PubMed] [Google Scholar]
- Meng X.; Cheng H.; Fujita S.-i.; Hao Y.; Shang Y.; Yu Y.; Cai S.; Zhao F.; Arai M. Selective hydrogenation of chloronitrobenzene to chloroaniline in supercritical carbon dioxide over Ni/TiO2: significance of molecular interactions. J. Catal. 2010, 269, 131–139. 10.1016/j.jcat.2009.10.024. [DOI] [Google Scholar]
- Chen J.; Yao N.; Wang R.; Zhang J. Hydrogenation of chloronitrobenzene to chloroaniline over Ni/TiO2 catalysts prepared by sol–gel method. Chem. Eng. J. 2009, 148, 164–172. 10.1016/j.cej.2008.11.046. [DOI] [Google Scholar]
- Machado R. M.; Heier K. R.; Broekhuis R. R. Developments in hydrogenation technology for fine-chemical and pharmaceutical applications. Curr. Opin. Drug Discovery Dev. 2001, 4, 745–755. [PubMed] [Google Scholar]
- Blaser H. U.; Malan C.; Pugin B.; Spindler F.; Steiner H.; Studer M. Selective hydrogenation for fine chemicals: recent trends and new developments. Adv. Synth. Catal. 2003, 345, 103–151. 10.1002/adsc.200390000. [DOI] [Google Scholar]
- Sheldon R. A.; Arends I. W.; Hanefeld U. Introduction: Green chemistry and catalysis. Green Chem. Catal. 2007, 1–47. 10.1002/9783527611003.ch1. [DOI] [Google Scholar]
- Chen H.; He D.; He Q.; Jiang P.; Zhou G.; Fu W. Selective hydrogenation of p-chloronitrobenzene over an Fe promoted Pt/AC catalyst. RSC Adv. 2017, 7, 29143–29148. 10.1039/C7RA04700B. [DOI] [Google Scholar]
- Lyu J.; Wang J.; Lu C.; Ma L.; Zhang Q.; He X.; Li X. Size-dependent halogenated nitrobenzene hydrogenation selectivity of Pd nanoparticles. J. Phys. Chem. C 2014, 118, 2594–2601. 10.1021/jp411442f. [DOI] [Google Scholar]
- Ma L.; Wang J.; Wang H.; Zhang Q.; Lu C.; He X.; Li X. High halogenated nitrobenzene hydrogenation selectivity over nano Ir particles. Chin. J. Chem. Eng. 2017, 25, 306–312. 10.1016/j.cjche.2016.08.005. [DOI] [Google Scholar]
- Vishwanathan V.; Jayasri V.; Basha P. M.; Mahata N.; Sikhwivhilu L.; Coville N. J. Gas phase hydrogenation of ortho-chloronitrobenzene (O-CNB) to ortho-chloroaniline (O-CAN) over unpromoted and alkali metal promoted-alumina supported palladium catalysts. Catal. Commun. 2008, 9, 453–458. 10.1016/j.catcom.2007.07.018. [DOI] [Google Scholar]
- Li F.; Liang J.; Zhu W.; Song H.; Wang K.; Li C. In-Situ Liquid Hydrogenation of m-Chloronitrobenzene over Fe-Modified Pt/Carbon Nanotubes Catalysts. Catalysts 2018, 8, 62. 10.3390/catal8020062. [DOI] [Google Scholar]
- Cárdenas-Lizana F.; Lamey D.; Kiwi-Minsker L.; Keane M. A. Molybdenum nitrides: a study of synthesis variables and catalytic performance in acetylene hydrogenation. J. Mater. Sci. 2018, 53, 6707–6718. 10.1007/s10853-018-2009-x. [DOI] [Google Scholar]
- Cárdenas-Lizana F.; Gomez-Quero S.; Perret N.; Kiwi-Minsker L.; Keane M. A. β-Molybdenum nitride: synthesis mechanism and catalytic response in the gas phase hydrogenation of p-chloronitrobenzene. Catal. Sci. Technol. 2011, 1, 794–801. 10.1039/c0cy00011f. [DOI] [Google Scholar]
- Cárdenas-Lizana F.; Lamey D.; Gómez-Quero S.; Perret N.; Kiwi-Minsker L.; Keane M. A. Selective three-phase hydrogenation of aromatic nitro-compounds over β-molybdenum nitride. Catal. Today 2011, 173, 53–61. 10.1016/j.cattod.2011.03.084. [DOI] [Google Scholar]
- Lyu J.; Wang J.; Lu C.; Ma L.; Zhang Q.; He X.; Li X. Size-dependent halogenated nitrobenzene hydrogenation selectivity of Pd nanoparticles. J. Phys. Chem. C 2014, 118, 2594–2601. 10.1021/jp411442f. [DOI] [Google Scholar]
- Delley B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. 10.1063/1.1316015. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Wang Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. 10.1103/PhysRevB.54.16533. [DOI] [PubMed] [Google Scholar]
- Delley B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. 10.1063/1.458452. [DOI] [Google Scholar]
- Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Altarawneh M.; Jaf Z.; Oskierski H.; Jiang Z.-T.; Gore J.; Dlugogorski B. Z. Conversion of NO into N2 over γ-Mo2N. J. Phys. Chem. C 2016, 120, 22270–22280. 10.1021/acs.jpcc.6b04107. [DOI] [Google Scholar]
- Halgren T. A.; Lipscomb W. N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. 10.1016/0009-2614(77)80574-5. [DOI] [Google Scholar]
- Laidler K. J.; King M. C. Development of transition-state theory. J. Phys. Chem. 1983, 87, 2657–2664. 10.1021/j100238a002. [DOI] [Google Scholar]
- He X.; Lyu J.; Zhou H.; Zhuang G.; Zhong X.; Wang J. G.; Li X. Density functional theory study of p-chloroaniline adsorption on Pd surfaces and clusters. Int. J. Quantum Chem. 2014, 114, 895–899. 10.1002/qua.24681. [DOI] [Google Scholar]
- Perret N.; Cárdenas-Lizana F.; Lamey D.; Laporte V.; Kiwi-Minsker L.; Keane M. A. Effect of crystallographic phase (β vs. γ) and surface area on gas phase nitroarene hydrogenation over Mo2N and Au/Mo2N. Top. Catal. 2012, 55, 955–968. 10.1007/s11244-012-9881-4. [DOI] [Google Scholar]
- Wang W.; Xu W.; Thapa K. B.; Yang X.; Liang J.; Zhu L.; Zhu J. Morpholine-Modified Pd/γ-Al2O3@ ASMA Pellet Catalyst with Excellent Catalytic Selectivity in the Hydrogenation of p-Chloronitrobenzene to p-Chloroaniline. Catalysts 2017, 7, 292. 10.3390/catal7100292. [DOI] [Google Scholar]
- Cárdenas-Lizana F.; Gómez-Quero S.; Keane M. A. Clean production of chloroanilines by selective gas phase hydrogenation over supported Ni catalysts. Appl. Catal., A 2008, 334, 199–206. 10.1016/j.apcata.2007.10.007. [DOI] [Google Scholar]
- Jaf Z. N.; Altarawneh M.; Miran H. A.; Jiang Z.-T.; Dlugogorski B. Z. Hydrodesulfurization of Thiophene over γ-Mo2N catalyst. Mol. Catal. 2018, 459, 21–30. 10.1016/j.mcat.2018.07.008. [DOI] [Google Scholar]
- Sheng T.; Qi Y.-J.; Lin X.; Hu P.; Sun S.-G.; Lin W.-F. Insights into the mechanism of nitrobenzene reduction to aniline over Pt catalyst and the significance of the adsorption of phenyl group on kinetics. Chem. Eng. J. 2016, 293, 337–344. 10.1016/j.cej.2016.02.066. [DOI] [Google Scholar]
- Luo Y.-R.Handbook of Bond Dissociation Energies in Organic Compounds; CRC press, 2002. [Google Scholar]
- Jaf Z. N.; Altarawneh M.; Miran H. A.; Jiang Z.-T.; Dlugogorski B. Z. Mechanisms governing selective hydrogenation of acetylene over γ-Mo2N surfaces. Catal. Sci. Technol. 2017, 7, 943–960. 10.1039/C6CY02110G. [DOI] [Google Scholar]
- Zhang L.; Jiang J.; Shi W.; Xia S.; Ni Z.; Xiao X. Insights into the hydrogenation mechanism of nitrobenzene to aniline on Pd3/Pt (111): a density functional theory study. RSC Adv. 2015, 5, 34319–34326. 10.1039/C5RA02389K. [DOI] [Google Scholar]
- Jauberteau I.; Bessaudou A.; Mayet R.; Cornette J.; Jauberteau J. L.; Carles P.; Merle-Méjean T. Molybdenum nitride films: crystal structures, synthesis, mechanical, electrical and some other properties. Coatings 2015, 5, 656–687. 10.3390/coatings5040656. [DOI] [Google Scholar]