

Abstract

An N-heterocyclic carbene-catalyzed intermolecular Stetter reaction of aromatic aldehydes with N-substituted itaconimides has been developed. A delicate balance between the Stetter reaction and the competing isomerization of the itaconimide double bond has been achieved in this operationally simple reaction to afford valuable new succinimide derivatives containing 1,4 and 1,5 dicarbonyl scaffolds in good to excellent yields. The reaction tolerates variable substituents on both aldehydes and N-substituted itaconimides.
Introduction
N-Heterocyclic carbenes (NHCs), a well-documented class of catalysts, have attracted immense attention due to their potential applications in the field of synthetic organic chemistry.1 During the last few years, efforts have been devoted to the development of novel synthetic methodologies in which NHC catalysts are involved as active organocatalysts or ligands for metal-catalyzed reactions. Consequently, recent research substantiates an increasing utilization of NHC toward the synthesis of natural products and useful bioactive molecules from simple feedstocks.2 In NHC-organocatalyzed reactions, the nucleophilic carbene transforms the aldehyde functionality into an acyl anion equivalent via polarity reversal (Umpolung), which enables aldehydes to react with various electrophiles. The nucleophilic addition of acyl anions to various Michael acceptors (Stetter reaction) has been illustrated as a powerful organocatalytic transformation, which offers an elegant approach to construct new carbon–carbon bonds.3 The Stetter reaction with various Michael acceptors provides 1,4-difunctionalized products, which constitute highly valuable building blocks in organic synthesis.4 NHC-catalyzed generation of active acyl anion intermediate and its intervention into new Michael acceptors is a challenging and desired synthetic transformation.3
This work endeavors to study the NHC-catalyzed conjugate addition of acyl anions to N-substituted itaconimides, representing an important and efficient synthetic method to construct valuable succinimide derivatives. The succinimide derivatives are significant compounds found in various natural products, which reveal a remarkable biological and pharmaceutical activity. Also, they have several important applications in agrochemicals, functional materials, and polymer sciences.5 Maleimides are considered to be the general synthon for the preparation of succinimide derivatives. Generally, nucleophilic additions to the active endocyclic polarized double bond or transition-metal-catalyzed C–H activation via olefin insertion are some of the strategies for the preparation of succinimide derivatives from maleimides.6 Apart from these methods, less attention has been given to itaconimide derivatives, which are considered as possible synthons for succinimide derivatives. The active exocyclic double bond and an enolizable amide bond of the N-substituted itaconimide have been recognized as important moieties for the construction of succinimide derivatives.5k Despite these advantages, one challenge that has to be addressed in the application of N-substituted itaconimides is a base-induced isomerization to 3-alkyl N-substituted maleimides,7 presumably due to their high thermodynamic stability.
Until now, few reports demonstrated Michael acceptor reactivity of N-substituted itaconimides (Figure 1). Tan and co-workers reported enantioselective 1–4 nucleophilic addition of phospine oxides and thiols with N-substituted itaconimides, catalyzed by chiral bicyclic guanidine, which acts as a strong chiral Brønsted base (Figure 1, eqs 1 and 2).8 Recently, Jiang et al. demonstrated an elegant approach for the addition–protonation reaction of azlactone with N-substituted itaconimides (Figure 1, eq 3).9 Although the Michael addition of nucleophiles such as heteroatomic anions and carbanions is known, to the best of our knowledge, transformation using nucleophilic acyl anions has not been reported. Notably, by taking advantage of the active exocyclic double bond of N-itaconimide, herein, we report its NHC-catalyzed Stetter reaction, avoiding the double-bond isomerization (Figure 1, eq 4).
Figure 1.
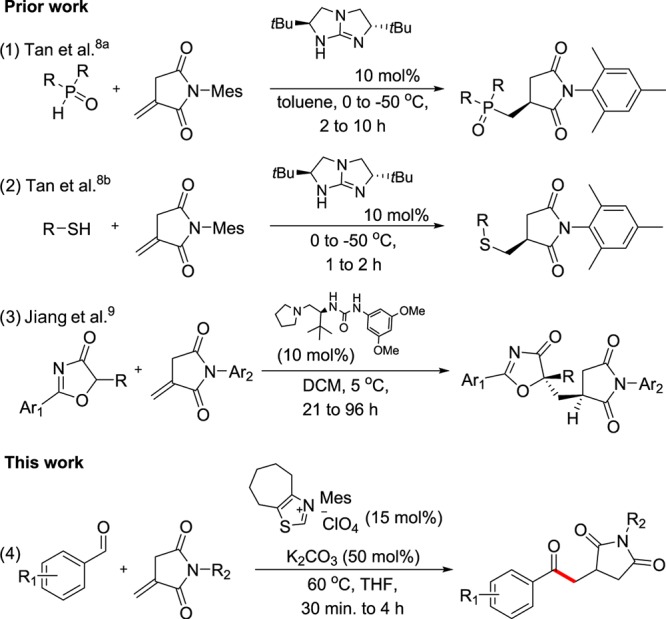
Prior and this work on Michael addition with N-substituted itaconimides.
Results and Discussion
We began our study by preparing the N-substituted itaconimide using a novel method. Recently, our group developed an expedient protocol for the synthesis of maleimide and succinimide derivatives by using a new dehydrating agent (NH4)2S2O8/dimethyl sulfoxide (DMSO).10 In continuation of that, herein, we applied the same protocol in the synthesis of N-aryl itaconimide derivatives. The developed reaction condition worked very well and tolerated various electron-withdrawing and electron-donating substituents on primary aromatic amines (Scheme 1). Our protocol is advantageous over the commonly used acetic anhydride-sodium acetate condition.11 The N-alkyl-substituted itaconimides were synthesized from maleimides by using the known literature procedure.12
Scheme 1. N-Aryl Itaconimides from Aromatic Amines and Itaconic Anhydride,

Reaction conditions: (i) amine (1 equiv, 1 g scale), anhydride (1.1 equiv), 1,4-dioxane (0.1 M), 25 °C, 30 min; (ii) APS (2 equiv), DMSO (2 equiv), 100 °C.
Isolated yields.
Our first effort for the Stetter reaction began by treating p-chlorobenzaldehyde (5) with N-phenyl itaconimide (4a) in the presence of 20 mol % thiazolydene-derived pre-NHC catalyst A and 20 mol % of K2CO3 in tetrahydrofuran (THF) at room temperature. To our delight, the reaction led to the expected product 6a in 55% yield. However, under this reaction condition, the base-facilitated isomerized product N-phenyl maleimide was also observed along with the Stetter product. To increase the yield of the Stetter product 6a and to suppress the formation of isomerized side product 7, four different reaction parameters were examined, which include pre-NHC catalysts, reaction temperature, bases, and solvents. First, different pre-NHC catalysts B–E were screened, but they appeared to be less effective as compared to catalyst A (Table 1, entries 2–5). All of the pre-NHC catalysts were screened with catalyst loading of 15 mol %. Further, performing the reaction at room temperature furnished 6a in diminished yield (Table 1, entry 6). Screening of different bases for the generation of active carbene intermediate from pre-NHC catalyst concludes that various bases such as Cs2CO3, NEt3, and DBU furnished the expected product in low yield with increased production of the side product 7 (Table 1, entries 7–9), but K2CO3 emerged as the best base for this transformation. Also, it was observed that lowering the catalyst loading and equivalent of base substantially decreases the yield of the product (Table 1, entry 10). Furthermore, we observed the formation of more side products with an increase in the equivalents of a base, which resulted in a lower yield of 6a (Table 1, entry 11). Solvent effects on this transformation were also studied, wherein other solvents such as acetonitrile, 1,4-dioxane, and toluene were found to be unfavorable for the current scenario (Table 1, entries 12–14). Next, the molar ratios of substrates 5 and 4a were also examined. Conducting the reaction with reduced equivalents of N-phenyl itaconimide showed a reduction in the yield (Table 1, entry 15). The optimized reaction condition (entry 1) afforded the expected Stetter product 6a up to 80% yield with 4% of 7.
Table 1. Optimization of Reaction Conditionsa.

| entry | variation from the standard condition | yield of 6a/7 (%)b |
|---|---|---|
| 1 | none | 80/4 |
| 2 | B instead of A | 25/22 |
| 3 | C instead of A | <1/19 |
| 4 | D instead of A | <1/27 |
| 5 | E instead of A | 27/34 |
| 6 | reaction at room temperature | 55/3 |
| 7 | Cs2CO3 instead of K2CO3 | 60/20 |
| 8 | NEt3 instead of K2CO3 | 50/40 |
| 9 | DBU instead of K2CO3 | 44/33 |
| 10 | 20 mol % of K2CO3, 10 mol % of NHC | 67/5 |
| 11 | 1 equiv of K2CO3 | 60/17 |
| 12 | CH3CN instead of THF | 10/26 |
| 13 | 1,4-dioxane instead of THF | 53/32 |
| 14 | toluene instead of THF | 20/15 |
| 15 | 1 equiv of 4a | 65/5 |
Reaction conditions: 5 (1.0 equiv, 0.35 mmol), 4a (1.5 equiv, 0.52 mmol), pre-NHC catalyst A (15 mol %), K2CO3 (50 mol%), solvent (2 mL), 4 h, under argon atmosphere.
Isolated yields.
With the optimized conditions in hand, we planned to study the substrate scope of this NHC-catalyzed Stetter reaction. Initially, we screened the itaconimides having various N-substituents by keeping p-chlorobenzaldehyde as the synthetic equivalent of the acyl anion. The developed reaction condition worked well for both N-aryl- and N-alkyl-substituted itaconimides. Gratifyingly, N-aryl itaconimides with a range of electron-withdrawing and electron-donating groups on the aromatic ring were well tolerated to furnish the expected Stetter products in 38–95% yields (Scheme 2, 6a–h). There was no drastic difference in the reactivity pattern associated with electron-donating or electron-withdrawing functional groups on the aromatic ring of N-aryl itaconimide. As mentioned above in the optimization study, the N-phenyl itaconimide 4a provided the expected product in 80% yield. Also, the developed protocol gave a good yield when tested on a higher scale (5.0 mmol). Moreover, N-aryl substituted with −OMe, −Me, and halo functional groups (Scheme 2, 6b–e) gave the corresponding product in good to moderate yields, whereas electron-withdrawing substituents −CF3, −NO2, and −CN gave Stetter product in moderate to good yields (Scheme 2, 6f–h). Moreover, N-alkyl itaconimides also worked well, leading to the desired products with moderate yields under the developed reaction condition (Scheme 2, 6i–k). The product 6k was obtained from 4-CN benzaldehyde in 1:1 d.r. We did not observe any chirality induction, probably because the chiral center in 4k is far away from the reaction center. Furthermore, we observed less than 5% isomerization of itaconimide to the corresponding methyl-substituted maleimides in all of the above substrates, but maleimides remained unreactive under the standard reaction condition.
Scheme 2. Variation in the Itaconimide N-Substituents,

Reaction conditions: 5 (1.0 equiv, 0.35 mmol), 4a–k (1.5 equiv, 0.52 mmol), pre-NHC catalyst A (15 mol %), K2CO3 (50 mol%), THF (2 mL), under argon atmosphere.
Isolated yields.
Reaction at 5.00 mmol scale.
After exploring the reactivity pattern of N-substituted itaconimides, we further intended to explore the scope of the reaction using substituted aldehydes by keeping N-phenyl itaconimide as the Michael acceptor. Both electron-withdrawing and electron-donating aldehydes worked well, leading to the formation of the desired products (Scheme 3, entries 8a–h) in moderate to good yields. The unsubstituted aldehyde substrate, benzaldehyde, provided the expected product 8a with diminished yield. The 4-fluoro and electron-donating substituent 3-OMe gave the Stetter products in moderate yields (Scheme 3, 8b and 8c). Moreover, it was observed that the aldehyde substrates with electron-donating substituents have a much longer reaction time for completion of the reaction as compared to the electron-withdrawing substituents. The developed reaction condition worked well with aromatic aldehydes having electron-withdrawing groups and gave corresponding products smoothly in good yields (Scheme 3, 8d–f). Furthermore, β-naphthaldehyde gave 8g in 41% yield. Interestingly, the protocol was also suitable for heterocyclic aldehyde and gave the desired product 8h in good yield (Scheme 3). However, the developed condition failed to give the expected products with butyraldehyde and cinnamaldehyde (Scheme 3, 8i and 8j).
Scheme 3. Variation in the Aldehyde Substrate,

Reaction conditions: 5a–h (1.0 equiv, 50 mg), 4a (1.5 equiv), pre-NHC catalyst A (15 mol %), K2CO3 (50 mol%), THF (2 mL), under argon atmosphere.
Isolated yields, N.D. = not detected.
Conclusions
In summary, we developed a convenient and hitherto unknown NHC-organocatalyzed Stetter reaction of aromatic aldehydes with N-substituted itaconimides affording valuable new succinimide derivatives in good to excellent yields under an operationally simple reaction condition with broad substrate scope. We are in the process of application of the developed protocol in the synthesis of small bioactive molecules and natural products. Furthermore, our next investigation involves the application of a developed protocol on itaconic anhydride/acid/ester and the development of an asymmetric variant of this reaction.
Experimental Section
General Information
All of the reagents and solvents were used as received from commercial sources unless and otherwise noted. THF was distilled over benzophenone-ketyl under an atmosphere of argon. All of the experiments were carried out under argon atmosphere. Precoated plates (silica gel 60 PF254, 0.25 or 0.5 mm) were utilized for thin-layer chromatography (TLC). Column chromatographic purifications were carried out on flash silica gel (240–400 mesh) using petroleum ether and ethyl acetate as eluents. The 1H, 13C, distortionless enhancement by polarization transfer (DEPT)-NMR spectra were recorded on 200/400/500, 50/100/125 MHz NMR spectrometer, respectively. Chemical shifts were reported as δ values from standard peaks. The multiplicities of signals were designated by the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants (J) are reported in hertz. High-resolution mass spectrometry (HRMS) was performed on a time-of-flight (TOF)/quadrupole-TOF mass spectrometer.
Experimental Procedures
General Experimental Procedure for the Preparation of N-Aryl Itaconimides (4a–h)
A solution of primary aromatic amine 1 (1 g, 1 equiv) and anhydride 2 (1.1 equiv) in 1,4-dioxane (0.1 M) was stirred at room temperature in a two-neck round-bottom flask equipped with a water condenser and a magnetic stirring bar. As soon as all of the amine converts to the corresponding amic acid 3 (monitored by TLC, ∼30 min), ammonium persulfate [(NH4)2S2O8] (2 equiv) and DMSO (2 equiv) were added and the reaction mixture was heated to 100 °C. Heating was continued at the same temperature for 10 h. The reaction mixture was cooled to room temperature and filtered through a cotton plug. The filtrate was evaporated under vacuum. The residue was dissolved in ethyl acetate and washed with dilute HCl, saturated aqueous NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum to furnish the corresponding known imides 4a–h7−9,11,13 in good to excellent yields.
General Experimental Procedure for the Preparation of N-Alkyl Itaconimides
N-Alkyl itaconimides were prepared according to reported procedures.12
General Experimental Procedure for the Preparation of 6a–k, 8a–h
A reaction mixture containing K2CO3 (50 mol %), NHC-precatalyst A (15 mol %), N-itaconimide 4a–k (1.5 equiv), and aromatic aldehyde 5, 5a–h (50 mg, 1.0 equiv) in THF (2 mL) under argon atmosphere was stirred by a magnetic stirring bar at 60 °C for 30 min to 4 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the crude reaction mixture was cooled to room temperature and filtered through a bed of celite. The residue was washed with ethyl acetate (5 mL × 3) and the combined filtrate was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using a solvent gradient of petroleum ether/ethyl acetate to furnish the desired products 6a–k, 8a–h in 20–95% yield.
Experimental Procedure for Large-Scale Preparation of 6a
A reaction mixture containing K2CO3 (104 mg, 50 mol %), NHC-precatalyst A (927 mg, 15 mol %), N-phenyl itaconimide 4a (1.1 g, 1.5 equiv), and aromatic aldehyde 5 (700 mg, 1.0 equiv) in THF (25 mL) under argon atmosphere was stirred by a magnetic stirring bar at 60 °C for 5 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the crude reaction mixture was cooled to room temperature, and filtered through a bed of celite. The residue was washed with ethyl acetate (20 mL × 3) and the combined filtrate was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using a solvent gradient of petroleum ether/ethyl (7:13) acetate to furnish the desired product 6a in 61% yield (980 mg).
Characterization of the Products
Compound 3-(2-(4-Chlorophenyl)-2-oxoethyl)-1-phenylpyrrolidine-2,5-dione (6a)
Reaction time: 4 h; Rf: 0.5 (7:13 EtOAc/pet. ether); white solid; 93 mg, 80% yield; mp = 151–153 °C. 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.3 Hz, 2H), 7.47–7.37 (m, 4H), 7.36–7.32 (m, 1H), 7.29 (d, J = 7.8 Hz, 2H), 3.57 (dd, J = 18.6, 3.4 Hz, 1H), 3.52 (dd, J = 18.6, 6.4 Hz, 1H), 3.35–3.25 (m, 1H), 3.09 (dd, J = 18.1, 9.3 Hz, 1H), 2.57 (dd, J = 18.1, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.65, 178.62, 175.46, 140.35, 134.17, 132.15, 129.47, 129.19, 129.14, 128.65, 126.60, 39.01, 35.72, 34.71. ESI HRMS: calcd for C18H14O3NCl [M + Na]+: 350.0554, found: 350.0556.
Compound 3-(2-(4-Chlorophenyl)-2-oxoethyl)-1-(4-methoxyphenyl)pyrrolidine-2,5-dione (6b)
Reaction time: 4 h; Rf: 0.5 (2:3 EtOAc/pet. ether); white solid; 67 mg, 53% yield; mp = 170–172 °C. 1H NMR (200 MHz, CDCl3) δ 7.83 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 7.20 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 3.76 (s, 3H), 3.60–3.50 (m, 2H), 3.36–3.20 (m, 1H), 3.08 (dd, J = 18.1, 9.5 Hz, 1H), 2.54 (dd, J = 18.1, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.68, 178.86, 175.73, 159.57, 140.36, 134.25, 129.47, 129.14, 127.82, 124.81, 114.55, 55.48, 39.07, 35.71, 34.71. ESI HRMS: calcd for C19H16ClNO4 [M + Na]+: 380.0660, found: 380.0656.
Compound 3-(2-(4-Chlorophenyl)-2-oxoethyl)-1-mesitylpyrrolidine-2,5-dione (6c)
Reaction time: 3 h; Rf: 0.5 (7:13 EtOAc/pet. ether); white solid; 84 mg, 64% yield; mp = 126–128 °C. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 6.91 (s, 1H), 6.89 (s, 1H), 3.58 (dd, J = 18.3, 3.4 Hz, 1H), 3.49 (dd, J = 18.7, 6.9 Hz, 1H), 3.37–3.30 (m, 1H), 3.09 (dd, J = 18.0, 9.1 Hz, 1H), 2.68 (dd, J = 17.9, 6.1 Hz, 1H), 2.23 (s, 3H), 2.17 (s, 3H), 2.01 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 195.32, 178.32, 175.31, 140.29, 139.34, 136.08, 135.09, 134.26, 129.49, 129.47, 129.22, 129.11, 127.57, 38.41, 36.22, 34.76, 21.07, 17.76. ESI HRMS: calcd for C21H20ClNO3 [M + H]+: 370.1204, found: 370.1200.
Compound 3-(2-(4-Chlorophenyl)-2-oxoethyl)-1-(3-fluorophenyl)pyrrolidine-2,5-dione (6d)
Reaction time: 2 h; Rf: 0.5 (7:13 EtOAc/pet. ether); white solid; 116 mg, 95% yield; mp = 123–125 °C. 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.3 Hz, 2H), 7.45–7.33 (m, 3H), 7.15–6.98 (m, 3H), 3.55 (d, J = 4.4 Hz, 2H), 3.22–3.33 (m, 1H), 3.08 (dd, J = 18.1, 9.8 Hz, 1H), 2.57 (dd, J = 18.1, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.65, 178.24, 175.03, 162.65 (d, J = 247.4 Hz), 140.45, 134.09, 133.45 (d, J = 10.0 Hz), 130.29 (d, J = 8.5 Hz), 129.48, 129.16, 122.29 (d, J = 3.1 Hz), 115.67 (d, J = 20.8 Hz), 114.22 (d, J = 23.9 Hz), 38.91, 35.67, 34.59. ESI HRMS: calcd for C18H13ClFNO3 [M + Na]+: 368.0460, found: 368.0454.
Compound 1-(4-Bromophenyl)-3-(2-(4-chlorophenyl)-2-oxoethyl)pyrrolidine-2,5-dione (6e)
Reaction time: 2 h; Rf: 0.5 (7:13 EtOAc/pet. ether); white solid; 116 mg, 80% yield; mp = 168–170 °C. 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 8.3 Hz, 2H), 7.40 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.8 Hz, 2H), 3.56 (d, J = 4.9 Hz, 2H), 3.33–3.22 (m, 1H), 3.07 (dd, J = 18.1, 9.8 Hz, 1H), 2.57 (dd, J = 18.6, 5.9 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 195.67, 178.29, 175.09, 140.47, 134.09, 132.36, 131.18, 129.47, 129.17, 128.15, 122.49, 38.89, 35.69, 34.60. ESI HRMS: calcd for C18H13BrClNO3 [M + H]+: 405.9840, found: 405.9833.
Compound 3-(2-(4-Chlorophenyl)-2-oxoethyl)-1-(3-(trifluoromethyl)phenyl)pyrrolidine-2,5-dione (6f)
Reaction time: 2 h; Rf: 0.5 (7:13 EtOAc/pet. ether); white solid; 106 mg, 76% yield; mp = 132–134 °C. 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.7 Hz, 2H), 7.63–7.5 (m, 2H), 7.57–7.52 (m, 2H), 7.40 (d, J = 8.7 Hz, 2H), 3.57 (d, J = 5.0 Hz, 2H), 3.35–3.27 (m, 1H), 3.10 (dd, J = 18.3, 9.9 Hz,1H), 2.60 (dd, J = 18.3, 5.8 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 195.68, 178.21, 174.96, 140.55, 134.11, 132.79, 131.71 (q, J = 32.4 Hz), 129.98, 129.75, 129.52, 129.22, 125.37 (d, J = 2.3 Hz), 124.56 (q, J = 272.7 Hz), 123.72 (d. J = 3.8 Hz), 38.91, 35.77, 34.63. ESI HRMS: calcd for C19H13ClF3NO3 [M + H]+: 396.0609, found: 396.0606.
Compound 3-(2-(4-Chlorophenyl)-2-oxoethyl)-1-(3-nitrophenyl)pyrrolidine-2,5-dione (6g)
Reaction time: 2 h; Rf: 0.5 (2:3 EtOAc/pet. ether); white solid; 50 mg, 38% yield; mp = 152–154 °C. 1H NMR (400 MHz, CDCl3) δ 8.26 (t, J = 2.0 Hz, 1H), 8.23–8.18 (m, 1H), 7.84 (d, J = 8.5 Hz, 2H), 7.75–7.70 (m, 1H), 7.62 (t, J = 8.0 Hz, 1H), 7.41 (d, J = 8.5 Hz, 2H), 3.64 (dd, J = 18.8, 6.0 Hz, 1H), 3.57 (dd, J = 18.8, 3.7 Hz, 1H), 3.36–3.27 (m, 1H), 3.11 (dd, J = 18.3, 9.6 Hz, 1H), 2.63 (dd, J = 18.3, 5.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.71, 178.00, 174.73, 148.51, 140.61, 133.97, 133.28, 132.56, 129.94, 129.52, 129.21, 123.25, 121.94, 38.83, 35.72, 34.52. ESI HRMS: calcd for C18H13ClN2O5 [M + Na]+: 395.0405, found: 395.0399.
Compound 4-(3-(2-(4-Chlorophenyl)-2-oxoethyl)-2,5-dioxopyrrolidin-1-yl)benzonitrile (6h)
Reaction time: 2 h; Rf: 0.5 (2:3 EtOAc/pet. ether); white solid; 104 mg, 83% yield; mp = 163–165 °C. 1H NMR (200 MHz, CDCl3) δ 7.82 (d, J = 8.3 Hz, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.51 (d, J = 8.3 Hz, 2H), 7.40 (d, J = 8.3 Hz, 2H), 3.62 (dd, J = 19.1, 5.9 Hz, 1H), 3.55 (dd, J = 18.6, 3.4 Hz, 1H), 3.35–3.23 (m, 1H), 3.08 (dd, J = 18.1, 9.8 Hz, 1H), 2.60 (dd, J = 18.1, 5.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.71, 177.92, 174.64, 140.59, 136.21, 133.96, 132.94, 129.48, 129.20, 127.10, 118.12, 112.10, 38.79, 35.68, 34.49. ESI HRMS: calcd for C19H13ClN2O3 [M + H]+: 353.0687, found: 353.0682.
Compound 1-Benzyl-3-(2-(4-chlorophenyl)-2-oxoethyl)pyrrolidine-2,5-dione (6i)
Reaction time: 4 h; Rf: 0.5 (2:3 EtOAc/pet. ether); white solid; 50 mg, 41% yield; mp = 155–157 °C. 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 7.0 Hz, 2H), 7.29–7.20 (m, 3H), 4.67 (d, J = 14.2 Hz, 1H), 4.62 (d, J = 14.2 Hz, 1H), 3.53 (dd, J = 18.1, 3.0 Hz, 1H), 3.29 (dd, J = 18.4, 8.0 Hz, 1H), 3.23–3.14 (m, 1H), 2.96 (dd, J = 18.4, 9.4 Hz, 1H), 2.38 (dd, J = 18.1, 5.3 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.48, 179.12, 175.95, 140.29, 135.74, 134.21, 129.43, 129.12, 128.72, 128.63, 127.89, 42.59, 38.95, 35.74, 34.76. ESI HRMS: calcd for C19H16ClNO3 [M + Na]+: 364.0711, found: 364.0711.
Compound 3-(2-(4-Chlorophenyl)-2-oxoethyl)-1-cyclohexylpyrrolidine-2,5-dione (6j)
Reaction time: 2 h; Rf: 0.5 (2:3 EtOAc/pet. ether); white solid; 24 mg, 20% yield; mp = 122–124 °C. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 8.3 Hz, 2H), 4.02–3.86 (m, 1H), 3.50 (dd, J = 18.1, 2.9 Hz, 1H), 3.32 (dd, J = 18.1, 7.3 Hz, 1H), 3.14–3.02 (m, 1H), 2.87 (dd, J = 18.1, 9.3 Hz, 1H), 3.32 (dd, J = 18.1, 5.4 Hz, 1H), 2.17–2.03 (m, 2H), 1.81–1.72 (m, 2H), 1.63–1.55 (m, 3H), 1.32–1.09 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 195.65, 179.61, 176.49, 140.21, 134.32, 129.43, 129.09, 51.91, 39.08, 35.29, 34.58, 28.76, 28.62, 25.86, 25.07. ESI HRMS: calcd for C18H20ClNO3 [M + Na]+: 356.1024, found: 356.1019.
Compound 4-(2-(2,5-Dioxo-1-((S)-1-phenylethyl)pyrrolidin-3-yl)acetyl)benzonitrile (6k)
Reaction time: 2 h; Rf: 0.5 (2:3 EtOAc/pet. ether); white solid; 69 mg, 52% yield; mp = 155–157 °C. 1H NMR (400 MHz, CDCl3, mixture of diastereomers) δ 7.94 (t, J = 8.7 Hz, 2H), 7.71 (d, J = 8.7 Hz, 1H), 7.70 (d, J = 8.7 Hz, 1H), 7.40 (d, J = 7.6 Hz, 2H), 7.30–7.24 (m, 2H), 7.21 (dd, J = 7.2, 1.5 Hz, 1H), 5.39 (q, J = 8.4 Hz, 1H), 3.52 (dd, J = 18.7, 3.4 Hz, 1H), 3.35 (dd, 18.7, 7.2 Hz, 0.5H), 3.31 (dd, J = 18.7, 7.6 Hz, 0.5H), 3.17–3.03 (m, 1H), 2.94–2.85 (m, 1H), 2.39–2.30 (m, 1H), 1.78 (d, J = 7.6 Hz, 1.5H), 1.77 (d, J = 7.2 Hz, 1.5H). 13C NMR (100 MHz, CDCl3) δ 195.54, 178.95, 178.87, 175.85, 175.78, 139.51, 139.43, 138.77, 132.62, 128.45, 128.39, 127.74, 127.50, 117.67, 116.99, 116.98, 50.50, 50.37, 39.26, 39.15, 35.35, 35.32, 34.51, 34.42, 16.48, 16.43. ESI HRMS: calcd for C21H18N2O3 [M + H]+: 347.1390, found: 347.1404.
Compound 3-(2-Oxo-2-phenylethyl)-1-phenylpyrrolidine-2,5-dione (8a)
Reaction time: 4 h; Rf: 0.5 (7:13 EtOAc/pet. ether); oil; 46 mg, 33% yield. 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 7.82, 2H), 7.54 (t, J = 7.3 Hz, 1H), 7.46–7.38 (m, 4H), 7.34 (d, J = 6.8 Hz, 1H), 7.30 (d, J = 7.8 Hz, 2H), 3.63 (dd, J = 18.6, 3.9 Hz, 1H), 3.55 (dd, J = 18.6, 6.4 Hz, 1H), 3.36–3.26 (m, 1H), 3.09 (dd, J = 18.1, 9.3 Hz, 1H), 2.57 (dd, J = 18.1, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 196.82, 178.76, 175.59, 135.89, 133.81, 132.23, 129.18, 128.79, 128.60, 128.07, 126.64, 39.08, 35.81, 34.80. ESI HRMS: calcd for C18H15NO3 [M + H]+: 294.1125, found: 294.1120.
Compound 3-(2-(3-Methoxyphenyl)-2-oxoethyl)-1-phenylpyrrolidine-2,5-dione (8b)
Reaction time: 4 h; Rf: 0.5 (2:3 EtOAc/pet. ether); oil; 47 mg, 40% yield. 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 7.3 Hz, 1H), 7.46–7.39 (m, 3H), 7.37–7.27 (m, 4H), 7.08 (dd, J = 7.8, 2.0 Hz, 1H), 3.79 (s, 3H), 3.62 (dd, J = 18.6, 3.4 Hz, 1H), 3.54 (dd, J = 18.6, 6.4 Hz, 1H), 3.35–3.26 (m, 1H), 3.09 (dd, J = 18.1, 9.8 Hz, 1H), 2.57 (dd, J = 18.1, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 196.70, 178.75, 175.59, 159.92, 137.22, 132.22, 129.78, 129.18, 128.61, 126.63, 120.72, 120.41, 112.13, 55.48, 39.18, 35.83, 34.77. ESI HRMS: calcd for C19H17NO4 [M + H]+: 324.1230, found: 324.1228.
Compound 3-(2-(4-Fluorophenyl)-2-oxoethyl)-1-phenylpyrrolidine-2,5-dione (8c)
Reaction time: 4 h; Rf: 0.5 (7:13 EtOAc/pet. ether); white solid; 55 mg, 44% yield; mp = 126–128 °C. 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 5.4 Hz, 1H), 7.91 (d, J = 5.4 Hz, 1H), 7.47–7.38 (m, 2H), 7.34 (d, J = 6.8 Hz, 1H), 7.29 (d, J = 7.8 Hz, 2H), 7.09 (t, J = 8.3 Hz, 2H), 3.64–3.46 (m, 2H), 3.36–3.25 (m, 1H), 3.09 (dd, J = 18.1, 9.3 Hz, 1H), 2.57 (dd, J = 18.6, 5.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.22, 178.67, 175.50, 166.11 (d, J = 255.9 Hz), 132.36 (d, J = 2.3 Hz), 132.19, 130.78 (d, J = 9.2 Hz), 129.18, 128.63, 126.62, 115.97 (d, J = 21.6 Hz), 38.96, 35.78, 34.75. ESI HRMS: calcd for C18H14FNO3 [M + H]+: 312.1030, found: 312.1028.
Compound Methyl 4-(2-(2,5-dioxo-1-phenylpyrrolidin-3-yl)acetyl)benzoate (8d)
Reaction time: 1.5 h; Rf: 0.5 (2:3 EtOAc/pet. ether); white solid; 67 mg, 63% yield; mp = 155–157 °C. 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 7.8 Hz, 2H), 7.95 (d, J = 7.8 Hz, 2H), 7.47–7.38 (m, 2H), 7.35 (d, J = 6.8 Hz, 1H), 7.30 (d, J = 7.8 Hz, 2H), 3.89 (s, 3H), 3.70–3.53 (m, 2H), 3.37–3.27 (m, 1H), 3.11 (dd, J = 18.1, 9.3 Hz, 1H), 2.58 (dd, J = 18.1, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 196.43, 178.54, 175.40, 165.99, 138.93, 134.52, 132.15, 129.98, 129.20, 128.66, 128.01, 126.60, 52.54, 39.40, 35.72, 34.70. ESI HRMS: calcd for C20H17NO5 [M + H]+: 52.1179, found: 352.1177.
Compound 3-(2-Oxo-2-(4-(phenylsulfonyl)phenyl)ethyl)-1-phenylpyrrolidine-2,5-dione (8e)
Reaction time: 30 min; Rf: 0.5 (3:2 EtOAc/pet. ether); white solid; 75 mg, 85% yield; mp = 179–181 °C. 1H NMR (400 MHz, CDCl3) δ 7.99 (apparent s, 4H), 7.88 (d, J = 7.8 Hz, 2H), 7.58–7.51 (m, 1H), 7.50–7.37 (m, 4H), 7.37–7.30 (m, 1H), 7.27 (d, J = 7.8 Hz, 2H), 3.64–3.48 (m, 2H), 3.36–3.25 (m, 1H), 3.08 (dd, J = 18.1, 9.8 Hz, 1H), 2.54 (dd, J = 18.1, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.82, 178.32, 175.24, 146.10, 140.55, 139.07, 133.76, 132.06, 129.51, 129.20, 128.89, 128.69, 128.13, 127.86, 126.56, 39.38, 35.64, 34.60. ESI HRMS: calcd for C24H19NO5S [M + H]+: 434.1057, found: 434.1051.
Compound 4-(2-(2,5-Dioxo-1-phenylpyrrolidin-3-yl)acetyl)benzonitrile (8f)
Reaction time: 30 min; Rf: 0.5 (1:1 EtOAc/pet. ether); white solid; 110 mg, 91% yield; mp = 187–189 °C. 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 8.3 Hz, 2H), 7.47–7.39 (m, 2H), 7.39–7.32 (m, 1H), 7.28 (d, J = 7.3 Hz, 2H), 3.66–3.51 (m, 2H), 3.38–3.28 (m, 1H), 3.11 (dd, J = 18.6, 9.8 Hz, 1H), 2.57 (dd, J = 18.1, 5.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.70, 178.32, 175.22, 138.65, 132.66, 132.06, 129.21, 128.71, 128.49, 126.55, 117.65, 117.08, 39.28, 35.61, 34.59. ESI HRMS: calcd for C19H14N2O3 [M + H]+: 319.1077, found: 319.1076.
Compound 3-(2-(Naphthalen-2-yl)-2-oxoethyl)-1-phenylpyrrolidine-2,5-dione (8g)
Reaction time: 4 h; Rf: 0.5 (7:13 EtOAc/pet. ether); white solid; 45 mg, 41% yield; mp = 106–108 °C. 1H NMR (500 MHz, CDCl3) δ 8.43 (s, 1H), 7.95 (dd, J = 8.8, 1.9 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.51 (t, J = 8.0 Hz, 1H), 7.43 (t, J = 8.0 Hz, 2H), 7.37–7.30 (m, 3H), 3.77 (dd, J = 18.3, 3.4 Hz, 1H), 3.68 (dd, J = 18.3, 6.9 Hz, 1H), 3.41–3.34 (m, 1H), 3.13 (dd, J = 18.3, 9.9 Hz, 1H), 2.63 (dd, J = 18.3, 5.7 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 196.76, 178.82, 175.60, 135.85, 133.26, 132.41, 132.26, 130.05, 129.61, 129.19, 128.87, 128.73, 128.61, 127.85, 127.04, 126.66, 123.47, 39.21, 35.94, 34.87; ESI HRMS: calcd for C22H17NO3 [M + H]+: 344.1281, found: 344.1278.
Compound 3-(2-Oxo-2-(pyridin-3-yl)ethyl)-1-phenylpyrrolidine-2,5-dione (8h)
Reaction time: 30 min; Rf: 0.5 (EtOAc); oil; 50 mg, 81% yield. 1H NMR (400 MHz, CDCl3) δ 9.11 (s, 1H), 8.74 (d, J = 4.3 Hz, 1H), 8.17 (d, J = 7.9 Hz, 1H), 7.49–7.25 (m, 6H), 3.67–3.50 (m, 2H), 3.41–3.26 (m, 1H), 3.09 (dd, J = 18.3, 9.8 Hz, 1H), 2.57 (dd, J = 18.3, 5.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 195.87, 178.40, 175.30, 154.11, 149.51, 135.35, 132.10, 131.22, 129.18, 128.65, 126.56, 123.78, 39.18, 35.53, 34.62. ESI HRMS: calcd for C17H14N2O3 [M + H]+: 295.1077, found: 295.1076.
Acknowledgments
M.M.A. thanks, CSIR-New Delhi, for the research fellowship. S.B.M. gratefully acknowledges generous financial support from DST-SERB, New Delhi.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01213.
1H NMR, 13C NMR, and DEPT spectra for all of the new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Nair V.; Vellalath S.; Babu B. P. Recent advances in carbon–carbon bond-forming reactions involving homoenolates generated by NHC catalysis. Chem. Soc. Rev. 2008, 37, 2691–2698. 10.1039/B719083M. [DOI] [PubMed] [Google Scholar]; b Biju A. T.; Kuhl N.; Glorius F. Extending NHC-catalysis: coupling aldehydes with unconventional reaction partners. Acc. Chem. Res. 2011, 44, 1182–1195. 10.1021/ar2000716. [DOI] [PubMed] [Google Scholar]; c Dominguez de Maria P.; Shanmuganathan S. Umpolung catalysis in benzoin-type and Stetter-type reactions: from enzymatic performances to bio-mimetic organocatalytic concepts. Curr. Org. Chem. 2011, 15, 2083–2097. 10.2174/138527211796150679. [DOI] [Google Scholar]; d Grossmann A.; Enders D. N-heterocyclic carbene catalyzed domino reactions. Angew. Chem., Int. Ed. 2012, 51, 314–325. 10.1002/anie.201105415. [DOI] [PubMed] [Google Scholar]; e Bugaut X.; Glorius F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. 10.1039/c2cs15333e. [DOI] [PubMed] [Google Scholar]; f Chen X.-Y.; Ye S. N-heterocyclic carbene-catalyzed reactions of C–C unsaturated bonds. Org. Biomol. Chem. 2013, 11, 7991–7998. 10.1039/c3ob41469h. [DOI] [PubMed] [Google Scholar]; g Ryan S. J.; Candish L.; Lupton D. W. Acyl anion free N-heterocyclic carbene organocatalysis. Chem. Soc. Rev. 2013, 42, 4906–4917. 10.1039/c3cs35522e. [DOI] [PubMed] [Google Scholar]; h De Sarkar S.; Biswas A.; Samanta R. C.; Studer A. Catalysis with N-heterocyclic carbenes under oxidative conditions. Chem. – Eur. J. 2013, 19, 4664–4678. 10.1002/chem.201203707. [DOI] [PubMed] [Google Scholar]; i Albanese D. C. M.; Gaggero N. N-heterocyclic carbene catalysis as a tool for gaining access to the 3,4-dihydropyran-2-one skeleton. Eur. J. Org. Chem. 2014, 2014, 5631–5640. 10.1002/ejoc.201402024. [DOI] [Google Scholar]; j Flanigan D. M.; Romanov-Michailidis F.; White N. A.; Rovis T. Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev. 2015, 115, 9307–9387. 10.1021/acs.chemrev.5b00060. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Tang W.; Du D. Access to spiro and fused indole derivatives from α,β-unsaturated aldehydes enabled by N-heterocyclic carbene catalysis. Chem. Rec. 2016, 16, 1489–1500. 10.1002/tcr.201600018. [DOI] [PubMed] [Google Scholar]; l Zhang C.-H.; Hooper J. F.; Lupton D. W. N-heterocyclic carbene catalysis via the α,β-unsaturated acyl azolium. ACS Catal. 2017, 7, 2583–2596. 10.1021/acscatal.6b03663. [DOI] [Google Scholar]
- Izquierdo J.; Hutson G. E.; Cohen D. T.; Scheidt K. A. A continuum of progress: applications of N-hetereocyclic carbene catalysis in total synthesis. Angew. Chem., Int. Ed. 2012, 51, 11686. 10.1002/anie.201203704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stetter H. Catalyzed addition of aldehydes to activated double bonds - A new synthetic approach. Angew. Chem., Int. Ed. 1976, 15, 639–647. 10.1002/anie.197606391. [DOI] [Google Scholar]; b Stetter H.; Kuhlmann H.. The Catalyzed Nucleophilic Addition of Aldehydes to Electrophilic Double Bonds. In Organic Reactions; Paquette L. A., Ed.; Wiley & Sons: New York, 1991; Vol. 40, pp 407–496. [Google Scholar]; c Read de Alaniz J.; Rovis T. The catalytic asymmetric intramolecular Stetter reaction. Synlett 2009, 2009, 1189–1207. 10.1055/s-0029-1216654. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yetra S. R.; Patra A.; Biju A. T. Recent advances in the N-heterocyclic carbene (NHC)-organocatalyzed Stetter reaction and related chemistry. Synthesis 2015, 47, 1357–1378. 10.1055/s-0034-1378692. [DOI] [Google Scholar]
- a Tomioka K.; Koga K. In Asymmetric Synthesis; Morrison J. D., Ed.; Academic Press: New York, 1983; Vol. 2, pp 201–224. [Google Scholar]; b Yoshikoshi A.; Miyashita M. Oxoalkylation of carbonyl compounds with conjugated nitro olefins. Acc. Chem. Res. 1985, 18, 284–290. 10.1021/ar00117a005. [DOI] [Google Scholar]; c Rosini G.; Ballini R. Functionalized nitroalkanes as useful reagents for alkyl anion synthons. Synthesis 1988, 1988, 833–847. 10.1055/s-1988-27726. [DOI] [Google Scholar]
- a Crider A. M.; Kolczynski T. M.; Yates K. M. Synthesis and anticancer activity of nitrosourea derivatives of phensuximide. J. Med. Chem. 1980, 23, 324–326. 10.1021/jm00177a024. [DOI] [PubMed] [Google Scholar]; b Isaka M.; Rugseree N.; Maithip P.; Kongsaeree P.; Prabpai S.; Thebtaranonth Y. Hirsutellones A–E, antimycobacterial alkaloids from the insect pathogenic fungus Hirsutella nivea BCC 2594. Tetrahedron 2005, 61, 5577–5583. 10.1016/j.tet.2005.03.099. [DOI] [Google Scholar]; c Uddin J.; Ueda K.; Siwu E. R. O.; Kita M.; Uemura D. Cytotoxic labdane alkaloids from an ascidian Lissoclinum sp.: Isolation, structure elucidation, and structure–activity relationship. Bioorg. Med. Chem. 2006, 14, 6954–6961. 10.1016/j.bmc.2006.06.043. [DOI] [PubMed] [Google Scholar]; d Li G.-Q.; Li Y.; Dai L.-X.; You S.-L. N-Heterocyclic carbene catalyzed ring expansion of 4-formyl-β-lactams: synthesis of succinimide derivatives. Org. Lett. 2007, 9, 3519–3521. 10.1021/ol0713537. [DOI] [PubMed] [Google Scholar]; e Rosenbauer E.-M.; Landfester K.; Musyanovych A. Surface-active monomer as a stabilizer for polyurea nanocapsules synthesized via interfacial polyaddition in inverse miniemulsion. Langmuir 2009, 25, 12084–12091. 10.1021/la9017097. [DOI] [PubMed] [Google Scholar]; f Cho H. P.; Engers D.; Venable D.; Niswender C.; Lindsley C.; Conn P.; Emmitte K.; Rodriguez A. A novel class of succinimide-derived negative allosteric modulators of metabotropic glutamate receptor subtype 1 provides insight into a disconnect in activity between the rat and human receptors. ACS Chem. Neurosci. 2014, 5, 597–610. 10.1021/cn5000343. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Wang L.; Ni Q.; Blümel M.; Shu T.; Raabe G.; Enders D. NHC-catalyzed asymmetric synthesis of functionalized succinimides from enals and α-ketoamides. Chem. – Eur. J. 2015, 21, 8033–8037. 10.1002/chem.201500661. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Lee J.; Zhou Z.-L.; Behrens S. H. Charging mechanism for polymer particles in nonpolar surfactant solutions: influence of polymer type and surface functionality. Langmuir 2016, 32, 4827–4836. 10.1021/acs.langmuir.6b00583. [DOI] [PubMed] [Google Scholar]; i Juriga D.; Nagy K.; Jedlovszky-Hajdu A.; Perczel-Kovach K.; Chen Y. M.; Varga G.; Zrínyi M. Biodegradation and osteosarcoma cell cultivation on poly(aspartic acid) based hydrogels. ACS Appl. Mater. Interfaces 2016, 8, 23463–23476. 10.1021/acsami.6b06489. [DOI] [PubMed] [Google Scholar]; j Schönherr H.; Feng C.; Shovsky A. Interfacial reactions in confinement: kinetics and temperature dependence of reactions in self-assembled monolayers compared to ultrathin polymer films. Langmuir 2003, 19, 10843–10851. 10.1021/la034887z. [DOI] [Google Scholar]; k Qiu S.; Tan C.-H.; Jiang Z. Highly chemo-, enantio-, and diastereoselective [4 + 2] cycloaddition of 5H-thiazol-4-ones with N-itaconimides. Beilstein J. Org. Chem. 2016, 12, 2293–2297. 10.3762/bjoc.12.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chauhan P.; Kaur J.; Chimni S. S. Asymmetric organocatalytic addition reactions of maleimides: a promising approach towards the synthesis of chiral succinimide derivatives. Chem. - Asian J. 2013, 8, 328–346. 10.1002/asia.201200684. [DOI] [PubMed] [Google Scholar]; b Miura W.; Hirano K.; Miura M. Copper-mediated oxidative coupling of benzamides with maleimides via directed C–H cleavage. Org. Lett. 2015, 17, 4034–4037. 10.1021/acs.orglett.5b01940. [DOI] [PubMed] [Google Scholar]; c Bettadapur K. R.; Lanke V.; Prabhu K. R. Ru (II)-catalyzed C–H activation: ketone-directed novel 1,4-addition of ortho C–H bond to maleimides. Org. Lett. 2015, 17, 4658–4661. 10.1021/acs.orglett.5b01810. [DOI] [PubMed] [Google Scholar]; d Sharma S.; Han S. H.; Oh Y.; Mishra N. K.; Lee S. H.; Oh J. S.; Kim I. S. Cross-coupling of acrylamides and maleimides under rhodium catalysis: Controlled olefin migration. Org. Lett. 2016, 18, 2568–2571. 10.1021/acs.orglett.6b00909. [DOI] [PubMed] [Google Scholar]; e Muniraj N.; Prabhu K. R. Cobalt(III)-catalyzed C–H activation: azo directed selective 1,4-addition of ortho C–H bond to maleimides. J. Org. Chem. 2017, 82, 6913–6921. 10.1021/acs.joc.7b01094. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liu H.; Fan Y.; Yang Y.; Jiang Z.; Tan C.-H. Bicyclic guanidine-catalyzed direct asymmetric allylic addition of N-aryl alkylidene-succinimides. Chem. – Eur. J. 2010, 16, 12534–12537. 10.1002/chem.201002183. [DOI] [PubMed] [Google Scholar]
- a Leow D.; Lin S.; Chittimalla S. K.; Fu X.; Tan C.-H. Enantioselective protonation catalyzed by a chiral bicyclic guanidine derivative. Angew. Chem., Int. Ed. 2008, 47, 5641–5645. 10.1002/anie.200801378. [DOI] [PubMed] [Google Scholar]; b Lin S.; Leow D.; Huang K.-W.; Tan C.-H. Enantioselective protonation of itaconimides with thiols and the rotational kinetics of the axially chiral C-N bond. Chem. – Asian J. 2009, 4, 1741–1744. 10.1002/asia.200900331. [DOI] [PubMed] [Google Scholar]
- Zhu B.; Lee R.; Li J.; Ye X.; Hong S.-N.; Qiu S.; Coote M. L.; Jiang Z. Chemoselective switch in the asymmetric organocatalysis of 5H-oxazol-4-ones and N-itaconimides: Addition-protonation or [4 + 2] cycloaddition. Angew. Chem., Int. Ed. 2016, 55, 1299–1303. 10.1002/anie.201507796. [DOI] [PubMed] [Google Scholar]
- Garad D. N.; Tanpure S. D.; Mhaske S. B. Radical-mediated dehydrative preparation of cyclic imides using (NH4)2S2O8–DMSO: Application to the synthesis of vernakalant. Beilstein J. Org. Chem. 2015, 11, 1008–1016. 10.3762/bjoc.11.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cava M. P.; Deana A. A.; Muth K.; Mitchell M. J. N-phenylmaleimide. Org. Synth. 1961, 41, 93. 10.15227/orgsyn.041.0093. [DOI] [Google Scholar]; b Pyriadi T. M.; Fraih M. Synthesis and polymerization of N-arylitaconimides: Free radically and anionically. J. Macromol. Sci., Chem. 1982, 18, 159–172. 10.1080/00222338208074415. [DOI] [Google Scholar]; c Oishi T.; Kawamoto T. Synthesis and polymerization of optically active N-[4-N′-(α-methylbenzyl)aminocarbonylphenyl]itaconimide. Polym. J. 1994, 26, 920–929. 10.1295/polymj.26.920. [DOI] [Google Scholar]; d Okada S.; Matyjaszewski K. Synthesis of bio-based poly(N-phenylitaconimide) by atom transfer radical polymerization. J. Polym. Sci., Part A: Polym. Chem. 2015, 53, 822–827. 10.1002/pola.27507. [DOI] [Google Scholar]
- Sippy K. B.; Anderson D. J.; Bunnelle W. H.; Hutchins C. W.; Schrimpf M. R. Preparation and characterization of N-(3-pyridinyl) spirocyclic diamines as ligands for nicotinic acetylcholine receptors. Bioorg. Med. Chem. Lett. 2009, 19, 1682–1685. 10.1016/j.bmcl.2009.01.099. [DOI] [PubMed] [Google Scholar]
- a Veverka M.; Kraľovičová E. Synthesis and biological activity of some derivatives related to 2-methylenebutanedioic acid. Collect. Czech. Chem. Commun. 1989, 54, 2731–2737. 10.1135/cccc19892731. [DOI] [Google Scholar]; b Veverka M. Transformations starting from biotechnologically available materials. 3. N-arylcitraconimides resulting from addition of amines to N-arylitaconimides. Chem. Papers 1992, 46, 116–119. [Google Scholar]; c Haga T.; Yamada S.; Mizukoshi S.; Ikeguchi M.. Algicides Containing 2-Methylenesuccinic Acid Imides. Jpn. Kokai Tokkyo Koho JP 03240705 A 19911028, Oct 28, 1991.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.