

Abstract
Malaria parasites have small extremely AT-rich genomes: microsatellite repeats (1–9 bp) comprise 11% of the genome and genetic variation in natural populations is dominated by repeat changes in microsatellites rather than point mutations. This experiment was designed to quantify microsatellite mutation patterns in Plasmodium falciparum. We established 31 parasite cultures derived from a single parasite cell and maintained these for 114–267 days with frequent reductions to a single cell, so parasites accumulated mutations during ∼13,207 cell divisions. We Illumina sequenced the genomes of both progenitor and end-point mutation accumulation (MA) parasite lines in duplicate to validate stringent calling parameters. Microsatellite calls were 99.89% (GATK), 99.99% (freeBayes), and 99.96% (HipSTR) concordant in duplicate sequence runs from independent sequence libraries, whereas introduction of microsatellite mutations into the reference genome revealed a low false negative calling rate (0.68%). We observed 98 microsatellite mutations. We highlight several conclusions: microsatellite mutation rates (3.12 × 10−7 to 2.16 × 10−8/cell division) are associated with both repeat number and repeat motif like other organisms studied. However, 41% of changes resulted from loss or gain of more than one repeat: this was particularly true for long repeat arrays. Unlike other eukaryotes, we found no insertions or deletions that were not associated with repeats or homology regions. Overall, microsatellite mutation rates are among the lowest recorded and comparable to those in another AT-rich protozoan (Dictyostelium). However, a single infection (>1011 parasites) will still contain over 2.16 × 103 to 3.12 × 104 independent mutations at any single microsatellite locus.
Keywords: microsatellite, mutation accumulation, Plasmodium falciparum, mutation rate, malaria
Introduction
The genome of Plasmodium falciparum malaria parasites is 80% AT rich. As a consequence, microsatellite repeats, defined here as strings of repeated motifs 1–9 bp, are extraordinarily common in malaria parasite genome. There are 132,449 microsatellites in the 3D7 reference genome (PlasmoDB, http://plasmodb.org/common/downloads/release-32/Pfalciparum3D7/; last accessed July 7, 2019): this equates to one microsatellite every 173-bp and 10.74% of the 23-Mb genome is composed of microsatellites (Gardner et al. 2002). These markers typically have multiple alleles and high heterozygosity in natural populations making them the dominant source of genomic variation in malaria parasites (Miles et al. 2016). In contrast, microsatellites constitute just 1–3% of the genome in organisms such as Drosophila or humans with AT/GC content closer to 50% (Lander et al. 2001; Subramanian and Kumar 2003).
Microsatellites are generally assumed to evolve neutrally and have been widely used as genetic markers for population genetic and epidemiological studies, including for malaria parasites (Anderson, Haubold, et al. 2000). However, the assumption of neutrality is questionable. Repeat mutation within promotors, introns, or coding regions can alter transcription through interference with transcription factor binding, changing spacing between regulatory elements, or by their effects on alternative splicing (Bagshaw 2017). It has been suggested that microsatellites may act as evolutionary capacitors fine-tuning and optimizing transcript levels. The effects on transcription may be extensive: 10–15% of heritability in human gene expression is estimated to result from associated cis-acting microsatellite loci (Gymrek 2017). Microsatellite repeat expansions are involved in at least 40 different human diseases, including fragile X syndrome or Freidrich’s ataxia (Groh, Silva, et al. 2014).
In many bacterial pathogens, loss or gain of repeats within promotors or coding sequences are involved in “phase shifts” resulting in changes in pathogenesis. For example, Campylobacter jejuni has 29 phase variable (or contingency loci) that are stochastically switched on or off by loss or gain of monomeric G/C repeats, which put genes in or out of frame (Lango-Scholey et al. 2016). In fact, repeat tracts are used for identifying genes involved in pathogenesis in bacteria (Siena et al. 2016; Aidley et al. 2018). Pathogens typically go through extreme bottlenecks during transmission from one host to another. In the case of bacteria, rapid mutation of phase variable loci may provide a mechanism for stochastic generation of phenotypic variation from a few colonizing bacteria. Similarly, a single malaria sporozoite inoculated from an infected mosquito can produce a human infection containing up to 1011 blood stage parasites. Rapid mutation of microsatellites that influence transcription has the potential to generate extensive phenotypic variation on which selection can act in blood stage malaria infections.
The extremely large number of repeat loci in malaria parasites and their potential impact on phenotypic variation make studying microsatellite evolution a priority. Scoring repeat variation using next-generation sequence data is problematic for several reasons: 1) current Illumina read length is similar to length of microsatellite arrays, which imposes an upper limit on what loci can be effectively scored and 2) amplification steps during library preparation typically generate “stutter,” generating a distribution of repeat array sizes which complicates scoring. Two groups have examined mutation rate in Plasmodium. Bopp et al. (2013) examined base substitution while a separate experiment examined both base substitution and indel mutations (Claessens et al. 2014; Hamilton et al. 2017). Although these studies inferred mutation rates from clone trees, we aimed to use a classical mutation accumulation (MA) experimental design in which multiple parasite clones derived from a single progenitor maintained over multiple generations with frequent bottlenecking to a single cell. A central aim of this work was to evaluate robustness of different methods for scoring spontaneous microsatellite or indel polymorphism in AT-rich malaria genomes in a MA experiment. We then aimed to evaluate the impact of repeat motif, array length and genomic context, gene function, and transcriptional activity on observed mutation rates.
Materials and Methods
MA Process
We generated 31 independent MA lines initiated from a single founder colony of P. falciparum 3D7 (fig. 1, supplementary fig. S1, Supplementary Material online). Plasmodium is a unicellular eukaryote with complex life cycles involving transmission by female Anopheles mosquitoes and human. The parasite is obligately sexual—male and female gametes must mate in the mosquito midgut. The diploid zygote goes through meiosis within 3 h of fertilization, and all the rest of life stages remain haploid. The MA experiment in this study only includes haploid human blood stage.
Fig. 1.
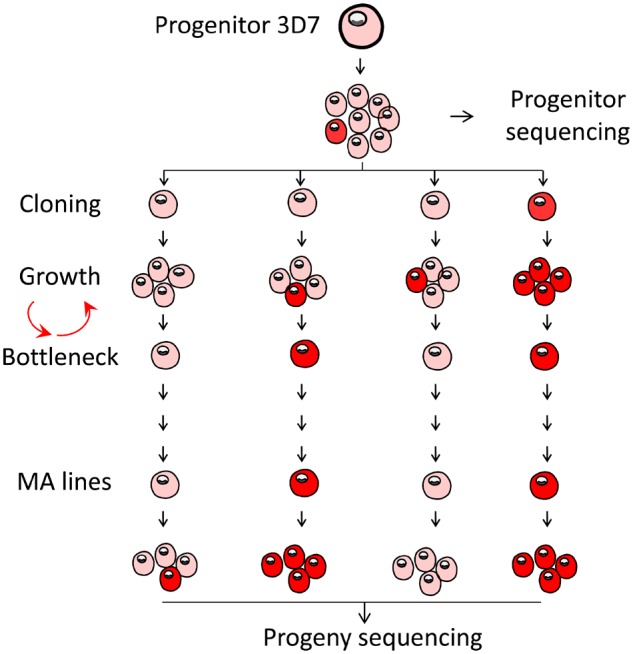
—Generating of MA lines with Plasmodium falciparum. The progenitor 3D7 parasite was dilution cloned prior to the experiment, expanded, and then dilution cloned again to found each MA line, with additional cloning steps at frequent (17–25-day) intervals. Transitions from pink to red shading indicate accumulation of mutations.
We cultured all parasite lines in complete media at 2% hematocrit and 37 °C, with chamber gassed daily and media changed every other day (Trager and Jenson 1978). After every 10.5 parasite cycles, we performed a bottlenecking process to ensure that MA is in an effective neutral fashion. Each bottleneck process started with a clonal dilution step; parasites were ultradiluted into a 96-well plate, to reach a theoretical concentration of 0.25 parasites per well, supplemented with 200 μl of complete media and 2% hematocrit. We identified positive wells with SYBR green I fluorescence assay and confirmed microscopically after 14–21 days postclonal dilution. We transferred the confirmed positive well into a 10-ml flask and cultured for about another week, or until the parasitaemia reached 2%. We then collected the parasites for DNA extraction or cryopreservation and started another round of bottlenecking. If there was no parasite colony surviving after a particular bottleneck, we would bring up the cryopreserved parasites from the previous bottleneck and again restart another round of parasite dilution and culture. We maintained the MA lines for 114–267 days (supplementary table S1, Supplementary Material online).
Whole-Genome Sequencing and Alignment
We extracted DNA with Qiagen DNA Mini kit, sheared about 1.5 µg DNA to ∼180 bp with Covaris S-series sonicator, and we prepared the sequence libraries with NEBnext library preparation. To reduce the sequencing biases of extremely AT-rich genome, we changed the DNA polymerase to Kapa HiFi DNA polymerase in the PCR enrichment step (Oyola et al. 2012). We genome sequenced all MA lines and 3D7 progenitor using Illumina Hiseq 2500 platform as described before (Cheeseman et al. 2015). Whole-genome sequencing reads for each libraries were individually mapped against the P. falciparum 3D7 reference genome (PlasmoDB, http://plasmodb.org/common/downloads/release-32/Pfalciparum3D7/; last accessed July 7, 2019) using alignment algorithm BWA mem (Li 2013) under the default parameters. The result pileup files were further converted to SAM format, sorted to BAM format, and deduplicated using Picard tools v2.0.1 (http://broadinstitute.github.io/picard/; last accessed July 7, 2019). We sequenced each sample twice as technical replicates, by equally dividing a single DNA extraction and performing two independent library preparation and Illumina sequencing.
Genotype Calling
We called base substitutions and small indels using three methods, including haplotype-based calling method-freeBayes (Garrison and Marth 2012), a local de novo assembly-based calling method-GATK HaplotypeCaller (Poplin et al. 2017), and a method designed for genotyping short tandem repeats-HipSTR (Willems et al. 2017). To reduce false positives, we excluded genotype callings at variable regions (subtelomeric repeats, hypervariable regions, and centromeres) of the P. falciparum genome (Miles et al. 2016) for all three methods. We applied postcall filtering to each callset as described below (supplementary table S2, Supplementary Material online).
Standard Filtering Parameters
FreeBayes
We first filtered the input aligned reads by requiring a mapping quality ≥30 and base quality ≥20. We excluded reads that did not fully span the variable window, when genotyped haplotypes comprised multinucleotide polymorphisms or complex variants, to try to detect mutations across repeats. We then generated a raw variant callset which included base substitutions, insertions and deletions, multinucleotide polymorphisms, and complex variants under a haploid model (-ploidy 1). We used hard filters (QUAL/AO > 10, SAF > 0, SAR > 0, RPL > 0, and RPR > 0) to remove low-quality variants. As freeBayes callset included short haplotypes, we normalized the output to show base substitutions and indels, for direct comparison to GATK and HipSTR genotypes.
GATK
We followed the best practice recommendations of Genome Analysis Toolkit GATK v3.7 and made slight adaptations for P. falciparum. Briefly, base quality score was recalled based on a set of verified known variants (Miles et al. 2016). Variants were first called independently for each MA line using HaplotypeCaller and then merged by GenotypeGVCFs, following default parameters but with –sample_ploidy 1. We applied filters to the original GATK genotypes using quality criteria QD > 2, FS < 60, MQ > 40, SOR < 3, GQ > 50, and DP ≥ 3 for base substitutions, and QD > 2.0, FS < 200, SOR < 10, GQ > 50, and DP ≥ 3 for indels. As the variant callsets from MA lines are too small, we did not perform recalibrate variant quality scores here.
HipSTR
We initially identified microsatellites with at least three repeats, repeat length ranging from 10 to 1,000 bp, repeat motif size of 1–9 bp from the P. falciparum 3D7 genome using the mreps program (Kolpakov et al. 2003). We only genotyped microsatellites located in the core genome for accuracy, and microsatellites shorter than 70 bp as genotyping longer microsatellites requires sequence reads longer than 100 bp (Willems et al. 2017). We performed microsatellite genotyping with HipSTR under the default parameters (Willems et al. 2017). Genotypes with any of the following characteristics—<two spanning reads, posterior <90%, >15% of reads with a flanking indel, or >15% of reads with a stutter artifact-were excluded from this callset.
Stringent Filtering Parameters
Accurate determination of mutation rate requires stringent filtering to remove false positive genotype calls. We used genotype concordance between the two independent sequencing runs for each sample to establish stringent filtering parameters to optimize call accuracy. We used the following statistic to calculate concordance:
where p is the concordance between two independent sequencing runs for an MA line, m is the number of genotype that were discordant in the two runs, and G is the total number of single nucleotide polymorphisms or microsatellite genotyped by both runs.
We first compared genotypes from two separate sequence runs, using callsets generated and filtered as described above (fig. 2, standard filter method). To further remove false positives, we further optimized the filter methods using two parameters, purity and diffPL genotype score. Purity is the percentage of reads that support the current genotype, whereas diffPL is the difference in likelihood between the reported and next best genotypes. We use conflict calls from two runs as false positive controls to identify appropriate threshold values for stringent filtering of callsets (supplementary fig. S2, Supplementary Material online). We filtered out genotype calls with purity <0.8, and removed loci where >50% samples examined failed the purity test. We applied the diffPL filter according to the threshold appropriate for each method (supplementary table S3, Supplementary Material online). The concordance approached 100% after stringent filter, both base substitutions and indels (fig. 2, stringent filter method).
Fig. 2.

—Comparison of different genotyping and filter methods. See text for parameters used for standard versus stringent filtering. Stringent filtering allowed high concordance calling in duplicate sequence runs for scoring both base substitutions and indels.
We then collapsed the filtered genotypes from the two sequence runs for each sample by merging identical calls, removing discordant calls and adding calls sequenced from only one run, and combined result from different methods to make the final callset. As a final check, we visually inspected this final genotypes with Integrative Genomics Viewer (https://software.broadinstitute.org/software/igv/; last accessed July 7, 2019) and manually removed possible errors (supplementary fig. S3, Supplementary Material online). We further verified the mutations by Sanger sequencing (supplementary tables S4 and S5, Supplementary Material online).
Consensus and Progenitor-Based Approach for Variant Discovery
We used a consensus approach to identify putative mutations in which each individual MA line or progenitor is compared with the consensus genotype of all the remaining lines. This approach identifies variants from large number of samples with low variance in coverage and is robust against sequencing or alignment errors in the reference genome (Lynch et al. 2008; Denver et al. 2009; Ossowski et al. 2010; Sung et al. 2015; Sun et al. 2017). We employed the consensus approach for our Plasmodium data set with several adjustments: The overall consensus base call is identified as the genotype with the maximum frequency through all the MA lines. We compared the overall consensus with genotypes of progenitor 3D7 and did not find any conflict. The individual consensus for each line is compared against the overall consensus. If the line-specific consensus has a call that differs from the overall consensus, and at least two other lines contained enough reads to be used in the comparison, the site was designated as a putative mutation for the discordant line.
Estimation of the False Negative Rate in Mutation Detection
We estimated the proportion of false negative mutation by introducing mutations into the P. falciparum 3D7 reference genome, running our pipeline, and calculating the fraction of the mutations called, similar to the approach described by Keightley et al. (2015). We first generated a “mock” Variant Call Format file containing 2,000 base substitutions and 3,000 microsatellite indels. For each single nucleotide polymorphism, we changed the reference base to a different randomly selected base; for each microsatellite with original repeat number x, we changed the repeat number to y, a randomly selected number between 0 and 2x (not equal to x) following the binomial distribution with parameter 0.5. These variations were translated into the reference genome sequence by GATK FastaAlternateReferenceMaker. We then mapped and reanalyzed the MA whole-genome sequencing data to this simulated genome using the same procedures as described above. For all the MA lines, we would expect to call the same base substitutions and microsatellite mutations as long as the locus was callable. The false negative rate was estimated as the number of mutations called as wildtype/number of callable mutations in all MA lines.
Mutation Rate Estimates
To calculate the base pair mutation rate per asexual cycle for the MA lines, we used the following equation:
where u is the mutation rate (per base per asexual generation [48 h]), m is the number of observed mutations for each MA line, n is the number of nucleotide sites analyzed (2.078 × 107 bp for core genome), and g is the number of asexual generations for each MA line. We estimated the 95% confidence intervals and standard error for proportions of polymorphic loci () by bootstrapping across loci with 1,000 replicates. We counted the number of days from when the progenitor 3D7 was cloned to the final cloning step for each of the MA lines. We then divided this number by the length of an asexual cycle (48 h) to determine the total number of asexual cycles for each MA line. We assumed 5 mitotic divisions per 48 h asexual cycle, because a single invading merozoite gives rise to up to 32 (25) daughter merozoites during this time. We adjusted the mutation rates by proportion of callable loci, genotype rate, and false negative rate estimated from synthetic mutations (Keightley et al. 2015).
To calculate base pair mutation rate for different substitution type, we divided the mutations according to type A:T -> G:C, G:C -> A:T, G:C -> T:A, G:C -> C:G, A:T -> C:G, and A:T -> T:A. The number of A/T and G/C nucleotides examined in the core genome were 16,898,216 and 3,883,857 bp. The size for coding, promoter (1-kb upstream of start codon ATG), intron, and intergenic region were 11,580,293, 4,356,657, 1,407,648, and 3,437,475 bp, respectively, in the core genome. The average GC content of these regions are coding DNA sequences (CDS) 23.13%, intergenic region 13.19%, intron 13.93%, and promoters 12.77%. We used the same equation for estimating the mutation rate of microsatellites but with n being to the number of microsatellites (123,722) within the core genome. We classified the microsatellite loci by repeat length, repeat motif size, and genomic locations to compare the impact of these features on mutation rates (supplementary table S6, Supplementary Material online).
Results
MA Lines
To investigate the mutation rate in the P. falciparum genome, we generated long-term MA lines starting with a randomly chosen single clone from laboratory adapted 3D7 (fig. 1) generated by dilution cloning. The MA lines passed through a single-cell bottleneck every 21(±4) days to minimize the strength of selection and to fix mutations within each line. Our MA experiment allowed the accumulation of mutations over an average of 170 (range: 114–267) days in 31 independent MA lines (supplementary table S1, Supplementary Material online). This equates to a total of 85 (57–134) 48-h asexual generations or 425 (285–668) mitotic divisions: in total 2,641.5 48-h asexual generations, or an estimated 13,207 mitotic divisions.
Compositions of Short Tandem Repeats in the P. falciparum Genome
We identified 132,449 perfect microsatellites from the P. falciparum 3D7 genome (http://plasmodb.org/common/downloads/release-32/Pfalciparum3D7/; last accessed July 7, 2019) with at least three repeats, 10–1,000 bp in length and 1–9 bp per repeat unit, which accounts for 10.74% of the whole genome. Among these microsatellites, 123,834 (93.50%) of them are located in the core genome (defined in Miles et al. [2016]). As genotyping microsatellites that exceed 70 bp in size invariably requires reads longer than 100 bp (Willems et al. 2017), and the genotype call rate declined for longer microsatellites, we restricted this analysis to microsatellites with a size range 10–70 bp. This accounts for 99.9% (123,722/123,834) of the core genome microsatellites and provides a good representation of microsatellites from the complete P. falciparum genome (supplementary fig. S4 and supplementary table S6, Supplementary Material online).
Microsatellites with a 1–6-bp repeat unit account for 98.16% of all the P. falciparum microsatellites. Of these, 37.68% (46,626 of 123,722) are homopolymeric tracts, and 35.48% (46,849/123,722) are dinucleotide repeats (supplementary fig. S4, Supplementary Material online). The microsatellites (94.64%) have a repeat unit containing A and T: this includes 99.94% (46,600 of 46,626) of homopolymeric tracts, 99.05% (43,490 of 43,903) of dinucleotides, and 82.38% (10,973 of 13,106) of trinucleotides.
Microsatellites are abundant in both coding and noncoding regions (supplementary fig. S5, Supplementary Material online). The total number of microsatellites located in CDS, intergenic, intronic, and promoter (within 1-kb upstream of the initiation codon ATG) regions are 19,912 (16.09%), 35,575 (28.75%), 19,086 (15.43%), and 49,149 (39.73%), respectfully. Microsatellite densities vary in different sequence categories. We observed 10.35, 13.56, and 11.28 microsatellites per kb in intergenic, intronic, and promoter regions, but only 1.72 microsatellites per kb in the CDS region (supplementary table S7, Supplementary Material online). These distributions are significantly different from random expectations (P < 2.2 × 10−16, Pearson’s chi-square test). The average repeat numbers for microsatellites in CDS region were also significantly lower than those located in noncoding regions (P < 2.2 × 10−16, two sample Wilcoxon rank sum test), with average repeat numbers of 12.0, 11.9, and 11.9 in intergenic, intronic, and promoter regions respectively but only 8.2 in CDS region (supplementary fig. S5, Supplementary Material online).
The repeat motifs of microsatellites were highly biased in both coding and noncoding regions (supplementary fig. S5 and supplementary table S7, Supplementary Material online). More than 78.06% of trinucleotides are located in the CDS region, whereas dinucleotides (1.89%), tetranucleotides (2.49%), or pentanucleotides (3.68%) are very rarely located in the CDS. The density of the trinucleotides is low through the whole genome and is comparable to frequencies of 7-, 8-, and 9-bp microsatellites (supplementary table S7, Supplementary Material online). However, trinucleotides were 2.72–3.73 times higher in CDS region than in all the other noncoding regions. The densities of dinucleotides, tetranucleotides, and pentanucleotides in noncoding regions were 50.40–77.13 times greater than those in the coding regions. The likely explanation for this is that nontrinucleotides are excluded from the coding region.
We also examined P. falciparum minisatellites defined as repeat arrays with least three repeat 10–30-bp units ranging from 30 to 1,000 bp in length. We identified 3,528 minisatellites from the 3D7 genome of which 3,294 (93.37%) are located in the core genome. Minisatelllites (61.35%) are located in gene coding region (supplementary fig. S6 and supplementary table S8, Supplementary Material online), which is much more than expected by chance (P = 1.19 × 10−5, Pearson’s chi-square test). Unlike microsatellites, the repeat number of minisatellites located in coding and noncoding regions is not significantly different (P = 0.035, two sample Wilcoxon rank sum test). As expected, there are only minisatellites with repeat unit multiples of three in the coding regions. Only 58.08% (1,913) of the minisatelllites are smaller than 70 bp, and therefore detectable by short-read Illumina sequencing.
Concordance of Genotype Calling in Duplicated Runs
We made two independent sequencing libraries for both the 3D7 progenitor and each of the 31 MA lines, with an average coverage depth of 91.8× and 2,141-Mb 101-bp paired-end reads sequenced for each library (supplementary table S1, Supplementary Material online). To reduce false positives due to alignment errors, we excluded highly variable genome regions (subtelomeric repeats, hypervariable regions, and centromeres) and only performed genotype calling in the 20,782-kb core genome (defined in Miles et al. [2016]). We called genotypes using three methods (GATK, freeBayes, and HipSTR). We filtered the initial mutation call set from each of the three methods using two approaches: 1) a standard method (using recommended filters) and 2) a stringent method, using optimized filtering parameters (fig. 2). We then computed genotype concordance for each replicate pair as (1 − [number of variants with a discordant genotype call]/[total number of variants with nonmissing genotype calls in both sequence runs]).
Under the standard filtering parameters, the average concordances were 99.16% (GATK) and 98.72% (freeBayes) for base substitution callings, and 98.70% (GATK), 98.80% (freeBayes), and 92.22% (HipSTR) for indel or microsatellite calls. These concordance rates are sufficiently accurate for scoring variation in populations or genetics crosses but are insufficiently accurate for scoring rare mutation in MA lines, because there will be a high proportion of false positives.
We therefore implemented more stringent filter methods (supplementary table S2, Supplementary Material online). We first explored the performance of different filters for optimizing genotyping calls. Two filtering parameters—the percentage of supporting reads to current genotype (purity) and the likelihood ratio for the called genotyped relative to the second best genotype (PLdiff)—were particularly effective for minimizing discordant genotype calls (fig. 2, supplementary fig. S2, Supplementary Material online). The concordance rates were dramatically improved, reaching 100% (GATK) and 100% (freeBayes) for base substitution callings, and 99.90% (GATK), 99.99% (freeBayes), and 99.95% (HipSTR) for indel or microsatellite calling (fig. 2): sufficiently accurate for rare mutations discovery in this study.
We further removed the remaining false positives by visual inspection of all the putative mutations detected (supplementary fig. S3, Supplementary Material online). We retained 13/20 (65.0%) of base substitutions identified from stringently filtered GATK calls and 13/15 (86.7%) of base substitutions identified from stringently filtered freeBayes calls. We retained 35/45 (77.8%) indels identified from our stringently filtered HipSTR calls set following visual inspection. Indel calls from GATK and freeBayes were retained at a higher rate than those from HipSTR (81/99 [81.8%] for GATK and 43/50 [86.0%] for freeBayes) after visual inspection (supplementary table S3, Supplementary Material online). To further verify the mutations, we randomly amplified and Sanger sequenced three base substitutions in three MA lines and the 3D7 progenitor, and four indel loci in five samples and 3D7 progenitor. All next-generation sequencing calls were confirmed by Sanger sequencing (supplementary tables S4 and S5, Supplementary Material online).
Mutations Observed in MA Experiment
We included 17 base substitutions and 106 indels from 31 MA lines in the mutation rate analysis (fig. 3). For the 17 remaining base substitutions: 9 were detected by both freeBayes and GATK, whereas 4 were detected by GATK only and 4 by freeBayes only (supplementary table S9 and supplementary fig. S7, Supplementary Material online). We detected 106 indels in the MA analysis. These indels included 81/106 (76.4%) identified by GATK, 40.6% by freeBayes, and 33.0% identified by HipSTR. Just 11/106 (10.4%) were identified using all three methods (supplementary table S10 and supplementary fig. S7, Supplementary Material online).
Fig. 3.

—The genome location of (A) base substitutions and (B) indels in the 31 independent MA lines. From the outer to inner circles: kilo base scale of 14 chromosomes; location of variable regions (subtelomeric repeats, hypervariable regions, and centromeres) of the Plasmodium falciparum genome are marked in red; position of each microsatellite mutation or base substitution in MA lines, with each ring representing the genome of an individual MA line; red dots from panel (B) indicate nonmicrosatellite mutations.
To estimate the false negative rates, we randomly introduced 2,000 base substitutions and 3,000 microsatellite mutations into the 3D7 reference genome (supplementary fig. S8 and supplementary table S11, Supplementary Material online). As we use 3D7 parasite as progenitor, for each MA lines there would be 5,000 synthetic mutations. Among the 2,000 base substitution loci, 1,988 (99.40%) were called by GATK and 1,975 (98.75%) by freeBayes. The base substitution genotype rate (the percentage of genotypes called) for this MA data set at callable loci was 97.61%. For microsatellite mutations, the callable loci and genotype rate were lower than base substitutions. We were able to call 94.70% (2,841/3,000) of the microsatellite loci with genotype rate of 72.04% by combining three methods (GATK, freeBayes, and HipSTR). The false negative rates for both base substitutions and microsatellite mutations were extremely low: 0.001% for base substitution and 0.683% for microsatellite. We adjusted the mutation rates by proportion of callable loci, genotype rate, and false negative rate as described in Keightley et al. (2015).
Base Substitutions
There were 17 base substitutions in 31 MA lines maintained for cumulative total of 5,283 culturing days (fig. 3A and supplementary table S9, Supplementary Material online), giving an estimated mutation rate to be 3.18 ± 0.74 × 10−10 base substitutions per site per asexual cycle.
We analyzed mutation spectra in the MA lines by considering the six nonstrand-specific base substitution types (fig. 4A). The Ts/Tv ratio (0.31) is not significantly different from the random expectation of 0.5, but the transition and transversion subtypes occurred at different rates. G:C -> A:T transitions had higher mutation rates, whereas A:T -> G:C transitions and A:T -> C:G transversions had lower rates compared with expectation (P < 0.05 for all comparisons, two-sided exact binomial test).
Fig. 4.

—Base substitutions rate. (A) Mutation spectrum across six base substitution types. (B) Mutation rate for different coding sequence categories. * indicates P value < 0.05.
We compared the distributions of mutations in coding sequence categories: CDS, promoter, intron, and intergenic regions. Mutation rates were not significantly different in these four functional sequence categories (fig. 4B), suggesting equal mutational susceptibilities for each of these categories in the MA-line genomes and further corroborating that selection has minimally affected the pool of mutations analyzed here. We observed six nonsynonymous mutations and one synonymous mutations in exon sequences. These numbers were not significantly different (P = 1, Pearson’s chi-square test) from the expected values based on the nonsynonymous to synonymous codon potential ratio estimated by Bopp et al. (2013).
Microsatellites, Insertions, and Deletions
We detected 106 indels in 31 MA lines. These are ∼6 times as common as base substitutions (supplementary table S10, Supplementary Material online). Indels (94.3% [100]) are found in arrays of tandem repeats: these include 98 microsatellites and 2 minisatellites. All the microsatellites and minisatellites involved loss or gain of complete repeat units. We also detected six indels, outside tandem repeats: all were deletions (average size = 18.3 bp, range = 15–26 bp) with homology sequences (10–26 bp) nearby (supplementary fig. S9, Supplementary Material online). We did not detect small indels (1–4 bp) outside microsatellite or larger indels that contained apparently random DNA sequences (supplementary table S10, Supplementary Material online). We found a similar pattern to the 3D7 MA lines analyzed by Hamilton et al. (2017), who observed 156 microsatellite indels, 7 minisatellite indels, and 1 nontandem repeat (with homology indel) among 164 indels. In comparison, 69.9% indels in humans are not associated with repeat sequences (Mills et al. 2006).
We analyzed the genomic distribution of the indels. Two-sixth nonvariable number tandem repeat (non-VNTR) indels and all (2/2) minisatellite indels are located in the coding region, whereas only 15/98 microsatellite changes are observed in coding region. We found a similar distribution of indels to Hamilton et al. (2017): they observed 1/1 non-VNTR indels, 7/7 minisatellite indels, and 8/156 microsatellites are located in coding region. The distribution of microsatellite indels is significantly different from minisatellite indels (P = 2.91 × 10−8, Pearson’s chi-square test), which may be due to restriction of microsatellite in coding region: 2,021/3,294 (61.35%) of the minisatelllites, but only 19,912/123,722 (16.09%) of the microsatellites are located in gene coding region. All (15/15) of the microsatellites indels in the coding region are divisible by 3 (supplementary table S10, Supplementary Material online). The noncoding region (6/83) contains fewer repeats evenly divisible by 3 compared with the coding region (P = 7.54 × 10−7, Pearson’s chi-square test).
Mode and Tempo of Microsatellite Mutations
We found 98 microsatellite mutations in our MA experiment. The overall mutation rate for P. falciparum microsatellite is 4.43 ± 0.37 × 10−7 per locus per asexual cycle, which ∼1,000 times higher than base substitution rate (3.18 ± 0.74 × 10−10 base substitutions per site per asexual cycle). Dinucleotide mutations are the most common microsatellite mutation (fig. 5A and B). As mutations for microsatellites bearing repeat motif size >6 are rarely detected, we compared mutation rate for microsatellites with homonucleotide to microsatellites with hexanucleotide repeat motifs (n = 96) (fig. 5C). The mutation rate increased as motif size increased. Hexanucleotides showed higher mutation rate compare to expectations that based on the assumption that all microsatellite have the same mutation rate (P = 7.36 × 10−3, two-sided exact binomial test), whereas the homopolymer mutation rate was lower than expected (P = 2.60 × 10−10, two-sided exact binomial test). We detected increased mutation rates as microsatellite repeat number increased (fig. 5D and E). The increasing trend is particularly obvious for dinucleotides (fig. 5F): mutation rate increases >20-fold as repeat number increases from 5–10 to 20–25, and fit to a one-parameter exponential model (supplementary fig. S10, Supplementary Material online, y = 0.40e0.20x, R2 = 0.98).
Fig. 5.

—Microsatellite mutation distributions. Spectrum of microsatellite mutations with different motif size, length (A), and repeat number (B). Mutations with different motif size were marked with different colors (see key). (C) Mutation rate for different motif sizes. (D) Composition of mutations by repeat number and motif size. (E) Mutation rate at different repeat number. (F) Dinucleotide mutation rate at different repeat number. The expected mutation rates (dotted lines) were estimated by assuming that all classes of microsatellites had the same mutation rate. Two-sided exact binomial test were used here for significance tests. * indicates P value < 0.05; ** indicates P value < 0.01.
The distribution of microsatellite sequences varies among genomic regions (fig. 6). The mutation rate in introns is significantly higher than the average (fig. 6A, P = 2.84 × 10−3, two-sided exact binomial test). There are no mononucleotide, dinucleotide, tetranucleotide, or pentanucleotide mutations detected in coding regions, and no trinucleotide mutation in noncoding region. There is no significant difference between genomic distribution of base substitutions and microsatellite mutations (P = 0.339, df = 3, Pearson’s chi-square test). The overall numbers in each sequence category do not differ significantly from expectation (P = 0.076, df = 3, Pearson’s chi-square test).
Fig. 6.

—Microsatellite mutation and genomic location. (A) Microsatellite mutation rate. (B) Distribution of mutations for different motifs sizes by genome location. (C) The distribution of microsatellite tracts by genome locations. The expected mutation rate was indicated by dotted line. Two-sided exact binomial test were used here for significance test. ** indicates P value < 0.01.
Among the 96 microsatellite mutations, there are 43 deletions (1–11 repeat units) and 53 insertions (1–8 repeat units). Although most mutations involve loss or gain of single repeat units, the distribution is significantly asymmetric, with 4.5 times more deletions than insertions showing a change of >5 repeat units (fig. 7). We investigated the effect of microsatellite repeat number on the observed bias toward repeat loss using dinucleotide mutations, the most abundant class of microsatellite mutations (fig. 7C and D). The size of insertions and deletions increased dramatically for long microsatellites. We observed mutations averaging 1.75 repeats for microsatellites with ≤15 repeats, but averaging 4.07 repeats for microsatellites with >15 repeats (P = 6.99 × 10−4, Wilcoxon rank sum test). Furthermore, the size of deletions was significantly larger than that of insertions (fig. 7D, P = 4.30 × 10−5, Wilcoxon rank sum test).
Fig. 7.

—Patterns of insertion and deletion in microsatellites and dependency on array length. (A) Mean repeat number associated with insertions or deletions for different motif sizes. For all motif sizes (except 4 bp) insertions are observed in microsatellites with fewer repeats than deletions: this is significant for the most abundant motif type (dinucleotides). (B) Size distribution of insertion and deletion events by motif type. The size distribution of indels is skewed and deletions tend to be larger than insertions. (C) The relationship between indel (±) size and repeat number with dinucleotides. Long repeat arrays show larger changes in repeat number than short repeat arrays and there is a significant excess of large repeat losses. Distribution of insertion and deletion size for short (≤15) and long (>15) repeats. (D) Deletions are significantly larger on average than insertions with dinucleotides (see also panel C).
Discussion
Scoring Methods for Indels and Microsatellites
Particularly stringent methods are needed for MA experiments, where numbers of mutations are expected to be quite modest. We used three different approaches to score microsatellite mutations, with stringent filtering to remove false positives. Our stringent methods filtered calls using purity and PLdiff resulted in 99.63–100% concordance between sequences generated from independent libraries. Even with almost perfect concordance between calls, visual inspection clearly indicated that some of the calls were incorrect. GATK called the most indels with an error rate of 18.2% on manual inspection. FreeBayes and HipSTR called fewer repeat mutations with 14.0% and 22.2% error rate, respectively. We removed 18% total calls following visual inspection. For comparison Hamilton et al. (2017) also removed 16/180 (8.9%) calls for the same reasons (Hamilton et al. 2017).
We found that different scoring approaches identify different subsets of mutations from the same data set. Of 106 indels identified (98 microsatellites and 8 indels), GATK identified the majority (81/106, 76.4%), followed by freeBayes 43/106 (40.6%) and HipSTR (33.0%). Importantly, only 11/106 calls were made by all three methods: Hence, we suggest that multiple methods are needed to provide the most complete inventory of indel mutations.
All indel mutations were associated with repeats or homologous regions (supplementary fig. S9, Supplementary Material online). Indels can be generated under multiple mechanisms, such as DNA biosynthesis errors (Garcia-Diaz and Kunkel 2006), DNA polymerase slippage (Klintschar et al. 2004; Forster et al. 2015), and repair of DNA double-strand breaks (Sallmyr and Tomkinson 2018). We speculate that the lack of small random indels in the 3D7 genome relative to humans may be due to the inefficiency of Plasmodium end-joining pathway (Kirkman et al. 2014).
Comparison with Other P. falciparum Mutation Rate Estimates
Indels and Microsatellites
Of the 106 variants identified, 98 were in microsatellites, with just 2 minisatellites and 6 in nonrepetitive tracts. This gives an overall microsatellite mutation rate of 4.43 (±0.37) × 10−7/asexual cycle. Our results both confirm and extend the analysis by Hamilton et al. (2017). They examined MA in 37 sublines from a 3D7 clone tree over a total of 203 days in culture (101.5 asexual generations). They found 164 indels, giving an overall microsatellite mutation rate of 4.45(±0.97) × 10−7, which is very similar to our estimate. Both our study and Hamilton et al. (2017) found that the microsatellite mutation rate was higher in introns compared with coding sequence, promotors, or intergenic regions. Microsatellites from the intronic regions are longer on average than those from CDS, intergenic, and promoter regions (fig. 6C, P < 2e-16, Analysis of variance [ANOVA]). When fitting both length and genomic locations into a binomial generalized linear model, length was a significant positive predictor (Z = 14.959, P < 2e-16) for microsatellite mutation, whereas genomic location had no influence (P > 0.05 for all regions). We conclude that the higher intronic mutation rates observed is driven by the longer microsatellites within introns rather than differences in selective constraints among the genome locations.
Base Substitutions
Our estimate of mutation rates (3.18 ± 0.74 × 10−10 base substitutions per site per asexual cycle) are similar to those from two previous experiments that have examined base substitution rates in P. falciparum. The mutation rate was 5.23 × 10−10 from MA lines (3 clones) built by Bopp et al. (2013), and 2.10 × 10−10 from MA lines (37 clones) generated by Hamilton et al. (2017), with the same calculation equation (see Materials and Methods). The base substitution rate was more than 1,000 times lower than microsatellite mutation rate. Due to the low mutation rate, base substitutions were observed in no more than one MA line, whereas some microsatellite mutations were detected in two or three MA lines (supplementary table S10, Supplementary Material online). Bopp et al. (2013) adjusted the mutation rate by assuming that lethal or deleterious nonsynonymous mutations that arise during long-term culture may not be detected. Their adjusted base substitution rate was 1.7 × 10−9 for 3D7 without drug selection. Using that method, our mutation rate becomes 8.14 × 10−10 base substitutions per site per asexual cycle. Thus, the 3D7 base substitution rates estimated by different laboratories are highly consistent.
Mode and Tempo of Microsatellite Mutation
Both the length of repeat arrays and the repeat motif have been shown to impact microsatellite mutation rate in multiple species (Ellegren 2004). Consistent with this, in P. falciparum, we see that large microsatellite motifs (4–6 bp) show higher mutation rates than motifs <4. There is also a strong relationship between microsatellite heterozygosity and repeat array length (Anderson, Su, et al. 2000). For dinucleotide AT repeats, the dominant repeat class in the P. falciparum genome, mutation rate increases from 1.43 × 10−7 per site per asexual cycle for 5–10 repeats to 3.06 × 10−6 for 20–25 repeats. There is a strong positive relationship between mutation rate and repeat number; this fits better to a one-parameter exponential model (supplementary fig. S10, Supplementary Material online, y = 0.40e0.20x, R2 = 0.98, P = 0.01; F value = 28.85, P = 0.03 compared with the liner model, ANOVA) as observed for dinucleotide repeats in humans (Ellegren 2000).
Stepwise mutation models, in which single repeat units are lost or gained from arrays, are frequently used to model microsatellite evolution. We found similar numbers of losses and gains of repeats (43 deletions vs. 53 insertions). This differs from Hamilton et al. (2017) who found significantly more insertions than deletions (56 deletions vs. 108 insertions). We found several features that fit poorly with a simple stepwise mutation model. Approximately half (59.2%) the mutations involved a single repeat unit, whereas the remainder involved multiple repeat units. Some of these length changes were large: we observed gains of 8 repeats and losses of 11 repeats. Interestingly, we found that mutational size was dependent on array length and highly asymmetric. Long arrays (≥15 repeats) showed larger mutational changes than smaller arrays (<15 repeats). Furthermore, these long arrays also tend to have larger deletions than insertions. This asymmetry is expected to limit large repeat expansions (Harr and Schlötterer 2000), such as those responsible for repeat associated disorders in humans (Groh, Lufino, et al. 2014). However, it is also possible that shorter read sequencing data limits our ability to score large insertions, resulting underestimation of this class mutations.
Mutation Frequency, Transcription, and Gene Essentiality
Several studies report an association between mutation rates and proximity to highly expressed genes (Chen and Zhang 2014; Zavodna et al. 2018). One possible interpretation is that mutation results from collisions between replication and transcription machinery, which will occur more frequently in highly expressed genes. To examine the roles of transcription, motif size, array length, genomic context, we fitted all these variables into a binomial family generalized linear model (supplementary table S12, Supplementary Material online). As expected motif size and array length were significant positive predictors of microsatellite mutuality. However, the interaction between transcription and genomic context with microsatellite mutuality was not significant. We also included mutagenesis index score (MIS) and mutant fitness score (MFS)—measures of gene essentiality from a piggyBac insertion study—from Zhang et al. (2018) in the full predictive model (supplementary table S12, Supplementary Material online), but detected no significant effects.
Do Microsatellite Mutation Impact Phenotype?
Microsatellite length changes are of particular interest if they impact phenotypes. Of the 106 indels observed (98 microsatellites, 2 minisatellites, and 6 indels not associated with repeats), 33 are found in promotors, 29 in introns, 25 in intergenic regions, and 19 within coding sequences. We examined two metrics for gene essentiality—the MIS and MFS scores (Zhang et al. 2018)—for genes with microsatellite mutations found in this study (supplementary table S10, Supplementary Material online). Ten of 38 genes were scored as essential in a piggyback mutagenesis screen (MIS < 0.2, MFS < −2), of which two contained coding indels and eight contained intronic indels. We suggest that at least some of the mutations observed will impact phenotype. Given that infected people may contain >1011 parasites, and an average mutation rate of 4.43 × 10−7/asexual cycle, we expect each infected person to contain an average of >40,000 mutations at each microsatellite locus in the parasite genome. If just a fraction of these mutations modify transcription, then microsatellites may be represent a potent source on genetic variation of which selection can act.
Comparisons of Mutation Rate with Other Species
Base Substitutions
To be comparable to other organisms, we translated our base substitution rate to per bp per cell division, assuming five cell divisions per P. falciparum asexual cycle (Arnot et al. 2011). This yielded an estimate of 6.36 × 10−11 per site per cell division. The per base pair per cell division mutation rates are 3.28 × 10−10 for bacteria Bacillus subtilis (Sung et al. 2015), 3.3 × 10−10 for yeast Saccharomyces cerevisiae (Lynch et al. 2008), 3.2 × 10−10 for Caenorhabditis elegans (Denver et al. 2009), 1.5 × 10−10 for Drosophila melanogaster (Haag-Liautard et al. 2007), and 1.0 × 10−10 for humans (Giannelli et al. 1999; Kondrashov 2003; Denver et al. 2009). Hence the estimated P. falciparum base mutation rate is comparable to other organisms.
Microsatellites
The mutation rates at a microsatellite loci vary according the number of repeated units, length (in base pairs), and the repeated motif (Lynch et al. 2008; Seyfert et al. 2008; Bhargava and Fuentes 2010). The mutation rates range from 10−8–10−3 per locus per cell divisions at different microsatellite catalogs for different organisms (Seyfert et al. 2008; Bhargava and Fuentes 2010). The strongest driver of microsatellite mutation is the array length: mutation rate increases as the microsatellite repeat number increases (fig. 8). The mutation rate for Plasmodium range from 2.16 (SE: 0.60) × 10−8 to 3.12 (1.12) × 10−7 per locus per cell division at different motif lengths (1–6 bp), and range from 3.06 (1.04) × 10−8 to 1.82 (0.44) × 10−7 at different repeat number (3–5 repeats to 20–25 repeats). As all the previous studies focus on mutations with di-, tri-, and/or tetra-nucleotide microsatellite mutations, to be comparable, we recalculated the total mutation rate of di-, tri-, and tetra-microsatellites at per locus per cell division scale in P. falciparum (fig. 8). Longer microsatellites have a lower genotyping rate with short-read next-generation sequencing based methods. To remove this possible bias, we adjusted the P. falciparum mutation rate by the average genotype rate for microsatellite repeats of different length. For comparison, we included both mutation rates before and after adjustment. The mutation rates both before and after adjustment are lower than those reported in human (Brinkmann et al. 1998; Hohoff et al. 2007; Lynch et al. 2008), C. elegans (Seyfert et al. 2008), and Saccharomycescerevisiae (Lynch et al. 2008). The adjustment increased the microsatellite mutation rate for P. falciparum to similar level as Dictyostelium discoideum (McConnell et al. 2007) and Drosophilamelanogaster (Schlötterer et al. 1998; Schug et al. 1998; Vázquez et al. 2000), which have unusually low microsatellite mutation rates. We conclude that the P. falciparum microsatellite mutation rate is at the low end of the spectrum compared with other species.
Fig. 8.
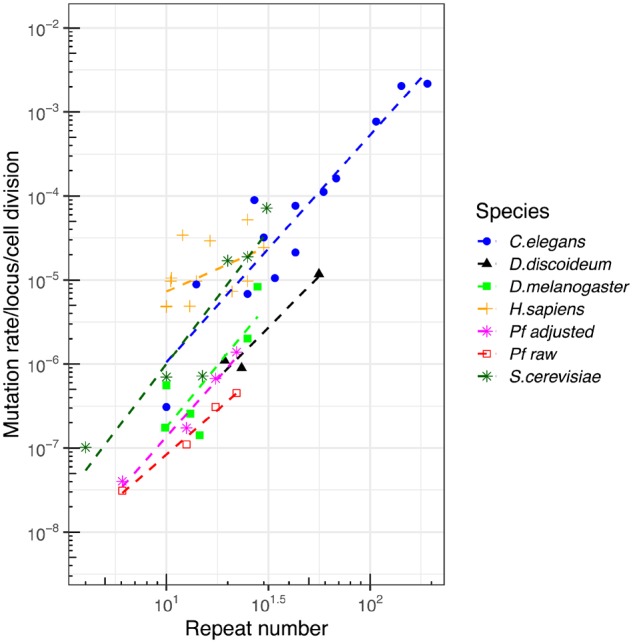
—Comparison of mutation rate with other species. We compared published data sets (see text for details) comparing array length and mutation rate across several species. Two lines are provided for our Plasmodium falciparum data set, showing unadjusted mutation rates, and mutation rates adjusted by genotyping rate (large microsatellite sequences have lower genotyping rate using short-read sequence). Plasmodium falciparum microsatellite mutation rates are at the low end of the spectrum seen in other organisms.
Mutation rates varies among species or strains within species, but relatively little is known about the factors that underlie this variation (Baer et al. 2007). Here, we address three possible explanations of the low microsatellite mutation rate in P. falciparum. First, differences among individuals or species in the ability to recognize and repair DNA damage or replication errors could potentially influence the mutation rate (Baer et al. 2007). Plasmodium spp. are known to lack nonhomologous end-joining pathway during DNA repair (Kirkman et al. 2014), which may influence the mutation rate. However, D. discoideum, which has retained nonhomologous end-joining pathway components (Pears and Lakin 2014), also showed low microsatellite mutation rate (McConnell et al. 2007). Second, mutation rates at meiosis are often assumed to be higher than those from mitosis (Magni and Von Borstel 1962; Rattray et al. 2015). The mutation rates for multicellular eukaryotes include both meiotic and mitotic mutations, but the mutation discussed here for D. discoideum and P. falciparum examine only mitotic mutations. Even though multicellular eukaryotes include both meiotic and mitotic mutations, there are many more mitotic divisions relative to meiotic divisions through the life cycle. Meiotic mutations should only account for very small proportion of the total mutations detected in the multicellular eukaryotes discussed here (human and C.elegans), which should not increase the overall mutation rate very much. Parallel meiotic and mitotic MA experiments in yeast (Nishant et al. 2010), revealed similar base substitution rates in MA experiments with meiotic division (1 meiosis per 20 mitosis) and without meiotic division. We suggest that the lack of meiotic division is unlikely to explain the low mutation rates observed in P. falciparum and D. discoideum. Third, the extreme AT-rich bias could possibly explain the low microsatellite mutation rate in both D. discoideum and P. falciparum. In the P. falciparum genome, there are insufficient microsatellites with motifs other than AT to compare mutation rates in AT and GC containing microsatellites, so we cannot examine the impact of AT bias directly. However, in humans, AC, AG, or GC repeats have similar mutation rates to AT repeats (Payseur et al. 2011) so AT-richness also seems an unlikely explanation. The reasons underlying the low microsatellite mutation rates observed in P. falciparum and D. discoideum are currently unresolved.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R37 AI048071 to T.J.C.A.) and the Research Facilities Improvement Program grant (C06 RR013556).
Data deposition: This project has been deposited at National Center for Biotechnology Information (NCBI) under the accession PRJNA521718 and PRJNA545178.
Literature Cited
- Aidley J, Wanford JJ, Green LR, Sheppard SK, Bayliss CD.. 2018. PhasomeIt: an ‘omics’ approach to cataloguing the potential breadth of phase variation in the genus Campylobacter. Microb Genomics. 4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TJ, et al. 2000. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 17(10):1467–1482. [DOI] [PubMed] [Google Scholar]
- Anderson TJ, Su XZ, Roddam A, Day KP.. 2000. Complex mutations in a high proportion of microsatellite loci from the protozoan parasite Plasmodium falciparum. Mol Ecol. 9(10):1599–1608. [DOI] [PubMed] [Google Scholar]
- Arnot DE, Ronander E, Bengtsson DC.. 2011. The progression of the intra-erythrocytic cell cycle of Plasmodium falciparum and the role of the centriolar plaques in asynchronous mitotic division during schizogony. Int J Parasitol. 41(1):71–80. [DOI] [PubMed] [Google Scholar]
- Baer CF, Miyamoto MM, Denver DR.. 2007. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 8(8):619.. [DOI] [PubMed] [Google Scholar]
- Bagshaw AT. 2017. Functional mechanisms of microsatellite DNA in eukaryotic genomes. Genome Biol Evol. 9(9):2428–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava A, Fuentes F.. 2010. Mutational dynamics of microsatellites. Mol Biotechnol. 44(3):250–266. [DOI] [PubMed] [Google Scholar]
- Bopp SE, et al. 2013. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 9(2):e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann B, Klintschar M, Neuhuber F, Hühne J, Rolf B.. 1998. Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat. Am J Hum Genet. 62(6):1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IH, et al. 2015. Pooled sequencing and rare variant association tests for identifying the determinants of emerging drug resistance in malaria parasites. Mol Biol Evol. 32(4):1080–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang J.. 2014. Yeast mutation accumulation experiment supports elevated mutation rates at highly transcribed sites. Proc Natl Acad Sci U S A. 111(39):E4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessens A, et al. 2014. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of Var genes during mitosis. PLoS Genet. 10(12):e1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci U S A. 106(38):16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. 2000. Heterogeneous mutation processes in human microsatellite DNA sequences. Nat Genet. 24(4):400.. [DOI] [PubMed] [Google Scholar]
- Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 5(6):435.. [DOI] [PubMed] [Google Scholar]
- Forster P, et al. 2015. Elevated germline mutation rate in teenage fathers. Proc R Soc B 282(1803):20142898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz M, Kunkel TA.. 2006. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem Sci. 31(4):206–214. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, et al. 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419(6906):498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G.. 2012. Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv: 1207.3907.
- Giannelli F, Anagnostopoulos T, Green P.. 1999. Mutation rates in humans. II. Sporadic mutation-specific rates and rate of detrimental human mutations inferred from hemophilia B. Am J Hum Genet. 65(6):1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Lufino MM, Wade-Martins R, Gromak N.. 2014. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10(5):e1004318.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Silva LM, Gromak N. 2014. Mechanisms of transcriptional dysregulation in repeat expansion disorders. Biochemical Society Transactions.42(4):1123–1128. [DOI] [PubMed]
- Gymrek M. 2017. A genomic view of short tandem repeats. Curr Opin Genet Dev. 44:9–16. [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C, et al. 2007. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445(7123):82. [DOI] [PubMed] [Google Scholar]
- Hamilton WL, et al. 2017. Extreme mutation bias and high AT content in Plasmodium falciparum. Nucleic Acids Res. 45:1889–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr B, Schlötterer C.. 2000. Long microsatellite alleles in Drosophila melanogaster have a downward mutation bias and short persistence times, which cause their genome-wide underrepresentation. Genetics 155(3):1213–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohoff C, et al. 2007. Y-chromosomal microsatellite mutation rates in a population sample from northwestern Germany. Int J Legal Med. 121(5):359–363. [DOI] [PubMed] [Google Scholar]
- Keightley PD, et al. 2015. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol. 32(1):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkman LA, Lawrence EA, Deitsch KW.. 2014. Malaria parasites utilize both homologous recombination and alternative end joining pathways to maintain genome integrity. Nucleic Acids Res. 42(1):370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klintschar M, et al. 2004. Haplotype studies support slippage as the mechanism of germline mutations in short tandem repeats. Electrophoresis 25(20):3344–3348. [DOI] [PubMed] [Google Scholar]
- Kolpakov R, Bana G, Kucherov G.. 2003. mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 31(13):3672–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS. 2003. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 21(1):12–27. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860.. [DOI] [PubMed] [Google Scholar]
- Lango-Scholey L, Aidley J, Woodacre A, Jones MA, Bayliss CD.. 2016. High throughput method for analysis of repeat number for 28 phase variable loci of Campylobacter jejuni strain NCTC11168. PLoS One 11(7):e0159634.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv: 1303.3997.
- Lynch M, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 105(27):9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni G, Von Borstel R.. 1962. Different rates of spontaneous mutation during mitosis and meiosis in yeast. Genetics 47(8):1097.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell R, Middlemist S, Scala C, Strassmann JE, Queller DC.. 2007. An unusually low microsatellite mutation rate in Dictyostelium discoideum, an organism with unusually abundant microsatellites. Genetics 177(3):1499–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles A, et al. 2016. Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res. 26(9):1288–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, et al. 2006. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 16(9):1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant K, et al. 2010. The baker’s yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genet. 6(9):e1001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, et al. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327(5961):92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyola SO, et al. 2012. Optimizing Illumina next-generation sequencing library preparation for extremely AT-biased genomes. BMC Genomics. 13:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payseur BA, Jing P, Haasl RJ.. 2011. A genomic portrait of human microsatellite variation. Mol Biol Evol. 28(1):303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pears CJ, Lakin ND.. 2014. Emerging models for DNA repair: Dictyostelium discoideum as a model for nonhomologous end-joining. DNA Repair (Amst). 17:121–131. [DOI] [PubMed] [Google Scholar]
- Poplin R, et al. 2017. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv: 201178.
- Rattray A, Santoyo G, Shafer B, Strathern JN.. 2015. Elevated mutation rate during meiosis in Saccharomyces cerevisiae. PLoS Genet. 11(1):e1004910.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallmyr A, Tomkinson AE.. 2018. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem. 293(27):10536.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C, Ritter R, Harr B, Brem G.. 1998. High mutation rate of a long microsatellite allele in Drosophila melanogaster provides evidence for allele-specific mutation rates. Mol Biol Evol. 15(10):1269–1274. [DOI] [PubMed] [Google Scholar]
- Schug MD, et al. 1998. The mutation rates of di-, tri-and tetranucleotide repeats in Drosophila melanogaster. Mol Biol Evol. 15(12):1751–1760. [DOI] [PubMed] [Google Scholar]
- Seyfert AL, et al. 2008. The rate and spectrum of microsatellite mutation in Caenorhabditis elegans and Daphnia pulex. Genetics 178(4):2113–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siena E, et al. 2016. In-silico prediction and deep-DNA sequencing validation indicate phase variation in 115 Neisseria meningitidis genes. BMC Genomics. 17(1):843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Kumar S.. 2003. Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genome Res. 13(5):838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, et al. 2017. Spontaneous mutations of a model heterotrophic marine bacterium. ISME J. 11(7):1713–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, et al. 2015. Asymmetric context-dependent mutation patterns revealed through mutation–accumulation experiments. Mol Biol Evol. 32(7):1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, Jenson JB.. 1978. Cultivation of malarial parasites. Nature 273(5664):621–622. [DOI] [PubMed] [Google Scholar]
- Vázquez JF, Pérez T, Albornoz J, Domínguez A.. 2000. Estimation of microsatellite mutation rates in Drosophila melanogaster. Genet Res. 76(3):323–326. [DOI] [PubMed] [Google Scholar]
- Willems T, et al. 2017. Genome-wide profiling of heritable and de novo STR variations. Nat Methods. 14(6):590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavodna M, Bagshaw A, Brauning R, Gemmell NJ.. 2018. The effects of transcription and recombination on mutational dynamics of short tandem repeats. Nucleic Acids Res. 46(3):1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, et al. 2018. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360(6388):eaap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.