

Abstract
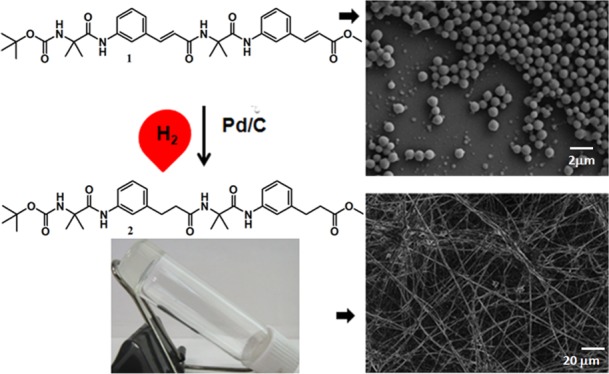
The effect of geometrically rigid trans α,β-unsaturated ε-amino acids on the structure, folding, and assembly of α,ε-hybrid peptide foldamers has been reported. From single-crystal diffraction analysis, the unsaturated tetrapeptide 1 has stapler-pin-like structure but without intramolecular hydrogen bond. The asymmetric unit has two molecules that are stabilized by multiple intermolecular hydrogen bonding interactions as well as π–π stacking interactions between the aromatic rings of 3-aminocinnamic acid. Peptide 1 does not form organogel. But on hydrogenation, peptide 1 provides the saturated α,ε-hybrid peptide foldamer 2, which forms instant gel in most of the aromatic solvents. The gel exhibits high stability. The unsaturated peptide 1 has porous microsphere morphology, but saturated analogue 2 has ribbonlike morphology. The gel has been used efficiently for removal of cationic organic pollutants from waste water.
Introduction
To mimic the structure and function of well-folded biopolymers, like peptides, proteins, and DNA, synthetic oligomers containing designer building blocks that exhibit distinct conformational characteristics have promoted many recent advances in foldamers’ study.1−3 The incorporation of noncoded β-, γ-, δ-, or ε-amino acids into folded structures with α-amino acid chains has been widely used in the design of foldamers having hybrid backbones.4−6 Previously, foldamer studies mainly focused on design and synthesis of new secondary motifs, a goal that is still alive, but recent trends have expanded to include functionality in foldamers.7−10 Introducing a conformational bias by incorporating conformationally rigid amino acid residues has been shown to increase the crystallinity of foldamers.11 This is reported by substantial solid state studies of peptides containing α-aminoisobutyric acid (Aib) or allied α,α-dialkylated amino acid residues.12,13 But the crystallographic analysis of hydrogen-bonded novel hybrid peptides has been possible in peptides containing stereochemically constrained unsaturated amino acids, where the auxiliary degrees of backbone torsional rotation have been restricted by unsaturation.14−16 For example, Gopi and co-workers have reported the unsaturated amino acid-triggered folding of polypeptides.17 But controlling molecular orientation and diverse degrees of self-assembly of unsaturated peptides in comparison to controlling those of their saturated analogues is still challenging.18
Herein, we have synthesized a tetrapeptide containing trans α,β-unsaturated ε-amino acid and α-aminoisobutyric acid. Although the 3-(3-aminophenyl)propionic acid has been extensively investigated in biology and material science,19,20 very little is known about the conformational properties of the unsaturated analogue 3-aminocinnamic acid. The natural existence and very good bioactivities of unsaturated amino acids motivated us to examine the conformational preferences of these amino acids in hybrid peptides. Aib is helicogenic and conformationally rigid. 3-Aminocinnamic acid is also geometrically constrained. So introduction of Aib and 3-aminocinnamic acid will be fascinating not only for molecular structure but also for directing molecular orientation (for adjustment of steric factors) and crystal packing (Figure 1). Interestingly, the α,ε-hybrid peptide 1 exhibits stapler-pin-like conformation in solid state. The two peptide molecules in the asymmetric unit are antiparallel and stabilized by multiple intermolecular hydrogen bonding interactions and π–π stacking interactions. Hybrid peptide 1 does not form gel in organic solvents. However, on hydrogenation, peptide 1 provides saturated analogue α,ε-hybrid peptide 2, which forms instant gel in most of the aromatic solvents. Field emission scanning electron microscopy (FE-SEM) reveals that peptide 1 has porous microsphere morphology but peptide 2 has ribbons-like morphology. Moreover, the organogel of α,ε-hybrid peptide 2 is used efficiently to remove organic pollutants from waste water.
Figure 1.

Chemical structures of α,ε-hybrid peptides 1 and 2.
Results and Discussion
The noncoded amino acid trans-3-aminocinnamic acid methyl ester was synthesized by the condensation reaction between 3-nitrobenzaldehyde and malonic acid in dimethylformamide–water medium at 120 °C for 6 h, followed by esterification with thionyl chloride in methanol at room temperature and finally the reduction of the nitro group with iron–acetic acid at 100 °C for 3 h (Scheme 1).
Scheme 1. Synthesis of Methyl (E)-3-Aminocinnamate 6.

The α,ε-hybrid peptide foldamer 1 was synthesized using traditional solution-phase peptide coupling methodology by dicyclohexylcarbodiimide (DCC) as a coupling reagent (Figure 2). Foldamer 2 has been synthesized by reduction of foldamer 1 with hydrogen and Pd/C catalyst in methanol (Figure 2). The disappearance of characteristic cinnamic acid signals and appearance of new signals for 3-phenylpropanoic acids in the 1H NMR spectrum indicates the formation of peptide 2 from peptide 1. The synthesized compounds and intermediates were purified by column chromatography and analyzed by 1H NMR, 13C NMR, Fourier transform infrared (FT-IR), and mass spectrometry.
Figure 2.

Reactions and conditions: (a) DCC, dry dichloromethane (DCM), 273 K, Et3N, 48 h, 48–58% (b) 1 N NaOH, MeOH, 300 K, 12 h, HCl, 83%.
First, we have investigated the assembly of α,ε-hybrid peptide foldamers 1 and 2 in solution using different spectroscopic experiments. The solution state UV/vis spectra shows no change of spectral positions (249 and 278 nm for π–π* transition), but intensity of absorption increases with increasing peptide 1 concentration in methanol (Figure 3a). For peptide 2, the typical absorption band in methanol solution at 244 nm increases with increasing peptide concentration (Figure 3b). The results suggest that irrespective of unsaturation and rigidity, the intermolecular interactions for both the peptides are similar.
Figure 3.
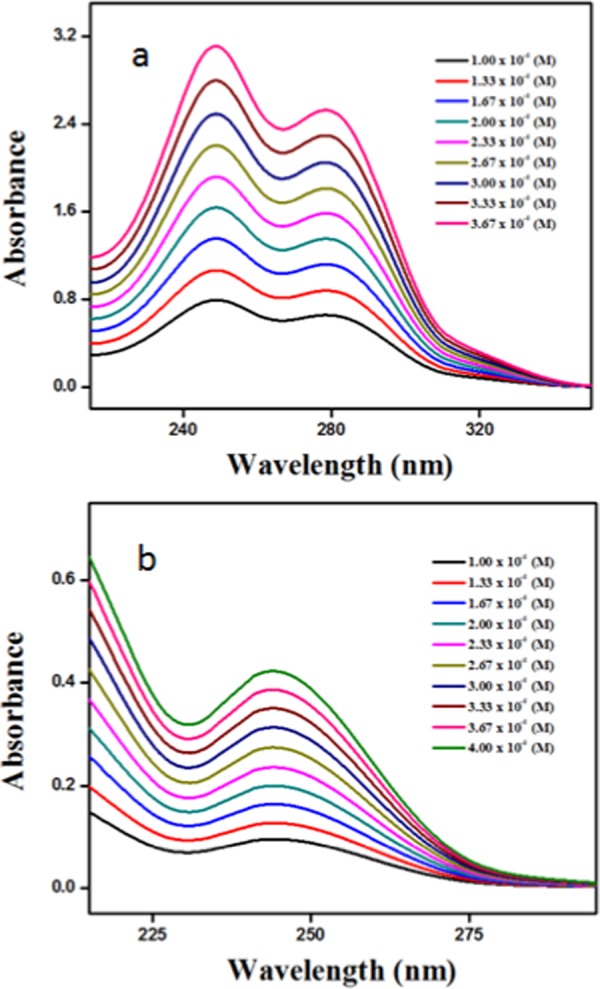
UV/vis spectra of (a) peptide 1 and (b) peptide 2 with increasing concentration in methanol.
FT-IR spectroscopy is a superb experiment to investigate the structure and self-assembly patterns of peptides. The unsaturated α,ε-hybrid peptide 1 exhibits N–H stretching vibrations at 3365 cm–1 and amide I and amide II exhibit peaks at 1677 and 1535 cm–1, respectively.21 However, the saturated α,ε-hybrid peptide 2 shows N–H stretching vibration at 3430 cm–1 and amide I and amide II show peaks at 1651 and 1584 cm–1, respectively (Figure 4).21 Hence, the peptide backbone changes on hydrogenation.
Figure 4.
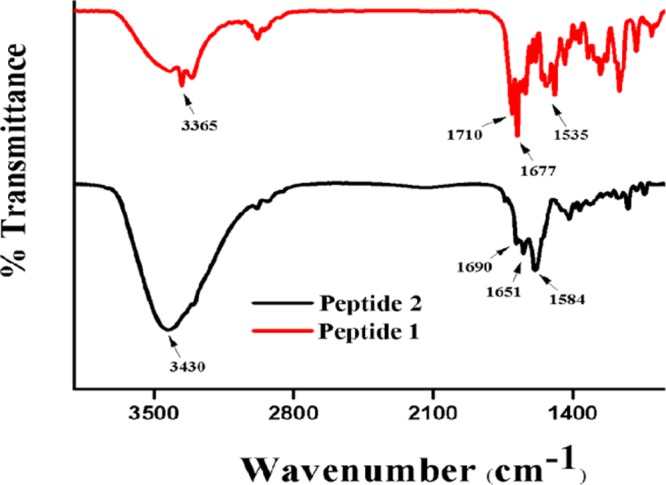
FT-IR spectra of peptides 1 and peptide 2.
To explore the molecular conformation and self-assembly pattern of the 3-aminocinnamic acid containing α,ε-hybrid peptide foldamer 1, single-crystal X-ray crystallography was performed. Light yellow color monoclinic crystals of peptide 1 were obtained from methanol–water solution by slow evaporation. There are two molecules of peptide 1 in the asymmetric unit (Figure 5) in antiparallel arrangement.22 The two molecules are stabilized by four intermolecular N–H···O hydrogen bonds between Aib NH and Boc C=O and 3-aminocinnamic acid NH and Aib C=O. A strong face to face π–π stacking interaction between 3-aminocinnamic acid moieties stabilized the dimer. The centroid to centroid distance is 3.621 Å (Figure 6). The α,ε-hybrid peptide foldamer 1 adopts stapler-pin-like conformation in solid state. The ϕ and ψ values of the Aib residues are in the helical region of the Ramachandran diagram. Table 1 shows the important backbone torsion angles of peptide 1. In higher order packing, the peptide 1 molecules self-assemble to form a porous structure (Figure 7) through intermolecular hydrogen bonding and π–π stacking interactions. The diameters of the pores are 4.4 and 6.1 Å. Table 2 shows the hydrogen bonding parameters of peptide 1. We have tried to crystallize peptide 2 from different solutions but failed to obtain X-ray quality crystal.
Figure 5.
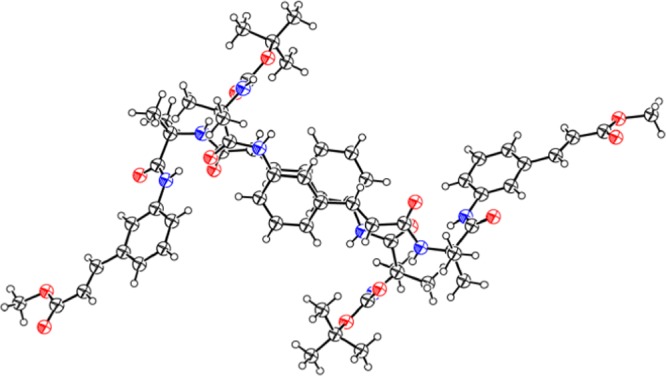
Oak Ridge thermal ellipsoid plot diagram of α,ε-hybrid peptide foldamer 1 showing antiparallel arrangement of molecules (50% probability).
Figure 6.
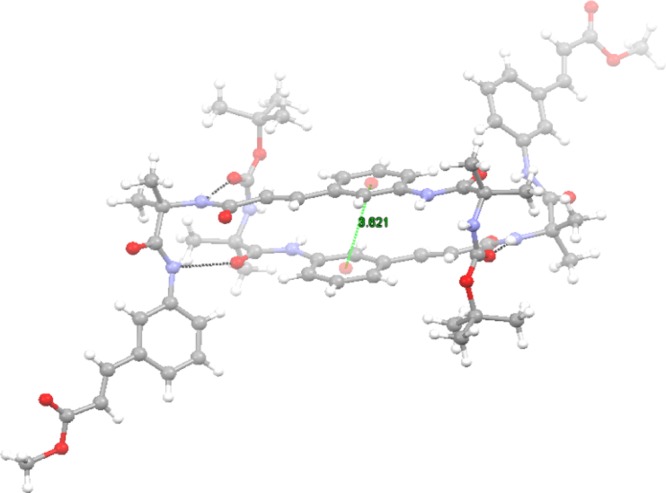
Dimer showing face to face π–π stacking. Intermolecular hydrogen bonds are shown as dotted lines.
Table 1. Important Backbone Torsion Angles (deg) for Peptide 1.
| Aib | ϕ1/deg | ψ1/deg | ϕ3/deg | ψ3/deg |
|---|---|---|---|---|
| A | –57.84 | –43.27 | 59.68 | 39.53 |
| B | 61.09 | 48.62 | –54.27 | –45.28 |
| cinnamic | ϕ2/deg | ψ2/deg | ϕ4/deg | ψ4/deg |
|---|---|---|---|---|
| A | 171.70 | –176.72 | –24.51 | –164.37 |
| B | –179.10 | –171.19 | 11.66 | –3.17 |
Figure 7.
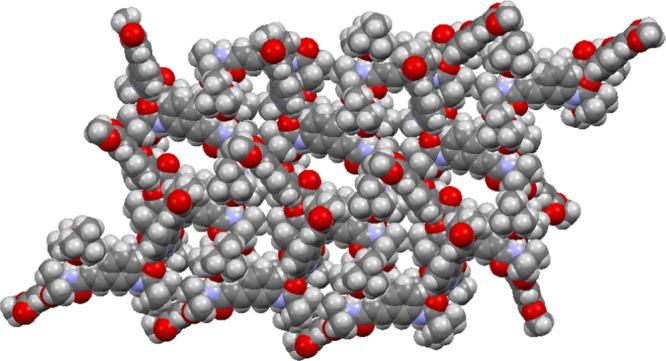
Higher order assembly of α,ε-hybrid peptide foldamer 1 to form porous structure.
Table 2. Hydrogen Bonding Parameters of Peptide 1a.
| D–H···A | D···H (Å) | H···A (Å) | D···A (Å) | D–H···A (deg) |
|---|---|---|---|---|
| N00C–H00C···O00A | 0.88 | 2.19 | 3.038(5) | 163a |
| N00D–H00D···O007 | 0.88 | 2.17 | 3.026(5) | 163 |
| N00E–H00E···O001 | 0.88 | 1.97 | 2.822(5) | 161b |
| N00F–H00F···O003 | 0.88 | 2.18 | 2.993(5) | 154 |
| N00F–H00F···O00D | 0.88 | 2.42 | 2.788(6) | 106 |
| N00G–H00G···O002 | 0.88 | 2.09 | 2.940(3) | 162 |
| N00I–H00I···O00E | 0.88 | 2.47 | 2.826(6) | 105 |
| N00I–H00I···O005 | 0.88 | 2.17 | 2.999(5) | 157b |
| N00J–H00J···N006 | 0.88 | 2.27 | 3.125(5) | 165 |
| N00J–H00J···N00G | 0.88 | 2.42 | 2.795(5) | 106 |
| N00K–H00J···N2 | 0.88 | 1.99 | 2.822(5) | 157 |
| N00K–H00K···N00C | 0.88 | 2.40 | 2.798(5) | 108 |
Symmetry equivalent: a = −x, 1 – y, 1 – z; b = 1 – x, −1/2 + y, 3/2 – z.
To determine the conformational properties of the peptides in solution, NMR experiments were performed. The variable temperature 1H NMR experiment of peptide 1 in CDCl3 exhibits very little shifts of the amide protons with increasing temperature, indicating hydrogen-bonded stable structure in solution (Figures S1 and S2, Supporting Information). Generally, incorporation of small amounts of hydrogen bond-accepting solvents like dimethyl sulfoxide (DMSO)-d6 in CDCl3 solution of peptide results in monotonic downfield shifts of exposed NH functions, leaving solvent-shielded NH functions almost unaffected.29 The effects of adding DMSO-d6 to CDCl3 solutions of peptide 1 are reported in Figure S3 in the Supporting Information. Figure S4 in the Supporting Information shows that Aib(1), Aib(3), and Aca(4) NHs are solvent exposed, as evident from their significant chemical shift changes (Δδ −0.23, 0.33, and −0.39, respectively) upon the addition of DMSO-d6 in CDCl3 solutions. Aca(2) NH exhibits very little chemical shift change (Δδ −0.09) for peptide 1 even at higher percentages of DMSO-d6. Therefore, the results obtained from the solution-state studies are consistent with the peptide 1 conformation in the crystal. Table S1 in the Supporting Information shows Δδ values of all NHs for peptide 1. On the other hand, temperature-dependent 1H NMR experiment of the saturated peptide 2 in CDCl3 also exhibits little shifts of the amide protons with increasing temperature, indicating hydrogen-bonded stable structure in solution. Whereas on the addition of DMSO-d6 to CDCl3 solutions of peptide 2 (Figure S4 in the Supporting Information), Aib(1) and Aib(3) NHs undergo significant chemical shift change (Δδ 0.66 and 1.24, respectively). Interestingly, AcaH(2) and AcaH(4) NHs exhibit minimal chemical shift change (Δδ −0.22 and −0.11, respectively) even at higher percentages of DMSO-d6. The results suggest that peptide 2 forms intramolecular hydrogen-bonded folded structure in CDCl3 solution.29Table S2 in the Supporting Information shows Δδ values of all NHs for peptide 2.
Considering the porous structure of α,ε-hybrid peptide foldamer 1, a wide range of organic solvents were tested to make gel by using the traditional heating–cooling technique and other methods.23 But the unsaturated tetrapeptide 1 did not form gel. On the other hand, the saturated peptide 2 was found to form gel (Figure 8) in most of the aromatic solvents like xylene, toluene, 1,2-dichlorobenzene, etc. The phase-selective gelation was confirmed by the inverted vial experiment (Figure 8).23 The transparent gel shows stability for 2–3 months at room temperature.
Figure 8.

(a) Inverted vial confirms the gelation of peptide 2 in xylene; (b) the phase-selective gelation of peptide 2 from xylene–water mixture.
The morphology of the α,ε-hybrid peptides was examined by field emission scanning electron microscopic (FE-SEM) measurements. For FE-SEM experiments, dilute solutions (0.5 mM) of corresponding peptides were drop casted on microscopic glass slides and finally dried under vacuum for 2 days. Figure 9 depicts the FE-SEM images of the peptides 1 and 2. From Figure 9a,b, the peptide 1 shows the polydisperse microspheres morphology. The average diameter of the microspheres is ca. 1 μm. From Figure 9c,d, the peptide 2 xerogel from xylene exhibits ribbons-like morphology. The diameter of the ribbons is ca. 500 nm and several micrometers in length.
Figure 9.

(a, b) FE-SEM images of unsaturated α,ε-hybrid peptide 1 showing polydisperse microspheres morphology. (c, d) FE-SEM images of the xerogel of saturated α,ε-hybrid peptide 2 from p-xylene showing ribbons-like entangled network.
The macroscopic character of the gel was obtained primarily from the rheology measurement.24 Both G′, a parameter for the elastic response of the gel (the storage modulus), and G″, a measure of the viscous response (the loss modulus), were examined at 25 °C as a function of time. For organogel (10 mg/mL) of peptide 2, the storage modulus (G′) was found to be approximately an order of magnitude higher than the loss modulus (G″), suggesting an elastic rather than a viscous sample (Figure 10). Such a rheological response is a characteristic, mainly shown by gel networks that have physical cross-links through weak co-operative interactions.24
Figure 10.

Rheology data of peptide 2 gel in xylene at 25 °C (10 mg/mL); (a) frequency sweep of the gel at a strain of 0.1%; (b) strain sweep of the gel at a frequency of 1 rad/s.
Presently, water pollution by organic dyes is a major problem for the global community. Most of these dyes are discharged from textile, cosmetic, and other industries. Even very low concentrations of dyes in the waste water are undesirable as well as toxic for living cells.25 Most of these dyes are nondegradable due to their robust chemical structures.26 In this context, gels can be used to purify water from dyes.27,28 For this purpose, 1 mM solution of dyes rhodamine 6G (cationic), methyl violet (cationic), methyl orange (anionic), and pyrocatechol violet (neutral) was prepared. Then, 2 mL of these solutions were placed in vials containing gel of α,ε-hybrid peptide 2 in xylene. The peptide 2 gel was found to absorb the cationic dyes selectively. The reason behind the absorption of only cationic dyes may be attributed to the presence of aromatic rings and the amide bonds that make the peptide neutral but electron rich. This electron-rich peptide can easily undergo attractive interactions with the cationic dyes but not with anionic or neutral dyes due to repulsive interactions. The absorption of dyes was very rapid, and this was studied by UV/vis spectroscopy (Figure 11). Within 24 h, more than 95% removal of the dyes was observed. The gel can also absorb a mixture of cationic dyes (rhodamine and methyl violet) selectively from waste water (a mixture of dyes, including cationic, anionic, and neutral dyes). The results obtained have been included in the Figure S5 Supporting Information. The peptide 2 gel can be reused several times for the cationic dye removal. For this purpose, first, we let the gel absorb the dyes (cationic) and then the gel–dye mixture was treated with deionized water.30 The mixture undergoes removal of the dyes.31 The process was repeated three times, and the removal (visually) of dyes occurred in each case.
Figure 11.

Dye removal studies using peptide 2 gel in xylene; (a) methyl violet and (b) rhodamine 6G.
Conclusions
In conclusion, we have discussed the effect of geometrically rigid trans α,β-unsaturated ε-amino acids on the conformation and assembly of α,ε-hybrid peptide foldamers. The unsaturated α,ε-hybrid peptide foldamer 1 has stapler-pin-like structure in solid state. Two molecules of α,ε-hybrid peptide foldamer 1 in the asymmetric unit are stabilized by multiple intermolecular hydrogen bonding interactions and π–π stacking interactions. Unsaturated α,ε-hybrid peptide foldamer 1 does not form organogel. But on hydrogenation, saturated α,ε-hybrid peptide foldamer 2 forms instant gel in most of the aromatic solvents. The gel exhibits high stability and can be used to remove cationic organic pollutants from waste water efficiently. These α,ε-hybrid peptide foldamers containing functionalizable carbon–carbon double bonds in peptide backbone may provide advanced functional materials by postsynthetic modification.
Experimental Section
General
All reagents were procured from SRL. m-Nitrobenzaldehyde and malonic acid were bought from Sigma chemicals.
Peptide Synthesis
The peptide 1 was synthesized by the traditional solution phase method.32 The amino acid C-terminus was protected by methyl ester formation. Coupling was promoted by dicyclohexylcarbodiimide (DCC). The peptide 2 was synthesized by hydrogenation of peptide 1 using hydrogen and Pd/C. The purification of products was done using column chromatography on silica gel (mesh size 100–200), with n-hexane–ethyl acetate solution as eluent. The reaction intermediates and final peptides were fully characterized by 1H NMR (400 and 500 MHz) spectroscopy, 13C NMR (125 MHz) spectroscopy, mass spectrometry, and FT-IR spectroscopy analysis. Further, X-ray crystallography was performed to characterize the peptide 1.
trans-3-Nitrocinnamic Acid 4
A mixture of m-nitrobenzaldehyde (4.53 g, 30 mmol), malonic acid (5.2 g, 50 mmol), piperidine (0.4 mL, 4 mmol), and dimethylformamide–water (46 mL, 20:3) was heated at 120 °C for 6 h, cooled, and poured into water (100 mL). The mixture was filtered, and the residue was washed by water (3 × 20 mL) and recrystallized from methanol. The crystals were taken in ethyl acetate and neutralized with a dilute solution of KHSO4. The ethyl acetate extract was dried by using anhydrous sodium sulfate and concentrated in vacuum.33 The pure compound was obtained as a yellow solid. Yield: 4.25 g (22 mmol, 73.34%). 1H NMR (400 MHz, DMSO-d6, δ ppm): 12.62 [1H, bs, COOH], 8.52–8.53 [1H, t, ArH], 8.22–8.25 [1H, m, ArH], 8.18–8.19 [1H, d, vinylic CH], 7.69–7.75 [2H, m, ArH], 6.73–6.77 [1H, d, vinylic CH]. 13C NMR (125 MHz, DMSO-d6, δ ppm): 167.07, 148.31, 141.42, 136.1, 133.97, 130.31, 124.32, 122.75, 122.28.
Methyl (E)-3-Nitrocinnamate 5
A solution of 4 (1.93 g, 10 mmol) in methanol (50 mL) was stirred and cooled in an ice-water bath. Thionyl chloride (2.5 mL, 34.46 mmol) was added drop wise and stirred continuously at room temperature for 6 h. Addition of water (100 mL) liberated the product, which was extracted with ethyl acetate and the ethyl acetate layer was washed with water (3 × 50 mL). The extract was then dried by using anhydrous sodium sulfate and concentrated under vacuum.33 The pure product was obtained as a yellow crystalline solid. Yield: 1.99 g (9.61 mmol, 96.1%). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.55 [1H, s, ArH], 8.23–8.25 [1H, m, ArH], 8.19–8.2 [1H, d, J = 7.88, ArH], 7.78–7.81 [1H, d, J = 16.08, vinylic CH], 7.69–7.72 [1H, m, ArH], 6.84–6.87 [1H, d, J = 16.08, vinylic CH], 3.75 [3H, s, OCH3]. 13C NMR (125 MHz, DMSO-d6, δ ppm): 166.31, 148.35, 142.13, 135.9, 134.08, 130.32, 124.55, 122.97, 120.79, 51.7.
Methyl (E)-3-Aminocinnamate 6
A mixture of 5 (1.04 g, 5 mmol), water (5 mL), acetic acid (20 mL), and Fe dust (15 g) in acetone (80 mL) was heated at 100 °C for 3 h and cooled; then, the solution was filtered through silica bed (230–400 mesh) and the filtrate was evaporated and diluted with water (50 mL). The aqueous layer was extracted by ethyl acetate (3 × 50 mL), and the extract was washed with 1 M sodium carbonate (3 × 50 mL) and brine (2 × 50 mL). The extract was dried by using anhydrous sodium sulfate and concentrated under vacuum to get the compound 6 as a pure product. Yield: 0.84 g, (4.74 mmol, 94.81%). 1H NMR (500 MHz, CDCl3, δ ppm): 7.58–7.62 [1H, d, J = 16.02, vinylic CH], 7.15–7.18 [1H, m, ArH], 6.91–6.93 [1H, d, J = 7.63, ArH], 6.82 [1H, bs, ArH], 6.69–6.71 [1H, m, ArH], 6.36–6.39 [1H, d, J = 16.02, vinylic CH], 3.79 [3H, s, OCH3], 3.69–3.76 [2H, b, NH2]. 13C NMR (125 MHz, CDCl3, δ ppm): 167.66, 146.95, 145.35, 135.58, 129.91, 118.82, 117.8, 117.26, 114.28, 51.77.
Boc-Aib-OH 7
2-Aminoisobutyric acid (5.2 g, 50 mmol), 1,4-dioxane (80 mL), water (30 mL), and 2 M NaOH (50 mL) were mixed and cooled in an ice-water bath. Di-tert-butylpyrocarbonate (13.8 mL, 60 mmol) was mixed and stirred continuously at room temperature for 6 h. Finally, the mixture was concentrated (50–60 mL) under vacuum, cooled in an ice-water bath, covered with 60 mL of ethyl acetate, and acidified using dilute solution of KHSO4, adjusting to pH 2–3. The aqueous solution was treated with ethyl acetate, and this process was repeated 2–4 times. The ethyl acetate layers were pooled, washed with water, and dried by using anhydrous sodium sulfate and concentrated in vacuum.33 The pure compound was obtained as a white solid. Yield: 9.04 g (44.48 mmol, 88.96%). 1H NMR (500 MHz, DMSO-d6, δ ppm): 12.13 [1H, bs, COOH], 6.97 [1H, bs, NH], 1.36 [9H, s, BOC], 1.29 [6H, s, Aib CβH]. 13C NMR (125 MHz, DMSO-d6, δ ppm): 176.16, 154.52, 77.62, 54.96, 28.18, 25.12.
Boc-Aib-Aca-OMe 8
Boc-Aib-OH (1.62 g, 8 mmol) was dissolved in 25 mL of dry DCM in an ice-water bath. Compound 6 (0.71 g, 4 mmol) was added to the solution, followed immediately by DCC (1.65 g, 8 mmol). The solution was cooled to room temperature and stirred for 48 h. DCM was evaporated, and the solid was mixed with ethyl acetate (50 mL), and dicyclohexylurea (DCU) was separated by filtration. The organic layer was cleaned with 2 M HCl (3 × 30 mL), brine (2 × 30 mL), and 1 M sodium carbonate (3 × 30 mL) and dried by using anhydrous sodium sulfate and concentrated in vacuum. The compound was purified by silica gel (100–200 mesh) using n-hexane–ethyl acetate (4:1) as eluent to yield peptide 8 as a white solid. Yield: 0.84 g (2.32 mmol, 58%). 1H NMR (400 MHz, CDCl3, δ ppm): 7.79 [1H, m, ArH], 7.63–7.67 [1H, d, J = 16.02, vinylic CH], 7.48–7.5 [1H, m, ArH], 7.3–7.34 [1H, m, ArH], 7.23–7.25 [1H, d, J = 7.63, ArH], 6.42–6.46 [1H, d, J = 16.02, vinylic CH], 4.94 [1H, bs, Ar NH], 3.79 [3H, s, OCH3], 1.7 [1H, s, Aib NH], 1.57 [6H, s, Aib CβH], 1.44 [9H, s, BOC]. 13C NMR (125 MHz, CDCl3, δ ppm): 172.94, 166.44, 155.91, 144.7, 135.37, 129.57, 123.95, 122.88, 121.65, 119.21, 118.51, 81.28, 58.06, 51.76, 28.41, 25.82.
Boc-Aib-Aca-OH 9
To 0.36 g (1 mmol) of compound 8, 10 mL of methanol and 4 mL of 2 M sodium hydroxide solution were added34 and stirred and the saponification reaction was monitored by thin layer chromatography (TLC). After 10 h, MeOH was dried under vacuum; the residue was mixed with 30 mL of water and treated with diethyl ether (2 × 30 mL). Finally, the pH of the aqueous layer was fixed at 2–3 using 1 M HCl and it was treated with ethyl acetate (3 × 30 mL). The extracts were pooled, dried by using anhydrous sodium sulfate, and concentrated under vacuum to obtain compound 9 as a white solid. Yield 0.29 g (0.83 mmol, 83%).
H2N-Aib-Aca-OMe 10
To 0.36 g (1 mmol) of compound 8, 5 mL of trifluoroacetic acid (TFA) was added and deprotection of BOC group was examined by TLC. After 6 h, TFA was removed under reduced pressure and the residue was neutralized by Et3N.
Boc-Aib-Aca-Aib-Aca-OMe 1
Compound 9 (0.29 g, 0.83 mmol) was mixed in 20 mL of dry DCM in an ice-water bath. Compound 10 was then added to the solution, followed immediately by 0.21 g (1 mmol) of DCC. The reaction was cooled to room temperature and stirred for 48 h. DCM was dried, and the product was mixed with ethyl acetate (40 mL), and dicyclohexylurea (DCU) was filtered off. The organic layer was treated with 2 M HCl (3 × 30 mL), brine (2 × 30 mL), and 1 M sodium carbonate (3 × 30 mL). The extract was then dried by using anhydrous sodium sulfate and concentrated under vacuum. The product was purified by silica gel (100–200 mesh) using n-hexane–ethyl acetate (3:1) as eluent to yield peptide 1 as a white solid. Yield: 0.24 g (0.4 mmol, 48.19%). 1H NMR (400 MHz, DMSO-d6, δ ppm): 9.52 [2H, s, Ar NH], 8.4 [1H, s, Aib NH], 8.07 [1H, bs, Aib NH], 7.91 [1H, b, ArH], 7.69–7.72 [1H, m, ArH], 7.57–7.61 [1H, d, J = 16.02, vinylic CH], 7.46 [1H, b, ArH], 7.29–7.39 [4H, m, ArH], 7.18–7.2 [1H, d, J = 7.92, vinylic CH], 6.92–7.02 [1H, m, ArH], 6.71–6.75 [1H, d, J = 15.26, vinylic CH], 6.48–6.52 [1H, d, J = 16.02, vinylic CH], 3.72 [3H, s, OCH3], 1.48 [6H, s, Aib CβH], 1.38 [9H, s, BOC], 1.22–1.25 [6H, m, Aib CβH]. 13C NMR (125 MHz, DMSO-d6, δ ppm): 173, 166.5, 164.49, 144.51, 139.91, 138.81, 135.02, 134.04, 128.91, 123.21, 122.77, 122.5, 122.08, 121.01, 119.26, 117.75, 78.19, 56.57, 51.49, 28.19, 25.04.
Boc-Aib-AcaH-Aib-AcaH-OMe 2
To the suspension of 1 (0.12 g, 0.2 mmol) in 10 mL of ethyl acetate, 50 mg of Pd/C (10%) was added and stirred vigorously for 8 h under hydrogen atmosphere. Progress of the reduction reaction was examined by TLC. After completion of the reaction, 50 mL of ethyl acetate was added into it and the solution was filtered through a silica bed and the filtrate was dried under vacuum to yield 0.115 g (0.193 mmol, 96.5%) of the pure peptide 2 as a white solid. 1H NMR (500 MHz, CDCl3, δ ppm): 9.25 [1H, s, Ar NH], 8.89 [1H, b, Ar NH], 7.63 [1H, b, ArH], 7.35 [1H, b, ArH], 7.27–7.29 [1H, m, ArH], 7.16–7.2 [1H, m, ArH], 7.09–7.1 [2H, m, ArH], 6.89–6.9 [2H, d, J = 6.48 ArH], 6.09 [1H, bs, Aib NH], 4.98 [1H, s, Aib NH], 3.66 [3H, s, OCH3], 2.88–2.91 [4H, m, 2CH2], 2.59–2.62 [2H, t, CH2], 2.48–2.51 [2H, t, CH2], 1.54 [6H, s, Aib CβH], 1.52 [6H, s, Aib CβH], 1.43 [9H, s, BOC]. 13C NMR (125 MHz, CDCl3, δ ppm): 173.49, 173.21, 172.34, 155.42, 141.42, 138.64, 138.51, 129.19, 129.05, 124.67, 124.02, 120.17, 119.76, 118.36, 117.99, 81.07, 58.8, 57.78, 51.74, 38.38, 35.78, 31.49, 31.05, 29.82, 28.4, 25.78, 25.45.
NMR Experiments
All NMR spectroscopy were done on a 400 MHz Jeol or 500 MHz Bruker spectrometer. Compound concentrations were in the 1–10 mM range in DMSO-d6 and CDCl3 solution.31
FT-IR Experiments
FT-IR spectroscopy in the solid state was performed with a Perkin Elmer Spectrum RX1 spectrophotometer using KBr disk method.
Absorption Spectroscopy
The absorption spectra of peptides were measured on a Perkin Elmer UV/vis spectrometer (Lambda 35) using a quartz cell having 1 cm path length.
Fluorescence Spectroscopy
The fluorescence spectra have been recorded on a Perkin Elmer fluorescent spectrometer (LS 55) using a quartz cell having 1 cm path length. Slits of 2.5/2.5 width were used.
Mass Spectrometry
Mass spectrometry was carried out on a Waters Corporation Q-Tof Micro YA263 high-resolution mass spectrometer by electrospray ionization (positive mode).
Field Emission Scanning Electron Microscopy
Field emission scanning electron microscopy (FE-SEM) was performed to examine the morphologies of the synthesized peptides. A drop of peptide solution was casted on a clean microscopic glass slide and dried under vacuum. The samples were gold-coated, and the images were captured in an FE-SEM apparatus (Jeol Scanning Microscope-JSM-6700F).
Gelation
The peptide 2 (5 mg) was mixed in 1 mL of solvent, and gel was obtained by heating–cooling technique.
Rheology Experiments
To examine the thixotropic behavior and mechanical strength of the gel, we have done rheological measurements on a MCR 102 rheometer (Anton Paar, Modular Compact Rheometer) by a steel parallel plate geometry having 40 mm diameter at 20 °C. The rheometer was attached to a Peltier circulator thermocube to control the temperature accurately. The storage modulus (G′) and loss modulus (G″) of the gel were then recorded by using the setup.31
X-ray Crystallography
Diffraction quality light yellow color crystals of peptide 1 were obtained from methanol–water solution by slow evaporation. Intensity data were collected with Mo Kα radiation by a Bruker APEX-2 CCD diffractometer. Data were processed using Bruker SAINT package. The structure solution and refinement were performed by SHELX97. Refinement of nonhydrogen atoms was performed using anisotropic thermal parameters.31 Crystal data of compound 1: C32H40N4O7, Mw = 592.68, P121/c1, a = 21.5402(19) Å, b = 17.4732(14) Å, c = 19.4743(15) Å, α = 90°, β = 114.956(3)°, γ = 90°, V = 6645.3(10) Å3, Z = 8, dm = 1.185 Mg/m3, K = 100, R1 = 0.0812, and wR2 = 0.1873 for 9841 data with I > 2σ(I). CCDC 1505412 contains the crystallographic data for peptide 1. The data was submitted at the Cambridge Crystallographic Data Centre with CCDC reference 1834467.
Acknowledgments
We acknowledge the CSIR, India for financial assistance (Project No. 02(0206)/14/EMR-II). This work is also supported by CSIR, India (fellowship to M.D., T.D., and D.P.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00832.
Synthesis and characterizations of peptides, 1H NMR, 13C NMR, and Figures S1–S17 (PDF)
Author Contributions
M.D. has synthesized the compounds. M.D., T.D., and D.P. have performed the experimental works. D.H. has done the analysis and written the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hecht S.; Huc I.. Foldamers: Structure, Properties, and Applications; Wiley-VCH: Weinheim, 2007. [Google Scholar]
- Gellman S. H. Foldamers: A Manifesto. Acc. Chem. Res. 1998, 31, 173–180. 10.1021/ar960298r. [DOI] [Google Scholar]
- Hill D. J.; Mio M. J.; Prince R. B.; Hughes T. S.; Moore J. S. A Field Guide to Foldamers. Chem. Rev. 2001, 101, 3893–4012. 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- Karle I. L.; Pramanik A.; Banerjee A.; Bhattacharya S.; Balaram P. ω-Amino Acids in Peptide Design. Crystal Structures and Solution Conformations of Peptide Helices Containing a β-Alanyl-γ-Aminobutyryl Segment. J. Am. Chem. Soc. 1997, 119, 9087–9095. 10.1021/ja970566w. [DOI] [Google Scholar]
- Roy R. S.; Gopi H. N.; Raghothama S.; Karle I. L.; Balaram P. Hybrid Peptide Hairpins Containing α- and ω-Amino Acids: Conformational Analysis of Decapeptides with Unsubstituted β-, γ-, and δ-Residues at Positions 3 and 8. Chem. - Eur. J. 2006, 12, 3295–3302. 10.1002/chem.200500742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R.; Vasudev P. J.; Ananda K.; Raghothama S.; Shamala N.; Karle I. L.; Balaram P. Hybrid Peptides: Expanding the β Turn in Peptide Hairpins by the Insertion of β-, γ-, and δ-Residues. Chem. - Eur. J. 2007, 13, 5917–5926. 10.1002/chem.200601562. [DOI] [PubMed] [Google Scholar]
- Daniels D. S.; Petersson E. J.; Qiu J. X.; Schepartz A. High-Resolution Structure of a β-Peptide Bundle. J. Am. Chem. Soc. 2007, 129, 1532–1533. 10.1021/ja068678n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson E. J.; Schepartz A. Toward β-Amino Acid Proteins: Design, Synthesis, and Characterization of a Fifteen Kilodalton β-Peptide Tetramer. J. Am. Chem. Soc. 2008, 130, 821–823. 10.1021/ja077245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. C.; Zuckermann R. N.; Dill K. A. Folding a Nonbiological Polymer into a Compact Multihelical Structure. J. Am. Chem. Soc. 2005, 127, 10999–11009. 10.1021/ja0514904. [DOI] [PubMed] [Google Scholar]
- Delsuc N.; Leger J. M.; Massip S.; Huc I. Proteomorphous Objects from Abiotic Backbones. Angew. Chem. 2007, 119, 218–221. 10.1002/ange.200603390. [DOI] [PubMed] [Google Scholar]
- Haldar D.; Schmuck C. Metal-free double helices from abiotic backbones. Chem. Soc. Rev. 2009, 38, 363–371. 10.1039/B803553A. [DOI] [PubMed] [Google Scholar]
- Aravinda S.; Shamala N.; Balaram P. Aib Residues in Peptaibiotics and Synthetic Sequences: Analysis of Nonhelical Conformations. Chem. Biodiversity 2008, 5, 1238–1262. 10.1002/cbdv.200890112. [DOI] [PubMed] [Google Scholar]
- Venkatraman J.; Shankaramma S. C.; Balaram P. Design of Folded Peptides. Chem. Rev. 2001, 101, 3131–3152. 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- Choi S. H.; Guzei I. A.; Spencer L. C.; Gellman S. H. Crystallographic Characterization of Helical Secondary Structures in α/β-Peptides with 1:1 Residue Alternation. J. Am. Chem. Soc. 2008, 130, 6544–6550. 10.1021/ja800355p. [DOI] [PubMed] [Google Scholar]
- Guo L.; Chi Y.; Almeida A. M.; Guzei I. A.; Parker B. K.; Gellman S. H. Stereospecific Synthesis of Conformationally Constrained γ-Amino Acids: New Foldamer Building Blocks That Support Helical Secondary Structure. J. Am. Chem. Soc. 2009, 131, 16018–16020. 10.1021/ja907233q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L.; Almeida A. M.; Zhang W.; Reidenbach A. G.; Choi S. H.; Guzei I. A.; Gellman S. H. Helix Formation in Preorganized β/γ-Peptide Foldamers: Hydrogen-Bond Analogy to the α-Helix without α-Amino Acid Residues. J. Am. Chem. Soc. 2010, 132, 7868–7869. 10.1021/ja103233a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M. G.; Thombare V. J.; Katariya M. M.; Veeresh K.; Raja K. M. P.; Gopi H. N. Non-classical helices with cis carbon-carbon double bonds in the backbone: structural features of α,γ-hybrid peptide foldamers. Angew. Chem., Int. Ed. 2016, 55, 7847–7851. 10.1002/anie.201602861. [DOI] [PubMed] [Google Scholar]
- Misra R.; Dey S.; Reja R. M.; Gopi H. N. Artificial β-Double Helices from Achiral γ-Peptides. Angew. Chem., Int. Ed. 2018, 57, 1057–1061. 10.1002/anie.201711124. [DOI] [PubMed] [Google Scholar]
- Cain B. F.; Atwell G. J.; Denny W. A. Potential antitumor agents. 23. 4′-(9-Acridinylamino)alkanesulfonanilide congeners bearing hydrophilic functionality. J. Med. Chem. 1977, 20, 987–996. 10.1021/jm00218a001. [DOI] [PubMed] [Google Scholar]
- Galan B. R.; Reback M. L.; Jain A.; Appel A. M.; Shaw W. J. Electrocatalytic Oxidation of Formate with Nickel Diphosphine Dipeptide Complexes: Effect of Ligands Modified with Amino Acids. Eur. J. Inorg. Chem. 2013, 2013, 5366–5371. 10.1002/ejic.201300751. [DOI] [Google Scholar]
- Moretto V.; Crisma M.; Bonora G. M.; Toniolo C.; Balaram H.; Balaram P. Comparison of the effect of five guest residues on the .beta.-sheet conformation of host (L-val)n oligopeptides. Macromolecules 1989, 22, 2939–2944. 10.1021/ma00197a010. [DOI] [Google Scholar]
- Banerjee A.; Maji S. K.; Drew M. G. B.; Haldar D.; Banerjee A. Amyloid-like fibril-forming supramolecular β-sheets from a β-turn forming tripeptide containing non-coded amino acids: the crystallographic signature. Tetrahedron Lett. 2003, 44, 335–339. 10.1016/S0040-4039(02)02570-4. [DOI] [Google Scholar]
- Maity S.; Jana P.; Haldar D. Fabrication of nanoporous material from a hydrophobic peptide. CrystEngComm 2011, 13, 3064–3071. 10.1039/c0ce00701c. [DOI] [Google Scholar]
- Baral A.; Roy S.; Dehsorkhi A.; Hamley I. W.; Mohapatra S.; Ghosh S.; Banerjee A. Assembly of an Injectable Noncytotoxic Peptide-Based Hydrogelator for Sustained Release of Drugs. Langmuir 2014, 30, 929–936. 10.1021/la4043638. [DOI] [PubMed] [Google Scholar]
- de Luna L. A. V.; da Silva T. H. G.; Nogueira R. F. P.; Kummrow F.; Umbuzeiro G. A. Aquatic toxicity of dyes before and after photo-Fenton treatment. J. Hazard. Mater. 2014, 276, 332–338. 10.1016/j.jhazmat.2014.05.047. [DOI] [PubMed] [Google Scholar]
- Robinson T.; McMullan G.; Marchant R.; Nigam P. Remediation of dyes in textile effluent: a critical review on current treatment technologies with a proposed alternative. Bioresour. Technol. 2001, 77, 247–255. 10.1016/S0960-8524(00)00080-8. [DOI] [PubMed] [Google Scholar]
- Okesola B. O.; Smith D. K. Applying low-molecular weight supramolecular gelators in an environmental setting–selfassembled gels as smart materials for pollutant removal. Chem. Soc. Rev. 2016, 45, 4226–4251. 10.1039/C6CS00124F. [DOI] [PubMed] [Google Scholar]
- Basak S.; Nandi N.; Paul S.; Hamley I. W.; Banerjee A. A tripeptide-based self-shrinking hydrogel for waste-water treatment: removal of toxic organic dyes and lead (Pb2+) ions. Chem. Commun. 2017, 53, 5910–5913. 10.1039/C7CC01774J. [DOI] [PubMed] [Google Scholar]
- Maji S. K.; Banerjee R.; Velmurugan D.; Razak A.; Fun H. K.; Banerjee A. Peptide Design Using ω-Amino Acids: Unusual Turn Structures Nucleated by an N-Terminal Single γ-Aminobutyric Acid Residue in Short Model Peptides. J. Org. Chem. 2002, 67, 633–639. 10.1021/jo010314k. [DOI] [PubMed] [Google Scholar]
- Cheng N.; Hu Q.; Guo Y.; Wang Y.; Yu L. Efficient and Selective Removal of Dyes Using Imidazolium-Based Supramolecular Gels. ACS Appl. Mater. Interfaces 2015, 7, 10258–10265. 10.1021/acsami.5b00814. [DOI] [PubMed] [Google Scholar]
- Nandi S. K.; Maji K.; Haldar D. Self-healing hydrogel from a dipeptide and HCl sensing. ACS Omega 2018, 3, 3744–3751. 10.1021/acsomega.8b00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar R.; Debnath M.; Maji K.; Haldar D. Solvent assisted structural diversity: supramolecular sheet and double helix of a short aromatic γ-peptide. RSC Adv. 2015, 5, 76257–76262. 10.1039/C5RA12831E. [DOI] [Google Scholar]
- Maity S.; Jana P.; Maity S. K.; Haldar D. Fabrication of Hollow Self-Assembled Peptide Microvesicles and Transition from Sphere-to-Rod Structure. Langmuir 2011, 27, 3835–3841. 10.1021/la104461m. [DOI] [PubMed] [Google Scholar]
- Ray S.; Takafuji M.; Ihara H. Amino-acid-based, lipid-directed, in situ synthesis and fabrication of gold nanoparticles on silica: a metamaterial framework with pronounced catalytic activity. Nanotechnology 2012, 23, 495301 10.1088/0957-4484/23/49/495301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.