

Abstract
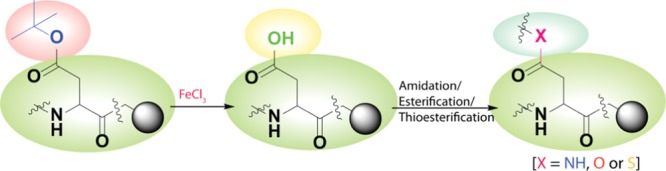
An efficient, convenient, and selective Lewis acid-based strategy for on-resin deprotection of the side chain tert-butyl-protected aspartic acid and glutamic acid of a peptide is achieved. The method is mild, cost-effective, and Fmoc chemistry compatible and allows on-resin incorporation of amides, esters, and thioesters in good yield. This method will find wide applicability in peptide and protein modification because it enriches the toolbox of orthogonal protection/deprotection techniques.
Introduction
Post-translational modification of protein, including phosphorylation, glycosylation, methylation, and lipidation, is a common phenomenon in a biosystem.1 Chemical modification of amino acids in peptides and proteins is essential and attractive for tuning selectivity and efficiency of drugs and understanding the molecular mechanism of their actions.2 Furthermore, site-specific introduction of biochemically active probes, such as a fluorescence probe or a high UV/visible absorbent, on peptides is often needed for monitoring protein conformation,3 structural analyses,4 and reducing immunogenicity. Such chemical modifications are possible by labeling or tagging a suitable moiety at the side chain functionality of amino acids (aspartic acid (Asp), glutamic acid (Glu), lysine (Lys), serine (Ser), etc.) in solution or via solid-phase peptide synthesis (SPPS) technology.5 For example, cysteine and lysine side chain modifications by installation of various electrophiles are well-known.6
SPPS technology is preferred over the synthesis in solution mainly to avoid chromatographic purification at each step among other operational advantages. Usually, Fmoc/tert-butyl (tBu) orthogonal protection technique-based SPPS is preferred over the tert-butyloxycarbonyl (Boc)/benzyl (Bzl) strategy to avoid highly corrosive trifluoroacetic acid (TFA) at each step of Boc deprotection and the extremely dangerous hydrofluoric acid for the final cleavage of the peptide from the solid support.7,8 On-resin site-specific peptide modification demands a third degree of orthogonality (terminal amine/side chain/resin). For example, benzyloxycarbonyl (Cbz)-protected Lys and Bzl-protected Asp or Glu, both at side chain, can be safely modified during Fmoc/tBu-based SPPS, as Cbz/Bzl can be removed using Pd-catalyzed hydrogenation9 keeping both base-labile Fmoc and acid-labile tBu/resin unaffected. However, side chain Bzl-protected Asp/Glu is at a risk of formation of not only aspartimide/glutamide peptides and piperidine derivatives but also 1,4-diazepine-2,5-dione-peptides, making the purification difficult and resulting in yield reduction.10 Similarly, Alloc/Allyl groups can be cleaved easily using excess phenylsilane and catalytic amount of tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] under an inert atmosphere11 and subsequently modified suitably.
Although efficient, this method is highly reagent-intensive and failure of complete oxygen removal may poison the palladium catalyst, leading to undesirable side reactions.12 The 4-{N-[1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl]amino}benzyl ester (Dmab)13 group was introduced in 1995 as an orthogonal protecting group in Fmoc/tBu chemistry to overcome the mentioned problems. Dmab contains the 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl group, which can be removed using 2% hydrazine within a second, but removal of the remaining aminobenzyl ester part is very slow. Payne et al. noticed that removal of the Dmab group from a resin-bound peptide needed 12 h in the presence of hydrazine and 20% N,N-diisopropylethylamine (DIPEA) in 9:1 (v/v) dimethylformamide (DMF)/H2O; however, in the presence of 5 mM NaOH, 3 h reaction time was enough.14 Ruczyński et al. observed that whereas side chain Dmab-protected Asp resulted in 72% aspartimide, 27% aspartyl methyl ester, and 12% aspartyl peptide, tBu-protected Asp yielded 100% aspartimide-free peptide.15 Similarly, Johnson et al. reported that pyroglutamide formation is more during the elongation of the peptide sequence in the presence of Dmab-protected Glu, whereas tBu-protected Glu did not produce the same.16 They also observed a side reaction due to slow 1,6 elimination of the 4-aminobenzyl ester group during hydrazine treatment. McMurray et al. introduced 2,4-dimethoxybenzyl (Dmb) group for on-resin head-to-tail cyclization.17 Dmb can be removed in the presence of 1% TFA for 30 min, but a less-acid-sensitive TFA/phenol (95:5, overnight)-cleavable aminomethylated polystyrene or a similar resin is required. Even the Fmoc/Fm group can be used for selective side chain modification, but this can be used only in Boc/Bzl-based SPPS.8 All of the three-dimensional orthogonal protection strategies have their own advantages and disadvantages. Therefore, development of a milder, cost-effective, and environmentally friendly method for the purpose is the need of the hour.
Most of the mentioned suitably protected Asp or Glu derivatives are either unavailable or costly. Therefore, we wanted to use relatively cheap and readily available Fmoc-Asp(OtBu)-OH or Fmoc-Glu(OtBu)-OH for on-resin peptide modification compatible to environmentally friendly Fmoc/tBu chemistry. The tBu group is a widely used acid-labile protecting group, which is usually cleaved during the final cleavage of the peptide from the resin by TFA.18 It can also be deprotected in the presence of protic acids, including HCl,19 H2SO4,20 HNO3,21 TFA,22 Lewis acids, for example; TiCl4;23 silyl triflates;24 ZnBr2 in CH2Cl2;25 and CeCl3·7H2O–NaI in acetonitrile.26 Most of these methods suffer from many disadvantages, including long reaction time, unsatisfactory yields, etc., and thus are not compatible to SPPS. More recently, Martín and Padrón et al.27 reported removal of the Boc group from N,N′-protected amino acids and amines using ferric chloride (FeCl3), which provided good yield (Scheme 1). However, these methods are restricted to solution-phase chemistry only. Also, the resins used for Fmoc chemistry are acid-labile. Therefore, selective removal of the tert-butyl group keeping the peptide intact on the resin is a challenge. We disclose herein an efficient environmentally friendly FeCl3-based method that can be applied on both the solution phase and Fmoc/tBu-based SPPS for side chain carboxylic acid group deprotection of Asp or Glu and subsequent modification, keeping the peptide intact on resin, including a commonly used methylbenzhydrylamine linker. The advantage of using FeCl3 in solid-phase tert-butyl ester hydrolysis is that it gives the desired product within 1.5 h and it can be easily removed from the resin by washing with DMF among others.
Scheme 1. Overview of the Work.

Results and Discussion
Optimization of the Cleavage Condition
To optimize the reaction conditions, we took Fmoc-Asp(OtBu)-OMe (A) as a model substrate along with various Lewis acids and solvents (Table 1). The isolated yield of the deprotected product increased, and the reaction time decreased gradually with an increment of the amount of FeCl3 used (entries 1–6, Table 1). FeCl3 (1.5 equiv) produced 80% (isolated by column chromatography) of the deprotected product at room temperature (rt) in dichloromethane (DCM), and this condition was accepted as optimum for reactions in solution.
Table 1. Optimization of the Reaction of Fmoc-Asp(OtBu)-OMea.

| entry | Lewis acids (equiv) | solvent | time | isolated yield (%) |
|---|---|---|---|---|
| 1 | FeCl3 (0.1) | DCM | 24 h | 20 |
| 2 | FeCl3 (0.5) | DCM | 24 h | 45 |
| 3 | FeCl3 (1) | DCM | 2 h | 70 |
| 4 | FeCl3 (1.5) | DCM | 1 h | 80 |
| 5 | FeCl3 (2) | DCM | 30 min | 80 |
| 6 | FeCl3 (5) | DCM | 15 min | 80 |
| 7 | FeCl3 (5) | MeCN | 6 h | 78 |
| 8 | FeCl3 (5) | DMF | 72 h | n.d.b |
| 9 | FeCl3 (5) | tetrahydrofuran (THF) | 72 h | n.d.b |
| 10 | FeCl3 (5) | MeOH | 72 h | n.d.b |
| 11 | ZnCl2 (5) | DCM | 72 h | 73 |
| 12 | ZnBr2 (5) | DCM | 24 h | 76 |
| 13 | CuCl2 (5) | DCM | 72 h | n.d.b |
Reaction condition: A (1 mmol), FeCl3, solvent (2 mL), rt.
Product spot could not be observed in thin-layer chromatography (TLC).
The reaction worked well in acetonitrile also, but 5 equiv of FeCl3 and 6 h time were required. No deprotection was observed till 72 h in DMF, THF, and methanol; the starting material was recovered back instead. Whereas ZnBr2 and ZnCl2 provided comparative yields at a longer reaction time (24 and 72 h, respectively, entries 11 and 12, Table 1), CuCl2 did not react till 72 h (entry 13, Table 1).
Compatibility with Other Protecting Groups
We found that 1.5 equiv of FeCl3 selectively removed tBu from A in the optimized condition without affecting the methyl group (Table 2, a preliminary time-dependent high-performance liquid chromatography (HPLC) analysis revealed the time of conversion of half of the substrate to be approximately 6–7 min; Figures S1–S7, Supporting Information). In similar experiments, the benzyl derivative of A (Fmoc-Asp(OBzl)-OMe (C), Figures S8–S12, Supporting Information), allyl ester of benzoic acid (allyl benzoate (D), Figures S13–S17, Supporting Information), and ethyl ester of benzoic acid (ethyl benzoate (E), Figures S18–S23, Supporting Information) were unaltered till 12 h. We also monitored the stability of other acid-sensitive protecting groups, for example, Trt and Pbf, by HPLC and observed that the Trt group got cleaved, whereas Pbf was intact (Fmoc-Gln(Trt)-OH (F), Figures S24–S28 and Fmoc-Arg(Pbf)-OH (G), Figures S29–S34, Supporting Information). Such HPLC analyses revealed that some protecting groups, such as Bzl, Cbz, All, Alloc, Pbf, ortho-nitrobenzenesulfonyl (o-NBS), Fmoc, Me, and Et are orthogonal to the tBu group with respect to the FeCl3-based cleavage condition. These groups can be used as side chain-protecting groups for suitable amino acids while applying the FeCl3-based cleavage condition during SPPS. Thus, this method indeed opens up a new avenue for orthogonal peptide modification, as desired. However, acid-sensitive groups, such as Boc (for, e.g., lysine), Trt (for O/N/S protection), and tert-butyl ethers (for serine, threonine, and tyrosine), do not resist the reaction conditions under which a tert-butyl ester is cleaved. On the other hand, the FeCl3-based condition can be applied for on-resin cleavage of the mentioned acid-labile groups in the case of Rink amide resin.
Table 2. Compatibility of Some of the Frequently Used Protecting Groups to FeCl3 Condition.
| entry | protecting group | tolerance | removal method | references |
|---|---|---|---|---|
| 1 | Trt | not stable | 1% TFA–DCM | (28) |
| 2 | Pbf | stable | 90% TFA/triisopropylsilane/H2O | (29) |
| 3 | Bzl | stable | hydrogenation | (9) |
| 4 | All/Alloc | stable | Pd(PPh3)4/PhSiH3 | (11) |
| 5 | o-NBS | stable | β-mercaptoethanol/1,8-diazabicyclo[5.4.0]undec-7-ene/DMF | (30) |
| 6 | Fmoc | stable | 20% piperidine/DMF | (7) |
| 7 | Et | stable | LiOH | (31) |
| 8 | Me | stable | LiOH | (31) |
Racemization Study
Next, we synthesized dl-Fmoc-Asp(OtBu)-OMe and l-Fmoc-Asp(OtBu)-OMe. Then, we applied our hydrolysis protocol on them and checked HPLC profiles of the products. dl-Fmoc-Asp(OH)-OMe exhibited two distinct peaks with retention times 17.45 and 20.57 min in HPLC (chiral column, isocratic gradient of 15% 2-propanol in hexane till 40 min), indicating the presence of two enantiomers. However, l-Fmoc-Asp(OH)-OMe exhibited a single peak at 20.83 min, indicating the presence of a single enantiomer (Figure 1). Comparison of these HPLC profiles indicated no detectable racemization caused by this method for enantiomerically pure substrates.
Figure 1.
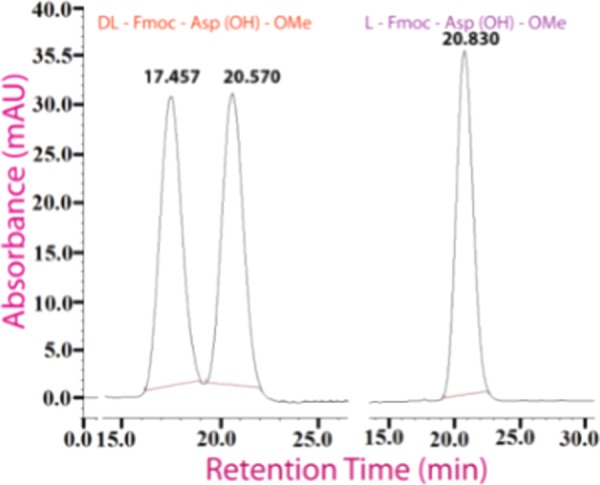
HPLC profile of the reaction products: dl-Fmoc-Asp-OMe (left) and l-Fmoc-Asp-OMe (right).
Introduction of Other Groups after Removal of tBu in SPPS
Next, we synthesized a broad range of model peptides containing various amino acid residues, including side chain tert-butyl ester-protected aspartic acid (Table 3). These peptides are regarded as synthetically difficult peptides as hydrophobic stretches in them are at a risk of on-resin aggregation32 but were important for our anti-Alzheimer’s drug design endeavor.33
Table 3. SPPS of Various Side Chain-Modified Peptides.

| entry | peptides | spectroscopic yield (%) | isolated yield (%) |
|---|---|---|---|
| 1 | Ac-D(X1)PFFA-NH2 | 94 | 40 |
| 2 | Ac-LFD(X2)PG-NH2 | 92 | 37 |
| 3 | Ac-LFD(X3)PG-NH2 | 93 | 38 |
| 4 | Ac-LFD(X4)PG-NH2 | 87 | 35 |
| 5 | Ac-AFLGD(X5)-NH2 | 86 | 32 |
| 6 | Ac-AFLGD(X6)-NH2 | 81 | 28 |
After stepwise synthesis of the whole peptide by the conventional method using (benzotriazole-1-yloxy) tris(dimethylamino)phosphonium hexafluorophosphate (BOP) as the coupling reagent following the Fmoc/tBu orthogonal protection7 technique on the Rink amide AM resin (loading 0.7 mmol/g), the N-terminus of the peptides was acetylated using acetic anhydride and N-methyl morpholine. Then, tBu was removed using FeCl3 (5 equiv; excess reagent was used to facilitate SPPS) in dichloromethane on-resin for 1.5 h from the side chain of the aspartic acid. After that, esterification, thioesterification, or amidation was performed in the presence of BOP (2.5 equiv), DIPEA (5 equiv), and the corresponding alcohol, thiol, or amine (3 equiv) (Scheme 2), respectively, on-resin.
Scheme 2. tert-Butyl Ester Hydrolysis and Side Chain Modification on a Solid Phase.

After completion of the synthesis, the peptides (Table 3) were cleaved from the resin, precipitated from cold ether, and dried. The obtained crude powder was subjected to HPLC analysis. Integration of the peak corresponding to the desired peptide represents its relative abundance with respect to the associated impurities (reported in Table 3 as spectroscopic yield). This is very important in peptide chemistry as it is related to the ease of chromatographic purification and efficiency of synthesis. Crude peptide mixtures were purified by preparative HPLC. White fluffy powders were obtained after lyophilization, and the isolated yields of purified products were calculated from them (reported in Table 3 as isolated yield).
The isolated peptides were characterized by mass spectrometry and one-dimensional (1D) [1H] and two-dimensional (2D) [1H, 1H] NMR spectroscopy. The variable-temperature-dependent 1H NMR experiments revealed that the chemical shift values of the amide-NHs were shifted during increment of temperature from 25 to 60 °C, which confirmed the position of the amide-NHs and the aromatic protons. The coexistence of the phenylalanine residue together with the benzylated thioester of the aspartic acid residue was clearly observed in the NMR spectra of 4 (Figure 2, a representative example). Aromatic protons of the side chain-protected thioester of aspartic acid resonated at 7.28 ppm, and benzylic SCH2 protons resonated at 4.13 ppm, whereas phenylalanine aromatic protons resonated at 7.19–7.15 ppm. Different amino acid residues and their 2D interactions could be identified easily from the 2D total correlation spectroscopy (TOCSY) and nuclear Overhauser enhancement spectroscopy (NOESY) spectra, respectively.
Figure 2.

(a) 1H NMR spectra of peptide 4 in dimethyl sulfoxide (DMSO)-d6. (b) Variable-temperature 1H NMR spectra, showing temperature-dependent amide-NH chemical shifts. (c) Two-dimensional TOCSY spectra, indicating the location of different amino acids. (d) Two-dimensional NOESY spectra, correlations between NH and aliphatic protons of amino acid residues.
Next, we wanted to verify whether the acid-cleavable resin (Rink amide) would be cleaved by FeCl3, which is also acidic in nature. For this, we synthesized a peptide (7, Supporting Information, p 10) that contains the same amino acid sequence as that of 3, except Fmoc-Asp(OBzl)-OH was used instead of Fmoc-Asp(OtBu)-OH. Peptide 7 serves as a reference because Asp(OBzl)-OH was incorporated directly, not from Asp(OtBu)-OH, whereas in the case of peptide 3, the cyclohexyl amine group was inserted after tBu cleavage from preinserted Asp(OtBu)-OH. After the final cleavage, yields (both spectroscopic and isolated) of 3 and 7 were comparable. This indicates that no considerable loss of peptide occurred due to FeCl3 treatment and subsequent modification. Furthermore, we analyzed the washing mixture after FeCl3 treatment but could not find any trace of the cleaved peptide in it. Therefore, we concluded that the Rink amide resin was not cleaved by FeCl3 treatment.
Compatibility with Other Protecting Groups in SPPS
An attractive feature of this protocol is its compatibility to various side chain-protecting groups (Table 2) that are frequently used in Fmoc-based SPPS. To verify this, a model peptide (8, Scheme 3) containing some of those protecting groups was synthesized and then we applied our method for removal of tert-butyl from Asp, followed by incorporation of an important cyclopropylamine34 moiety onto it on a solid support. Analysis after cleavage of the peptide from the solid support revealed that the All, Alloc, o-NBs, and Fmoc groups remained unchanged. In peptide 8, we attached the side chain carboxylic acid group of the first Asp to the Rink amide resin to selectively address each of the Asp residue present in it after cleavage from the resin.
Scheme 3. Model Peptide Containing Various Protecting Groups during SPPS.

One of the advantages of this new strategy is that it enables introduction of multiple groups at the side chains of the tert-butyl ester-protected aspartic acids or glutamic acids directly on a peptidic resin. After introducing one substituent onto the first aspartic acid or glutamic acid, the peptide chain can be elongated and a second substituent can be introduced onto another aspartic or glutamic acid residue. We chose a partial sequence of the α-synuclein peptide, that is, α-synuclein (110–115), to demonstrate this. The α-synuclein peptide is known as the main culprit for Parkinson’s disease and also belongs to the class of difficult sequences. We synthesized a model peptide containing a thioester, an amide, and an ester group on two glutamic acids and an aspartic acid, respectively, all on-resin (Scheme 4).
Scheme 4. Esterification/Thioesterification and Amidation Reaction Performed on α-Synuclein (110–115).

Aspartimide or glutamide formation under basic conditions is a common problem in usual peptide synthesis. Therefore, proline was used as the preceding residue of the Asp or Glu residues in peptides (1–4, 7, and 8), eliminating the possibility of aspartimide or glutamide side product formation. However, for the synthesis of 9 (Scheme 4), proline or N-alkylated amino acid was not used as other amino acids often follow Asp or Glu in biological system. However, we used 20% piperidine/DMF with 5% formic acid35 for Fmoc cleavage because aspartimide or glutamide formation is usually higher in this step. Moreover, there is a chance of aspartimide formation at the time of conversion of the deprotected carboxylic acid to esters or thioesters, as coupling reagents activate the carboxylic acid group. Furthermore, benzylic ester and thiobenzyl ester (end products) also are prone to aspartimide formation under basic conditions. The amount of added base was carefully maintained not to exceed the required amount to suppress aspartimide formation. With all of these precautions, we were delighted to obtain 32% as the spectroscopic yield and 7% as the isolated yield for 9, which is good for such an aspartimide-formation-prone difficult sequence.
Fluorophores, such as dansyl chloride, are widely used in protein conformational studies.36 To demonstrate that such an important moiety can be introduced on resin by our protocol easily, we have synthesized 10, as depicted in Scheme 5. We have obtained 72% spectroscopic yield and 21% isolated yield of 10.
Scheme 5. Fluorophore Attachment on Peptide 10.

Plausible Mechanistic Pathway
A plausible mechanism of deprotection of tert-butyl ester outlined on the basis of the existing literature27,37 is shown in Scheme 6. The Lewis acid Fe(III) first coordinates to the oxygen atoms of the tert-butyl ester group and forms a complex. When iron(III) is coordinated with the oxygen atoms of the tert-butyl ester, the electron density on the oxygen atom gets shifted toward Fe. Then, the carbon–oxygen bond (C–O) becomes week and easily breaks down to form isobutene and the desired deprotected carboxylic acid.
Scheme 6. Plausible Mechanistic Pathway of the Reaction.

Conclusions
In summary, we have developed an efficient, convenient, Lewis acid-based deprotection strategy for modification of tert-butyl ester containing a side chain of aspartic acid and glutamic acid during solid-phase peptide synthesis. The reaction condition is mild and tolerated by many common side chain-protecting groups. The applicability of this approach is in introducing various nucleophiles (both electron-withdrawing and electron-donating groups) and flurophores on the side chain of aspartic acid and glutamic acid using standard coupling methods on-resin without cleaving the peptide from resin. Most importantly, this new method eliminates the effort, cost, time, and waste generation for modification of the amino acids separately, when such amino acid derivatives are not commercially available. One of the most abundant and nontoxic metals, Iron, is used as a strong Lewis acid to deprotect carboxylic acid in a greener manner.
Experimental Section
General Information
All chemicals were purchased from commercial sources. Crude peptides were dissolved in CH3CN/H2O and purified by a Thermo Scientific Dionex UltiMate 3000 Rapid Separation LC system using a C18 Thermo Scientific column or by RP-HPLC (Waters 600E) using a C18-μ Bondapak column at a flow rate of 5 mL/min. A binary solvent system was used: solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in CH3CN). A UV detector was used with an option of dual detection at 214 and 254 nm. A total run time of 20 min was used, and gradient used for purification was 5–100% CH3CN for 18 min followed by 100% CH3CN till 20 min. The purity of the peptides was confirmed using an analytical HPLC system with an analytical column at a flow rate of 1 mL/min, linear gradient of 5–100% CH3CN over 18 min in a total run time of 20 min. Otherwise, specific conditions are mentioned for special cases. Dual wavelength was selected at 214 and 254 nm. The mass of the peptide samples was analyzed on a quadrupole time-of-flight system, in electrospray ionization (ESI)-positive mode, equipped with MassHunter work station software. All 2D NMR spectra were recorded on 600 MHz at 298 K using DMSO-d6 solvent. One-dimensional [1H] spectra were recorded with 16 scans, and 2D [1H, 1H] total correlation spectroscopy (TOCSY), 2D [1H, 1H] nuclear Overhauser enhancement spectroscopy (NOESY), and correlated spectroscopy spectra were recorded with NS = 32 scans, relaxation delay = 2 s, acquisition time = 0.1556 s, spectral width = 6578.9 Hz, and acquired size = 1024 × 217 in both dimensions (F1 and F2). The mixing times are 0.08 s (for TOCSY) and 0.6 s (for NOESY). Chemical shifts were referenced to residual DMSO-d6 at δ = 2.5 ppm in 1H NMR and δ = 39.5 ppm in 13C NMR.
General Procedure for the Removal of tert-Butyl Ester
A stirred solution of tert-butyl ester (1 mmol) in 2 mL of dichloromethane was treated with 1.5 equiv of FeCl3 at room temperature and stirred for 1 h. After completion of the reaction (as indicated by TLC), the solution was diluted with water and extracted with dichloromethane. The organic layer was concentrated under reduced pressure and purified by column chromatography using silica gel and the ethyl acetate–hexane mixture as the eluent.
General Procedure for Peptide Syntheses
Syntheses of the peptides were carried out by the solid-phase peptide synthesis method on the Rink amide AM resin (loading 0.7 mmol/g) following the standard Fmoc/tBu orthogonal protection strategy. The syntheses were performed manually on a Stuart blood tube rotator. The resin was taken into a 2 mL frit-fitted plastic syringe and swollen in dichloromethane (DCM) for 2 h followed by DMF for 1 h. Fmoc amino acids (2 equiv), coupling reagent (2.5 equiv; BOP), and base (5 equiv; DIPEA) were used. Each coupling step was monitored by Kaiser’s test, and in cases of incomplete acylation, coupling cycles were repeated, followed by capping with acetic anhydride (2 equiv) and N-methyl imidazole (3 equiv). Fmoc deprotection was performed with 20% piperidine in DMF mixture for 21 min (7 min × 3). The final peptide was cleaved from the solid support using a cleavage cocktail (90% TFA, 5% DCM, and 5% H2O) for 3 h. After cleavage from the resin, the crude peptide was precipitated by cold diethyl ether followed by centrifugation to achieve crude solid peptide.
Kinetics of Removal of tert-Butyl Ester (A)
To understand the reaction kinetics, we performed HPLC studies at a fixed time interval. We took 1 mmol Fmoc-Asp(OtBu)-OMe (A) and 1.5 equiv of FeCl3 in DCM medium. We took 10 μL of the reaction mixture in an Eppendorf vial at 2 min interval. Then, 5 μL of DIPEA was added to it for quenching the reaction. After that, the solvent was evaporated by passing N2 gas into it and the reaction mixture was diluted by adding HPLC-grade acetonitrile (500 μL) and filtered via 0.22 μm filter paper. The sample (20 μL) from that Eppendorf vial was injected in the HPLC system and the HPLC profile was checked. An overlay of the HPLC profiles revealed a gradual conversion of the tert-butyl ester (A) (tR = 8.6 min) to its free carboxylic acid analogue (B) (tR = 6.8 min). At 6 min, both (A) and (B) existed in 50:50 ratio, and at 20 min, in 95:5 ratio. Then, we checked the HPLC profile up to 1 h, but from 20 min to 1 h, conversion was very slow. After 1 h, we observed 98% conversion from (A) to (B) (see Figures S1–S7, Supporting Information).
Kinetics of Removal of Benzyl Ester (C)
Like tert-butyl ester, we took Fmoc-Asp(OBzl)-OMe (C) for checking whether benzyl ester will be cleaved in the presence of FeCl3. For this purpose, we took 1 mmol (C) and 1.5 equiv of FeCl3 in DCM medium. We took 10 μL of the reaction mixture in an Eppendorf vial from the main bulk after 10 min. Then, 5 μL of DIPEA was added to this for quenching the reaction. After that, the solvent was evaporated from that Eppendorf vial by passing N2 gas and then the reaction mixture was diluted by adding HPLC-grade acetonitrile (500 μL). The sample (20 μL) from this was injected in the HPLC system, and the HPLC profile was checked. From HPLC diagrams, we could not detect any deprotected product till 12 h (see Figures S8–S12, Supporting Information).
Kinetics of Removal of the Allyl Group from Allyl Benzoate (D)
Allyl benzoate (1 mmol; D) was dissolved in DCM, 1.5 equiv of FeCl3 was added to it, and the reaction was kept for 12 h. After 1 h, we took 10 μL of the reaction mixture in an Eppendorf vial and added 5 μL of DIPEA to it for quenching the reaction. Then, the solvent was evaporated by passing N2 gas and the reaction mixture was diluted by adding HPLC-grade acetonitrile (500 μL). The sample (20 μL) from that Eppendorf vial was injected in the HPLC system. After 3, 6, 8, and 12 h, we had done the same treatment. No deprotected free carboxylic acid peak was obtained till 12 h reaction in HPLC (see Figures S13–S17, Supporting Information).
Kinetics of Removal of the Ethyl Ester Group from Ethyl Benzoate (E)
Like for allyl benzoate, a similar experiment had been done on ethyl benzoate, but we could not obtain any free carboxylic acid peak on the HPLC profile (see Figures S18–S23, Supporting Information).
Kinetics of Removal of the Trt Group from Fmoc-Gln(Trt)-OH (F)
Fmoc-Gln(Trt)-OH (1 mmol) was taken in a round-bottom flask and dissolved in DCM, then FeCl3 (1.5 equiv) was added, and the reaction was kept for 4 h. The reaction mixture (10 μL) was taken in an Eppendorf vial after 45 min and then 5 μL of DIPEA was added to it for quenching the reaction. After that, the solvent was evaporated and the reaction mixture was diluted by adding HPLC-grade acetonitrile (500 μL). The solvent (20 μL) was injected in the HPLC system. After 4 h, we obtained fully deprotected amide product, indicated in the HPLC profile (see Figures S24–S28, Supporting Information).
Kinetics of Removal of the Pbf Group from Fmoc-Arg(Pbf)-OH (G)
Fmoc-Arg(Pbf)-OH (1 mmol; G) was dissolved in DCM, then 1.5 equiv of FeCl3 was added to it, and the reaction was kept for 12 h. After 1 h, 10 μL of the reaction mixture was taken in an Eppendorf vial and 5 μL of DIPEA was added to it for quenching the reaction, followed by evaporation of the solvent. Then, it was diluted by adding 500 μL of HPLC-grade acetonitrile, and 20 μL of the sample from that Eppendorf was injected in the HPLC system. HPLC profiles indicated that there was no deprotected product peak till 12 h reaction (see Figures S29–S34, Supporting Information).
Fmoc-Asp-OMe (B)
1H NMR (DMSO-d6, 300 MHz) δ 2.78–2.55 (2H, m); 3.61 (3H, s); 4.32–4.19 (3H, m); 4.44–4.37 (1H, m); 7.35–7.30 (2H, t, J = 7.5 Hz); 7.44–7.39 (2H, t, J = 7.5 Hz); 7.71–7.68 (2H, d, J = 7.2); 7.84 (1H, br); 7.90–7.87 (2H, d, J = 7.8 Hz). 13C NMR (DMSO-d6, 150 MHz) δ 35.8, 46.6, 50.4, 52.2, 65.7, 120.1, 125.2, 127.1, 127.6, 140.7, 143.7, 143.8, 155.8, 171.4, 171.7. high-resolution mass spectrometry (ESI): calcd [M + H]+ 370.1212, found m/z 370.1223.
Peptide 1
The synthesis of peptide 1 was carried out as previously described using Fmoc-Asp(OtBu)-OH, Fmoc-Pro-OH, Fmoc-Phe-OH, and Fmoc-Ala-OH on the Rink Amide AM resin (loading 0.7 mmol/g). After acetylation of the N-terminus of the peptide, the resin was washed three times with DCM. Then, the tert-butyl ester group present on the side chain of the aspartic acid was cleaved using ferric chloride (5 equiv) in dichloromethane for 1.5 h. After completion of the reaction, the resin was washed with dimethylformamide (DMF) 10 times (1 mL × 10 min) to remove the excess FeCl3. Then, the free carboxylic acid was coupled using 4-methoxy benzyl alcohol (3 equiv), BOP (2.5 equiv), and DIPEA (5 equiv) for 3 h. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 600 MHz) δ 1.20 (3H, s); 1.73–1.33 (4H, m); 1.84 (3H, s); 2.76–2.59 (2H, m); 3.09–2.85 (4H, m); 3.60–3.56 (2H, m); 3.71 (3H, s); 4.20–4.19 (1H, m); 4.25 (1H, br); 4.53 (1H, br); 4.43 (1H, br); 5.01 (1H, br); 6.90 (2H, br); 7.26–7.01 (14H, m); 7.99 (1H, br s); 8.00 (1H, br s); 8.47 (1H, br s). 13C NMR (DMSO-d6, 150 MHz) δ 18.2, 22.2, 23.8, 28.8, 36.4, 36.9, 37.3, 46.8, 47.3, 48.1, 53.5, 53.8, 55.4, 59.9, 114.3, 122.6, 126.3, 127.9, 128.1, 129.1, 129.2, 137.6, 143.7, 156.9, 169.0, 169.2, 170.0, 170.3, 170.4, 170.9, 173.9. ESI-mass spectrometry (MS): calcd [M + H]+ 743.3326, found m/z 743.3404. HPLC: retention time (tR) = 11.90 min. Isolated pure product 21 mg (yield: 40% w.r.t. resin loading).
Peptide 2
The synthesis of peptides 2 and 3 was carried out as previously described using Fmoc-Asp(OtBu)-OH, Fmoc-Pro-OH, Fmoc-Phe-OH, Fmoc-Leu-OH, and Fmoc-Gly-OH on the Rink Amide AM resin (loading 0.7 mmol/g). After acetylation of the N-terminus of the peptide, the resin was washed three times (1 mL × 3 min) with DCM. Then, the tert-butyl ester group present on the side chain of the aspartic acid was cleaved using ferric chloride (5 equiv) in dichloromethane for 1.5 h. After completion of the reaction, the resin was washed with dimethylformamide (DMF) 10 times (1 mL × 10 min) to remove the excess FeCl3. Then, the free carboxylic acid was coupled using β-naphthol (peptide 2), cyclohexyl amine (peptide 3) (3 equiv), BOP (2.5 equiv), and DIPEA (5 equiv) for 3 h. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 600 MHz) δ 0.81–0.77 (6H, m); 1.34–1.32 (2H, m); 1.50–1.48 (1H, m); 1.78 (1H, br); 1.81 (3H, s); 2.02–1.86 (3H, m); 2.87–2.84 (1H, m); 3.04–2.95 (2H, m); 3.26–3.22 (1H, m); 3.6 (2H); 4.28–4.22 (2H, m); 5.11–5.09 (1H, m); 7.11 (2H, br s); 7.31–7.20 (6H, m); 7.55–7.51 (3H, m); 7.85–7.84 (1H, d, J = 7.8 Hz); 7.93–7.91 (2H, t, J = 7.8 Hz); 7.98–7.97 (2H, br); 8.15 (1H, br); 8.69 (1H, br). 13C NMR (DMSO-d6, 150 MHz) δ 21.6, 22.4, 22.9, 24.3, 24.1, 29.1, 36.1, 37.3, 40.4, 41.9, 46.9, 47.9, 51.0, 53.5, 60.3, 118.5, 121.4, 125.7, 125.9, 126.3, 126.4, 126.5, 126.6, 127.8, 128.0, 129.2, 134.0, 137.3, 146.3, 168.6, 169.2, 169.3, 170.6, 171.0, 171.5, 171.9. HPLC: retention time (tR) = 12.08 min. ESI-MS: calcd [M + H]+ 715.3377, found m/z 715.3649. Isolated pure product 18 mg (yield: 37%, w.r.t. resin loading).
Peptide 3
1H NMR (DMSO-d6, 600 MHz) δ 0.84–0.78 (6H, m); 1.11–1.08 (4H, m); 1.22–1.21 (3H, m); 1.36–1.32 (2H, t, J = 7.2 Hz); 1.52–1.49 (2H, m); 1.71–1.53 (5H, m); 1.81 (3H, s); 1.92–1.87 (2H, m); 2.08–2.04 (1H, m); 2.44–2.41 (2H, m); 2.71–2.67 (1H, m); 2.80–2.77 (1H, m); 2.95–2.92 (1H, m); 3.48–3.42 (2H, m); 3.65 (2H, br); 4.23–4.20 (2H, m); 4.51–4.48 (1H, m); 4.80–4.76 (1H, m); 6.91 (1H, br s); 7.22–7.12 (6H, m); 7.75–7.74 (1H, d, J = 7.8 Hz); 7.97–7.96 (1H, d, J = 7.8 Hz); 8.03–8.01 (1H, d, J = 7.8 Hz); 8.11 (1H, br); 8.41–8.40 (1H, d, J = 7.8 Hz). 13C NMR (DMSO-d6, 150 MHz) δ 21.5, 22.4, 22.9, 24.1, 24.5, 25.1, 29.1, 32.1, 32.3, 37.3, 37.5, 40.4, 42.1, 47.0, 47.3, 47.7, 51.0, 53.1, 60.3, 126.3, 127.9, 129.2, 137.2, 168.8, 169.3, 170.2, 170.4, 171.0, 171.4, 171.7. HPLC: retention time (tR) = 10.45 min. ESI-MS: calcd [M + H]+ 670.3850, found m/z 670.4146. Isolated pure product 18 mg (yield: 38% w.r.t. resin loading).
Peptide 4
The synthesis of peptide 4 was carried out as previously described using Fmoc-Asp(OtBu)-OH, Fmoc-Pro-OH, Fmoc-Phe-OH, Fmoc-Leu-OH, and Fmoc-Gly-OH on the Rink Amide AM resin (loading 0.7 mmol/g). After acetylation of the N-terminus of the peptide, the resin was washed three times (1 mL × 3 min) with DCM. Then, the tert-butyl ester group present on the side chain of the aspartic acid was cleaved using ferric chloride (5 equiv) in dichloromethane for 1.5 h. After completion of the reaction, the resin was washed with dimethylformamide (DMF) 10 times (1 mL × 10 min) to remove the excess FeCl3. Then, the free carboxylic acid was coupled using benzyl mercaptan (3 equiv), BOP (2.5 equiv), and DIPEA (5 equiv) for 3 h. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 600 MHz) δ 0.84–0.78 (6H, m); 1.34–1.33 (2H, m); 1.50–1.48 (1H, m); 1.81 (3H, s); 1.91–1.77 (2H, m); 2.0–1.91 (2H, m); 2.78–2.85 (2H, m); 2.95–3.11 (2H, m); 3.60–3.54 (4H, m); 4.13 (2H, s); 4.21 (2H, br); 4.47 (1H, br); 4.94 (1H, br); 7.01 (1H, br); 7.09 (1H, br); 7.28–7.15 (10H, m); 7.85 (1H, br); 7.98 (1H, br); 8.05 (1H, br); 8.48 (1H, br). 13C NMR (DMSO-d6, 150 MHz) δ 21.6, 22.4, 22.9, 24.3, 24.1, 29.0, 32.4, 37.3, 40.4, 41.9, 44.4, 46.8, 47.6, 51.0, 53.4, 60.4, 126.3, 127.1, 128.0, 128.5, 128.7, 129.2, 137.3, 137.4, 168.6, 169.4, 170.5, 171.1, 171.5, 171.9, 195.5. ESI-MS: calcd [M + H]+ 695.3149, found m/z 695.3088. HPLC: retention time (tR) = 11.90 min. Isolated pure product 18 mg (yield: 35% w.r.t. resin loading).
Peptide 5
The synthesis of peptides 5 and 6 was carried out as previously described using Fmoc-Asp(OtBu)-OH, Fmoc-Ala-OH, Fmoc-Phe-OH, Fmoc-Leu-OH, and Fmoc-Gly-OH on the Rink Amide AM resin (loading 0.7 mmol/g). After acetylation of the N-terminus of the peptide, the resin was washed three times (1 mL × 3 min) with DCM. Then, the tert-butyl ester group present on the side chain of the aspartic acid was cleaved using ferric chloride (5 equiv) in dichloromethane for 1.5 h. After completion of the reaction, the resin was washed with dimethylformamide (DMF) 10 times (1 mL × 10 min) to remove the excess FeCl3. Then, the free carboxylic acid was coupled using 4-methylbenzenethiol (peptide 5), methyl phenylalaninate (peptide 6) (3 equiv), BOP (2.5 equiv), and DIPEA (5 equiv) for 3 h. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 600 MHz) δ 0.85–0.78 (6H, m); 1.07–1.06 (3H, d, J = 7.2 Hz); 1.50–1.44 (2H, m); 1.56–1.53 (1H, m); 1.79 (3H, s); 2.84–2.78 (1H, m); 2.97–2.91 (1H, m); 3.13–3.02 (2H, m); 3.76–3.66 (1H, m); 4.17–4.12 (1H, q, J = 7.2 Hz); 4.24–4.19 (1H, m); 4.49–4.43 (1H, m); 4.63–4.57 (1H, m); 7.28–7.16 (10H, m); 7.34 (1H, br); 7.99 (2H, br); 8.14–8.04 (3H, m). 13C NMR (DMSO-d6, 100 MHz) δ 17.6, 20.7, 21.3, 22.3, 22.9, 23.8, 36.6, 40.3, 42.0, 44.5, 48.4, 49.2, 51.3, 53.6, 123.5, 126.2, 127.9, 129.1, 129.9, 134.3, 137.5, 139.3, 168.6, 169.6, 171.0, 171.7, 172.4, 172.5. ESI-MS: calcd [M + H]+ 669.2992, found m/z 669.3103. HPLC: retention time (tR) = 11.36 min. Isolated pure product 15 mg (yield: 32% w.r.t. resin loading).
Peptide 6
1H NMR (DMSO-d6, 400 MHz) δ 0.87–0.80 (6H, m); 1.08–1.07 (3H, d, J = 7.2 Hz); 1.49–1.45 (2H, m); 1.57–1.56 (1H, m); 1.79 (3H, s); 2.48–2.42 (1H, m); 2.60–2.55 (1H, m); 3.06–2.77 (4H, m); 3.55 (3H, s); 3.72–3.62 (2H, m); 7.18–4.14 (1H, m); 4.28–4.23 (1H, m); 4.49–4.39 (3H, m); 7.13 (1H, br); 7.12 (1H, br); 7.28–7.14 (10H, m); 8.06–7.99 (5H, m); 8.41–8.40 (1H, d, J = 7.6 Hz). 13C NMR (DMSO-d6, 100 MHz) δ 17.9, 21.5, 22.5, 23.1, 24.0, 36.9, 40.6, 42.2, 48.4, 49.5, 51.3, 51.9, 53.8, 53.9, 126.3, 126.7, 128.1, 128.4, 129.1, 129.3, 137.1, 137.8, 168.5, 169.5, 169.8, 171.0, 172.0, 172.5, 172.6, 172.9. ESI-MS: calcd [M + H]+ 724.3592, found m/z 724.3728. HPLC: retention time (tR) = 6.79 min. Isolated pure product 14 mg (yield: 28% w.r.t. resin loading).
Peptide 7
The synthesis of peptide 7 was carried out as previously described using Fmoc-Asp(OBzl)-OH, Fmoc-Pro-OH, Fmoc-Phe-OH, Fmoc-Leu-OH, and Fmoc-Gly-OH, on the Rink Amide AM resin (loading 0.7 mmol/g). After the synthesis of the whole peptide sequence, the N-terminus of the peptide was acetylated. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 600 MHz) δ 0.83–0.77 (6H, m); 1.34–1.31 (2H, t, J = 7.2 Hz); 1.50–1.46 (1H, m); 1.78–1.74 (2H, m); 1.80 (3H, s); 2.0–1.87 (2H, m); 2.60–2.56 (1H, m); 2.81–2.78 (1H, m); 2.87–2.83 (1H, m); 2.98–2.96 (1H, m); 3.45–3.41 (1H, m); 3.55–3.52 (1H, m); 3.62–3.57 (2H, m); 4.23–4.20 (2H, m); 4.50–4.46 (1H, m); 4.91–4.88 (1H, m); 5.09–5.04 (2H, m); 7.00 (1H, br); 7.10 (1H, br); 7.18–7.15 (3H, m); 7.23–7.21 (2H, m); 7.38–7.31 (5H, m); 7.86–7.84 (1H, d, J = 7.8 Hz); 7.97–7.96 (1H, d, J = 7.8); 8.06 (1H, br); 8.47–8.46 (1H, d, J = 7.8 Hz). 13C NMR (DMSO-d6, 150 MHz) δ 21.6, 22.4, 22.9, 24.1, 24.3, 29.1, 35.8, 37.3, 40.5, 42.0, 46.8, 47.5, 51.1, 53.4, 65.9, 60.3, 126.3, 128.0, 128.1, 128.2, 128.4, 129.3, 135.9, 137.3, 168.9, 169.4, 170.2, 170.4, 171.1, 171.5, 171.9. ESI-MS: calcd [M + H]+ 679.3377, found m/z 679.3343. HPLC: retention time (tR) = 11.37 min. Spectroscopic yield = 96%. Isolated pure product 21 mg (yield: 45% w.r.t. resin loading).
Peptide 8
The synthesis of peptide 8 was carried out as previously described using Fmoc-Asp(OH)-OAll, Fmoc-Lys(o-NBS)-OH, Fmoc-Lys(Alloc)-OH, Fmoc-Pro-OH, and Fmoc-Asp(OtBu)-OH, on the Rink Amide AM resin (loading 0.7 mmol/g). At first, Fmoc-Asp(OH)-OAll was coupled with the resin and then the peptide synthesis was continued using the conventional coupling method. Then, the tert-butyl ester group present on the side chain of the aspartic acid was cleaved using ferric chloride (5 equiv) in dichloromethane for 1.5 h. After completion of the reaction, the resin was washed with dimethylformamide (DMF) 10 times (1 mL × 10 min) to remove the excess FeCl3. Then, the free carboxylic acid was coupled using cyclopropylamine (5 equiv), BOP (2.5 equiv), and DIPEA (3 equiv) for 3 h. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 600 MHz) δ 0.56–0.32 (4H, m); 1.40–1.17 (9H, m); 1.86–1.62 (7H, m); 2.38–2.36 (2H, m); 2.62–2.54 (2H, m); 2.94–2.81 (4H, m); 3.73–3.63 (2H, m); 4.30–4.06 (5H, m); 4.43–4.42 (2H, d, J = 4.8 Hz); 4.51–4.50 (2H, d, J = 4.8 Hz); 4.62–4.56 (2H, m); 5.15–5.13 (2H, d, J = 10.2 Hz); 5.28–5.23 (2H, m); 5.90–5.80 (2H, m); 6.93 (1H, br); 7.17 (1H, br); 7.44–7.30 (5H, m); 7.75–7.68 (3H, m); 7.89–7.84 (4H, m); 8.06–8.05 (2H, m); 8.13 (1H, br); 8.18 (1H, br). ESI-MS: calcd [M + H]+ 1171.4692, found m/z 1171.4759. HPLC: retention time (tR) = 8.70 min. Spectroscopic yield = 71%. Isolated pure product 20 mg (yield: 24% w.r.t. resin loading).
Peptide 9
The synthesis of peptide 9 was carried out as previously described using Fmoc-Asp(OtBu)-OH, Fmoc-Gly-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, and Fmoc-Glu(OtBu)-OH, on the Rink Amide AM resin (loading 0.7 mmol/g). After coupling of Fmoc-Asp(OtBu)-OH with the resin, we had started the tert-butyl ester cleavage and coupled the free carboxylic acid on the side chain of aspartic acid with benzyl alcohol (5 equiv), BOP (2.5 equiv), and DIPEA (3 equiv) for 3 h. Then, Fmoc-Glu(OtBu)-OH was coupled with the main backbone of the peptide sequence and treated similarly as describe above. Then, we obtained the side chain-coupled amide (using benzyl amine (5 equiv)) product. Then, the peptide sequence was continued, and after the N-terminus of the peptide was acetylated, we did deprotection of tert-butyl ester on the side chain of the second glutamic acid present in the peptide sequence. To obtain thioester-containing side chain-protected second glutamic acid, we have taken benzyl mercaptan (5 equiv), BOP (2.5 equiv), and DIPEA (3 equiv) for 3 h. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 400 MHz) δ 0.80–0.73 (8H, m); 0.85–0.84 (3H, d, J = 6 Hz); 1.06–1.00 (1H, m); 1.22 (3H, s); 1.34 (1H, br); 1.48–1.43 (2H, m); 1.57 (1H, br); 1.69 (1H, br); 1.82 (4H, br); 1.94–1.89 (2H, m); 2.05 (3H, s); 2.20–2.17 (2H, t, J = 7.2 Hz); 2.66–2.60 (2H, m); 2.83–2.77 (1H, m); 3.45 (1H, s); 3.76–3.63 (2H, m); 4.09 (2H, s); 4.25–4.17 (4H, m); 4.56-4.50 (1H, q, J = 7.6 Hz, 6.4 Hz); 5.05 (2H, s); 7.33–7.21 (14H, m); 7.79–7.77 (1H, d, J = 8.4 Hz); 7.97–7.96 (1H, d, J = 6.8 Hz); 8.08–8.02 (2H, m); 8.15–8.14 (1H, d, J = 7.2 Hz). 13C NMR (DMSO-d6, 150 MHz) δ 11.0, 15.3, 21.4, 22.5, 23.0, 24.1, 27.3, 27.8, 29.0, 32.1, 32.2, 35.9, 36.6, 42.1, 49.3, 51.4, 52.0, 52.7, 57.1, 65.7, 118.1, 126.7, 127.2, 127.9, 128.0, 128.3, 128.4, 128.6, 128.7, 135.9, 137.8, 139.1, 168.9, 169.8, 169.9, 170.0, 171.1, 171.3, 171.6, 171.8, 172.1, 172.3, 197.6. ESI-MS: calcd [M + H]+ 1001.4728, found m/z 1001.4726. HPLC: retention time (tR) = 8.29 min. Spectroscopic yield = 32%. Isolated purified product yield 7% w.r.t. resin loading.
Peptide 10
The synthesis of peptide 10 was carried out as previously described using Fmoc-Asp(OtBu)-OH, Fmoc-Pro-OH, Fmoc-Phe-OH, Fmoc-Leu-OH, and Fmoc-Gly-OH on the Rink Amide AM resin (loading 0.7 mmol/g). After acetylation of the N-terminus of the peptide, the resin was washed three times (1 mL × 3 min) with DCM. Then, the tert-butyl ester group present on the side chain of the aspartic acid was cleaved using ferric chloride (5 equiv) in dichloromethane for 1.5 h. After completion of the reaction, the resin was washed with dimethylformamide (DMF) 10 times (1 mL × 10 min) to remove the excess FeCl3. Then, the free carboxylic acid was coupled by one side of ethylene diamine (3 equiv), BOP (2.5 equiv), and DIPEA (5 equiv) for 3 h. After washing with DMF (1 mL × 5 min), the other side of diethylene amine was coupled with dansyl chloride (3 equiv) using base DIPEA (5 equiv) for 3 h. After completion of the reaction, the peptide-anchored resin was washed several times with DMF followed by DCM. Then, the peptide was cleaved from the resin by the cleavage cocktail. TFA was evaporated. The crude product was precipitated by cold diethyl ether, purified by preparative HPLC, and analyzed and characterized by HPLC, mass spectrometry, and 1D and 2D NMR spectroscopy.
1H NMR (DMSO-d6, 600 MHz) δ 0.83–0.78 (6H, m); 1.32 (2H, br s); 1.49 (1H, br); 1.80 (3H, s); 1.85 (2H, br); 2.08 (1H, br); 2.33 (1H, br); 2.63 (1H, br); 2.81–2.77 (2H, m); 2.85 (6H, s); 3.07–2.92 (3H, m); 3.45–3.43 (1H, m); 4.19 (4H, br); 4.47 (1H, br); 4.74 (1H, br); 6.91 (1H, br); 7.07 (1H, br); 7.31–7.12 (6H, m); 7.62 (2H, br); 7.76 (1H, br); 8.12–7.96 (5H, m); 8.29 (1H, br); 8.35 (1H, br); 8.46 (1H, br). 13C NMR (DMSO-d6, 150 MHz) δ 21.5, 22.4, 22.9, 24.1, 29.1, 37.3, 37.4, 40.4, 41.6, 42.1, 42.1, 45.2, 47.0, 47.3, 51.1, 53.2, 60.3, 115.5, 119.5, 123.1, 126.3, 127.9, 128.3, 129.0, 129.3, 136.5, 137.2, 158.2, 169.4, 170.0, 170.2, 170.3, 171.1, 171.5, 171.8. ESI-MS: calcd [M + H]+ 864.4000, found m/z 864.4067. HPLC: retention time (tR) = 10.68 min. Spectroscopic yield = 72%. Isolated pure product 13 mg (yield: 21% w.r.t. resin loading).
Acknowledgments
We are thankful to Central Instruments Facility (CIF), IITG, for NMR and the Department of Biotechnology, Govt. of India, for financial support (twinning program for the North Eastern Region, sanction no. BT/PR16164/NER/95/88/2015). We are also thankful to Dr. D. Das, Department of Chemistry, IITG for lyophilization facility and DST FIST program support for ESI-MS facility.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01143.
Synthetic schemes and 2D NMR data; copies of the HPLC profiles; HPLC spectra for racemization study; copies of characterization spectra for all of the synthesized peptides (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wells L.; Vosseller K.; Hart G. W. Glycosylation of Nucleocytoplasmic Proteins: Signal Transduction and O-GlcNAc. Science 2001, 291, 2376–2378. 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- Liu W. R.; Wang Y.-S.; Wan W. Synthesis of proteins with defined posttranslational modifications using the genetic noncanonical amino acid incorporation approach. Mol. BioSyst. 2011, 7, 38–47. 10.1039/C0MB00216J. [DOI] [PubMed] [Google Scholar]
- Hancock J. F.; Paterson H.; Marshall C. J. A Polybasic Domain or Palmitoylation Is Required in Addition to the CAAX Motif to Localize p21ras to the Plasma Membrane. Cell 1990, 63, 133–139. 10.1016/0092-8674(90)90294-O. [DOI] [PubMed] [Google Scholar]
- Marks J. R.; Placone J.; Hristova K.; Wimley W. C. Spontaneous Membrane-Translocating Peptides by Orthogonal High-Throughput Screening. J. Am. Chem. Soc. 2011, 133, 8995–9004. 10.1021/ja2017416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- a Calce E.; Leone M.; Monfregola L.; De Luca S. Chemical Modifications of Peptide Sequences via S-Alkylation Reaction. Org. Lett. 2013, 15, 5354–5357. 10.1021/ol402637d. [DOI] [PubMed] [Google Scholar]; b Wollack J. W.; Zeliadt N. A.; Mullen D. G.; Amundson G.; Geier S.; Falkum S.; Wattenberg E. V.; Barany G.; Distefano M. D. Multifunctional Prenylated Peptides for Live Cell Analysis. J. Am. Chem. Soc. 2009, 131, 7293–7303. 10.1021/ja805174z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin I.; Beyerman M.; Bienert M. Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. 10.1038/nprot.2007.454. [DOI] [PubMed] [Google Scholar]
- Muttenthaler M.; Albericio F.; Dawson P. E. Methods, setup and safe handling for anhydrous hydrogen fluoride cleavage in Boc solid-phase peptide synthesis. Nat. Protoc. 2015, 10, 1067–1083. 10.1038/nprot.2015.061. [DOI] [PubMed] [Google Scholar]
- Bajwa J. S. Chemoselective Deprotection of Benzyl Esters in the Presence of Benzyl Ethers, Benzyloxymethyl Ethers and N-Benzyl Groups by Catalytic Transfer Hydrogenation. Tetrahedron Lett. 1992, 33, 2299–2302. 10.1016/S0040-4039(00)74195-5. [DOI] [Google Scholar]
- Süli-Vargha H.; Schlosser G.; Ilaš J. 1, 4-Diazepine-2, 5-dione ring formation during solid phase synthesis of peptides containing aspartic acid β-benzyl ester. J. Pept. Sci. 2007, 13, 742–748. 10.1002/psc.885. [DOI] [PubMed] [Google Scholar]
- Grieco P.; Gitu P. M.; Hruby V. J. Preparation of ‘side-chain-to side-chain’ cyclic peptides by Allyl and Alloc strategy: potential for library synthesis. J. Pept. Res. 2001, 57, 250–256. 10.1111/j.1399-3011.2001.00816.x. [DOI] [PubMed] [Google Scholar]
- a Grieco P.; Han G.; Weinberg D.; MacNeil T.; Van der Ploeg L. H. T.; Hruby V. J. Design and Synthesis of Highly Potent and Selective Melanotropin Analogues of SHU9119 Modified at Position 6. Biochem. Biophys. Res. Commun. 2002, 292, 1075–1080. 10.1006/bbrc.2002.6739. [DOI] [PubMed] [Google Scholar]; b Guo H.; Gallazzi F.; Miao Y. Gallium-67-Labeled Lactam Bridge-Cyclized Alpha-MSH Peptides with Enhanced Melanoma Uptake and Reduced Renal Uptake. Bioconjugate Chem. 2012, 23, 1341–1348. 10.1021/bc300191z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W. C.; Bycroft B. W.; Evans D. J.; White P. D. A Novel 4-Aminobenzyl Ester-based Carboxy-protecting Group for Synthesis of Atypical Peptides by Fmoc-But Solid-phase Chemistry. J. Chem. Soc., Chem. Commun. 1995, 2209–2210. 10.1039/c39950002209. [DOI] [Google Scholar]
- Conroy T.; Jolliffe K. A.; Payne R. J. Efficient use of the Dmab protecting group: applications for the solid-phase synthesis of N-linked glycopeptides. Org. Biomol. Chem. 2009, 7, 2255–2258. 10.1039/b821051a. [DOI] [PubMed] [Google Scholar]
- Ruczyński J.; Lewandowska B.; Mucha P.; Rekowski P. Problem of aspartimide formation in Fmoc-based solid-phase peptide synthesis using Dmab group to protect side chain of aspartic acid. J. Pept. Sci. 2008, 14, 335–341. 10.1002/psc.941. [DOI] [PubMed] [Google Scholar]
- Johnson T.; Liley M.; Cheeseright T. J.; Begum F. Problems in the synthesis of cyclic peptides through use of the Dmab protecting group. J. Chem. Soc., Perkin Trans. 1 2000, 2811–2820. 10.1039/b001694m. [DOI] [Google Scholar]
- McMurray J. S. Solid Phase Synthesis of a Cyclic Peptide Using Fmoc Chemistry. Tetrahedron Lett. 1991, 32, 7679–7682. 10.1016/0040-4039(91)80563-L. [DOI] [Google Scholar]
- a Greene T. W.; Wuts P. G. M.. Protective Groups in Organic Synthesis, 3rd ed.; John Wiley and Sons: New York, 1999. [Google Scholar]; b Kocienski P. J.Protecting Groups; Thieme: Stuttgart, 1994. [Google Scholar]
- Gibson F. S.; Bergmeier S. C.; Rapoport H. Selective Removal of an N-BOC Protecting Group in the Presence of a tert-Butyl Ester and Other Acid-Sensitive Groups. J. Org. Chem. 1994, 59, 3216–3218. 10.1021/jo00090a045. [DOI] [Google Scholar]
- Strazzolini P.; Misuri N.; Polese P. Efficient cleavage of carboxylic tert-butyl and 1-adamantyl esters, and N-Boc-amines using H2SO4 in CH2Cl2. Tetrahedron Lett. 2005, 46, 2075–2078. 10.1016/j.tetlet.2005.01.129. [DOI] [Google Scholar]
- Strazzolini P.; Dall’Arche M. G.; Giumanini A. G. Nitrolysis of Carboxylic t-Butyl and 1-Adamantyl Esters. Tetrahedron Lett. 1998, 39, 9255–9258. 10.1016/S0040-4039(98)02079-6. [DOI] [Google Scholar]
- Srinivasan N.; Yurek-George A.; Ganesan A. Rapid deprotection of N-Boc amines by TFA combined with freebase generation using basic ion-exchange resins. Mol. Diversity 2005, 9, 291–293. 10.1007/s11030-005-4386-8. [DOI] [PubMed] [Google Scholar]
- Valencic M.; van der Does T.; de Vroom E. Titanium Tetraehloride Promoted Hydrolysis of Cephalosporin tert-Butyl Esters. Tetrahedron Lett. 1998, 39, 1625–1628. 10.1016/S0040-4039(97)10857-7. [DOI] [Google Scholar]
- Jones A. B.; Villalobos A.; Linde R. G. II; Danishefsky S. J. A Formal Synthesis of FK-506. Exploration of Some Alternatives to Macrolactamization. J. Org. Chem. 1990, 55, 2786–2797. 10.1021/jo00296a042. [DOI] [Google Scholar]
- Kaul R.; Brouillette Y.; Sajjadi Z.; Hansford K. A.; Lubell W. D. Selective tert-Butyl Ester Deprotection in the Presence of Acid Labile Protecting Groups with Use of ZnBr2. J. Org. Chem. 2004, 69, 6131–6133. 10.1021/jo0491206. [DOI] [PubMed] [Google Scholar]
- Marcantoni E.; Massaccesi M.; Torregiani E.; et al. Selective Deprotection of N-Boc-Protected tert-Butyl Ester Amino Acids by the CeCl3,7H2O–NaI System in Acetonitrile. J. Org. Chem. 2001, 66, 4430–4432. 10.1021/jo010010y. [DOI] [PubMed] [Google Scholar]
- López-Soria J. M.; Pérez S. J.; Hernández J. N.; Ramírez M. A.; Martín V. S.; Padrón J. I. A practical, catalytic and selective deprotection of a Boc group in N,N′-diprotected amines using iron(III)-catalysis. RSC Adv. 2015, 5, 6647–6651. 10.1039/C4RA12143K. [DOI] [Google Scholar]
- Barlos K.; Mamos P.; Papaioannou D.; Patrianakou S.; Sanida C.; Schäfer W. Einsatz von Trt- und Fmoc-Gruppen zum Schutz polyfunktioneller α-Aminosauren. Liebigs Ann. Chem. 1987, 1987, 1025–1030. 10.1002/jlac.198719870868. [DOI] [Google Scholar]
- Carpino L. A.; Shroff H.; Triolo S. A.; Mansour E.-S. M. E.; Wenschuh H.; Albericio F. The 2,2,4,6,7-Pentamethyldihydrobenzofuran-5-sulfonyl Group (Pbf) as Arginine Side Chain Protectant. Tetrahedron Lett. 1993, 34, 7829–7832. 10.1016/S0040-4039(00)61487-9. [DOI] [Google Scholar]
- Poreddy A. R.; Schall O. F.; Marshall G. R.; Ratledge C.; Slomczynska U. Solid-Phase Synthesis of Methyl Carboxymycobactin T 7 and Analogues as Potential Antimycobacterial Agents. Bioorg. Med. Chem. Lett. 2003, 13, 2553–2556. 10.1016/S0960-894X(03)00473-6. [DOI] [PubMed] [Google Scholar]
- Dayal B.; Salen G.; Toome B.; Tint G. S.; Shefer S.; Padia J. Lithium hydroxide/aqueous methanol: mild reagent for the hydrolysis of bile acid methyl esters. Steroids 1990, 55, 233–237. 10.1016/0039-128X(90)90021-3. [DOI] [PubMed] [Google Scholar]
- Bacsa B.; Horváti K.; Bõsze S.; Andreae F.; Kappe C. O. Solid-Phase Synthesis of Difficult Peptide Sequences at Elevated Temperatures: A Critical Comparison of Microwave and Conventional Heating Technologies. J. Org. Chem. 2008, 73, 7532–7542. 10.1021/jo8013897. [DOI] [PubMed] [Google Scholar]
- a Nadimpally K. C.; Paul A.; Mandal B. Reversal of Aggregation Using β-Breaker Dipeptide Containing Peptides: Application to Aβ(1–40) Self-Assembly and Its Inhibition. ACS Chem. Neurosci. 2014, 5, 400–408. 10.1021/cn500064z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paul A.; Nadimpally K. C.; Mondal T.; Thalluri K.; Mandal B. Inhibition of Alzheimer’s amyloid-β peptide aggregation and its disruption by a conformationally restricted α/β hybrid peptide. Chem. Commun. 2015, 51, 2245–2248. 10.1039/C4CC09063B. [DOI] [PubMed] [Google Scholar]
- Chanthamath S.; Nguyen D. T.; Shibatomi K.; Iwasa S. Highly Enantioselective Synthesis of Cyclopropylamine Derivatives via Ru(II)-Pheox-Catalyzed Direct Asymmetric Cyclopropanation of Vinylcarbamates. Org. Lett. 2013, 15, 772–775. 10.1021/ol303404c. [DOI] [PubMed] [Google Scholar]
- Michels T.; Dolling R.; Haberkorn U.; Mier W. Acid-Mediated Prevention of Aspartimide Formation in Solid Phase Peptide Synthesis. Org. Lett. 2012, 14, 5218–5221. 10.1021/ol3007925. [DOI] [PubMed] [Google Scholar]
- Weber G. Polarization of the Fluorescence of Macromolecules. 2. Fluorescent conjugates of ovalbumin and bovine serum albumin. Biochem. J. 1952, 51, 155–167. 10.1042/bj0510155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navath R. S.; Pabbisetty K. B.; Hu L. Chemoselective deprotection of N-Boc group in amino acids and peptides by bismuth(III) trichloride. Tetrahedron Lett. 2006, 47, 389–393. 10.1016/j.tetlet.2005.11.003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.