

Abstract
The protein cereblon serves as a substrate receptor of a ubiquitin ligase complex that can be tuned toward different target proteins by cereblon-binding agents. This approach to targeted protein degradation is exploited in different clinical settings and has sparked the development of a growing number of thalidomide derivatives. Here, we probe the chemical space of cereblon binding beyond such derivatives and work out a simple set of chemical requirements, delineating the metaclass of cereblon effectors. We report co-crystal structures for a diverse set of compounds, including commonly used pharmaceuticals, but also find that already minimalistic cereblon-binding moieties might exert teratogenic effects in zebrafish. Our results may guide the design of a post-thalidomide generation of therapeutic cereblon effectors and provide a framework for the circumvention of unintended cereblon binding by negative design for future pharmaceuticals.
Introduction
Thalidomide had a changeful career. Marketed as a sedative and hypnotic, it was considered a safe agent to alleviate morning sickness of pregnant women, until it had to be understood that a sudden wave of severe birth defects had to be ascribed to its administration. However, because of its immunomodulatory, anti-inflammatory, and anti-angiogenic properties, thalidomide continued raising attention as a promising agent in the treatment of a growing range of clinical conditions over the years after its withdrawal,1−4 sparking the development of derivatives as anti-cancer agents, commonly referred to as immunomodulatory drugs (IMiDs). Especially lenalidomide, the most prominent IMiD, has been highly successful in the treatment of multiple myeloma and other B cell malignancies but also in myelodysplastic syndrome.5
Thalidomide and related IMiDs mediate their effects by binding to the protein cereblon, a substrate receptor of the CRL4A E3 ubiquitin ligase complex.6 The IMiD-binding site is situated in the C-terminal domain, the thalidomide-binding domain of human cereblon, which is also termed CULT domain.7 Other domains of cereblon or components of the E3 ligase complex are not involved in IMiD binding. The architecture of the binding pocket is highly conserved across species, from bacterial to mammalian cereblon proteins.8−10 It contains three strictly conserved tryptophan residues that form a rectangular cage upon ligand binding—in unliganded state, the thalidomide-binding domain was reported to be unfolded to large extents.11 Thalidomide and its derivatives have their cereblon-binding moiety, a glutarimide ring, slotted into this cage, while the remainder of the molecule protrudes from the binding pocket. This protruding moiety contributes to the molecular interface formed by the surface around the folded binding pocket and thereby modulates the substrate spectrum of the cereblon-CRL4A E3 ubiquitin ligase complex. The binding of effector molecules and also mutation of the binding pocket can thereby preclude the recognition and processing of endogenous substrates, as shown for the homeobox transcription factor MEIS2,9 ion channels,12 and the amyloid precursor protein.13
Contrariwise, the molecular interface formed upon IMiD binding mediates the recognition and ubiquitination of neosubstrates, such as the B cell-specific zinc-finger transcription factors IKZF1 and IKZF3,14−16 casein kinase 1A1 (CK1α),17 and the translation termination factor GSPT1,18 which are targeted via different IMiDs. The identification of these neo-substrates provided a rationale for the efficacy of IMiDs in multiple myeloma (IKFZ1 and IKFZ3), 5q-deletion associated myelodysplastic syndrome (CK1α), and acute myeloid leukemia (GSPT1). Notably, these different neo-substrates are recognized and bound via a common structural degron, a zinc finger motif for IKFZ1 and IKFZ3 or a zinc finger-like loop for CK1α and GSPT1. This degron is selectively recognized by the molecular interface formed around the IMiD-binding pocket, with a specificity that is fine-tuned by the protruding moiety of the bound IMiD.18,19 Furthermore, beyond classical IMiDs, targeted degradation via cereblon is currently investigated in a so-called PROTAC (proteolysis targeting chimera) approach. PROTACs are designed small molecules with a binding moiety for a specific target protein linked to a moiety that is recognized by an E3 ubiquitin ligase complex, providing a simple means of proximity-induced ubiquitination. Using thalidomide as a cereblon recruiting moiety, a number of PROTACs for cereblon-targeting have been developed to recruit the cancer targets BCR-ABL,20 BRD4,21 BRD9,22 SIRT2,23 and ERK1/224 for degradation.
For a better understanding of cereblon-mediated substrate recognition and potential teratogenicity, we here aim to characterize the chemical and structural requirements for cereblon effector molecules. Inspired by structural similarity to glutarimide, we have previously verified that the uracil moiety of uridine is bound in the same way and causes the same teratogenic effects in zebrafish as thalidomide.10 These results implied also other compounds with structurally related groups as potential cereblon effectors. In fact, several pharmaceuticals were classified as teratogens merely because of their structural similarity to thalidomide. We have previously established cereblon isoform 4 from Magnetospirillum gryphiswaldense (MsCI4), a bacterial single-domain homolog to the thalidomide-binding domain of human cereblon, as a model system,10,11 and developed an in-vitro Förster resonance energy transfer (FRET) assay for the identification and characterization of cereblon effectors.25 We now use this assay to delineate the chemical space of cereblon binding in a rational approach driven by structural similarity to thalidomide. Representative binders are tested in vivo in zebrafish, and the binding mode of most classes of compounds is elucidated by crystallography at high resolution. Our results delineate a simple pharmacophore for thalidomide-like cereblon binding. The resulting list of potential and verified binders comprises a wide spectrum of different classes of small molecules and commonly used pharmaceuticals, constituting the metaclass of cereblon effectors.
Results
Generalized Pharmacophore-Based Nomenclature
In the initial characterization of the thalidomide-binding domain, we found that substitutions at the glutarimide or uracil ring are compatible with cereblon binding only in certain positions.10 As the IUPAC numbering of the positions differs between the different binding moieties, we use a simplified nomenclature based on the position occupied within the binding pocket (Figure 1). This nomenclature outlines a simple pharmacophore with up to 6 positions a–f, depending on ring size. Position a is that of the branched nitrogen in uridine and respectively the branched glutarimide carbon in thalidomide, which faces to the outside of the binding pocket. Positions b, c, and d correspond to the outward-facing carbonyl, the NH group, and the inward-facing carbonyl group, respectively.
Figure 1.
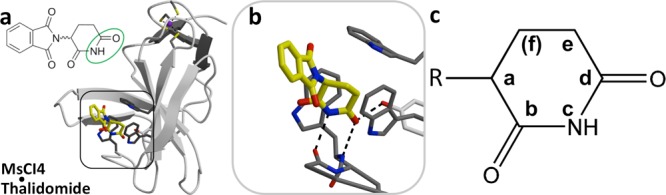
Binding of effector molecules. (a) Structure of the thalidomide-bound cereblon isoform 4 from M. gryphiswaldense (MsCI4) rendered from PDB 4V2Y. (b) Close-up of the binding site, indicating hydrogen bonds formed within the pocket. (c) Pharmacophore-based nomenclature describing the ring-positions a–f within the pocket.
Minimal Requirements for Basal Binding Moieties
The two known binding groups glutarimide and uracil are both cyclic imides, with the functional group R–CO–NH–CO–R′ occupying positions b, c, and d. It seemed therefore likely that also other such imides would bind to cereblon. Remarkably, a common mode of teratogenicity of imides had been suggested by Gary Shull in 1984, long before the connection to cereblon was established, albeit in another biological context.26 In co-crystal structures, the glutarimide and uracil moieties form the same two crucial hydrogen bonds within the binding pocket; one between the NH group in position c and a backbone carbonyl, and another one between the carbonyl in position d and a backbone NH group. As judged from the available structures10 and sequence conservation,7,10 these are the only conserved hydrogen bonds that are formed in cereblon·thalidomide complexes across all species. This suggests that only one of the imide carbonyl groups is required and therefore implies lactams, cyclic rings with the functional group R–NH–CO–R′, as minimal binders. Consequently, we composed our starting set of test compounds of both imides and lactams of reasonable ring size but also included their basal cyclic secondary amines without carbonyl groups.
To test for the binding of the different compounds, we had previously developed a FRET-based assay. It relies on the out-competition of a reporter ligand, which forms a FRET pair with the tryptophans of the MsCI4 binding pocket, by a test ligand. In this assay, different cereblon proteins show the same differential affinities to test compounds on a relative scale,25 such that Ki values obtained for MsCI4 can be translated for the human protein semi-quantitatively. For the comparison of Ki values between different studies, it is important to note that the affinities of isolated thalidomide-binding domains are generally reduced about 100-fold as compared to full-length human cereblon.27 Using the FRET assay, we now found that at least one carbonyl group is necessary for binding (Figure 2), whereas the affinity is increased by a second carbonyl group for both 5- and 6-membered rings. Generally, the affinity for 5-membered rings was higher than that of their 6-membered counterparts. Further, while 4-membered basal β-lactam was bound with an affinity comparable to its 5-membered counterpart, 7-membered basal ε-lactam (caprolactam, Table S1) showed no detectable binding, indicating that 7-membered rings are beyond the size-limit. In further tests, we could verify the necessity of the NH group of the lactams and imides for binding: neither 4-, 5-, or 6-membered cyclic anhydrides, lactones nor ketones, with the respective functional groups R–CO–O–CO–R′, R–O–CO–R′, or R–CO–R′, showed any detectable binding (Table S1).
Figure 2.

Minimal binding requirements for basic binding moieties. (a) At least one carbonyl group is necessary for binding. For both 5- and 6-membered rings, imides showed higher affinities than lactams, whereas basic heterocyclic amines did not bind. Maleimide bound with reduced affinity as compared to its saturated counterpart succinimide. (b) Compounds with heteroatoms in position a showed reduced affinity. Notably, the unsaturated uracil bound with a higher affinity than the saturated dihydrouracil, showing the opposite trend as the succinimide–maleimide pair. Experimental Ki values for MsCI4 are listed in shaded boxes, with the intensity of red increasing with affinity.
We subsequently tested the effect of an additional heteroatom within the rings and found that they systematically lower the affinity for both 5- and 6-membered rings, with sulfur having a larger impact than a second NH group in position a. Conversely, comparing the affinities of saturated and unsaturated rings did not reveal a clear trend for the compounds tested (Figure 2).
Effect of Substitutions
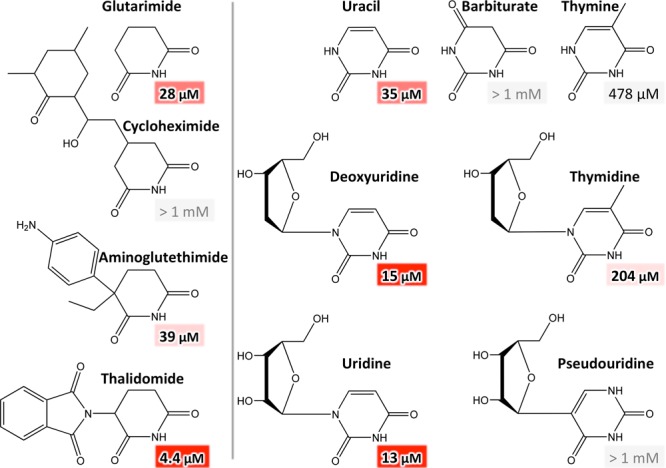
To sample the effect of possible substituents, we assayed a variety of commercially available substances based on the identified basal binding moieties with different modifications. We first probed the effect of substitutions that do not impair the imide moiety, leaving positions b, c, and d unaltered. Therefore, for 6-membered rings, we probed the effect of substituents and modifications in positions a, e, and f (Figure 3). In comparison to glutarimide, we tested the compounds thalidomide and aminoglutethimide, both branched in position a, as well as cycloheximide, branched in position f. Notably, while the affinity of thalidomide, which carries a single substituent at position a, is significantly improved over the basal glutarimide, the affinity for aminoglutethimide, which is double branched in position a, is slightly lowered. On the contrary, the large position-f substituent in cycloheximide abolished binding, as could be expected from the tight steric constraints within the binding pocket.
Figure 3.
Substitutions in positions a, e, and f in 6-membered rings derived from glutarimide (left) and uracil (right). Compounds with single substitutions in position a showed increased affinity, whereas aminoglutethimide, double branched in a, showed reduced affinity; substitutions in f abrogated binding. Binding of pseudouridine was not detectable, implying that a polar group in position e abolishes binding. Although any substitutions in position e are expected to abolish binding for steric reasons, the affinities for thymine and thymidine, both single branched in position e, were not negligible.
In comparison to uracil, we assayed the compounds barbiturate, uridine, deoxyuridine, pseudouridine, thymine, and thymidine. In barbiturate, the carbonyl group in position f abolished binding as observed for the larger substituent in cycloheximide. Uridine and deoxyuridine, both N-glycosides of uracil, showed enhanced affinity as compared to uracil, whereas pseudouridine, a C-glycoside of uracil, showed no detectable binding. Pseudouridine has the sugar attached to the uracil C,5 which potentially renders the uracil N1 in position e for steric reasons (Figure 3). This implies that an NH group in position e, representing a buried unsatisfied polar group, abolishes binding. Surprisingly, we observed very low but measurable affinities for thymine and thymidine. On the basis of our previous experiments, in which thymidine did not cause the same chemical shift changes as uridine in NMR measurements and did not show teratogenic effects in vivo,10 these were expected to be undetectable. Consequently, the interactions of thymine and thymidine with the thalidomide-binding pocket observed in the FRET assay are seemingly not equivalent to those of uridine or thalidomide, possibly leading to an incomplete folding of the pocket.
For 5-membered rings, we assayed compounds based on succinimide (pyrrolidine-2,5-dione), hydantoin (imidazolidine-2,4-dione), oxazolidine-2,4-dione, and γ-butyrolactam (Figure 4). For the succinimide derivative ethosuximide, double-branched at position a, the affinity was lower as for succinimide. Interestingly, for the corresponding hydantoin derivative 5-ethyl-5-methylhydantoin, no binding was detectable. This can, as for pseudouridine, most probably be accredited to the additional NH group that would render a buried unsatisfied polar group in position e, as the double-branched position would need to occupy position a for steric reasons. Consistently, the analogous oxazolidinedione derivative 5,5-dimethyloxazolidine-2,4-dione showed no detectable binding, presumably as the strongly electronegative oxygen is not compatible with position e. In contrast, N-methylhydantoin, which is single-branched at the additional heteroatom of the hydantoin ring, binds even tighter than its parent compound. Last, as a derivative of γ-butyrolactam, we aimed to test rolipram. Because of solubility issues (see Materials and Methods), we were not able to assess a binding constant, but could verify its binding via crystallography (see below).
Figure 4.
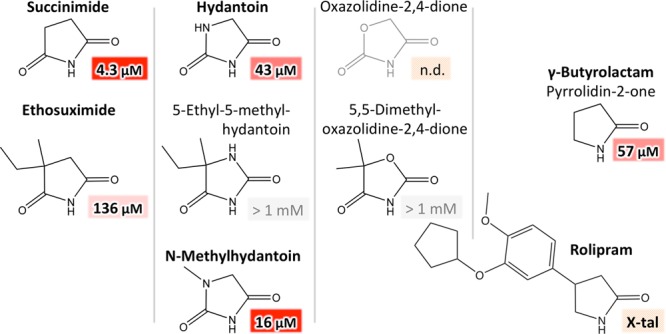
Substitutions in positions a and e in 5-membered rings. Derivatives of succinimide, hydantoin, oxazolidinedione, and γ-lactam were assayed. In agreement with 6-membered rings, single substitutions in a lead to increased, while double substitutions in a lead to reduced affinity. Also, polar groups or branching in position e are detrimental. Oxazolidinedione was not available; binding of rolipram could only be verified by crystallography.
Finally, we assayed the effect of substitutions in positions b and d, the positions of the nonobligatory and the mandatory carbonyl group, respectively (Figure 5). In both positions, imino groups were detrimental for binding, possibly because of their major tautomers, in which the hydrogen would be abstracted from the ring’s NH group, thereby preventing one of the two hydrogen bonds with the protein backbone. In contrast, thiol groups behaved differently in the two positions. While a thiol in position d was detrimental, it led to a threefold increase in affinity in position b. In the same position, an exchange to a methyl group led to slightly reduced affinity.
Figure 5.
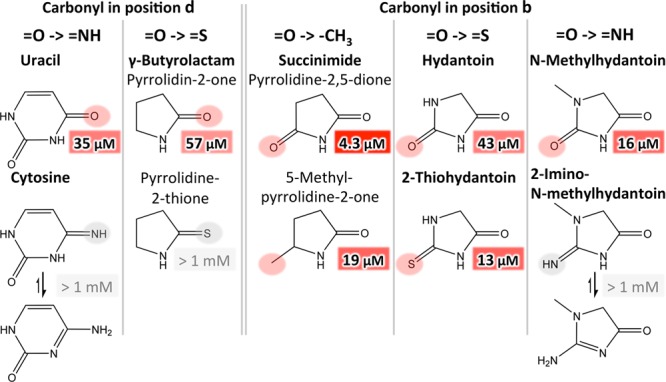
Substitutions in positions b and d. In both b and d, imino groups are detrimental for binding. In d, also a thiocarbonyl group is detrimental. In b, a thiocarbonyl group leads to increased, while a methyl group leads to reduced affinity. For cytosine and 2-imino-N-methylhydantoin, the major tautomers are shown on the bottom.
Noncyclic Compounds
Inspired by Shull’s paper,26 we also assayed the linear imide acetylurea. It indeed bound to MsCI4, albeit with a poor Ki of 522 μM (Figure S1). We assume that it binds in a conformation mimicking cyclic imides, with the primary amine in position a, the imide group occupying positions b, c, and d, and the methyl group in position e. This assumption is supported by the fact that the derivative phenylacetylurea, which is substituted at the acetylurea methyl group, did not bind in our assay. We therefore speculated that the acetamide group (CH3CONH2) could be the minimal binding moiety, so that acetylated primary amines could be generally bound in this mode. This could suggest posttranslational acetylation as a possible protein modification recognized by cereblon. However, we could not detect any binding for basal acetamide, N-(2-aminoethyl)acetamide, N-(3-hydroxypropyl)acetamide, or 2-azaniumyl-6-acetamidohexanoate (N-ε-acetyllysine) (Figure S1).
Crystal Structures Confirm a Common Binding Mode for Representative Compounds
In a next step, we characterized the binding of representative compounds via X-ray crystallography. To this end, we grew crystals of the MsCI4·thalidomide complex10 and exchanged the bound thalidomide for other ligands via soaking, for lactams and imides with 5- and 6-membered rings and with different substitutions in positions a and b. This approach was indeed highly selective for cereblon effectors—attempts to exchange thalidomide for nonbinders resulted in unfolding of the thalidomide-binding pocket as reported for related “washing”-experiments.11 The soaking experiments resulted in MsCI4·ligand crystal structures capturing the binding mode of aminoglutethimide, ethosuximide, rolipram, γ-butyrolactam, δ-valerolactam, glutarimide, hydantoin, 2-thiohydantoin, and thiazolidine-2,4-dione at resolutions between 1.55 and 2.3 Å (Figure 6). Together with the previously reported MsCI4·deoxyuridine structure, the available structures now exhibit the accommodation of eight different binding moieties: glutarimide, uracil, succinimide, hydantoin, 2-thiohydantoin, thiazolidine-2,4-dione, γ-butyrolactam, and δ-valerolactam. The structures verify a number of assumptions we made on the basis of the binding studies: (I) The comparison of the binding modes of thalidomide, aminoglutethimide, and glutarimide shows that the substituents in position a do not influence the basic binding mode of the basal binding moiety. (II) The structures with ethosuximide, hydantoin, 2-thiohydantoin, and thiazolidine-2,4-dione verify that imides with 5-membered rings have the same basic binding mode as 6-membered rings. (III) The binding mode of γ-butyrolactam, δ-valerolactam and rolipram verifies that lactams have a binding mode analogous to imides, with their carbonyl group occupying position d. (IV) The binding modes of hydantoin, 2-thiohydantoin, and thiazolidine-2,4-dione show that 5-membered rings with further heteroatoms are oriented such that the heteroatom, nitrogen or sulfur, occupies position a. (V) The structure with 2-thiohydantoin shows that a thiol group in position b is spatially accommodated like a carbonyl group. The latter observation further suggests that methyl substituents as in 5-methyl-pyrrolidin-2-one can be accommodated in position b in the same way. Importantly, in a direct comparison with the crystal structure of the human thalidomide-binding domain, none of the protruding moieties of the effectors is interacting or in contact with any nonconserved amino acid side chain, such that their binding modes are not influenced by species-specific differences (Figure S2).
Figure 6.

Crystallographic characterization of effector molecules. The binding of representative compounds was studied at 1.2–2.3 Å resolution. For all, the binding mode was the same as for thalidomide in Figure 1. Individual compounds are shown with an FO–FC omit map contoured at the indicated contrast levels. For 2-thiohydantoin and thiazolidine-2,4-dione, a second omit map at higher contrast level in black clearly identifies the sulfur. Superpositions of the compounds within the binding pocket are shown on the right, from different orientations. The top row shows the unbranched compounds and the bottom row shows the a-substituted compounds identified in this study, together with the previously reported thalidomide and deoxyuridine.10 The PDB accession codes of the new co-structures are 5OH1, 5OH2, 5OH3, 5OH4, 5OH7, 5OH8, 5OH9, 5OHA, and 5OHB.
Minimal Core Compounds are Active in Vivo
To verify that the identified binders, especially the basal compounds without protruding moieties, also bind eukaryotic cereblon in vivo, we assayed the effects of representative compounds in a zebrafish assay. The assay was initially used to show that the teratogenic effect of thalidomide was mediated via cereblon and can thus also be used to assess the teratogenic potential of substances. The treatment of zebrafish embryos with thalidomide resulted in the development of stunted pectoral fins, an effect that could be rescued by the injection of mRNA encoding a thalidomide-insensitive cereblon mutant.6 We previously adopted this assay to show that uridine had the same effect on fin formation10 and now used it to test the effect of minimal core compounds, that is, effectors with no or only small protruding moieties. We therefore tested glutarimide, hydantoin, succinimide, and ethosuximide in comparison to the nonbinder barbiturate (Figure 7). As observed for thalidomide and uridine, a higher proportion of stunted fins was observed in presence of each effector, and in all cases, the effect was alleviated when thalidomide-insensitive cereblon mutant mRNA was injected. As in previous experiments, injection of wild-type cereblon mRNA was less alleviating.10 Notably, the results do not show a clear correlation between the affinity of the compounds and the severity of the teratogenic effect. The strongest effect was achieved with glutarimide, which has an affinity ranking in the midfield of the tested compounds. Furthermore, while the results of glutarimide, succinimide, and ethosuximide are well comparable to those of thalidomide and uridine,10 the effect of hydantoin seems attenuated, hinting at an influence of differences in bioavailability and metabolic stability of the compounds. A difference in the magnitude of the rescue effects of the thalidomide-insensitive cereblon mutant mRNA for the different compounds may also point at further toxicological effects in addition to cereblon binding for some compounds. However, no effect was seen in the closely related nonbinder barbiturate. These results indicate that the compounds identified to bind MsCI4 in vitro indeed also bind eukaryotic cereblon in vivo. They further indicate that already minimal binders without protruding moieties exert effects similar to thalidomide in zebrafish, implying that ligand-induced folding of the binding pocket is already sufficient to modulate cereblon substrate specificity.
Figure 7.

Zebrafish pectoral fin deformity resulting from treatment with cereblon effectors. The images illustrate the diagnostic criteria of pectoral fin malformations at 75 h post fertilization. The effectors glutarimide, hydantoin, succinimide, and ethosuximide cause developmental defects as evoked by thalidomide. These effects can be alleviated by injecting zcrbnYW/AA mRNA, encoding a thalidomide-insensitive cereblon mutant. In comparison, barbiturate does not cause any effects. The total number of fish in each experiment is given below the bars, while the bars indicate the percentages of these fish fulfilling the criteria for no, mild, medium, or severe effects.
Discussion
Metaclass of Cereblon Effectors
Our results allow for the formulation of a simple pharmacophore that describes a metaclass of cereblon effectors. Noteworthy binding was only found for cyclic binding moieties, with lactams being the most basic form. Overall, 5-membered rings have a higher affinity than 4- or 6-membered rings, whereas 7-membered rings are too large to bind. This results in a simple pharmacophore model that describes up to 6 positions of a ring, positions a–f (Figure 1). For binding, an unmodified lactam group occupying positions c and d is mandatory. In position e and—for 6-membered rings—position f, heteroatoms or branching substitutions abolish binding. Consequently, only positions a and b are open for modifications, and the affinity of compounds can be modulated by such modifications. In position a, the incorporation of a heteroatom reduces affinity. This could not be tested for position b, as there were no suitable test compounds available. In both positions, single substituents increase the affinity, while compounds double-branched in position a show reduced affinity. The metaclass comprises compounds from different classes of common small molecules, covering several pharmaceuticals (Figure 8 and Table S1).
Figure 8.
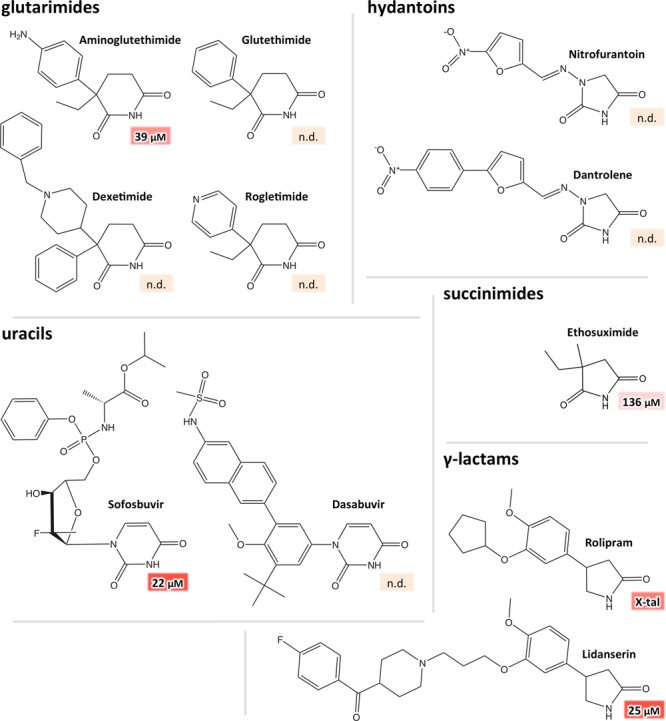
Comparison of different pharmaceuticals verified or predicted as cereblon effectors. For glutarimides, the predicted binders glutethimide and its derivatives aminoglutethimide, rogletimide, and dexetimide are compared. Exemplarily, the affinity was determined for the freely available aminoglutethimide. Because of their almost identical structure, differing only in the most distal end of their protruding moiety, glutethimide and rogletimide are expected to bind with a virtually identical affinity. We further tried to assay the structurally slightly more complex dexetimide but could not overcome solubility issues. From the uracils dasabuvir and sofosbuvir only the latter could be assessed in the FRET assay, revealing an affinity comparable to uridine. Dasabuvir was not tractable due to solubility issues. For the same reason, the two hydantoins nitrofurantoin and dantrolene could not be assessed. As a succinimide-based drug, the affinity of ethosuximide was determined. From the γ-lactams rolipram and lidanserin, only the latter could be assessed in the FRET assay, revealing an affinity comparable to uridine. Rolipram was not tractable in the FRET assay but binding was confirmed via crystallography.
For 6-membered rings, the δ-lactam piperidin-2-one poses the minimal binding moiety. In pharmaceuticals, piperidin-2-ones substituted in positions a and b are most widely represented in the form of glutarimides. For basic glutarimide, we verified binding in vitro and in vivo. Prominent glutarimides substituted in position a are the known cereblon effectors thalidomide, pomalidomide, and lenalidomide but also the hypnotic sedative glutethimide and its derivatives. As a representative, we verified the binding of the derivative aminoglutethimide, an antisteroid drug used in the treatment of Cushing’s syndrome28 and of breast and prostate cancer. For having modifications only in the protruding moiety outside the binding pocket, also the other derivatives depicted in Figure 8, the anticholinergic drug dexetimide and the aromatase inhibitor and anticancer agent rogletimide are likely binders. We note that the identification of anticancer agents as binders does not necessarily imply a functional connection. For pyrimidine-based uracils, which have a second amine embedded within the ring, we showed the binding of basal uracil and its saturated analog dihydrouracil. The additional amino group has to occupy position a, which is therefore the only position that may be branched in cereblon-binding uracils. This is well illustrated by the discrimination between the N-glycoside uridine and the C-glycoside pseudouridine. The verification of uridine and deoxyuridine as binders in the FRET assay also substantiates the in-vivo effect we reported for uridine.10 Uracil solely—or bound to a ribose analogue—is often found as a building block of antiviral drugs. This implies pharmaceuticals such as dasabuvir and sofosbuvir, both approved for the treatment of hepatitis C infections, as potential binders. While dasabuvir was intractable in our assay, we could determine the affinity of sofosbuvir, which is comparable to that of uridine (Figure 8).
Analogously, for 5-membered rings, the γ-lactam pyrrolidin-2-one poses the basic binding moiety, which is found in several pharmaceuticals. A prominent example is the selective phosphodiesterase-4 inhibitor and potential antidepressant drug rolipram,29 containing a γ-lactam moiety substituted in position a, for which we verified binding. Lidanserin, which acts as a 5-HT2A and α1-adrenergic receptor antagonist and was developed as an antihypertensive agent,30 is based on the same scaffold. Expectedly, we could verify its binding with an affinity in the range of most other effectors single-branched in a (Figure 8). The minimal imide binding moiety built from a 5-membered ring is succinimide, for which we verified binding in-vitro and in-vivo. As a succinimide derivative substituted in a, we demonstrated both in-vitro and in-vivo binding for ethosuximide, a prominent anticonvulsant considered as the first choice drug for treating absence seizures.31 Also the unsaturated succinimide counterpart maleimide, a common building block in organic synthesis, could be verified as a binder, albeit with lower affinity. For counterparts with an additional heteroatom, we could show binding of thiazolidine-2,4-dione and hydantoin. Here, as for the uracils, the heteroatom has to occupy position a, which allows only substitutions in this position. Consequently, we could confirm the binding of N-methylhydantoin. It is highly likely that the N-substituted hydantoin nitrofurantoin, an antibiotic commonly used in the treatment of urinary tract infections, and the muscle relaxant dantrolene are also cereblon binders, albeit both were intractable in the FRET assay (Figure 8). For the thiohydantoins, in which the carbonyl group in position b is replaced by a thiocarbonyl group, the same considerations apply as for basal hydantoins, with increased overall affinity in the assay. We expect the same binding mode for the structurally similar oxazolidinediones, representing the core structure of a variety of anticonvulsant drugs, but suitable compounds for testing were not available to us.
Finally, the minimal 4-membered ring, the β-lactam azetidin-2-one is the smallest compound for which we verified binding in the assay. Owing to its rectangular geometry, substitutions in a could potentially abolish binding, which remains to be tested. However, position b is expected to be open for modifications as in the larger γ- and δ-lactams.
On Cereblon and Teratogenicity
Given that the thalidomide-binding domain is found in both prokaryotes and eukaryotes, as single-domain protein and embedded within multi-domain proteins, localized within the cell and potentially secreted to the extracellular milieu,7 it seems highly likely that it serves a universal function. The strict conservation of the thalidomide-binding site across species in both sequence and structure strongly suggests that it recognizes a ligand or structural motif that is universal in all domains of life. So far, apart from uridine, the only identified natural interactors of the thalidomide-binding domain are endogenous substrate proteins. Their binding mode is structurally not yet characterized, but thought to be mutually exclusive to the binding of the known effectors.
Our work strengthens the idea that cereblon effectors are potential teratogens. However, experimental work on teratogenicity is complicated by the fact that not all species are susceptible to thalidomide teratogenicity to the same extent. While zebrafish and chicks appear to be affected in a similar way as humans,6 rodents were reported to be mostly insensitive to thalidomide.32 For this reason, teratogenicity studies in animals cannot be directly translated to humans. Consequently, it is unclear if all cereblon effectors could have teratogenic effects in human, although they appear to generally induce teratogenicity in zebrafish.
It is a long-standing question if teratogenicity is caused by an endogenous substrate that is precluded from ubiquitination or by a neo-substrate that is recruited for ubiquitination. As the known endogenous substrate proteins do not possess chemical moieties covered by the metaclass of cereblon effectors, their binding mode is conceivably different and might induce another yet unobserved conformation of the thalidomide-binding domain than that induced by the effectors. For zebrafish, it has been shown that cereblon knock-down causes similar effects as thalidomide treatment, and that teratogenicity can be rescued via overexpression of a cereblon mutant deficient in thalidomide binding.6 This suggests that cereblon must be present to recognize one or more endogenous substrates to avoid teratogenicity, and that thalidomide interferes with this recognition. We have now shown that already the smallest effectors are sufficient to induce folding of the thalidomide-binding pocket and to invoke teratogenicity in zebrafish. Without a protruding moiety, these smallest effectors cannot contribute directly to the recognition of neo-substrates. Consequently, while different effectors mediate the recognition of different neo-substrates, their least common denominator is seemingly the preclusion of endogenous substrates. This in turn suggests that teratogenicity might be ascribed to endogenous substrates.
Implications for Medicinal Chemistry
The modulation of the cereblon-CRL4A E3 ligase ubiquitination behavior is already exploited in different clinical settings. Apart from the classical IMiDs that recruit certain neo-substrates to cereblon, targeted protein degradation mediated by IMiD-derived PROTACs via cereblon is an especially promising avenue. For both approaches, our structural und functional characterization yields important insights. So far, IMiDs and IMiD-derived PROTACs were based on thalidomide. Our results allow for a more rational design of future cereblon effectors based on optimized minimal binding moieties: they allow for the design of effectors of lower molecular weight, with less hydrogen bond donors, and acceptors, and improved water solubility, which facilitates the formal compliance with the classical “rule of five” of druglikeness.33 Although the basic mechanisms are not fully understood yet, the choice of binding moieties not derived from thalidomide might have the potential to circumvent cereblon-mediated teratogenicity. Incidentally, our work implies a strategy for the circumvention of cereblon binding via negative design for future pharmaceuticals.
Materials and Methods
FRET Assay
The FRET-based binding assay was performed using the MsCI4WW/FF protein as described previously.25 Briefly, the assay is based on the reporter ligand MANT-uracil, which has a MANT fluorophore coupled to a uracil ring as a binding moiety. The MANT fluorophore forms a FRET pair with the tryptophans of the thalidomide-binding pocket, and out-competition of MANT-uracil by a test compound is detected by loss of the FRET effect. For each test compound, titration series with 16 concentrations were evaluated. As dimethyl sulfoxide (DMSO) was found to have a detrimental effect in the assay, the final DMSO concentration had to be kept below an upper limit of 0.03%.25 Hence, wherever possible, stock solutions of test compounds were prepared in water. This was not possible for aminoglutethimide, dasabuvir, dantrolene, dexetimide, lidanserin, nitrofurantoin, and rolipram. For two of these seven compounds, aminoglutethimide and lidanserin, this could be circumvented by using acetonitrile as an alternative solvent.25 For the other five compounds, the FRET assay could not be conducted. IC50 values were determined by applying a log(dose) response curve on averaged data obtained from three independent measurements and Ki values calculated according to the Cheng–Prusoff equation. All substances are summarized in Table S1 together with their Ki values for MsCI4; the individual curves are presented in Figure S3.
Crystallography
For structural ligand binding studies with wild-type MsCI4, orthorhombic MsCI4·thalidomide co-crystals were reproduced according to ref (10), with a reservoir solution containing 100 mM sodium acetate pH 4.6 and 15% (w/v) poly(ethylene glycol) (PEG) 20 000. These crystals contain 3 monomers of MsCI4·thalidomide in the asymmetric unit in space group P212121. To exchange thalidomide for other ligands, crystals were transferred into droplets of reservoir solution supplemented with 15% (v/v) PEG 300 and 10 mM of either aminoglutethimide, γ-butyrolactam, ethosuximide, glutarimide, hydantoin, thiazolidinedione, 2-thiohydantoin, or δ-valerolactam, or with solid rolipram. After 40 h of soaking, these crystals were loop-mounted and flash-cooled in liquid nitrogen. Data were collected at 100 K and a wavelength of 1 Å on a PILATUS 6M-F detector at beamline X10SA of the Swiss Light Source (PSI, Villigen, Switzerland). Diffraction images were indexed, integrated, and scaled using XDS.34 All structures were solved on the basis of the MsCI4·thalidomide coordinates (PDB: 4V2Y) and finalized by cyclic manual modeling with Coot35 and refinement with REFMAC5.36 Data collection and refinement statistics are summarized together with PDB accession codes in Table S2. All molecular depictions were prepared using MolScript,37 BobScript,38 and Raster3D.39
In-Vivo Zebrafish Assay
The zebrafish pectoral fin deformity assay was performed as described previously.10 The length of the pectoral fins was measured using self-developed software, which allows curvature determination in a semi-automatic fashion. The deformity was classified as “no effect” for fins exceeding 85% of the control length, “mild” for fins corresponding to 75–85% of the control length, “medium” for fins shortened to 60–75%, and “severe” for fins shorter than 60% of the control length. In first experiments, we tested concentrations of 200 and 400 μM thalidomide to compare the phenotypes. As a phenotype was not clearly identifiable at 200 μM in our experimental setup, we settled on a concentration of 400 μM, which is also consistent with the experiments of Ito et al.6 Glutarimide, hydantoin, succinimide, and ethosuximide were all tested at this concentration, in comparison to the nonbinder barbiturate. The experiments were performed in direct succession of our experiments with thalidomide and nucleosides, by the same person and under identical experimental conditions,10 in accordance with institutional guidelines as defined by Institutional Animal Care and Use Committee for U.S. institutions.
Acknowledgments
We are grateful to the staff of beamline X10SA of the Swiss Light Source (PSI, Villigen, Switzerland) for excellent technical support. This work was supported by institutional funds of the Max Planck Society.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00959.
Figure with noncyclic compounds tested; figure on residue conservation in the vicinity of the binding pocket; binding curves for all compounds tested; Ki values for all compounds tested; and crystallographic data collection and refinement statistics (PDF)
Accession Codes
The crystal structures described in this work have been deposited in the Protein Data Bank under the accession codes 5OH1, 5OH2, 5OH3, 5OH4, 5OH7, 5OH8, 5OH9, 5OHA, and 5OHB. The authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
† Laboratory of Biophysical Chemistry of Macromolecules, Ecole polytechnique fédérale de Lausanne, CH-1015 Lausanne, Switzerland.
The authors declare no competing financial interest.
Supplementary Material
References
- Sheskin J. Thalidomide in the Treatment of Lepra Reactions. Clin. Pharmacol. Ther. 1965, 6, 303–306. 10.1002/cpt196563303. [DOI] [PubMed] [Google Scholar]
- Sampaio E. P.; Sarno E. N.; Galilly R.; Cohn Z. A.; Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med. 1991, 173, 699–703. 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amato R. J.; Loughnan M. S.; Flynn E.; Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 4082–4085. 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslett P. A. J.; Corral L. G.; Albert M.; Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J. Exp. Med. 1998, 187, 1885–1892. 10.1084/jem.187.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett J. B.; Dredge K.; Dalgleish A. G. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat. Rev. Cancer 2004, 4, 314–322. 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- Ito T.; et al. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- Lupas A. N.; Zhu H.; Korycinski M. The Thalidomide-Binding Domain of Cereblon Defines the CULT Domain Family and Is a New Member of the β-Tent Fold. PLoS Comput. Biol. 2015, 11, e1004023 10.1371/journal.pcbi.1004023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain P. P.; et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M. D.; et al. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J. Struct. Biol. 2014, 188, 225–232. 10.1016/j.jsb.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Hartmann M. D.; Boichenko I.; Coles M.; Lupas A. N.; Alvarez B. H. Structural dynamics of the cereblon ligand binding domain. PLoS One 2015, 10, e0128342 10.1371/journal.pone.0128342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; et al. CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat. Commun. 2014, 5, 3924. 10.1038/ncomms4924. [DOI] [PubMed] [Google Scholar]
- Del Prete D.; Rice R. C.; Rajadhyaksha A. M.; D’Adamio L. Amyloid Precursor Protein (APP) May Act as a Substrate and a Recognition Unit for CRL4CRBNand Stub1 E3 Ligases Facilitating Ubiquitination of Proteins Involved in Presynaptic Functions and Neurodegeneration. J. Biol. Chem. 2016, 291, 17209–17227. 10.1074/jbc.M116.733626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi A. K.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. Br. J. Haematol. 2014, 164, 811–821. 10.1111/bjh.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 2016, 535, 252–257. 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- Petzold G.; Fischer E. S.; Thomä N. H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 2016, 532, 127–130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem. Int. Ed. 2016, 55, 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard D.; et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chem. Int. Ed. 2017, 56, 5738–5743. 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiedel M.; et al. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2017, 61, 482. 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Lebraud H.; Wright D. J.; Johnson C. N.; Heightman T. D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boichenko I.; Deiss S.; Bär K.; Hartmann M. D.; Alvarez B. H. A FRET-Based Assay for the Identification and Characterization of Cereblon Ligands. J. Med. Chem. 2016, 59, 770–774. 10.1021/acs.jmedchem.5b01735. [DOI] [PubMed] [Google Scholar]
- Shull G. E. Differential inhibition of protein synthesis: a possible biochemical mechanism of thalidomide teratogenesis. J. Theor. Biol. 1984, 110, 461–486. 10.1016/s0022-5193(84)80187-3. [DOI] [PubMed] [Google Scholar]
- Akuffo A. A.; et al. Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J. Biol. Chem. 2018, 293, 6187–6200. 10.1074/jbc.M117.816868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross B. A.; Mindea S. A.; Pick A. J.; Chandler J. P.; Batjer H. H. Medical management of Cushing disease. Neurosurg. Focus 2007, 23, E10 10.3171/foc-07/09/e10. [DOI] [PubMed] [Google Scholar]
- Kehr W.; Debus G.; Neumeister R. Effects of rolipram, a novel antidepressant, on monoamine metabolism in rat brain. J. Neural. Transm. 1985, 63, 1–12. 10.1007/bf01249580. [DOI] [PubMed] [Google Scholar]
- Schroder G.; et al. Pharmacological profile of a new potent 5-hydroxytryptamine (5-HT2) alpha 1-receptor antagonist. Arzneimittelforschung 1988, 38, 765–770. [PubMed] [Google Scholar]
- Drugs used in generalized seizures; Katzung B., Ed.; Lange Medical Books/McGraw-Hill, 2003. [Google Scholar]
- Fratta I. D.; Sigg E. B.; Maiorana K. Teratogenic Effects of Thalidomide in Rabbits, Rats, Hamsters, and Mice. Toxicol. Appl. Pharmacol. 1965, 7, 268–286. 10.1016/0041-008x(65)90095-5. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. 10.1107/s0021889893005588. [DOI] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Lebedev A.; Wilson K. S.; Dodson E. J. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 247–255. 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- Kraulis P. J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991, 24, 946–950. 10.1107/s0021889891004399. [DOI] [Google Scholar]
- Esnouf R. M. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 938–940. 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- Merritt E. A.; Bacon D. J. [26] Raster3D: Photorealistic molecular graphics. Methods Enzymol. 1997, 277, 505–524. 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.