

Abstract
An efficient Pd-catalyzed cross-coupling reaction of phenylboronic acids and benzyl carbonates was developed, producing diarylmethanes. Benzyl acetates could also be used as coupling partners instead of benzyl carbonates, affording diarylmethanes in comparable yields. This reaction can be conducted under air atmosphere without any care for moisture and oxygen. The ester function showed an intermediate reactivity between chloro and bromo groups. This property facilitated the selective synthesis of diverse (benzyl)biphenyls by successive Suzuki–Miyaura coupling reactions using bromo- and chloro-substituted benzyl esters with two types of boronic acids.
Introduction
The diphenylmethane framework is often found in polymers,1 dyes,2 and bioactive compounds.3 Although simple diphenylmethane derivatives are easily prepared by acid-catalyzed benzylation,4 this protocol cannot afford substituted diphenylmethanes because of several limitations of Friedel–Crafts alkylation. This problem is easily solved by using the Suzuki–Miyaura coupling reaction, facilitating the synthesis of substituted unsymmetrical diphenylmethanes. In such cases, benzyl halides are widely used as a coupling partner of an organic boron compound; however, it is necessary to convert the precursor benzyl alcohol to the corresponding benzyl halide beforehand.5 During the recent developments of synthetic methods for diphenylmethanes, much attention has been paid to other coupling partners: benzyl ethers6 and benzyl esters such as acetates,7 pivalates,8 carbamates,9 carbonates,10 sulfonates,11,12 sulfones,13 and phosphates.14 However, these methods require special catalysts and inert gas atmosphere, preventing the practical synthesis of diphenylmethanes. Therefore, we aimed to develop a more convenient approach to diphenylmethane derivatives from easily available boronic acid derivatives and benzyl acetates (or carbonates) under air atmosphere12,15 using a simple catalyst prepared from a commercially available Pd source and phosphine ligand. Furthermore, the selective synthesis of (benzyl)biphenyls was achieved via the successive Suzuki–Miyaura coupling reactions using bromo- and chloro-substituted benzyl esters with two types of boronic acids.
Results and Discussion
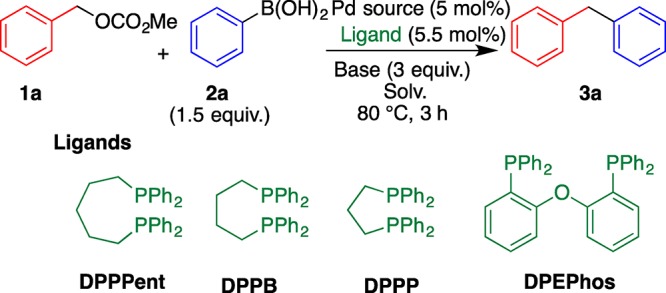
The cross-coupling reaction of benzyl methyl carbonate (1a) with phenylboronic acid (2a) to form diphenylmethane (3a) under air atmosphere was selected as the model reaction to optimize the reaction conditions, and phosphine ligands, Pd sources, bases, and solvents were screened (Table 1). Among the Pd sources, PdCl2 showed a higher reactivity than Pd(OAc)2 and [Pd(η3-C3H5)Cl]2 (entries 1–3); thus, PdCl2 was used for subsequent optimization because of its low-cost and easy-to-use property. A phosphine ligand was crucial for the success of this reaction (entries 3–8). Although the monophosphine coordination Pd complex did not work, the bidentate phosphine ligands exhibited a high catalytic activity and bis[2-(diphenylphosphino)phenyl] ether (DPEPhos) afforded 3a in the highest yield. NaHCO3 was found to be a more effective base than K2CO3 and Na2CO3 (entries 8–10). This reaction was also influenced by the solvent (entries 10–14). Polar protic solvents such as t-BuOH and EtOH were suitable solvents to increase the yield of 3a up to 79% (entry 14).
Table 1. Optimization of Reaction Conditions.
| entry | Pd source | ligand | base | solvent | yield/%a |
|---|---|---|---|---|---|
| 1 | [Pd]b | DPPPent | K2CO3 | DMF | 27 |
| 2 | Pd(OAc)2 | DPPPent | K2CO3 | DMF | 26 |
| 3 | PdCl2 | DPPPent | K2CO3 | DMF | 32 |
| 4 | PdCl2 | K2CO3 | DMF | 0 | |
| 5 | PdCl2 | PPh3c | K2CO3 | DMF | 13 |
| 6 | PdCl2 | DPPB | K2CO3 | DMF | 24 |
| 7 | PdCl2 | DPPP | K2CO3 | DMF | 21 |
| 8 | PdCl2 | DPEPhos | K2CO3 | DMF | 41 |
| 9 | PdCl2 | DPEPhos | Na2CO3 | DMF | 35 |
| 10 | PdCl2 | DPEPhos | NaHCO3 | DMF | 56 |
| 11 | PdCl2 | DPEPhos | NaHCO3 | PhMe | 18 |
| 12 | PdCl2 | DPEPhos | NaHCO3 | EtOAc | 0 |
| 13 | PdCl2 | DPEPhos | NaHCO3 | t-BuOH | 67 |
| 14 | PdCl2 | DPEPhos | NaHCO3 | EtOH | 79 |
GC yield (average of two runs).
[Pd(η3-C3H5)Cl]2 (2.5 mol %).
11 mol %.
With optimized conditions in hand, coupling reactions of other benzyl carbonates 1b–f and phenylboronic acids 2a–d were performed to obtain unsymmetrical diphenylmethanes (Table 2). This reaction was influenced by the electronic nature of the substituent on benzyl carbonate 1 (entries 1–5). When electron-rich benzyl ester 1b was used, the reaction efficiently afforded diphenylmethane 3b in an excellent yield (entry 1). Benzyl esters 1c and 1d showed a reactivity similar to that of 1a (entries 2 and 3). Notably, chloro-substituted benzyl carbonate 1d also afforded diarylmethane 3d in 68% yield without any detectable byproduct caused by an oxidative addition of Pd(0) to the C–Cl bond (entry 3). In the cases of 1e and 1f bearing an electron-withdrawing trifluoromethyl group, the yields of 3e and 3f decreased (entries 4 and 5). By contrast, the electronic nature of boronic acids did not affect this reaction; both the electron-rich and electron-poor boronic acids 2b and 2c exhibited almost the same reactivity, affording the corresponding products 3g and 3h in high yields (entries 6 and 7). Next, the steric effect of the ortho-substituent was investigated (entries 8–10). Although sterically hindered carbonate 1f decreased the reaction efficiency, boronic acid 2d afforded diphenylmethane 3i without any influence of the ortho-substituent.
Table 2. Cross-Coupling Reactions Using Other Carbonates 1 and Boronic Acids 2.
| carbonate |
boronic
acid |
product |
||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | yield/%a | |||
| 1 | 4-MeO | 1b | H | 2a | 3b | 97 |
| 2 | 4-Me | 1c | H | 2a | 3c | 73 |
| 3 | 4-Cl | 1d | H | 2a | 3d | 68 |
| 4 | 4-CF3 | 1e | H | 2a | 3e | 55 |
| 5 | 4-NO2 | 1f | H | 2a | 3f | 44 |
| 6 | 4-MeO | 1b | 4-MeO | 2b | 3g | 83 |
| 7 | 4-MeO | 1b | 4-CF3 | 2c | 3h | 80 |
| 8 | 2-Me | 1g | H | 2a | 3i | 41 |
| 9 | 4-MeO | 1b | 2-Me | 2d | 3j | 72 |
| 10 | 2-Me | 1g | 2-Me | 2d | 3k | 32 |
Isolated yield.
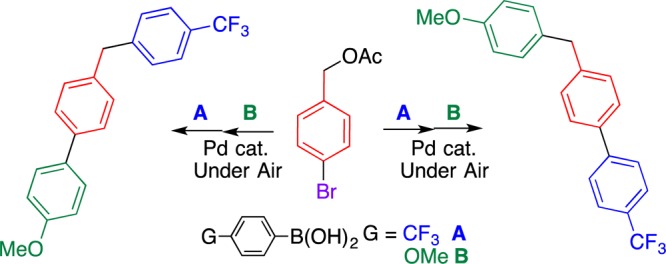
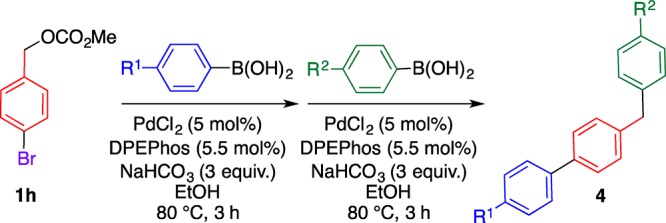
The tolerance of a chloro group of 1d under the coupling conditions prompted us to study the selective synthesis of (benzyl)biphenyls 4 (Scheme 1). Diphenylmethane 3d obtained from chlorobenzyl carbonate 1d and 2a underwent the coupling reaction with 4-methoxyphenylboronic acid (2b), affording (4-benzyl)biphenyl 4a in a moderate yield. On the other hand, when bromobenzyl carbonate 1h was reacted with 2a under the same conditions, the coupling reaction occurred on the benzene ring, affording biphenyl 5.16 Different types of (benzyl)biphenyl 4b were prepared by subsequent coupling reactions with 2b. Thus, the order of reactivity for the coupling reaction is C–Br > C–O > C–Cl, facilitating the selective C–C bond formation at the desired position.17 Different reactivities of the C–Br and C–O bonds of 3h facilitate the one-pot synthesis of several types of (benzyl)biphenyls 4c–g in moderate to high yields by two sequential coupling reactions by changing the reaction order with boronic acids 2 (Table 3), that is, the first coupling reaction mainly occurs at the C–Br bond, and the second reaction occurs at the C–O bond. These results provide important insights into the molecular design and elaborate synthesis of (benzyl)biphenyls and their analogues.
Scheme 1. Selective Synthesis of Differently Substituted (Benzyl)biphenyls 4a and 4b.
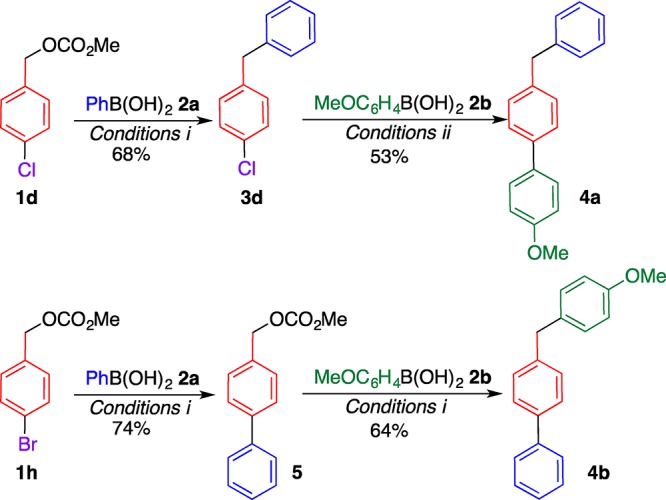
Each reaction was conducted using boronic acid 2 (1.5 equiv), PdCl2 (5 mol %), DPEPhos (5.5 mol %), and NaHCO3 (3 equiv) in ethanol. Condition i: 80 °C, 3 h. Condition ii: 100 °C, 2 d.
Table 3. One-Pot Synthesis of (Benzyl)biphenyls 4 by Changing the Order of Reaction with Boronic Acids 2.
| product |
||||
|---|---|---|---|---|
| entry | R1 | R2 | yield/% | |
| 1 | H | CF3 | 4c | 74 |
| 2 | CF3 | H | 4d | 80 |
| 3 | MeO | CF3 | 4e | 54 |
| 4 | CF3 | MeO | 4f | 47 |
| 5 | Me | MeO | 4g | 76 |
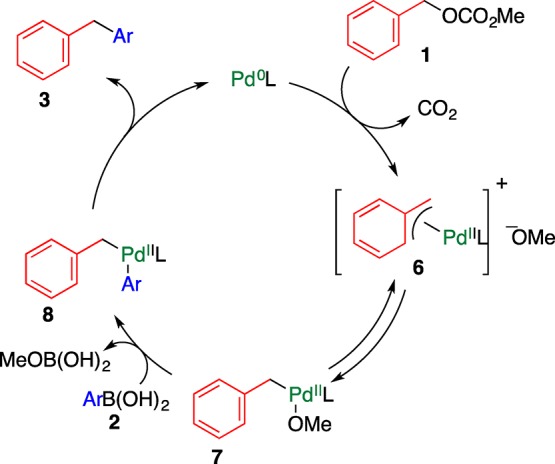
A plausible reaction mechanism is shown in Scheme 2. The reaction is initiated by the oxidative addition of the Pd species to the C–O bond, accompanied by the elimination of CO2, affording cationic (η3-benzyl)Pd(II) 6.10 After σ-complex 7 is formed under equilibrium with 6, intermediate 8 is formed by the transmetalation with arylboronic acid. The subsequent reductive elimination furnishes the cross-coupling product 3 and regenerates the Pd(0) species. When an electron-withdrawing group was introduced on the benzyl group of 1, the reaction did not proceed efficiently because cationic intermediate 6 is destabilized.
Scheme 2. Plausible Mechanism for the Coupling Reaction Using Benzyl Carbonate 1.
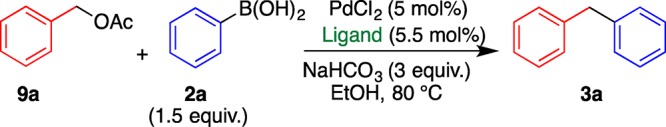
Considering the above results, the Suzuki–Miyaura coupling reaction was studied using more easily available benzyl acetates 9 instead of carbonates 1. When acetate 9a was subjected to the reaction with phenylboronic acid (2a) under the optimized conditions determined for carbonate 1a, the reaction proceeded similarly, affording diphenylmethane (3a) in a considerably low yield (Table 4, entry 1). This result indicates that the reactivity of 9a is lower than that of 1a. Indeed, the yield increased to 72% by prolonging the reaction time to 24 h. In this reaction, bidentate phosphine ligands were also effective; DPPPent showed the best performance (entries 1–5).
Table 4. Study on the Ligand for the Cross-Coupling of Benzyl Acetate (9a) and Phenylboronic Acid (2a)a.
| yield/% |
|||
|---|---|---|---|
| entry | ligand | after 3 h | after 1 d |
| 1 | DPEPhos | 23 | 72 |
| 2 | PPh3b | 10 | 21 |
| 3 | DPPP | 22 | 53 |
| 4 | DPPB | 26 | 67 |
| 5 | DPPPent | 34 | 84 |
GC yield (average of two runs).
11 mol %.
The reaction rate was significantly influenced by the type of solvent (Table 5). Polar protic solvents such as EtOH and MeOH were effective to complete the reaction within 1 day (entries 1, 7, and 8). Indeed, while the reaction did not proceed in toluene, adding a small amount of EtOH accelerated the reaction (entries 5 and 6). The reaction proceeded in alcoholic media even at 60 °C, even though a longer reaction time was necessary (entries 7 and 8).
Table 5. Study on the Solvent for the Cross-Coupling Reaction of Benzyl Acetate (9a) and Phenylboronic Acid (2a).
| yield/%a |
||||
|---|---|---|---|---|
| entry | solv. | temp/°C | after 1 d | after 3 d |
| 1 | EtOH | 80 | 72 | 72 |
| 2 | DMF | 80 | 3 | 8 |
| 3 | 1,4-dioxane | 80 | 6 | 6 |
| 4 | EtOAc | 80 | 3 | 12 |
| 5 | PhMe | 80 | 0 | 0 |
| 6 | PhMeb | 80 | 11 | 17 |
| 7 | EtOH | 60 | 26 | 75 |
| 8 | MeOH | 60 | 29 | 79 |
GC yield (average of two runs).
0.2 mmol EtOH was added.
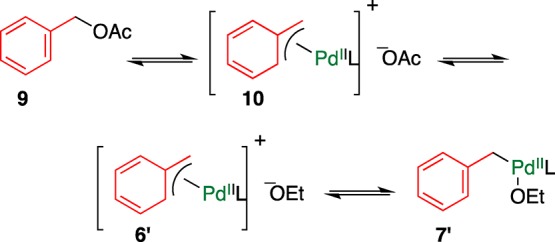
The coupling reaction of benzyl acetate 9 proceeds in a manner similar to that of benzyl carbonate 1, as shown in Scheme 2. In this mechanism, the formation of intermediate complexes 6 and 7 seems to be crucial. In the case of carbonate 1, the formation of 6 accompanied by decarboxylation is an irreversible process, facilitating the subsequent coupling reaction. Conversely, acetate 9 is regenerated under equilibrium even though cationic (η3-benzyl)Pd(II) 10 is formed; this is a probable reason for the less reactivity of acetate 9 than that of carbonate 1. When EtOH was used as a solvent, intermediates 6′ and 7′ were efficiently formed under the biased equilibrium, accelerating the reaction (Scheme 3).
Scheme 3. Plausible Mechanism for Forming Intermediate 7′ from Benzyl Acetate 9.
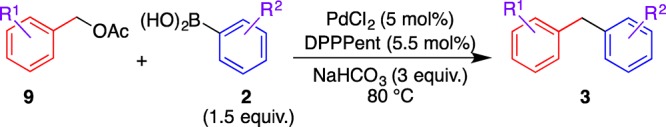
Other benzyl acetates 9b–e were subjected to the coupling reaction with arylboronic acids 2a–d under the optimized conditions (Table 6). The reactions showed substituent effect similar to that observed in the reactions of benzyl carbonates 1 shown in Table 2, that is, although the electron-rich acetates efficiently underwent the coupling reaction, the yield of 3 decreased in the case of electron-poor acetates (entries 1–3). Furthermore, the use of an ortho-substituent decreased the yield of the reaction (entry 4). On the other hand, this reaction was not influenced by the electronic nature of boronic acid 2 (entries 5–7).
Table 6. Cross-Coupling of Other Acetates 9 with Boronic Acids 2.
| acetate |
boronic
acid |
product |
||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | yield/%a | |||
| 1 | 4-MeO | 9b | H | 2a | 3b | 93 |
| 2 | 4-Cl | 9c | H | 2a | 3d | 77 |
| 3 | 4-CF3 | 9d | H | 2a | 3e | 56 |
| 4 | 2-Me | 9e | H | 2a | 3i | 32 |
| 5 | 4-MeO | 9b | 4-MeO | 2b | 3g | 87 |
| 6 | 4-MeO | 9b | 4-CF3 | 2c | 3h | 91 |
| 7 | 4-MeO | 9b | 2-Me | 2d | 3j | 81 |
Isolated yield.
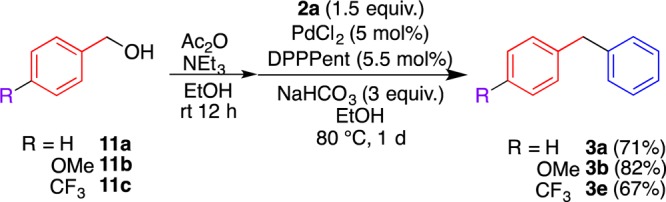
Easy access to benzyl acetates enabled the direct transformation of benzyl alcohols 11 to diphenylmethanes 3 in one pot (Scheme 4). Benzyl alcohol 11a afforded 3a in a comparable yield upon the sequential treatment with acetic anhydride in the presence of triethylamine followed by the Pd-catalyzed coupling reaction with boronic acid 2a; the precursor benzyl acetate 9a was not isolated. This protocol was applicable to both electron-rich and electron-poor benzyl alcohols 11b and 11c, furnishing the corresponding diphenylmethanes 3b and 3e, respectively, without a significant decrease in the yields.
Scheme 4. Direct One-Pot Conversion from Benzyl Alcohols 11 to Diarylmethanes 3.
4-Bromobenzyl acetate 9f also served as the precursor of (benzyl)biphenyls 4e and 4f (Scheme 5). The less reactivity of 9f than 1h facilitated a more selective coupling reaction at the C–Br bond than that at the C–O bond, thus affording 4e and 4f in higher yields, respectively. Because (benzyl)biphenyls bearing both electron-donating and electron-withdrawing groups have not been synthesized except for several examples,18 this protocol is a new synthetic tool for such compounds.
Scheme 5. One-Pot Synthesis of Two Kinds of (Benzyl)biphenyls 4e and 4f by Changing the Order of Reaction with Boronic Acids 2b and 2c.
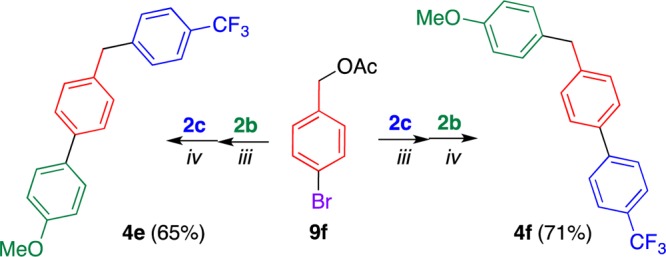
Each reaction was conducted using boronic acid 2 (1.5 equiv), PdCl2 (5 mol %), DPPPent (5.5 mol %), and NaHCO3 (3 equiv) with heating at 80 °C in ethanol. Condition iii: for 3 h. Condition iv: for 1 d.
Conclusions
Two synthetic methods were developed for diphenylmethanes 3 by Suzuki–Miyaura coupling using benzyl esters such as benzyl carbonates 1 and benzyl acetates 9. In both the cases, the Pd catalyst was generated from commercially available PdCl2 and bidentate bis(phosphine)s, and the reaction was conducted in air atmosphere. This is advantageous from practical viewpoints compared to the conventional methods. It is also possible to synthesize diphenylmethanes 3 in one pot from benzyl alcohols 11 by sequential acetylation and coupling reactions. Furthermore, diverse (benzyl)biphenyls 4 were successfully synthesized by utilizing different reactivities of the C–Br and C–O bonds.
Experimental Section
General Procedure of the Suzuki–Miyaura Coupling Reaction
To a solution of PdCl2 (1.8 mg, 10 μmol), DPEPhos (5.9 mg, 11 μmol), NaHCO3 (50.2 mg, 0.6 mmol), and phenylboronic acid 2a (36.6 mg, 0.3 mmol) in ethanol (1.0 mL), benzyl carbonate 1a (33.2 mg, 0.2 mmol) was added, and the resultant mixture was heated in a screw-capped sealed tube at 80 °C for 3 h. After filtration using a Celite pad, the filtrate was extracted with hexane (10 mL × 3). The combined organic layer was washed with brine (10 mL × 1), dried over MgSO4, and concentrated under reduced pressure. The residue was treated with flash column chromatography (EtOAc/hexane = 90/10) to afford the coupling product 3a (26.5 mg, 0.158 mmol, 79%).
When other conditions and substrates were employed, the experiments were conducted in a similar way.
4′-Phenyl-4-[(4-trifluoromethylphenyl)methyl]-1,1′-biphenyl (4c)
White solid; mp 87–88 °C. 1H NMR (CDCl3, 400 MHz): δ 7.67–7.23 (m, 13H), 4.07 (s, 2H); 13C NMR (CDCl3, 100 MHz): δ 145.1, 140.9, 139.5, 139.2, 129.6, 129.4, 128.9, 127.5, 127.2, 127.1, 125.6, 125.5, 41.4; IR (neat) 2359, 2253, 1793, 1617, 1487, 1382, 1325, 1129, 908, 740 cm–1; HRMS (EI, magnetic field) calcd for C20H15F3, 312.1126; found, 312.1123.
4′-(4-Methoxyphenyl)-4-[(4-trifluoromethylphenyl)methyl]-1,1′-biphenyl (4e)
White solid; mp 127–128 °C. 1H NMR (CDCl3, 400 MHz): δ 7.56–7.47 (m, 6H), 7.33 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 4.05 (s, 2H), 3.84 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 159.2, 145.3, 139.1, 138.5, 133.4, 129.3, 128.7, 128.4, 128.1, 127.1, 125.5, 123.0, 114.3, 55.4, 41.4; IR (neat) 3446, 2360, 2341, 1610, 1041, 907, 732 cm–1; HRMS (EI, magnetic field) calcd for C21H17F3O, 342.1231; found, 342.1231.
4-(4-Methoxyphenyl)-4′-[(4-trifluoromethylphenyl)methyl]-1,1′-biphenyl (4f)
White solid; mp 88–90 °C. 1H NMR (CDCl3, 400 MHz): δ 7.66–7.25 (m, 10H), 7.14 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 3.97 (s, 2H), 3.79 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 158.2, 144.6, 141.9, 137.5, 132.9, 130.0, 130.0, 129.6, 129.5, 127.3, 125.8, 123.1, 114.1, 55.4, 40.8; IR (neat) 3446, 2359, 2342, 1576, 1042, 907, 733 cm–1; HRMS (EI, magnetic field) calcd for C21H17F3O, 342.1231; found, 342.1230.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01450.
1H and 13C NMR spectra for benzyl(biphenyl)s 4c, 4e, and 4f (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Li W.; Liu F.; Wei L.; Zhao T. J. Appl. Polym. Sci. 2007, 104, 3903–3908. 10.1002/app.26051. [DOI] [Google Scholar]; b Akiike T.; Kakutani K. Jpn. Kokai Tokkyo Koho, JP 2006143765, 2006.; c Nikitenko S. I.; Koltypin Y.; Pickup D. M.; Van-Eck E. R. H.; Gedanken A. Ultrason. Sonochem. 2003, 10, 11–15. 10.1016/s1350-4177(02)00102-5. [DOI] [PubMed] [Google Scholar]
- a Xie B.-B.; Xia S.-H.; Chang X.-P.; Cui G. Phys. Chem. Chem. Phys. 2016, 18, 403–413. 10.1039/c5cp05312a. [DOI] [PubMed] [Google Scholar]; b Prosposito P.; Zhang H.; Glasbeek M. J. Sol-Gel Sci. Technol. 2011, 60, 347–351. 10.1007/s10971-011-2506-8. [DOI] [Google Scholar]
- a Nambo M.; Kurihara D.; Yamada T.; Nishiwaki-Ohkawa T.; Kadofusa N.; Kimata Y.; Kuwata K.; Umeda M.; Ueda M. Plant Cell Physiol. 2016, 57, 2255–2268. 10.1093/pcp/pcw140. [DOI] [PubMed] [Google Scholar]; b Chiellini G.; Nesi G.; Sestito S.; Chiarugi S.; Runfola M.; Espinoza S.; Sabatini M.; Bellusci L.; Laurino A.; Cichero E.; Gainetdinov R. R.; Fossa P.; Raimondi L.; Zucchi R.; Rapposelli S. J. Med. Chem. 2016, 59, 9825–9836. 10.1021/acs.jmedchem.6b01092. [DOI] [PubMed] [Google Scholar]; c Pham T. T. M.; Sylvestre M. J. Bacteriol. 2013, 195, 3563–3574. 10.1128/jb.00161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Eisenreich W.; Ladyzhynsky N. S.; Li D.; Schultz L.; Wang Z.; Macha S.; Barta A. PCT Int. Appl., WO 2010092126, 2010.
- a Taylor S. F. R.; Sá J.; Hardacre C. ChemCatChem 2011, 3, 119–121. 10.1002/cctc.201000337. [DOI] [Google Scholar]; b Li J.-H.; Liu W.-J.; Yin D.-L. Synth. Commun. 2004, 34, 3161–3165. 10.1081/scc-200028600. [DOI] [Google Scholar]; c Mukaiyama T.; Kamiyama H.; Yamanaka H. Chem. Lett. 2003, 32, 814–815. 10.1246/cl.2003.814. [DOI] [Google Scholar]; d Zhou D.-Q.; Wang C.-M.; Yang J.-H.; Huang M.-Y.; Jiang Y.-Y. Polym. Adv. Technol. 2002, 13, 169–172. 10.1002/pat.169. [DOI] [Google Scholar]
- a Bedford R. B.; Brenner P. B.; Carter E.; Carvell T. W.; Cogswell P. M.; Gallagher T.; Harvey J. N.; Murphy D. M.; Neeve E. C.; Nunn J.; Pye D. R. Chem.—Eur. J. 2014, 20, 7935–7938. 10.1002/chem.201402174. [DOI] [PubMed] [Google Scholar]; b Sun Y.-Y.; Yi J.; Lu X.; Zhang Z.-Q.; Xiao B.; Fu Y. Chem. Commun. 2014, 50, 11060–11062. 10.1039/c4cc05376a. [DOI] [PubMed] [Google Scholar]; c Zhang Y.-Q. J. Chem. Res. 2013, 37, 375–376. 10.3184/174751913x13687900303985. [DOI] [Google Scholar]; d Norbe S. M.; Monterio A. L. Tetrahedron Lett. 2004, 45, 8225–8228. 10.1016/j.tetlet.2004.09.020. [DOI] [Google Scholar]; e Doucet H.; Santelli M.; Chahen L. Synlett 2003, 1668–1672. 10.1055/s-2003-40994. [DOI] [Google Scholar]
- a Tobisu M.; Zhao J.; Kinuta H.; Furukawa T.; Igarashi T.; Chatani N. Adv. Synth. Catal. 2016, 358, 2417–2421. 10.1002/adsc.201600336. [DOI] [Google Scholar]; b Tobisu M.; Yasutome A.; Kinuta H.; Nakamura K.; Chatani N. Org. Lett. 2014, 16, 5572–5575. 10.1021/ol502583h. [DOI] [PubMed] [Google Scholar]
- a Stewart G. W.; Maligres P. E.; Baxter C. A.; Junker E. M.; Krska S. W.; Scott J. P. Tetrahedron 2016, 72, 3701–3706. 10.1016/j.tet.2016.02.030. [DOI] [Google Scholar]; b Kuwano R.; Yokogi M. Chem. Commun. 2005, 5899–5901. 10.1039/b513372f. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Fan X.-H.; Zhang L.-P.; Yang L.-M. RSC Adv. 2015, 5, 15338–15340. 10.1039/c4ra16452k. [DOI] [Google Scholar]
- Wang X.-X.; Luo M.-J.; Lu J.-M. Org. Biomol. Chem. 2015, 13, 11438–11444. 10.1039/c5ob01782c. [DOI] [PubMed] [Google Scholar]
- a Ohtake Y.; Emura T.; Nishimoto M.; Takano K.; Yamamoto K.; Tsuchiya S.; Yeu S.-Y.; Kito Y.; Kimura N.; Takeda S.; Tsukazaki M.; Murakata M.; Sato T. J. Org. Chem. 2016, 81, 2148–2153. 10.1021/acs.joc.5b02734. [DOI] [PubMed] [Google Scholar]; b Kuwano R.; Yokogi M. Org. Lett. 2005, 7, 945–947. 10.1021/ol050078q. [DOI] [PubMed] [Google Scholar]
- Wu G.; Xu S.; Deng Y.; Wu C.; Zhao X.; Ji W.; Zhang Y.; Wang J. Tetrahedron 2016, 72, 8022–8030. 10.1016/j.tet.2016.10.031. [DOI] [Google Scholar]
- Wang X.-X.; Xu B.-B.; Song W.-T.; Sun K.-X.; Lu J.-M. Org. Biomol. Chem. 2015, 13, 4925–4930. 10.1039/c4ob02675f. [DOI] [PubMed] [Google Scholar]
- Nambo M.; Keske E. C.; Rygus J. P. G.; Yim J. C.-H.; Crudden C. M. ACS Catal. 2017, 7, 1108–1112. 10.1021/acscatal.6b03434. [DOI] [Google Scholar]
- a Liu K.; Rao W.; Parikh H.; Li Q.; Guo T. L.; Grant S.; Kellogg G. E.; Zhang S. Eur. J. Med. Chem. 2012, 47, 125–137. 10.1016/j.ejmech.2011.10.031. [DOI] [PubMed] [Google Scholar]; b McLaughlin M. Org. Lett. 2005, 7, 4875–4878. 10.1021/ol0517271. [DOI] [PubMed] [Google Scholar]
- Synthesis of diphenylmethanes under air is also reported by other groups:Endo K.; Ishioka T.; Ohkubo T.; Shibata T. J. Org. Chem. 2012, 77, 7223–7231. 10.1021/jo3015165. [DOI] [PubMed] [Google Scholar]
- Ohsumi M.; Kuwano R. Chem. Lett. 2008, 37, 796–797. 10.1246/cl.208.796. [DOI] [Google Scholar]
- A (benzyl)biphenyl derivative was prepared using the different reactivity between C–Cl and C–F bonds:Champagne P. A.; Benhassine Y.; Desroches J.; Paquin J.-F. Angew. Chem., Int. Ed. 2014, 53, 13835–13839. 10.1002/anie.201406088. [DOI] [PubMed] [Google Scholar]
- a Vernekar S. K. V.; Liu Z.; Nagy E.; Miller L.; Kirby K. A.; Wilson D. J.; Kankanala J.; Sarafianos S. G.; Parniak M. A.; Wang Z. J. Med. Chem. 2015, 58, 651–664. 10.1021/jm501132s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li Y.; Zhao Y.; Liu Z.; Wang R. J. Chem. Inf. Model. 2011, 51, 1474–1491. 10.1021/ci200036m. [DOI] [PubMed] [Google Scholar]; c Rottländer M.; Knochel P. Synlett 1997, 1084–1086. 10.1055/s-1997-1529. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.