

Abstract
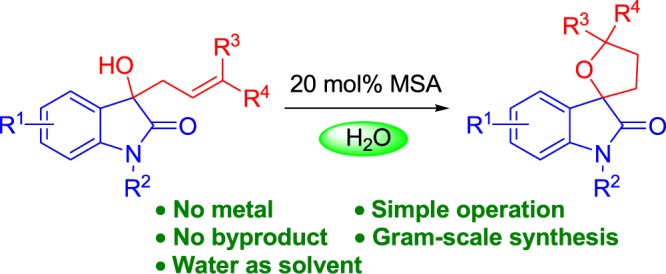
Water is an ideal solvent for chemical transformations in environmentally friendly and sustainable processes. An operationally simple, metal-free, and green approach to the synthesis of oxindole-fused spirotetrahydrofurans from 3-allyl-3-hydroxy-2-oxindoles in water is described. This method requires only cheap methanesulfonic acid as a catalyst and eliminates the need of complicated reagents. This atom- and step-economical transformation is highly efficient, which provides a new approach for the construction of oxindole-fused spirotetrahydrofuran molecules with potential pharmaceutical interest.
Introduction
Spirooxindoles have become important synthetic targets due to their promising biological activities in various therapeutic fields.1 As an important subtype of spirooxindoles, the 3,2′-tetrahydrofuryl spirooxindole unit constitutes the core structures of a variety of alkaloids and pharmaceuticals with pronounced and diverse bioactivity profiles (Figure 1).2,3 Moreover, oxindoles with a five-membered ring fused at the C-3 position are considered as potential candidates for drug discovery among spirooxindoles and thus have attracted significant attention from chemists.4 Several synthetic methodologies have been developed for the construction of such skeletons.5−11 However, all of them were carried out in organic solvents rather than in the lowest-cost and most common and clean resource, water. If water could be served as a solvent for the synthesis of those spirooxindole derivatives, it would be even more easy and ideal from the viewpoint of green chemistry.12 Moreover, the approaches involve intermolecular processes, which often require excess substrates and/or relatively expensive supporting ligands and face low selectivity and competitive reaction issues compared with their intramolecular counterparts.13
Figure 1.

Bioactive natural products containing a 3,2′-tetrahydrofuryl spirooxindole scaffold.
For instance, in 2007, the Schreiber group investigated an elegant approach to synthesizing 3,2′-tetrahydrofuryl spirooxindoles via Lewis acid-mediated annulations of isatins in CH2Cl2.5 Despite great innovation, this synthesis required 3–5 steps and involved the use of macrobead-bound crotylsilanes, which are not easy to synthesize. Later, Franz and co-workers applied the Sc-catalyzed asymmetric [3 + 2] allylsilane annulation reaction to the synthesis of 3,2′-tetrahydrofuryl spirooxindoles in CH2Cl2; this method involved the presynthesis of the ScCl3(THF)3 complex and required the assistance of several other reagents, such as NaSbF6 and TMSCl.6 Another representative approach to synthesizing 3,2′-tetrahydrofuryl spirooxindoles was the catalytic asymmetric 1,6-addition of 3-hydroxy-2-oxindoles to linear 2,4-dienals in CH2Cl2, which was presented by Melchiorre and co-workers in 2013.7 Although this protocol also provides a straightforward access to the desired spirooxindoles, it needs an additional step to access suitable 3-hydroxy-2-oxindole precursors and four steps to form another synthetic precursor 2,4-dienals. Besides the above representative methods, several similar synthetic strategies giving access to 3,2′-tetrahydrofuryl spirooxindoles have also been disclosed.8−10 However, issues still exist, such as using toluene,8 tetrahydrofuran (THF),9 or CHCl310 as the solvents and the requirement of complicated reagents or catalysts, and the scope might be impaired by the unavailability of particular substrates such as substituted allenoates,8 vinyl cyclopropanes,9 and ynones.10 Other groups also made significant contributions for the synthesis of 3,2′-tetrahydrofuryl spirooxindoles.11 However, the chemistry has been investigated with limited substrate scope.
Given the importance of 3,2′-tetrahydrofuryl spirooxindoles and our interest in the synthesis of oxindole derivatives,14 we sought to develop an easy and clean synthesis of this interesting scaffold that addresses the above challenges. Specifically, we hoped to identify conditions that would (1) enable the use of water, which is cheap, readily available, and nontoxic, as a solvent, (2) proceed with metal-free catalysts in an intramolecular process, and (3) not require complicated and expensive reagents, leading to an organic waste-minimized protocol. Herein, we report the results of our investigation of the acid-catalyzed intramolecular cyclization reaction of allylated oxindoles in water. This reaction is an environmentally benign process, and no byproduct is produced in this process. To the best of our knowledge, the given approach represents the first example of using water as the solvent in an intramolecular manner for the synthesis of 3,2′-tetrahydrofuryl spirooxindoles and provides the clearest and most atom-economical processes as alternatives to traditional intermolecular reactions.
Results and Discussion
We began our studies by choosing 3-prenyl-3-hydroxy-2-oxindole 1a as the model substrate, which can be easily prepared from the reaction of isatin with prenylzinc according to the procedure described previously by our group.14 The results are presented in Table 1. First, benzenesulfonic acid (BSA) was tested as the reaction catalyst. We found that the reaction proceeded very slowly to give a low yield of desired product 2a (entry 1). However, allylated oxindole 1a was found to cyclize much faster when catalyzed by p-toluene sulfonic acid (p-TsOH) in refluxing THF to afford product 2a in 55% yield (entry 2). In contrast to the previously reported intermolecular reaction for the preparation of 3,2′-tetrahydrofuryl spirooxindoles, which required the addition of a variety of additives or metal-based catalysts to reach high reactivity of the transformations, this intramolecular hydroalkoxylation would be preferable in terms of simplicity and atom-economy. However, this transformation required 30 h to proceed with full conversion and resulted in the formation of dehydration product 3a. To improve the reaction efficiency, we continued our investigation with other possible catalysts and solvents for cyclization reactions (entries 3–5). The results showed that this cyclization of 1a can be accelerated dramatically in 1,4-dioxane when acid p-TsOH was replaced by methanesulfonic acid (MSA), which gave products 2a in 68% yield after 8 h (entry 5). Water offers practical advantages over organic solvents and has a similar boiling point to that of 1,4-dioxane, which prompted us to explore the possibility of using water as a solvent for the synthesis of 2a. Fortunately, we found that desired product 2a could be readily obtained in 80% yield in the presence of MSA in water and dehydration product 3a was not observed (entry 6). A decrease in the catalyst loading to 10 mol % resulted in a slightly lower product yield and slightly longer reaction time (entry 7). To our great delight, when the reaction was conducted under an air atmosphere, the result was the same as that under a nitrogen atmosphere (entry 8). To simplify the operation, we decided to perform our process under an air atmosphere. Although other acids such as H2SO4 and TfOH were also effective for the intramolecular process, none of them gave higher reaction efficiency than that of MSA (entries 9 and 10). Therefore, the optimized conditions for this cyclization reaction are as follows: 20 mol % MSA as the catalyst and H2O as the solvent at reflux under air. It must be mentioned that dehydration product 3a was not observed in all cases in which water was used as the solvent.
Table 1. Optimization of the Reaction Conditionsa.

| entry | catalyst (mol %) | solvent | time (h) | 2a (%)b | 3a (%)b |
|---|---|---|---|---|---|
| 1 | BSA (20) | THF | 48 | 42 | 4 |
| 2 | p-TsOH (20) | THF | 30 | 55 | 8 |
| 3 | p-TsOH (20) | 1,4-dioxane | 16 | 60 | 5 |
| 4 | MSA (20) | THF | 12 | 65 | trace |
| 5 | MSA (20) | 1,4-dioxane | 8 | 68 | trace |
| 6 | MSA (20) | H2O | 5 | 80 | ND |
| 7 | MSA (10) | H2O | 6 | 75 | ND |
| 8c | MSA (20) | H2O | 5 | 82 | ND |
| 9 | H2SO4 (20) | H2O | 6 | 70 | ND |
| 10 | TfOH (20) | H2O | 8 | 65 | ND |
Reactions were carried out with 1a (0.5 mmol) in refluxing solvent (10 mL).
Isolated yield.
Reaction was performed under air.
With conditions for this transformation established, we next investigated the scope and limitations of this method with various 3-prenyl-3-hydroxy-2-oxindoles 1b–w (Table 2). The initial investigation of the reaction was focused on varying the substituents on the phenyl ring of 2-oxindoles with a free N–H group. We targeted these N-free substrates because many oxindole-containing bioactive compounds do not carry a protecting group at N-1.15 Therefore, from the synthetic point of view, the use of N-unprotected oxindole is preferred.16 Pleasingly, the reaction worked well for N-unprotected 2-oxindoles 1b–i, in which the phenyl ring was functionalized with electron-donating or -withdrawing groups at various positions. The outcome of the reaction did not depend much on the nature of the substituents because no obvious yield changes were exhibited for the generation of 2b–i (entries 1–8). For example, N-unprotected oxindole substrates with a weak electron-donating methyl (1b) and a strong electron-donating methoxy (1c) group at the C-5 position led to corresponding products 2b and 2c in 70 and 72% yields, respectively (entries 1 and 2). Halogens on the phenyl ring of oxindoles 1d–i were also compatible in this reaction, with corresponding products 2d–i obtained in 70–76% yields (entries 3–8). However, the reaction time varied with the introduction of halogens at different positions of the phenyl ring of the oxindole core. For example, the 4- and 5-Br-substituted oxindoles (1f and 1g) gave cyclized products 2f and 2g in 6 h (entries 5 and 6), whereas the 6- and 7-Br-substituted oxindoles (1h and 1i) required 8 and 12 h to reach complete conversion of the starting material into desired products 2h and 2i, respectively (entries 7 and 8). The results indicated that the presence of a substituent on the C-6 or C-7 position of oxindoles appears to show a slight deleterious effect on reactivity. Additionally, the influence of various N-protecting groups of oxindoles was investigated (entries 9–22). Small substituents such as methyl groups on the N-atom of oxindoles (1j–p) reacted well to afford desired products 2j–p in 6–12 h (entries 9–15). Other groups, such as ethyl, allyl, or benzyl groups, in place of methyl groups on the N-atom were also tolerated (entries 16–22). However, these substrates took a longer reaction time for completion of the reaction. For example, when the N-methyl group (1j, entry 9) was changed into an ethyl group (1q, entry 16), the reaction time increased to 40 h. A similar tendency was observed when the N-methyl protecting group at the oxindole framework was replaced by an N-allyl group (1j vs 1t, 1k vs 1u, 1o vs 1v). In the reaction with N-benzyl-substituted substrate 1w, full conversion was observed after 70 h (entry 22). Nevertheless, corresponding products 2q–w could still be obtained in good yields. The best explanation of the increasing reaction time with the increasing hydrophobicity of alkyl substituents might be that stronger hydrophobicity reduced the solubility of oxindoles in water, thereby requiring a longer time for completion. All of the products were identified via NMR and high-resolution mass spectrometry (HRMS). The structure of product 2o was further determined by single-crystal X-ray analysis (see the Supporting Information).
Table 2. Substrate Scopea.

| entry | 1 | R1, R2 | time (h) | 2 | yield (%)b |
|---|---|---|---|---|---|
| 1 | 1b | 5-Me, H | 6 | 2b | 70 |
| 2 | 1c | 5-MeO, H | 6 | 2c | 72 |
| 3 | 1d | 5-F, H | 6 | 2d | 70 |
| 4 | 1e | 5-Cl, H | 6 | 2e | 73 |
| 5 | 1f | 4-Br, H | 6 | 2f | 76 |
| 6 | 1g | 5-Br, H | 6 | 2g | 74 |
| 7 | 1h | 6-Br, H | 8 | 2h | 75 |
| 8 | 1i | 7-Br, H | 12 | 2i | 74 |
| 9 | 1j | H, Me | 6 | 2j | 81 |
| 10 | 1k | 5-Me, Me | 12 | 2k | 76 |
| 11 | 1l | 5-MeO, Me | 8 | 2l | 72 |
| 12 | 1m | 5-F, Me | 8 | 2m | 71 |
| 13 | 1n | 5-Cl, Me | 10 | 2n | 82 |
| 14 | 1o | 5-Br, Me | 8 | 2o | 78 |
| 15 | 1p | 4-Br, Me | 12 | 2p | 80 |
| 16 | 1q | H, Et | 40 | 2q | 84 |
| 17 | 1r | 5-Me, Et | 35 | 2r | 92 |
| 18 | 1s | 5-Br, Et | 40 | 2s | 73 |
| 19 | 1t | H, allyl | 35 | 2t | 76 |
| 20 | 1u | 5-Me, allyl | 35 | 2u | 70 |
| 21 | 1v | 5-Br, allyl | 48 | 2v | 84 |
| 22 | 1w | H, benzyl | 70 | 2w | 73 |
Reactions were carried out at the 0.5 mmol scale and catalyzed by 20 mol % MSA in H2O (10 mL) at reflux.
Isolated yield.
To assess the efficiency and potential for applications of this method, a gram-scale synthesis of spirocylic oxindole 2a was performed under the standard conditions (Scheme 1). Pleasingly, the reaction could be scaled easily to 5 mmol and likely even larger without significantly compromising the yield. Specifically, when 1.13 g of 1a was utilized, 0.79 g of product 2a was obtained in 70% yield (small scale 82%, entry 8 in Table 1).
Scheme 1. Scaled-Up Synthesis.

Mechanistically, we envisioned that these intramolecular reactions might occur through acid-catalyzed electrophilic addition (Scheme 2). Initially, MSA was used as the catalyst and a proton is added to the less substituted terminal alkene carbon so as to form carbocation I. The formation of the Markovnikov intermediate is favored because carbocation I is significantly stabilized by its three alkyl substituents. Then, the positively charged part of I combines with the oxygen atom in the hydroxy group that is electron-rich to form intermediate II. Finally, cyclized product 2 is formed from intermediate II via deprotonation.
Scheme 2. Proposed Reaction Mechanism.

A reviewer suggested to test this hypothesis by performing the model reaction in the absence of an acidic catalyst. Thus, a control experiment was carried out by refluxing 1a in the absence of MSA. Under the standard conditions, the reaction of 1a in water in the absence of MSA for 6 h did not lead to any cyclized product 2a and 1a was recovered in almost quantitative yield, suggesting that the proposed mechanism is applicable to explain the observations in the present study.
Because of the success described above, other 3-allyl-3-hydroxy-2-oxindoles, such as 3′-ethyl-3′-phenyl-substituted allyl compound 4, were also investigated (Scheme 3). To our delight, these types of compounds were also competent substrates in this reaction. With a slight change in the conditions (using a small amount of dioxane as a co-solvent), the corresponding 3,2′-tetrahydrofuryl spirooxindole 5 was obtained in synthetically useful yield by simply treating compound 4 with MSA.
Scheme 3. Reaction Using Other 3-Allyl-3-hydroxy-2-oxindoles.

Finally, we are pleased to report our achievement involving the use of acenaphthylenone instead of oxindole in this reaction to produce related spiro-product 7 (Scheme 4). Tetrahydrofuran spirooxindole derivatives possess attractive biological activities.2,3,5 We believe that the development of an efficient approach to synthesizing their analogues would be useful for structure–activity relationship studies. As expected, spiro-product 7 was obtained with excellent yield when substrate 6 was subjected to the same reaction albeit a longer reaction time was required compared to that for oxindole substrates 1.
Scheme 4. Synthesis of Spiro Furanyl Acenaphthylenone Derivative 7.

Conclusions
In conclusion, we have developed a simple and sustainable MSA-catalyzed method for synthesizing 3,2′-tetrahydrofuryl spirooxindole derivatives from 3-allyl-3-hydroxy-2-oxindole derivatives. In this synthesis process, water was used as the solvent and the reaction was carried out under open air. Furthermore, this method requires the addition of only one reagent, whereas previous methods for synthesizing 3,2′-tetrahydrofuryl spirooxindole generally require the addition of two or more reagents. Therefore, the approach is not only green and economic but also facile and easy to scale up. As such, this environmentally friendly method to synthesizing 3,2′-tetrahydrofuryl spirooxindole derivatives holds promising potential for future applications in both academic and industrial research.
Experimental Section
General Methods
1H NMR and 13C NMR spectra were recorded at 400 and 100 MHz in CDCl3 with chemical shift (δ) given in ppm relative to tetramethylsilane as the internal standard. Multiplicities were indicated: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), etc; coupling constants (J) were given in hertz (Hz). High-resolution mass spectra (HRMS) were recorded using electrospray ionization (ESI) and time-of-flight mass analysis.
General Procedure for the Synthesis of Spiro-Compounds
To a solution of allylated alcohols (0.5 mmol) in 10 mL of H2O was added MSA (0.1 mmol). The reaction mixture was heated to reflux until thin-layer chromatography monitoring showed complete consumption of the substrate. After refluxing, the mixture was cooled to room temperature and extracted with ethyl acetate (3 × 5.0 mL). The combined organic layer was washed with brine, dried with MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/ethyl acetate = 5:1) to afford the corresponding spiro-products.
5,5-Dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2a)
Yellow oil (87 mg, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.28 (s, 1H), 7.23 (td, J = 7.7, 1.3 Hz, 1H), 7.06 (td, J = 7.6, 1.0 Hz, 1H), 6.83 (d, J = 7.7 Hz, 1H), 2.53–2.36 (m, 2H), 2.34–2.21 (m, 1H), 2.15–2.02 (m, 1H), 1.55 (s, 3H), 1.49 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.2, 140.5, 131.6, 129.4, 123.8, 123.0, 110.0, 84.6, 84.2, 38.5, 37.0, 29.1, 28.7. HRMS (ESI): m/z calcd for C13H15NNaO2 [M + Na]+: 240.1000; found: 240.1007.
5,5,5′-Trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2b)
Pale yellow oil (81 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.09 (d, J = 1.7 Hz, 1H), 7.03 (m, 1H), 6.72 (d, J = 7.8 Hz, 1H), 2.48–2.38 (m, 2H), 2.34 (s, 3H), 2.30–2.25 (m, 1H), 2.12–2.05 (m, 1H), 1.55 (s, 3H), 1.50 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.3, 138.0, 132.5, 131.5, 129.6, 124.5, 109.7, 84.5, 84.4, 38.5, 37.0, 29.2, 28.7, 21.1. HRMS (ESI): m/z calcd for C14H18NO2 [M + H]+: 232.1338; found: 232.1312.
5′-Methoxy-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2c)
Brown oil (89 mg, 72% yield). 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 6.88 (s, 1H), 6.75 (d, J = 2.4 Hz, 2H), 3.80 (s, 3H), 2.49–2.36 (m, 2H), 2.29–2.22 (m, 1H), 2.10–2.04 (m, 1H), 1.54 (s, 3H), 1.48 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.6, 156.1, 134.0, 133.0, 113.6, 111.1, 110.5, 84.7, 84.7, 55.9, 38.5, 37.1, 29.2, 28.6. HRMS (ESI): m/z calcd for C14H17NNaO3 [M + Na]+: 270.1106; found: 270.1136.
5′-Fluoro-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2d)
Colorless oil (82 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 7.01 (dd, J = 7.7, 2.6 Hz, 1H), 6.92 (td, J = 8.8, 2.6 Hz, 1H), 6.79 (dd, J = 8.5, 4.2 Hz, 1H), 2.49–2.35 (m, 2H), 2.29–2.22 (m, 1H), 2.10–2.03 (m, 2H), 1.54 (s, 3H), 1.48 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.8, 159.4 (J = 239.5 Hz), 136.5 (J = 2.2 Hz), 133.3 (J = 7.2 Hz), 115.6 (J = 23.4 Hz), 111.6 (J = 24.4 Hz), 110.9 (J = 7.8 Hz), 85.0, 84.6 (J = 1.7 Hz), 38.4, 37.1, 29.1, 28.6. HRMS (ESI): m/z calcd for C13H15FNO2 [M + H]+: 236.1087; found: 236.1092.
5′-Chloro-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2e)
Colorless oil (92 mg, 73% yield). 1H NMR (400 MHz, CDCl3) δ 8.97 (s, 1H), 7.23 (d, J = 2.1 Hz, 1H), 7.20 (dd, J = 8.2, 2.1 Hz, 1H), 6.80 (d, J = 8.2 Hz, 1H), 2.49–2.35 (m, 2H), 2.30–2.23 (m, 1H), 2.11–2.05 (m, 1H), 1.54 (s, 3H), 1.49 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.5, 139.2, 133.4, 129.3, 128.2, 124.2, 111.3, 85.1, 84.4, 38.4, 37.1, 29.1, 28.6. HRMS (ESI): m/z calcd for C13H14ClNNaO2 [M + Na]+: 274.0611; found: 274.0612.
4′-Bromo-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2f)
Yellow oil (112 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 7.17 (d, J = 8.1 Hz, 1H), 7.08 (t, J = 7.9 Hz, 1H), 6.78 (d, J = 7.6 Hz, 1H), 2.88–2.80 (m, 1H), 2.56–2.49 (m, 1H), 2.32–2.23 (m, 2H), 1.54 (s, 3H), 1.49 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.0, 143.0, 130.8, 127.5, 127.4, 119.5, 109.2, 86.3, 85.9, 38.0, 32.8, 29.9, 29.4. HRMS (ESI): m/z calcd for C13H15BrNO2 [M + H]+: 296.0286; found: 296.0309.
5′-Bromo-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2g)
Brown oil (109 mg, 74% yield). 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H), 7.38–7.35 (m, 2H), 6.74 (dd, J = 7.8, 0.8 Hz, 1H), 2.50–2.36 (m, 2H), 2.30–2.24 (m, 1H), 2.11–2.05 (m, 1H), 1.54 (s, 3H), 1.49 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.3, 139.7, 133.7, 132.2, 127.0, 115.5, 111.8, 85.1, 84.3, 38.4, 37.1, 29.1, 28.6. HRMS (ESI): m/z calcd for C13H15BrNO2 [M + H]+: 296.0286; found: 296.0270.
6′-Bromo-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2h)
Brown oil (111 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 7.19 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 8.0 Hz, 1H), 7.03 (s, 1H), 2.45–2.37 (m, 2H), 2.28–2.23 (m, 1H), 2.09–2.07 (m, 1H), 1.54 (s, 3H), 1.47 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.5, 142.0, 130.6, 125.8, 125.0, 122.8, 113.7, 84.9, 84.0, 38.5, 37.0, 29.1, 28.6. HRMS (ESI): m/z calcd for C13H15BrNO2 [M + H]+: 296.0286; found: 296.0261.
7′-Bromo-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2i)
Light red oil (109 mg, 74% yield). 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.22 (d, J = 7.4 Hz, 1H), 6.96 (t, J = 7.8 Hz, 1H), 2.48–2.38 (m, 2H), 2.31–2.24 (m, 1H), 2.09–2.05 (m, 1H), 1.54 (s, 3H), 1.47 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 178.8, 139.8, 133.0, 132.0, 124.2, 122.6, 103.1, 85.2, 85.0, 38.4, 37.2, 29.1, 28.6. HRMS (ESI): m/z calcd for C13H15BrNO2 [M + H]+: 296.0286; found: 296.0261.
1′,5,5-Trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2j)
Yellow oil (94 mg, 81% yield). 1H NMR (400 MHz, CDCl3) δ 7.35–7.22 (m, 2H), 7.09 (td, J = 7.5, 1.0 Hz, 1H), 6.79 (dd, J = 8.2, 1.0 Hz, 1H), 3.17 (s, 3H), 2.46–2.39 (m, 2H), 2.32–2.24 (m, 1H), 2.11–2.07 (m,1H), 1.54 (s, 3H), 1.48 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 178.0, 143.5, 131.1, 129.4, 123.4, 123.0, 108.1, 84.5, 83.9, 38.7, 36.7, 29.1, 28.7, 26.1. HRMS (ESI): m/z calcd for C14H17NNaO3 [M + Na]+: 254.1157; found: 254.1168.
1′,5,5,5′-Tetramethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2k)
Yellow oil (93 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 7.08 (s, 1H), 7.06 (s, 1H), 6.65 (d, J = 7.7 Hz, 1H), 3.12 (s, 3H), 2.44–2.37 (m, 2H), 2.33 (s, 3H), 2.26–2.20 (m, 1H), 2.08–2.01 (m, 1H), 1.52 (s, 3H), 1.46 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 178.0, 141.1, 132.5, 131.0, 129.6, 124.2, 107.9, 84.4, 84.0, 38.7, 36.7, 29.2, 28.7, 26.1, 21.1. HRMS (ESI): m/z calcd for C15H19NNaO2 [M + Na]+: 268.1313; found: 268.1338.
5′-Methoxy-1′,5,5-trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2l)
Light red oil (94 mg, 72% yield). 1H NMR (400 MHz, CDCl3) δ 6.90 (d, J = 2.5 Hz, 1H), 6.80 (dd, J = 8.4, 2.6 Hz, 1H), 6.67 (d, J = 8.4 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.43–2.37 (m, 2H), 2.28–2.22 (m, 1H), 2.08–2.03 (m, 1H), 1.52 (s, 3H), 1.46 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.7, 156.3, 136.9, 132.5, 113.2, 111.3, 108.4, 84.6, 84.2, 55.9, 38.6, 36.8, 29.2, 28.6, 26.2. HRMS (ESI): m/z calcd for C15H19NNaO3 [M + Na]+: 284.1263; found: 284.1284.
5′-Fluoro-1′,5,5-trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2m)
Pale yellow oil (88 mg, 71% yield). 1H NMR (400 MHz, CDCl3) δ 7.04–6.96 (m, 2H), 6.70 (dd, J = 8.4, 4.0 Hz, 1H), 3.14 (s, 3H), 2.46–2.39 (m, 2H), 2.29–2.20 (m, 1H), 2.10–2.03 (m, 1H), 1.53 (s, 3H), 1.46 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.7, 159.5 (J = 239.6 Hz), 139.3 (J = 1.9 Hz), 132.8 (J = 7.4 Hz), 115.5 (J = 23.4 Hz), 111.6 (J = 24.5 Hz), 108.7 (J = 7.9 Hz), 84.8, 83.9 (J = 1.8 Hz), 38.6, 36.8, 29.1, 28.5, 26.2. HRMS (ESI): m/z calcd for C14H17FNO2 [M + H]+: 250.1243; found: 250.1277.
5′-Chloro-1′,5,5-trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2n)
Colorless oil (109 mg, 82% yield). 1H NMR (400 MHz, CDCl3) δ 7.27–7.24 (m, 2H), 6.70 (d, J = 8.1 Hz, 1H), 3.14 (s, 3H), 2.45–2.38 (m, 2H), 2.29–2.22 (m, 1H), 2.09–2.04 (m, 1H), 1.52 (s, 3H), 1.46 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.5, 142.0, 132.8, 129.2, 128.3, 124.0, 109.1, 84.9, 83.7, 38.5, 36.8, 29.1, 28.6, 26.2. HRMS (ESI): m/z calcd for C14H16ClNNaO2 [M + Na]+: 288.0767; found: 288.0761.
5′-Bromo-1′,5,5-trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2o)
Pale yellow oil (121 mg, 78% yield). 1H NMR (400 MHz, CDCl3) δ 7.41 (dd, J = 8.2, 2.0 Hz, 1H), 7.37 (d, J = 2.0 Hz, 1H), 6.66 (d, J = 8.2 Hz, 1H), 3.13 (s, 3H), 2.45–2.38 (m, 2H), 2.29–2.22 (m, 1H), 2.09–2.04 (m, 1H), 1.52 (s, 3H), 1.47 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.4, 142.5, 133.2, 132.2, 126.7, 115.6, 109.6, 84.9, 83.7, 38.5, 36.8, 29.1, 28.6, 26.2. HRMS (ESI): m/z calcd for C14H16BrNNaO2 [M + Na]+: 332.0262; found: 332.0259.
4′-Bromo-1′,5,5-trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2p)
Yellow oil (124 mg, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.18–7.11 (m, 2H), 6.70 (dd, J = 7.4, 1.3 Hz, 1H), 3.12 (s, 3H), 2.86–2.78 (m, 1H), 2.56–2.48 (m, 1H), 2.28–2.21 (m, 2H), 1.58 (s, 3H), 1.56 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.6, 145.8, 130.8, 127.5, 127.1, 119.4, 107.2, 86.2, 85.5, 38.2, 32.7, 29.8, 29.4, 26.2. HRMS (ESI): m/z calcd for C14H17BrNO2 [M + H]+: 310.0443; found: 310.0430.
1′-Ethyl-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2q)
Yellow oil (103 mg, 84% yield). 1H NMR (400 MHz, CDCl3) δ 7.28–7.22 (m, 2H), 7.03 (t, J = 7.5 Hz, 1H), 6.77 (d, J = 7.8 Hz, 1H), 3.67 (q, J = 7.2 Hz, 2H), 2.39–2.36 (m, 2H), 2.26–2.19 (m, 1H), 2.07–2.00 (m, 1H), 1.51 (s, 3H), 1.45 (s, 3H), 1.22 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 177.5, 142.5, 131.3, 129.3, 123.5, 122.7, 108.2, 84.4, 83.9, 38.6, 36.8, 34.5, 29.1, 28.6, 12.5. HRMS (ESI): m/z calcd for C15H20NO2 [M + H]+: 246.1494; found: 246.1511.
1′-Ethyl-5,5,5′-trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2r)
Light red oil (120 mg, 92% yield). 1H NMR (400 MHz, CDCl3) δ 7.11–7.07 (m, 2H), 6.69 (d, J = 7.8 Hz, 1H), 3.69 (q, J = 7.2 Hz, 2H), 2.44–2.37 (m, 2H), 2.35 (s, 3H), 2.28–2.21 (m, 1H), 2.10–2.03 (m, 1H), 1.54 (s, 3H), 1.48 (s, 3H), 1.24 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 177.5, 140.1, 132.3, 131.3, 129.5, 124.4, 108.0, 84.4, 84.1, 38.6, 36.9, 34.6, 29.2, 28.7, 21.1, 12.5. HRMS (ESI): m/z calcd for C16H22NO2 [M + H]+: 260.1651; found: 260.1642.
5′-Bromo-1′-ethyl-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2s)
Colorless oil (126 mg, 73% yield). 1H NMR (400 MHz, CDCl3) δ 7.41–7.37 (m, 2H), 6.68 (d, J = 8.1 Hz, 1H), 3.68 (q, J = 7.1 Hz, 2H), 2.45–2.37 (m, 2H), 2.28–2.19 (m, 1H), 2.10–2.03 (m, 1H), 1.53 (s, 3H), 1.47 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 177.0, 141.5, 133.5, 132.1, 126.9, 115.3, 109.8, 84.9, 83.7, 38.5, 37.0, 34.7, 29.1, 28.5, 12.4. HRMS (ESI): m/z calcd for C15H18BrNNaO2 [M + Na]+: 346.0419; found: 346.0402.
1′-Allyl-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2t)
Yellow oil (98 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 (dd, J = 7.3, 1.2 Hz, 1H), 7.28–7.24 (m, 1H), 7.10–7.06 (m, 1H), 6.79 (d, J = 7.8 Hz, 1H), 5.84 (dt, J = 10.4, 5.3 Hz, 1H), 5.28–5.21 (m, 2H), 4.37–4.23 (m, 2H), 2.47–2.41 (m, 2H), 2.34–2.26 (m, 1H), 2.12–2.07 (m, 1H), 1.55 (s, 3H), 1.49 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.8, 142.6, 131.4, 131.1, 129.3, 123.5, 123.0, 117.6, 109.0, 84.6, 83.9, 42.3, 38.6, 37.0, 29.1, 28.7. HRMS (ESI): m/z calcd for C16H20NO2 [M + H]+: 258.1494; found: 258.1468.
1′-Allyl-5,5,5′-trimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2u)
Yellow oil (103 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 1.6 Hz, 1H), 7.06–7.04 (m, 1H), 6.68 (d, J = 7.9 Hz, 1H), 5.83 (dd, J = 10.4, 5.1 Hz, 1H), 5.26–5.20 (m, 2H), 4.34–4.20 (m, 2H), 2.48–2.39 (m, 2H), 2.35 (s, 3H), 2.29–2.24 (m, 1H), 2.11–2.05 (m, 1H), 1.55 (s, 3H), 1.50 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.8, 140.2, 132.5, 131.5, 131.0, 129.5, 124.2, 117.4, 108.8, 84.5, 84.0, 42.3, 38.6, 37.0, 29.2, 28.7, 21.1. HRMS (ESI): m/z calcd for C17H21NNaO2 [M + Na]+: 294.1470; found: 294.1474.
1′-Allyl-5′-bromo-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2v)
Yellow oil (141 mg, 84% yield). 1H NMR (400 MHz, CDCl3) δ 7.38–7.34 (m, 2H), 6.65 (d, J = 8.1 Hz, 1H), 5.78 (dd, J = 10.3, 5.2 Hz, 1H), 5.23–5.19 (m, 2H), 4.32–4.19 (m, 2H), 2.46–2.38 (m, 2H), 2.31–2.21 (m, 1H), 2.10–2.03 (m, 1H), 1.52 (s, 3H), 1.47 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.2, 141.6, 133.2, 132.1, 131.0, 126.8, 117.8, 115.6, 110.6, 85.0, 83.7, 42.3, 38.5, 37.1, 29.1, 28.6. HRMS (ESI): m/z calcd for C16H19BrNO2 [M + H]+: 336.0599; found: 336.0578.
1′-Benzyl-5,5-dimethyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (2w)
Yellow oil (120 mg, 73% yield). 1H NMR (400 MHz, CDCl3) δ 7.34–7.28 (m, 6H), 7.18 (td, J = 7.7, 1.2 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.68 (d, J = 7.8 Hz, 1H), 4.97–4.79 (m, 2H), 2.52–2.48 (m, 2H), 2.38–2.29 (m, 1H), 2.15–2.10 (m, 1H), 1.60 (s, 3H), 1.52 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 178.2, 142.5, 135.8, 131.1, 129.3, 128.8, 128.8, 127.6, 127.2, 127.2, 123.5, 123.1, 109.2, 84.6, 84.0, 43.7, 38.6, 37.2, 29.2, 28.8. HRMS (ESI): m/z calcd for C20H21NNaO2 [M + Na]+: 330.1470; found: 330.1497.
5-Ethyl-5-phenyl-4,5-dihydro-3H-spiro[furan-2,3′-indolin]-2′-one (5)
Yellow oil (70 mg, 48% yield). 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 0.5H), 8.59 (s, 0.5H), 7.57–7.52 (m, 2H), 7.44–7.37 (m, 3H), 7.30–7.09 (m, 2H), 6.98–6.97 (m, 1H), 6.83 (dd, J = 7.7, 6.0 Hz, 1H), 2.84–2.75 (m, 1H), 2.65–2.16 (m, 3H), 2.11–2.06 (m, 2H), 0.84 (dt, J = 14.4, 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 179.8, 179.3, 146.0, 145.5, 140.7, 140.7, 131.4, 131.1, 129.5, 129.4, 128.6, 128.3, 128.0, 127.8, 126.6, 126.4, 125.9, 125.7, 124.7, 124.0, 123.0, 122.8, 110.1, 110.0, 91.1, 90.5, 84.3, 84.2, 38.5, 37.4, 36.5, 36.4, 36.2, 35.9, 9.0, 8.8. HRMS (ESI): m/z calcd for C19H19NNaO2 [M + Na]+: 316.1313; found: 316.1313.
5′,5′-Dimethyl-4′,5′-dihydro-2H,3′H-spiro[acenaphthylene-1,2′-furan]-2-one (7)
Yellow oil (111 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.1 Hz, 1H), 7.95 (d, J = 7.0 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.76–7.70 (m, 2H), 7.60 (d, J = 6.9 Hz, 1H), 2.57–2.31 (m, 3H), 2.27–2.12 (m, 1H), 1.59 (s, 3H), 1.55 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 205.4, 141.4, 141.3, 131.5, 131.0, 130.5, 128.9, 128.3, 125.0, 121.6, 119.5, 87.7, 84.5, 39.0, 36.7, 29.2, 28.5. HRMS (ESI): m/z calcd for C17H16NaO2 [M + Na]+: 275.1048; found: 275.1068.
Acknowledgments
This research was supported by the Science and Technology Support Program of Jiangsu Province (BE2017643) and by PAPD of Jiangsu Higher Education Institutions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01279.
Author Contributions
§ J.-H.Z. and R.-B.W. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Galliford C. V.; Scheidt K. A. Angew. Chem., Int. Ed. 2007, 46, 8748. 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]; b Santos M. M. M. Tetrahedron 2014, 70, 9735. 10.1016/j.tet.2014.08.005. [DOI] [Google Scholar]; c Yu B.; Zheng Y.-C.; Shi X.-J.; Qi P.-P.; Liu H.-M. Anti-Cancer Agents Med. Chem. 2016, 16, 1315. 10.2174/1871520615666151102093825. [DOI] [PubMed] [Google Scholar]; d Ye N.; Chen H.; Wold E. A.; Shi P.-Y.; Zhou J. ACS Infect. Dis. 2016, 2, 382. 10.1021/acsinfecdis.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ball-Jones N. R.; Badillo J. J.; Franz A. K. Org. Biomol. Chem. 2012, 10, 5165. 10.1039/c2ob25184a. [DOI] [PubMed] [Google Scholar]
- a Jimenez J. I.; Huber U.; Moore R. E.; Patterson G. M. L. J. Nat. Prod. 1999, 62, 569. 10.1021/np980485t. [DOI] [PubMed] [Google Scholar]; b Buttachon S.; Chandrapatya A.; Manoch L.; Silva A.; Gales L.; Bruyère C.; Kiss R.; Kijjoa A. Tetrahedron 2012, 68, 3253. 10.1016/j.tet.2012.02.024. [DOI] [Google Scholar]; c Zhou M.; Miao M.-M.; Du G.; Li X.-N.; Shang S.-Z.; Zhao W.; Liu Z.-H.; Yang G.-Y.; Che C.-T.; Hu Q.-F.; Gao X.-M. Org. Lett. 2014, 16, 5016. 10.1021/ol502307u. [DOI] [PubMed] [Google Scholar]; d Ma X.; Peng J.; Wu G.; Zhu T.; Li G.; Gu Q.; Li D. Tetrahedron 2015, 71, 3522. 10.1016/j.tet.2015.03.050. [DOI] [Google Scholar]; e Wu B.; Chen G.; Liu Z.-g.; Pei Y. Rec. Nat. Prod. 2015, 9, 271. [Google Scholar]
- a Heindel N. D.; Minatelli J. A. J. Pharm. Sci. 1981, 70, 84. 10.1002/jps.2600700117. [DOI] [PubMed] [Google Scholar]; b Wu J.-S.; Zhang X.; Zhang Y.-L.; Xie J.-W. Org. Biomol. Chem. 2015, 13, 4967. 10.1039/C5OB00256G. [DOI] [PubMed] [Google Scholar]; c Rana S.; Blowers E. C.; Tebbe C.; Contreras J. I.; Radhakrishnan P.; Kizhake S.; Zhou T.; Rajule R. N.; Arnst J. L.; Munkarah A. R.; Rattan R.; Natarajan A. J. Med. Chem. 2016, 59, 5121. 10.1021/acs.jmedchem.6b00400. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Qiu L.; Wang D.; Lei Y.; Gao L.; Liu S.; Li J.; Hu W. Eur. J. Org. Chem. 2016, 2016, 2671. 10.1002/ejoc.201600315. [DOI] [Google Scholar]
- Yu B.; Yu D.-Q.; Liu H.-M. Eur. J. Med. Chem. 2015, 97, 673. 10.1016/j.ejmech.2014.06.056. [DOI] [PubMed] [Google Scholar]
- Franz A. K.; Dreyfuss P. D.; Schreiber S. L. J. Am. Chem. Soc. 2007, 129, 1020. 10.1021/ja067552n. [DOI] [PubMed] [Google Scholar]
- Hanhan N. V.; Ball-Jones N. R.; Tran N. T.; Franz A. K. Angew. Chem., Int. Ed. 2012, 51, 989. 10.1002/anie.201105739. [DOI] [PubMed] [Google Scholar]
- Silvi M.; Chatterjee I.; Liu Y.; Melchiorre P. Angew. Chem., Int. Ed. 2013, 52, 10780. 10.1002/anie.201305870. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Liu Z.; An Y.; Jiang R.; Zhang X.; Li C.; Jia X.; Li J. J. Org. Chem. 2016, 81, 9158. 10.1021/acs.joc.6b01711. [DOI] [PubMed] [Google Scholar]
- Mei L.-y.; Wei Y.; Xu Q.; Shi M. Organometallics 2013, 32, 3544. 10.1021/om400473p. [DOI] [Google Scholar]
- a Yang L.; Xie P.; Li E.; Li X.; Huang Y.; Chen R. Org. Biomol. Chem. 2012, 10, 7628. 10.1039/c2ob26338f. [DOI] [PubMed] [Google Scholar]; b Lian Z.; Shi M. Eur. J. Org. Chem. 2012, 2012, 581. 10.1002/ejoc.201101338. [DOI] [Google Scholar]
- a Lee S.; Hartwig J. F. J. Org. Chem. 2001, 66, 3402. 10.1021/jo005761z. [DOI] [PubMed] [Google Scholar]; b Savitha G.; Niveditha S. K.; Muralidharan D.; Perumal P. T. Tetrahedron Lett. 2007, 48, 2943. 10.1016/j.tetlet.2007.02.045. [DOI] [Google Scholar]; c Jithender E.; Katsuyama I.; Zhang D.; Johnson S.; Tanaka F. Tetrahedron Lett. 2015, 56, 735. 10.1016/j.tetlet.2014.12.094. [DOI] [Google Scholar]; d Jayakumar S.; Kumarswamyreddy N.; Prakash M.; Kesavan V. Org. Lett. 2015, 17, 1066. 10.1021/acs.orglett.5b00034. [DOI] [PubMed] [Google Scholar]
- a Li C.-J.; Chen L. Chem. Soc. Rev. 2006, 35, 68. 10.1039/B507207G. [DOI] [PubMed] [Google Scholar]; b Chanda A.; Fokin V. V. Chem. Rev. 2009, 109, 725. 10.1021/cr800448q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Smith G. M.; Carpenter J. D.; Marks T. J. J. Am. Chem. Soc. 1986, 108, 6805. 10.1021/ja00281a059. [DOI] [Google Scholar]; b Mehta G.; Muthusamy S. Tetrahedron 2002, 58, 9477. 10.1016/S0040-4020(02)01187-0. [DOI] [Google Scholar]; c Nishiwaki N. Synthesis 2016, 48, 1286. 10.1055/s-0035-1561359. [DOI] [Google Scholar]
- Zhao L.-M.; Zhang A.-L.; Zhang J.-H.; Gao H.-S.; Zhou W. J. Org. Chem. 2016, 81, 5487. 10.1021/acs.joc.6b00836. [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Brennan M. K. Synthesis 2009, 2009, 3003. 10.1055/s-0029-1216975. [DOI] [Google Scholar]; b Wang Y.; Lu H.; Xu P.-F. Acc. Chem. Res. 2015, 48, 1832. 10.1021/acs.accounts.5b00217. [DOI] [PubMed] [Google Scholar]; c Zhou L.; Yang J.-S.; Wu X.; Zou J.-H.; Xu X.-D.; Tu G.-Z. Heterocycles 2005, 65, 1409. 10.3987/COM-04-10315. [DOI] [Google Scholar]; d Cao X.-F.; Wang J.-S.; Wang X.-B.; Luo J.; Wang H.-Y.; Kong L.-Y. Phytochemistry 2013, 96, 389. 10.1016/j.phytochem.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Young I. S.; Baran P. S. Nat. Chem. 2009, 1, 193. 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.