

Abstract
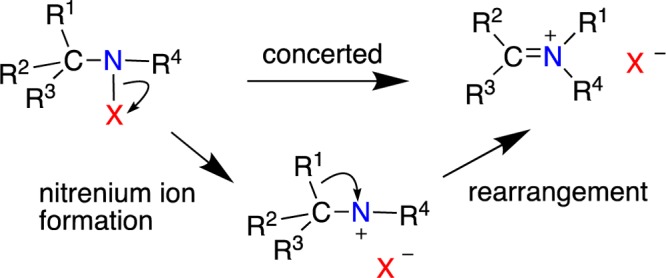
Nitrenium ion species are examined using computational methods (DFT, MP2, coupled-cluster, and a composite method, CBS-APNO) with a particular emphasis on nonaromatic species (i.e., those lacking an aromatic or heteroaromatic ring in direct conjugation with the formal nitrenium ion center.) The substitution of the N+ center with alkyl, alkoxy, vinyl, acyl, and sulfonyl, among others, was evaluated. For these species, three properties are considered. (1) The stability of the nitrenium ions to unimolecular isomerizations such as 1,2 alkyl or H shifts; to the extent that the singlet states could be characterized as discrete minima on the potential energy surface (PES), (2) the effect of the substituents on singlet–triplet energy splitting as well as (3) the relative stabilities of the nitrenium ions as defined by N-hydration enthalpies (RR′N+ + H2O → RR′NOH2+). Nearly all simple alkyl and di-alkyl nitrenium ion singlet states are predicted to rearrange without detectable barriers, largely through 1,2 H or alkyl shifts. Methyl and N,N-dimethylnitrenium ion singlet states could be characterized as formal minima on the PES. However, these species show small or insignificant barriers to isomerization. Disubstituted nitrenium ions that include an alkyl group and a conjugating substituent such as alkoxyl, vinyl, or phenyl show meaningful barriers to isomerization and are thus predicted to possess nontrivial lifetimes in solution. Alkyl groups substantially stabilize the singlet state relative to the situation in the parent nitrenium ion NH2+ to the point where the two states are nearly degenerate. Other groups that interact with the nitrenium ion center decrease the ΔEst in the order formoyl < vinyl < phenyl < alkoxy ∼ sulfonyl < cyclopropyl ∼ cyclobutyl. The latter two substituents interact strongly with the (singlet) nitrenium ion center through the formation of a nonclassical bonding reminiscent of the bisected cyclopropylcarbinyl ion case for carbocations. When singlet-state stability is evaluated in the context of N-hydration enthalpies, it is found that the ordering is acyl < vinyl < alkoxyl < phenyl < cyclopropyl and cyclobutyl.
1. Introduction
Nitrenium ions, which are reactive intermediates characterized by a di-coordinate positively charged nitrogen, have been the focus of investigations over several decades.1−5 Experimental studies based upon chemical trapping,2,6−13 laser flash photolysis,14−22 mass spectrometry,23,24 and electrochemistry25,26 have made it clear that arylnitrenium ions (i.e., those with one or two aromatic rings directly conjugated to the formal nitrenium ion nitrogen) have sufficient stability that they can survive in solution long enough to form stable adducts with various external trapping agents. Recent nitrenium ion investigations have emphasized the arylnitrenium ions undoubtedly because of the involvement of such species in DNA-damaging reactions originating from the metabolism of arylamines and aromatic nitro compounds.8,17,27−33
The current report, however, deals with nonaromatic nitrenium ions. It considers aliphatic, alicyclic, acyl, vinyl, and alkoxy substituted nitrenium ions and evaluates their stabilities toward the unimolecular decay processes. The specific species considered here are listed, in rough order of complexity, as structures 1–43 in Chart 1. There has been recent interest in using highly stabilized nitrenium ions (analogous to N-heterocyclic carbenes) in catalysis, metal binding, etc. However, these highly stabilized examples are directly addressed by this report.34−38 Like carbenium ions, the extant literature on alkylnitrenium ions generally suggests that these intermediates are susceptible to 1,2-shifts of hydride or alkyl groups.5,39−44 Indeed, much of the early literature was concerned with determining whether products from presumed nitrenium ion precursors arise from bona fide equilibrated nitrenium ion intermediates or from concerted reactions where a thermally equilibrated nitrenium ion is not formed.4,5,40
Chart 1.

Three considerations motivated this study. First, there is growing interest in the use of nitrenium ions in synthetic chemistry. For example, Wardrop45 and others have achieved useful cyclization reactions via N-acyl-N-alkoxynitrenium ions. More generally, the prospect of using nitrenium ions in electrophilic substitution reactions remains an attractive one.46−50 Thus, the predictions of lifetimes and the properties of these species have the potential to direct experimental efforts toward more productive directions. Second, numerous biological, environmental, and synthetic reactions involve the oxidation of amines or reduction of nitro compounds. Such processes could, at least in principle, proceed via nitrenium ion intermediates. Improved estimates of nitrenium ion stabilities can help identify or exclude the involvement of these intermediates in such processes. Finally, the question of alkylnitrenium ion intermediates is of considerable historical interest. The late Paul Gassman recognized that cyclic nitrenium ions such as 14 could share many of the same properties as the corresponding carbenium ions, including the possibility of nonclassical structures.40 The nitrenium ions, however, are potentially even more complex because of the possibility of existing in either a triplet or a singlet state. At the time it was not known which was the ground state, but qualitative orbital analysis suggested that the two states would be very close in energy. Formation of the corresponding amine, along with rearrangement products, led Gassman and his co-workers to conclude that (a) the nitrenium ion was formed as a discrete intermediate from heterolysis of the chloroamine and (b) that this nitrenium ion could convert to the triplet state competitively with the rearrangement. The triplet state was thought to be responsible for the formation of the amine, via sequential H atom abstractions).
The triplet state mechanism proposed by Gassman was not universally embraced. Indeed, the formation of amines is exceptional, rather than typical, in solvolysis of potential alkylnitrenium ion precursors. Hoffman’s attempt to generate the same nitrenium ion using a different leaving group generated only rearrangement products.43 On this basis, it was argued that the amine products observed by Gassman were caused by a homolysis pathway, rather than by a free nitrenium ion. More recently, an attempt to generate the 2,2,6,6-tetramethylpiperidinyl nitrenium ion 11 produced rearrangement products but no other products that could be definitively attributed to the formation of a nitrenium ion (Scheme 1).6
Scheme 1.

The decades that followed Gassman’s study have seen substantial improvements in theoretical and computational methods to the point where they are capable of examining the stability and existence of hypothetical intermediates. In the longstanding question of classical versus nonclassical bonding in the 2-norbornyl cation,51−54 calculations, which were later confirmed by X-ray crystallography, confirm delocalized sigma bonding for this intermediate.
There have been a few previous computational studies on nitrenium ions that are related to the present work. First, the parent system, NH2+, has received considerable computational attention and it is now clear that the triplet state is the ground state by a substantial amount, having an adiabatic energy gap of 29.9 kcal/mol relative to the closed-shell (n2) singlet.55−60 In contrast to the parent nitrene (NH), the bent structure of NH2+ breaks the degeneracy of the nonbonding orbitals, resulting in a closed shell singlet state. Indeed, to the extent that multireference calculations are available,57,58,61 nitrenium ions appear to possess closed shell singlet states. Gonzales et al.62 demonstrated that the singlet is stabilized relative to the triplet state when the H atoms are replaced with halogens or CN.
Of relevance to the rearrangement chemistry discussed below is a density functional theory (DFT)-based study of methylphenylnitrenium ion 23, which found a barrier of 19–22 kcal/mol for a 1,2-hydride shift to form a stable iminium ion.63 This is qualitatively consistent with solution phase experimental data, which showed that the same nitrenium ion could rearrange competitively with bimolecular trapping by added nucleophiles.64 DFT methods have been used to study rearrangement processes in few selected examples of alkylnitrenium ions. For example, Cramer65 found that the three-member aziridenium ion is stable in the triplet state, but that the singlet state rearranges, without an apparent barrier, to form a ring-opened dimethyleneiminium ion. An early ab initio study by Ford and Herman,66 focusing on methyl and dimethylnitrenium ions, found that the singlet states of these ions rearrange without a detectable barrier (at the HF/6-31G* level) to form the corresponding iminium ions. The triplet states, in contrast, showed significant barriers to hydrogen migration.
The following computations show that the most simple, free alkylnitrenium ions possess either no or extremely low barriers to isomerization in the singlet state. In most cases, including Gassman’s nitrenium ion, such species are predicted to undergo barrierless 1,2 alkyl or hydride shifts. The triplets do show significant barriers to isomerization and could, in principle, live long enough to participate in various bimolecular reactions. Substitution with even modestly effective pi-conjugating groups such as carbonyls, vinyl, phenyl, and alkoxy groups stabilizes the singlet state and the barriers to isomerization are sufficient to allow these species to participate in bimolecular trapping reactions.
2. Results and Discussion
2.1. Geometries of Nitrenium Ions
Those nitrenium ions showing potential energy minima for both the singlet state and the triplet state are listed in Table 1. As noted in previous studies,3,59,67−72 both singlets and triplets are bent about the R–N–R′ bond angle, with the sp2 singlets preferring bond angles of ca. 105–115°, absent sterically demanding substituents. The triplets are more nearly linear with typical bond angles in the range of 145–155°.
Table 1. R1NR2 Bond Angles for Singlet and Triplet Nitrenium Ions.
| number | R1 | R2 | singlet R1–N–R2 angle | triplet R1–N–R2 angle | notes |
|---|---|---|---|---|---|
| 1 | H | H | 107.5 | 151.2 | |
| 2 | CH3 | H | 113.6 | 147.4 | SMD (singlet unstable in gas) |
| 3 | CH3 | CH3 | 119.8 | 148.9 | |
| 4 | CH3CH2 | H | (rearranges) | 146.1 | |
| 5 | iPr | H | (rearranges) | 144.5 | |
| 6 | tBu | H | (rearranges) | 144.5 | |
| 7 | tBu | tBu | (rearranges) | 151.5 | |
| 8 | (azirenyl) | (azirenyl) | (rearranges) | 77.5 | triplet C–C bond 1.72 Å |
| 9 | (endo-azetidinyl) | (endo-azetidinyl) | (rearranges) | 103.6 | |
| 10 | (endo-pyrolidnyl) | (endo-pyrolidnyl) | (rearranges) | 122.7 | |
| 11 | (endo-TMP) | (endo-TMP) | (rearranges) | 138.9 | |
| 12 | (bicyclohexyl) | (bicyclohexyl) | (rearranges) | 92 | |
| 13 | (bicyclophepty) | (bicycloheptyl) | (rearranges) | 117.4 | |
| 14 | (Gassman) | (Gassman) | (rearranges) | 117.2 | |
| 15 | c-Pr | H | 113.5 | 137.6 | |
| 16 | c-Pr | CH3 | 119.7 | 143.9 | |
| 17 | c-Pr | tBu | 122.1 | 142.7 | |
| 18 | c-Bu | H | 112.8 | 140.3 | |
| 19 | CH2=CH | H | 112.7 | 147.7 | |
| 20 | CH2=CH | CH3 | 119.3 | 150.7 | |
| 21 | CH2=CH | tBu | 121.7 | 148.3 | |
| 22 | Ph | H | 112.4 | 134.7 | |
| 23 | Ph | CH3 | 124.1 | 148.7 | triplet planar |
| 24 | Ph | tBu | 131.5 | 143.3 | triplet perpendicular |
| 25 | CH3O | H | 105.7 | 123.6 | long C–O bond 1.53 Å triplet triplet nonplanar |
| 26 | CH3O | CH3 | 112.5 | 127.7 | triplet nonplanar |
| 27 | CH3O | tBu | 113.8 | 125.6 | triplet nonplanar |
| 28 | HCO | H | 114.3 | 127.6 | triplet nonplanar singlet N–O bonding |
| 29 | CH3CO | H | 113.1 | 120.1 | singlet N–O bonding |
| 30 | CH3CO | CH3 | (rearranges) | 131.3 | |
| 31 | HCO | CH=CH2 | 116.6 | 139.9 | singlet vinyl and CO not coplanar |
| 32 | CH3CO | OCH3 | 107.3 | 114.5 | triplet: long (CO)–N bond, 1.91 Å triplet (CO)–N 1.66 Å |
| 33 | (betalactam) | (betalactam) | (rearranges) | (rearranges) | |
| 34 | (gammalactam) | (gammalactam) | (rearranges) | (rearranges) | |
| 35 | (succinimide) | (succinimide) | (rearranges) | 105.1 | |
| 36 | CH2=CHCH2 | H | 113 | 129.6 | triplet vinyl out-of-plane singlet aziridinylcarbinyl cation ish by M062x others have shift |
| 37 | CH2–CHCH2CH2 | H | (rearranges) | 110.5 | triplet seems to have interaction between vinyl and N+ |
| 38 | CH2=CHCH2CH2CH2 | H | (rearranges) | (rearranges) | |
| 39 | CH2F | H | (rearranges) | 145.6 | |
| 40 | CF3 | H | (rearranges) | 148.7 | long 1.58 Å C–N bond triplet |
| 41 | HPO2 | H | (rearranges) | 122 | NH syn to P=O triplet |
| 42 | H3Si | H | (rearranges) | 179.9 | |
| 43 | HSO2 | H | 115.4 | 115.9 | singlet cyclizes O–N dist 1.54 Å |
Figure 1 shows the specific geometries for the singlet and triplet states of dimethyl 3 and vinyl 19 nitrenium ions. Dimethylnitenium ion singlet 3S has a C–N–C bond angle of 119.8°, indicative of nitrogen having an sp2 hybridized nonbonding orbital. Indeed, a natural bond order (NBO) analysis provides a value of sp2.06 for the nonbonding orbital that lies in the CNC plane. In contrast, the triplet state shows a more obtuse bond angle of 148.9° and slightly longer C–N bond distances of 1.40 Å. For this state, the NBO analysis confirms that the two semioccupied orbitals are relatively unhybridized: the out-of-plane nonbonding molecular orbital (NBMO) is 99.97% p and the in-plane NBMO is sp5.24 (i.e., ca. 84% p). The singlet state shows significant hyperconjugative interactions between the N+ center and the C–H bonds of the methyl groups. Specifically, the N–C bond distances are 1.37 Å, making them significantly shorter than the typical C–N single bond distances of 1.47 Å. Additionally, one of the C–H bonds on both methyls has an elongated C–H distance of 1.14 Å (vs typical values of 1.09 Å) and a compressed N–C–H angle of 93°. These parameters are also consistent with the view of hyperconjugation stabilizing the singlet state (relative to NH2+).
Figure 1.
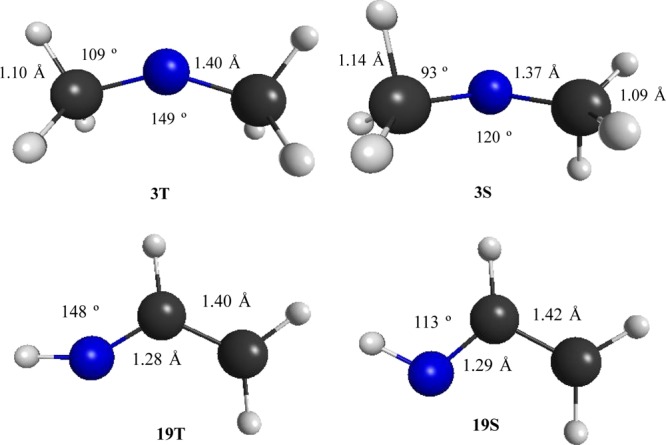
Equilibrium geometries (M06-2X) for (top) singlet 3S and triplet 3T dimethylnitrenium ion and (bottom) singlet 19S and triplet 19T vinylnitrenium ion along with selected geometric parameters.
The relevant orbitals are depicted in Figure 2, which shows an NBO representation of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for both spin states of the dimethylnitrenium ion (3S). Both the HOMO of the singlet and the lower singly occupied molecular orbital (SOMO) are symmetric with respect to the C–N–C plane and are largely localized on the nitrogen. The LUMO of the singlet and the higher energy SOMO are both antisymmetric with respect to the C–N–C plane. In the case of the triplet, the SOMO – 2 is consistent with a p-orbital localized mainly on the nitrogen. However, the singlet LUMO shows significant delocalization on to the adjacent C–H bonds, consistent with significant hyperconjugative interactions from the alkyl substituents inferred from the geometries.
Figure 2.
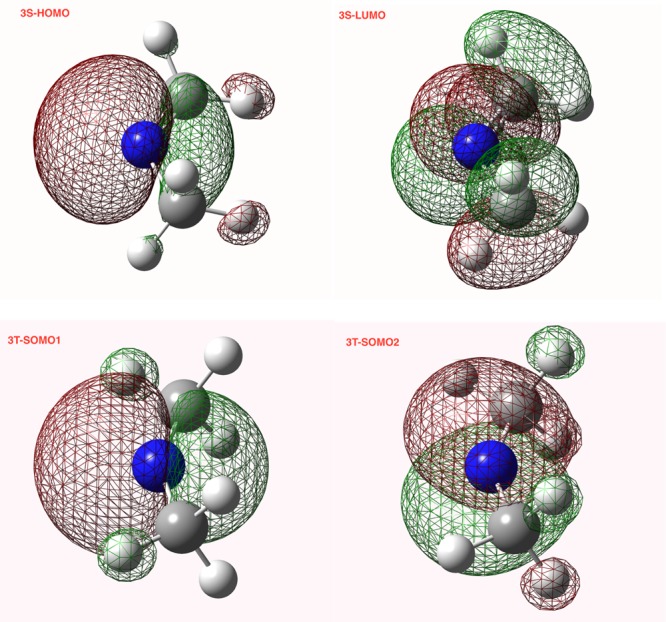
NBO representations of the frontier orbitals for the singlet (3S) and triplet (3T) dimethylnitrenium ion.
The geometries of the vinylnitrenium ion (19) singlet and triplet states are shown in detail as Figure 1. For this example, the singlet and triplet states show similar geometries, aside from the CNH bond angle. Both states are dominated by an allyl cation-like conjugation of the C=C group with the N+, as confirmed by C–C bond distances of 1.42 Å (19S) and 1.40 Å (19T) as well as C–N bond distances of 1.29 Å (19S) and 1.28 Å (19T). The effect of increasing steric pressure from branched alkyl substituents is to destabilize the singlet state and shift ΔEst in the direction of the triplet state. Methyl substitution increases the RNC bond angle from 112.7° (19S R=H) to 119.3° (20S R=CH3) to 121.6° (21S R=C(CH3)3).
Cyclopropyl (15–17) and cyclobutyl (18) substituents show interesting nonclassical structures in the singlet states. The geometry of the exocyclopropylnitrenium 15 ion is shown in Figure 3. This structure features significant shortening of the distal ring C2–C3 bond (1.38 Å) as well as that of the C1–N bond. This is accompanied by lengthening of the proximal C1–C2 and C1–C3 bonds. In all, this structure is analogous to the so-called “bisected cyclopropylcarbinyl cation,”73,74 which is one of the fluxional structures observed experimentally when cyclopropylmethyl cations are generated.75−77 In contrast, the triplet shows little sigma delocalization, adopting a structure that shows more modest cyclopropyl bond length distortions of 1.46 Å for the distal C2–C3 bonds and 1.59 Å for the proximal C1–C2,3 bonds.
Figure 3.
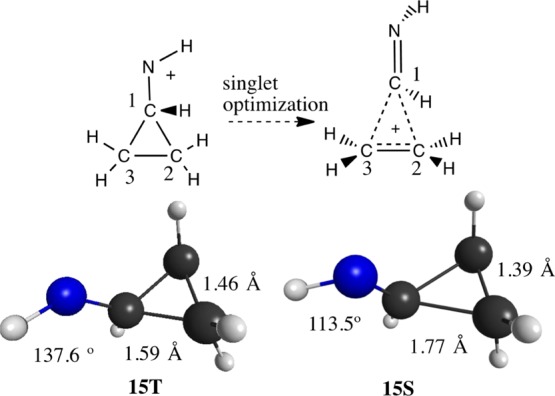
Exo-cyclopropylnitrenium ion triplet (15T) and singlet (15S) states along with selected geometric parameters.
The singlet exo-cyclobutylnitrenium ion also minimizes to a structure that exhibits nonclassical bonding. In this case, proximal C2–C1 breaks (2.59 Å), accompanied by formation of what can be characterized as a C1=N double bond (1.25 Å), along with a two-electron, three-center bond from C4···C3 (1.69 Å) and C4···C2 (1.80 Å). In the meantime, the distal C3–C2 bond shortens to nearly standard alkene length (1.39 Å). In contrast, the triplet shows a more classical structure having ring C–C bond distances of 1.54–1.61 Å (Figures 4).
Figure 4.
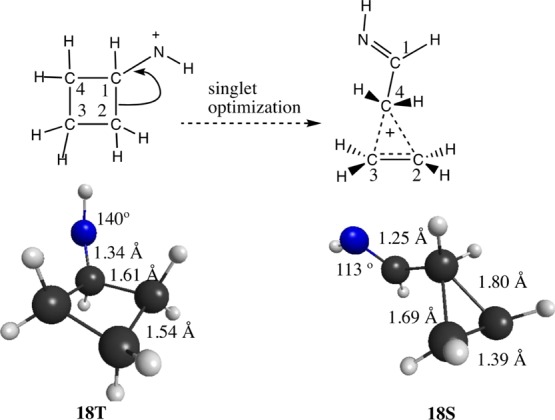
Exo-cyclobutylnitrenium ion triplet (18T) and singlet (18S) states along with the selected geometric parameters.
The acyl substituents, formyl 28 and acetyl 29, also develop a distorted structure to stabilize their singlet states. In this case, the carbonyl O moves closer to the N (1.60 Å), forming an acute O–C–N bond angle of 77°. Indeed, 28S could be regarded as a cyclized oxaziridinyl cation. In contrast, the triplet state 28T shows a more classical geometry, having an N–O distance of 2.27 Å and an obtuse N–C–O angle of 125.7° (Figure 5). For the cases where the remaining N-substituent is methoxy 31S or vinyl 32S, this distortion is not present. The corresponding N–C–O bond angle for singlet N-formyl-N-vinylnitrenium ion (32S), for example, is 117°, implying that the vinyl group provides sufficient stabilization and this distortion is avoided. Similar distortions have been seen for singlet acylnitrenes78 and acylcarbenes.79
Figure 5.
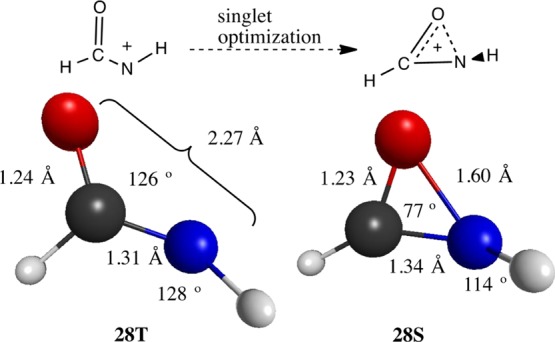
Formylnitrenium ion triplet (28T) and singlet (28S) states along with the selected geometric parameters.
The singlet sulfonylnitrenium ion 43S shows distortions analogous to those seen in the acylnitrenium ions. In this case, one of the O atoms on sulfur bends (96.2° in the triplet to 57.5° in the singlet) toward the N+ to stabilize the unfilled p-orbital on N. As shown in Figure 6 the O–N distance decreases from 2.40 Å in the triplet to 1.54 Å in the singlet. Similar distortions have been observed for singlet sulfonylnitrenes.80
Figure 6.
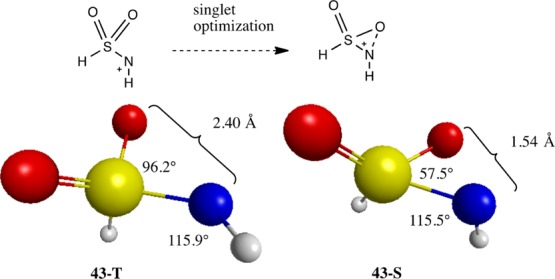
Sulfonylnitrenium ion triplet (43T) and singlet (43S) states along with the selected geometric parameters.
The allylnitrenium ion 36S also distorts its structure to avoid forming a true singlet nitrenium ion (Figure 7). In this case, a shallow minimum (Ea = +0.19 kcal/mol) was located, which features electron donation from the vinylic group into the vacant p orbital on N. This structure could alternatively be considered as a bisected 2-aziridinyl carbinyl cation. Given the relatively short N+–vinyl bond distance (1.48 Å), it is not clear if this species should be classified as a true nitrenium ion. This intermediate is connected by a very small barrier [classically 0.19 kcal/mol, negative when zero-point vibrational energy (ZPVE) is considered] to vinylformiminium ion 36-iso. Both 36 and cyclopropylnitrenium ion 15 are part of the interesting and complex C3H6N+ potential energy surface (PES) which is described in detail elsewhere.69
Figure 7.
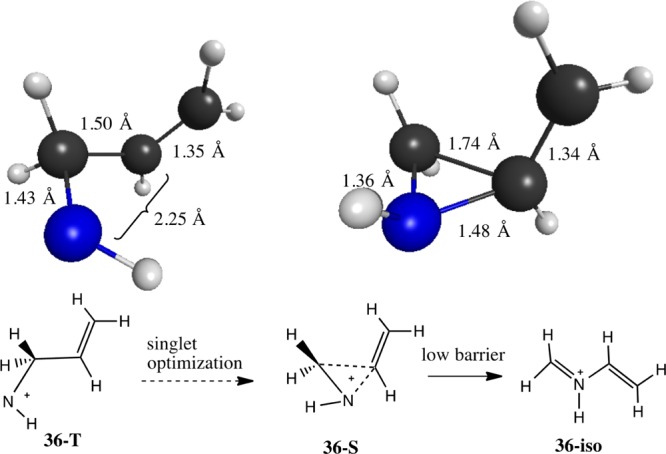
Allylnitrenium ion triplet (36T) and singlet (36S) states along with the selected geometric parameters.
2.2. Singlet–Triplet Energy Gaps
Table 2 lists the singlet–triplet energy gaps for those nitrenium ions having singlet states computed using M06-2X, M06-2X-SMD, and, for selected examples, using the composite CBS-APNO method. As seen in previous experiments and calculations, the parent system 1 is a triplet ground state with a singlet–triplet energy gap of ca. 30 kcal/mol. The present calculations are in accord with the earlier finding.
Table 2. Singlet–Triplet Energy Gaps (kcal/mol, Including ZPVE) for Nitrenium Ions Having Stable Singlet Statesa.
| R1 | R2 | M06-2X gas | M06-2X SMD-H2O | CBS-APNO | |
|---|---|---|---|---|---|
| 1 | H | H | 34.7 | 30.61 | 29.5 |
| 3 | CH3 | CH3 | 2.10 | –3.05 | 2.07 |
| 28 | HCO | H | –2.50 | –2.88 | –11.27 |
| 2 | CH3 | H | na | –6.67 | –11.38 |
| 21 | CH2=CH | tBu | –3.85 | –6.83 | |
| 20 | CH2=CH | CH3 | –4.88 | –8.30 | |
| 31 | CH2=CH | HCO | –5.10 | –10.27 | |
| 19 | CH2=CH | H | –5.67 | –9.94 | –9.08 |
| 24 | Ph | tBu | –7.97 | –8.97 | |
| 32 | CH3CO | CH3O | –9.12 | –13.23 | |
| 29 | CH3CO | H | –9.60 | –10.48 | |
| 32 | HCO | CH3O | –13.8 | –16.47 | |
| 23 | Ph | CH3 | –14.6 | –16.23 | |
| 22 | Ph | H | –19.76 | –21.80 | |
| 17 | cyclopropyl | tBu | –24.3 | –26.72 | |
| 25 | CH3O | H | –27 | –26.82 | –29 |
| 16 | cyclopropyl | CH3 | –27.2 | –28.13 | |
| 27 | CH3O | tBu | –27.6 | –27.48 | |
| 43 | HSO2 | H | –29.1 | –30.4 | |
| 34 | CH2=CH | CH3O | –30 | –28.68 | |
| 36 | CH2=CHCH2 | H | –40.5 | na | |
| 26 | CH3O | CH3 | –30.1 | –30.01 | |
| 15 | cyclopropyl | H | –33.0 | –33.37 | –35.2 |
| 18 | cyclobutyl | H | –36.7 | –44.75 |
Negative values denote singlet ground states.
Intuitively, alkyl substitution should have two competing effects on ΔEst. First, hyperconjugation (i.e., interaction of filled C–C or C–H sigma orbitals with the nominally unfilled p orbital on the N+ center) should increase the energy splitting of the NBMOs on the nitrenium ion, favoring the singlet state. On the other hand, steric pressures ought to force wider N–C–R bond angles, which would favor the triplet. Obviously, the latter effect would be most apparent with alpha-branched alkyl groups (e.g., tert-butyl) and in the cases of disubstituted (secondary) nitrenium ions. Conversely, monosubstitution with primary alkyl or methyl groups is expected to minimize the triplet-favoring steric effect while maintaining at least some measure of hyperconjugative stabilization of the singlet state.
The calculations shown in Table 2 indicated that the electronic effect is dominant, and that steric effects also play a smaller, but not insignificant role. Dimethylnitrenium ion 3, like the parent system NH2+, is predicted to have a triplet ground state in the gas phase. However, unlike the substantial 29.5 kcal/mol energy gap seen for NH2+, the gap for 3 diminishes by ca. 28 kcal/mol toward the singlet state. Indeed, the subtler effects of aqueous solvation are sufficient to invert the state ordering for the dimethyl species, making it a singlet, albeit with a very small energy gap. The monomethylnitrenium singlet state 2S could only be characterized in an aqueous solvent. However, its gap of −6.7 kcal/mol compared with −3.1 kcal/mol for the dimethyl 3 is consistent with the intuitive expectations. Monosubstitution allows for electronic stabilization of the singlet, while minimizing its destabilization through steric pressures.
Unfortunately, simple mono- or disubstituted alkyl nitrenium ions with higher degrees of branching could not be characterized. However, it was possible to examine the effects of alkyl branching in disubstituted nitrenium ions where the remaining substituent provided some stabilization through π-conjugation. Specifically, the effects of H, CH3, and t-butyl substitution were examined in the cases where the remaining substituent was a vinyl, phenyl, methoxy, or cyclopropyl group 15–17, 19–27. These calculations are also in accord with the picture of competing electronic and steric effects.
For the phenyl, vinyl, and cyclopropyl groups, the differential effect of N-alkyl substitution is primarily steric. Thus, methyl and tert-butyl increasingly favor the triplet state relative to the H analogue. In the case of a relatively small vinyl group, the effect is modest; the change of ΔEst is ca. 3 kcal/mol through the series 19–21. With the larger cyclopropyl 15–17 and phenyl groups 22–24, the change in more dramatic. The tert-butyl group shifts ΔEst ca. 11 kcal/mol toward the triplet in the phenyl series (24) and ca. 9 kcal/mol in the cyclopropyl series (17).
The smaller methoxy group imposes less steric pressure on the N+ center (25–27). As a result, the methyl group is able to slightly (ca. 3 kcal/mol) favor the singlet state relative to the H analogue, whereas with tert-butyl the steric effects slightly outweigh the electronic stabilization and the gap shifts back by about 1 kcal/mol toward the triplet relative to the H analogue.
Most of the nonalkyl substituents examined in this study, including acyl, vinyl, phenyl, methoxy 25, cyclpropyl 15, and cyclobutyl 18, similarly stabilize the singlet state relative to the parent system. Aromatic groups have been examined extensively elsewhere using slightly different methods. The examples included here (22–24) are provided for comparison purposes. The ΔEst values and geometries for the aromatic series from M06-2X agree with previous reports with only some minor, quantitative differences. A single phenyl ring 22 stabilizes the singlet state through positive charge delocalization and the gap calculated in this study is −19.8 kcal/mol.
The vinyl group would be expected to behave in a qualitatively similar way. The singlet should be stabilized by charge delocalization through the pi system. Interestingly, the effect is much more modest compared with the effects of phenyl substitution, with a vinyl group generating a ΔEst of only −5.7 kcal/mol compared with −19.5 kcal/mol for phenyl 22.
Alkoxyl groups, being strong pi-electron donors, are expected to substantially stabilize the singlet state, and the M06-2X calculations support this intuitive expectation. The methoxynitrenium 25 ion has a ΔEst of −27 kcal/mol, indicating that this substituent is more effective than a simple phenyl ring in terms of favoring the singlet state.
Unexpectedly, cyclopropyl 15 and cyclobutyl groups 18 have an even more substantial effect on ΔEst. These substituents provide ΔEst values of <−30 kcal/mol. This is attributed to the ability of the singlet states to form the nonclassical (sigma-delocalized) bonding with the small ring substituents (Figures 3 and 4). Similar effects occur in the analogous carbocations. The bicyclobutonium ion is a textbook example of the so-called “banana bonds” in the cyclopropyl ring, delocalizing a positive charge as effectively as, or more than, a pi bond. The corresponding triplet states are unable to form these types of bonds and show structures similar to classically bonded triplets.
Sulfonyl 43 and acyl 28, 29 substituents are, in most contexts, considered to be electron-withdrawing groups. However, in the case of the nitrenium ions, these substituents provide ΔEst values that favor the singlet state. The formyl substituent 28 has an effect that is comparable to a methyl group with ΔEst = −2.9 kcal/mol. An acetyl shows a larger effect with ΔEst = −10 kcal/mol, exceeding the stabilization afforded by a vinyl group and sulfonyl ΔEst = −30 kcal/mol, far exceeding the effect of a phenyl or methoxy substituent. As with the cyclopropyl and cyclobutyl groups, singlet stabilization in these cases can be traced to unusual bonding available to the singlet but not triplet states. For these three examples, the N+–X–O bonds are highly acute and the N+–O distances are small. Thus, the formylnitrenium ion can be plausibly described as an oxaziridinyl cation. Or alternatively, filled–unfilled interactions from the formal O lone pair with the singlet N-based LUMO stabilizes the singlet relative to the triplet state.
Aqueous solvation has a modest, but largely consistent, effect on ΔEst. In most examples, solvation favors the singlet by 1–4 kcal/mol.
2.3. Relative N-Hydration Affinities
In order to assess the ability of various substituents to stabilize singlet nitrenium ions, the enthalpies for series of isodesmic hydration reactions were computed. Specifically, we compared the enthalpies for H2O transfer from NH2OH2+ to the nitrogen centers of the singlet nitrenium ions listed in Table 3 using the M06-2X-SMD model with the aqueous solvation parameters. The latter provides the relative enthalpies as defined in eq 1. The absolute N-hydration enthalpy changes were then computed for the parent system, NH2+, using the composite model chemistry method CBS-APNO/SMD, CBS-APNO, a basis set extrapolation procedure that is designed to give a highly accurate thermochemistry. The latter provides a value of ΔHhyd = −76.51 kcal/mol.
Table 3. Hydration Enthalpies (M06-2x-SMD) for Singlet Nitrenium Ions.

| entry | R1 | R2 | ΔHrel (kcal/mol, 298 K) | ΔHhyd (kcal/mol, 298 K) | |
|---|---|---|---|---|---|
| 1 | H | H | 1 | 0.00 | –76.51a |
| 2 | H–CO | H | 28 | 29.90 | –46.61 |
| 3 | CH3CO | H | 25 | 41.36 | –35.15 |
| 4 | CH3 | H | 2 | 58.60 | –17.91 |
| 5 | CH3 | CH3 | 3 | 64.87 | –11.64 |
| 6 | H–CO | CH=CH2 | 31 | 70.36 | –6.15 |
| 7 | CH3CO | OCH3 | 33 | 74.67 | –1.84 |
| 8 | CH2=CH– | H | 19 | 75.02 | –1.49 |
| 9 | H–CO | OCH3 | 32 | 75.38 | –1.13 |
| 10 | CH3O | H | 25 | 76.64 | 0.13 |
| 11 | CH3O | C(CH3)3 | 27 | 79.53 | 3.02 |
| 12 | CH3O | CH3 | 26 | 80.23 | 3.72 |
| 13 | CH2=CH– | C(CH3)3 | 21 | 82.17 | 5.66 |
| 14 | CH2=CH– | CH3 | 20 | 82.81 | 6.30 |
| 15 | Ph | C(CH3)3 | 24 | 83.79 | 7.28 |
| 16 | Ph | H | 22 | 84.30 | 7.79 |
| 17 | Ph | CH3 | 23 | 88.30 | 11.79 |
| 18 | cyclopropyl | H | 15 | 91.85 | 15.34 |
| 19 | cyclopropyl | CH3 | 16 | 98.41 | 21.90 |
| 20 | cyclobutyl | H | 18 | 99.26 | 22.75 |
| 21 | cyclopropyl | C(CH3)3 | 17 | 101.45 | 24.94 |
A CBS-APNO calculation using an SMD-simulated aqueous solvation.
2.4. Barriers to Rearrangements
Singlet alkylnitrenium ions are isomeric with iminium ions derived from a 1,2 alkyl or H shift from the N-alkyl group to N+. Because the iminium ion is significantly more stable, it could be anticipated that the rearrangement step will be very fast. In fact, historically speaking, there had been considerable debate as to whether free singlet nitrenium ions could exist as discrete intermediates, or if the formation of the iminium ions would be concerted with their formation.
Relatively few simple alkylnitrenium ions could be characterized as minima on the singlet PES. Therefore, we investigated the barriers associated with 1,2 alkyl and H atom migration for those nitrenium ions that could be characterized as singlets. These include the dimethylnitrenium ion 3, the methylnitrenium ion 2 in aqueous solution, and several other nitrenium ions where the remaining substituent was capable of stabilizing the singlet state through resonance electron donation.
All of the alkylnitrenium ions examined here show low barriers to isomerization via 1,2-alkyl or H shifts to the N+ center. Transition states for the unstabilized monomethyl 2 and dimethylnitrenium ions 3 are very low. In the case of monomethyl 2, no minimum could be located in the gas phase, and there was a small 7.2 kcal/mol barrier in the aqueous solution. Dimethyl 3 has even lower barriers, 0.11 and 0.04 kcal/mol, respectively, in the gas phase and aqueous solution. These formal barriers are unlikely to have any experimental relevance. Attempts to explore these rearrangements with higher homologues (ethyl, isopropyl, etc.) were unsuccessful as it was not possible to locate singlet nitrenium ion minima (see below).
It was, however, possible to examine the effects of increasing alkyl branching on isomerization barriers in the cases where the remaining N-substituent stabilized the singlet state via resonance interaction. Specifically, methyl and tert-butyl groups were each combined with methoxy and vinyl N-substituents (entries 3 and 4 for vinyl as well as entries 5 and 6 for methoxy). In both cases, the barrier for alkyl migration (tert-butyl groups) is substantially smaller than the comparable barrier for H atom migration (from the methyl groups) (Table 4). Thus, it is reasonable to infer that in the mono- and dialkyl cases, alkyl migrations would have smaller barriers than seen in the mono- or dimethylnitrenium ions. As the barriers in the methyl cases are very small to begin with, larger alkyl groups would be expected to show rearrangements without barriers or with trivial barriers.
Table 4. Calculated Barriers (M06-2X, kcal/mol) for Alkyl or Hydrogen Migrations for Various Singlet Nitrenium Ions in the Gas Phase and Aqueous Solution.
| entry | R1 | R2 | Strct. | ΔE⧧ gas (kcal/mol) | ΔEiso gas (kcal/mol) | ΔE⧧ water (kcal/mol) | ΔEiso (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 1 | CH3 | H | 2 | na | na | 7.2 | –95.0 |
| 2 | CH3 | CH3 | 3 | 0.11 | –72.5 | 0.04 | –65.4 |
| 3 | CH2=CH | CH3 | 20 | 13.8 | –50.0 | 12.6 | –48.4 |
| 4 | CH2=CH | C(CH3)3 | 21 | 1.4 | –56.8 | 3.5 | –53.4 |
| 5 | CH3O | CH3 | 26 | 24.1 | –20.6 | 19.5 | –28.4 |
| 6 | CH3O | C(CH3)3 | 27 | 8.1 | –38.8 | 6.6 | –38.1 |
| 7 | Ph | CH3 | 23 | 22.8 | –39.0 | 17.8 | –42.5 |
| 8 | cyclopropyl | CH3 | 16 | 4.9 | –40.5 | 4.9 | –36.1 |
2.5. Nitrenium Ions Showing No Local Minima as Singlets
Table 5 shows schematic representations of input and optimized geometries for 23 simple alkyl, dialkyl, cycloalkyl, and acyl alkyl nitrenium ions. None of these examples converged to local minima at the starting nitrenium ion-like geometries. Structures were minimized at the M06-2X level, and using the triplet geometries, at the SMD-M06-2X and MP2 levels as well. Except where noted, each of these optimizations converged on the same structures, which were verified as local minima through vibrational frequency analysis of the reported structure. Similarly, unless otherwise stated, each of these examples did converge to a triplet minimum near the input geometry.
Table 5. Schematic Depiction of Nitrenium Ions That Do Not Show Discrete Singlet Minima (M06-2X) and Their Converged Structures.

In most cases, the singlet minima are structures that involve 1,2 alkyl or H atom shifts from the alkyl substituent to the nitrenium ion center, creating iminium ion structures. Notably, Gassman’s nitrenium ion 14 minimizes directly to the same alkyl shift isomer that was experimentally trapped by nucleophiles. Similarly, the 2,2,6,6-tetramethylpiperidinyl nitrenium ion 11 minimized to the methyl shift isomer, consistent with experimental reports.6
For the aziridinyl nitrenium ion 8, the minimized structure involves ring-opening to form a bis-methyleneiminium ion, 8S-iso. This confirms earlier calculations that had used different methodologies.65 An early experimental study by Gassman et al.81 found that solvolysis of N-chloroaziridines produced products consistent with the concerted formation of 8S-iso (Scheme 2).
Scheme 2.

A few examples resulted in net elimination reactions. For example, methylacetylnitrenium ion 30 minimizes to an acetylcation/formimine molecular complex. Methyl- (2) and ethyl- (3) nitrenium ions minimized to molecular complexes of H2 and the conjugated acids of HCN or CH3CN. The endo-succinimidylnitrenium ion 35 is similarly predicted to eliminate ethylene to form an interesting heterocummulene cation OCNCO+, which has been characterized by mass spectrometry and X-ray crystallography.82,83
In contrast, optimization of these same species as triplets readily converged on local minima with characteristic nitrenium ion structures. The one exception to this was the triplet state of the 1-pent-4-enylnitrenium ion (38). The corresponding singlet cyclizes through a formal insertion of the N+ into the proximal vinylic C–H bond. Interestingly, the triplet also minimizes to a diradical structure wherein the N+ adds to the vinylic group in a 5-endo-trig fashion.
2.6. Concerted Dehydration/Rearrangement
Although these geometry optimizations provide a fairly consistent picture of unstable singlet states for alkylnitrenium ions, one should be cautious about overinterpreting the results. First, while the algorithm employed in the optimizations has proven to be extremely robust and reliable at locating minima, it does not explore the entirety of the phase space. Thus, there is always a possibility, if a remote one, that some unlocated minimum exists for a given singlet nitrenium ion. Second, the geometry minimization procedures are not the same as a molecular dynamics simulation. Thus, the singlet minima reported in Table 5 do not necessarily reflect products that would result from attempted formation of the corresponding singlet nitrenium ion under chemically realistic conditions. The latter would generally be formed through some sort of adiabatic bond heterolysis and/or oxidation process, rather than being instantaneously born at some arbitrary geometry. In those cases, the pathway that is followed would presumably be dependent on the nature of the leaving group or oxidizing species.
A comprehensive study of all possible oxidation and heterolysis processes that could make nitrenium ions was deemed beyond the scope of this study. However, in order to identify the possible products from such pathways, one such process was considered. Specifically, the heterolysis of O-protonated hydroxylamines was examined (Figure 8). In these computations, the structures of the O-protonated conjugate acids for N-methylhydroxylamine, N-tert-butylhydroxylamine, and the corresponding derivative of Gassman’s nitrenium ion 14 were minimized at the M06-2X-SMD level using aqueous solvation. For each of these structures, the N–O bond length was held constant at increasing increments of 0.04 Å from its equilibrium value, whereas all other geometric parameters were allowed to relax at each increment. In each case, this heterolysis scan resulted in a product where the corresponding alkyl group or H atom migrated in concert with the loss of the water molecule. The results of these scans provided starting geometries, which could be then optimized to the transition states for the corresponding heterolysis reaction as well as product complexes where the water was bound to the product.
Figure 8.

Concerted dehydration/rearrangement for O-protonated hydroxylamines.
Shown in Figures 9–11 are reactants, products, and transition states for the dehydration reactions for the N–H2O adducts of methylnitrenium ion 2, tert-butylnitrenium ion 5, and Gassmans’ nitrenium ion 14. In each case, the transition state features significant movement of the beta alkyl or H atom. Of particular interest is the case of the methynitrenium ion. As shown in Table 5, this structure optimizes to H2 and HCN–H+ when the singlet is modeled in the gas phase. However, the predicted product under heterolysis conditions is the iminium ion resulting from the 1,2-shift, rather than the H2 elimination product. As might be expected, the more substituted alkyl group in 5-OH2+ follows a similar concerted dehydration/alkyl migration pathway. However, in this case, the barrier is a bit lower and the overall driving force is larger, presumably due to relief of steric strains in the reactants and stabilization of the C=N bond in the product through alkyl substitution.
Figure 9.
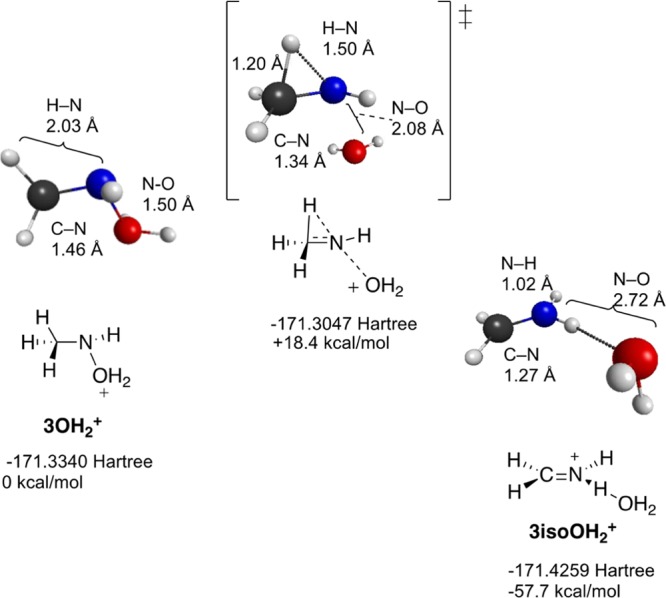
Stationary points in the singlet state dehydration of CH3NHOH2+ (3OH2+) illustrating loss of water concerted with H migration.
Figure 11.
Stationary points with selected geometric parameters from the concerted dissociation/rearrangement of 33-OH2+. A single transition state connects the protonated hydroxamic acid 33-OH2 to the ring-contracted product.
Figure 10.
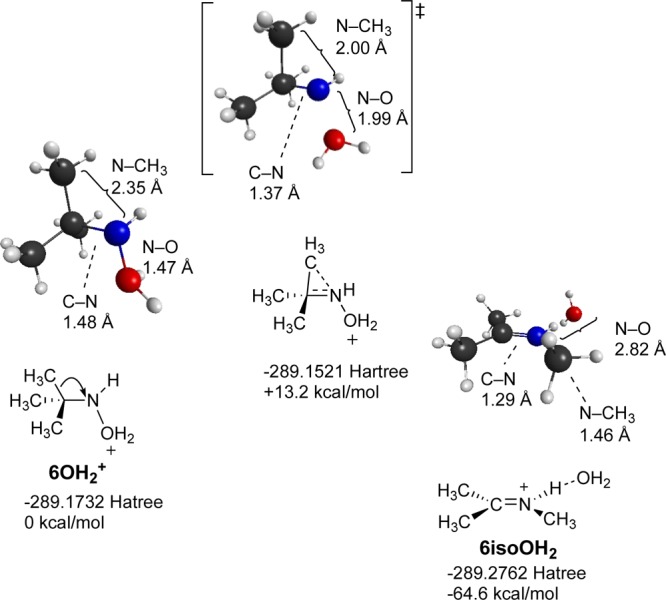
Stationary points in the singlet state dehydration of tBuNHOH2+ (6OH2+) illustrating loss of water concerted with CH3 migration.
The endocyclic acylnitrenium ions (derived from lactams) 33 and 34 minimize to interesting ring-opened structures in their triplet states. The singlet states minimize to highly strained, protonated, unsaturated lactams. In contrast, Lesard et al.48,84,85 demonstrated alkyl migrations resulting in synthetically useful ring contractions of N-chlorolactams and related derivatives (i.e., potential nitrenium ion precursors). An example of these is provided in Scheme 3. Development of the positive charge on the nitrogen results in the migration of either the carbonyl-adjacent group (C3) to the nitrogen, providing a carbonyliminium ion, or the migration of the carbon one atom removed from the nitrogen (C5) to the latter, forming an alkylidene iminium ion.
Scheme 3.

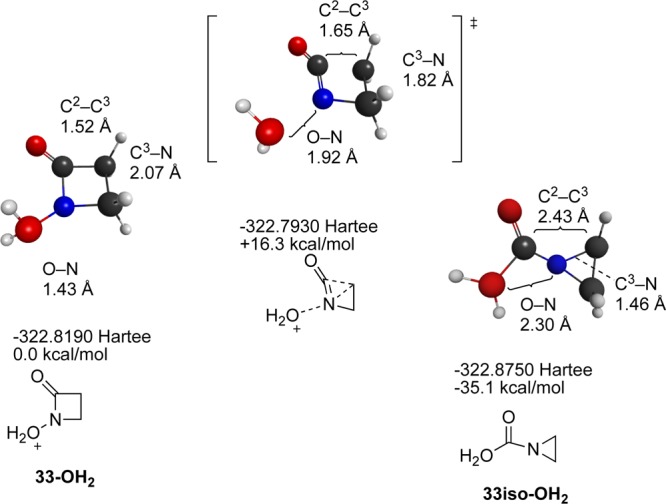
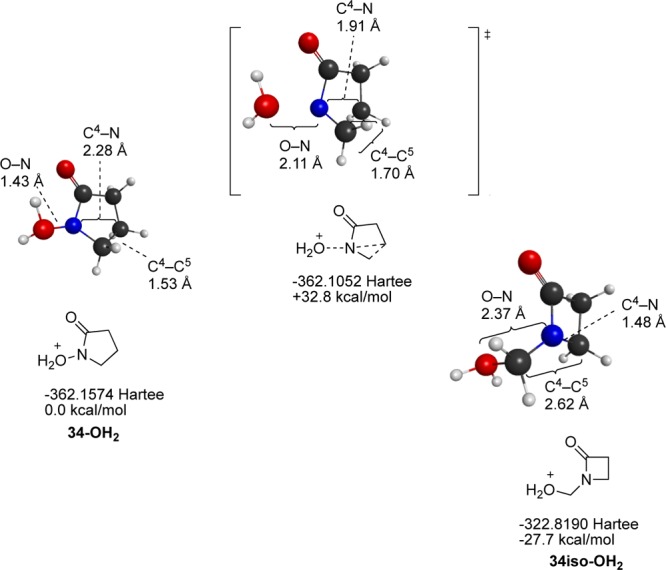
To resolve this apparent discrepancy, heterolyses of two lactam-derived nitrenium ions, 33-OH2+ and 34-OH2+ were examined (Scheme 4). In both cases, low energy transition states leading to ring contraction products were located. Interestingly, 33-OH2+ dissociates with concerted formation of the ring-contracted carboxylic acid, whereas the larger 34-OH2+ dissociates to form the corresponding hydroxymethyl derivative (Figure 12). Although this study did not consider specific species and leaving groups examined in the earlier report, it does provide a picture that is broadly consistent with these experimental findings. Alkyl migration, rather than H migration, is preferred.
Scheme 4.
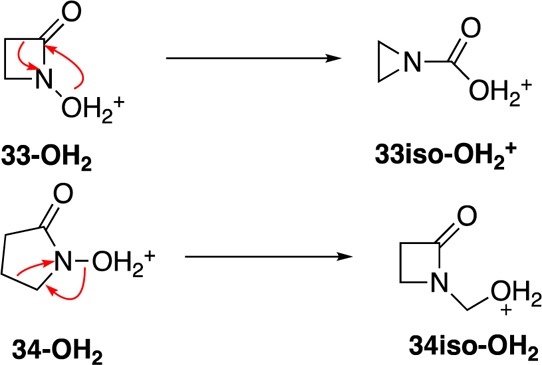
Figure 12.
Stationary points in the dissociation/recombination of 34-iso-OH2. A single transition state connects the protonated hydroxamic acid 34-OH2 to the ring-contracted product.
Also examined was Gassman’s bicyclic nitrenium ion 14 (Figure 13). As described above, attempts to characterize the isolated singlet state of this species result in collapse to the 1,2-alkyl shift product (14-iso). Because of the historic interest in 14, calculations on its formation via heterolysis were carried out in order to determine if some intermediate might form, which would allow for the proposed intersystem crossing to 14T. Results from the N–OH2 bond scan and minimization process, summarized in Figure 11, show that this is not the case. The N-hydrate undergoes a very low barrier (3.3 kcal/mol) rearrangement to an isomer where the 1,2-alkyl shift occurs concertedly with the migration of the H2O to the neighboring carbon. Attempts to locate a minimum analogous to the H-bonded complexes seen in the case of 2 and 5 were unsuccessful. Minimizations starting at these approximate geometries converged on 14-iso-H2O. A similar study on 13-H2O, a derivative of the Gassman nitrenium ion, lacking the methyl groups, provided similar results. The barrier for this example was calculated to be 5.8 kcal/mol.
Figure 13.
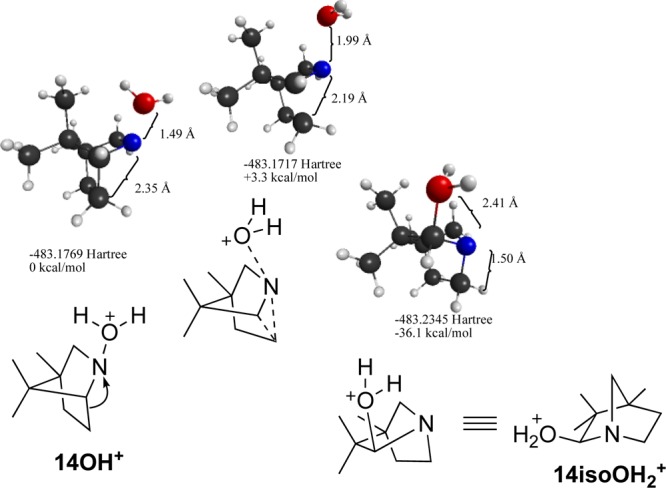
Stationary points from the concerted heterolytic dissociation and alkyl migration of Gassman’s nitrenium ion 14-OH2+.
This computational finding is consistent with earlier experimental results of Hoffman et al.43 as well as Gassman and Hartman,86 both of whom found that attempts to generate 14S (using arylsulfonate leaving groups, rather than water as modeled here) resulted in direct formation of rearrangement products analogous to 14-isoOH2+. Apparently, the formation of the anti-Bredt iminium ion 14-iso from complete dissociation of the leaving group (H2O, in the present study) is unfavorable, and a concerted migration of the leaving group to the neighboring carbon is preferred.
3. Conclusions
The calculations described here, along with earlier experimental studies, consistently indicate that simple aliphatic or endoalicyclic nitrenium ions are not stable and should rearrange without a barrier in the singlet state. In the two cases (2S and 3S) that do show formal minima, the small barriers to rearrangement are not likely to have any chemical significance. Although the triplets are kinetically stable, the projected singlet–triplet energy gaps either favor the singlet or are small enough that facile intersystem crossing is likely to defeat any attempt at isolating or detecting the triplets. Absent compelling evidence to the contrary, detection of apparent nitrenium ion products from alkylamine oxidation should be attributed to concerted processes. Exceptions to this generalization are the cyclobutyl and cyclopropyl substituted cases. These form interesting structures having nonclassical σ-bonding, similar to the corresponding carbenium ions.
The situation changes when even modestly effective π-donating substituents are attached to the nitrenium ion center. Vinyl, aryl, alkoxy, cyclobutyl, and cyclopropyl groups all stabilize the singlet state and create barriers to rearrangement when the remaining substituent is an alkyl group. This stabilization is also seen in increasing N-hydration enthalpies (Table 3) and ΔEst values that increasingly favor the singlet state (Table 4). In those cases, formation along with detection and/or chemical trapping of a discrete nitrenium ion species should be possible, albeit technically challenging. Acyl substituents have a very modest stabilizing effect on the singlet state as measured by the calculated N-hydration enthalpies and ΔEst, but are not sufficiently stabilizing to inhibit migration processes in cases where the remaining substituent is an alkyl group. The relatively simple substitutions considered here are expected to result in singlet ground states where ΔEst decreases in the order alkyl ∼ acyl < vinyl < phenyl < alkoxy ∼ sulfonyl < cyclopropyl ∼ cyclobutyl. Simple arylnitrenium ions are also expected to be ground-state singlets. However, it has been shown that with complex substitution patterns (e.g., meta donors or antiaromatic systems), certain arylnitrenium ions can become ground-state triplets.87−89 Thus, future studies will be directed at identifying situations where the non-aryl substituents studied here might result in unusual electronic properties.
4. Computational Methods
Geometries of singlet- and triplet-state nitrenium ions and related species described herein were optimized using varying levels of theory such as the hybrid density functional M06-2X90 using the 6-311G(d,p)91 basis set for gas phase calculation, abbreviated below as M06-2X. This combination of functional and basis set was chosen because, in cases where comparisons could be made, it gave reasonable agreement with more accurate composite methods (e.g., CBS-APNO), but at a significantly lower cost and could thus be applied to larger species. M06-2X-SMD refers to the same calculations carried out using the implicit continuum solvation model “SMD” developed by Truhlar and co-workers.92 Unless otherwise noted, such calculations were carried out using the parameters for aqueous solvation. MP2 refers to second-order Møller–Plesset perturbation theory. Where possible, more accurate calculations were carried out using a basis set extrapolation method CBS-APNO,93 which has been shown to provide excellent thermochemical data for cationic intermediates.94 Singlet–triplet energy splittings (ΔEst) are adiabatic values that include unscaled ZPVE corrections for both the singlet and triplet states. All the reported calculations were done using the Gaussian 09 software package.95 Analysis of orbitals and their hybridization was obtained using the NBO method of Weinhold.96
The nitrenium ions considered in this study are shown in Chart 1. For each numbered structure, X, X-S refers to the nitrenium ion singlet state, X-T refers to the nitrenium ion’s triplet state, X-isoN refers to the isomers of the nitrenium ion (generally, these result from 1,2 alkyl shifts). The subscript N provides for situations where more than one isomer can be formed. X-OH2+ refers to the N-hydration adduct (i.e., O-protonated N-hydroxylamine analogue).
Acknowledgments
This work was supported by the National Science Foundation CHE1112018.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01038.
Cartesian coordinates for structures 1–43 (PDF)
The author declares no competing financial interest.
Supplementary Material
References
- Falvey D. E.Electronic Properties Of Nitrenium Ions. In Nitrenes and Nitrenium Ions; Falvey D. E., Gudmundsdottir A. D., Eds.; John Wiley And Sons, Inc.: New York, 2013; pp 191–216. [Google Scholar]
- Novak M.; Rajagopal S.. N-Arylnitrenium Ions. In Advances in Physical Organic Chemistry; Academic Press Ltd: London, 2001; Vol. 36, pp 167–254. [Google Scholar]
- Falvey D. E. Singlet And Triplet States In The Reactions Of Nitrenium Ions. J. Phys. Org. Chem. 1999, 12, 589–596. . [DOI] [Google Scholar]
- Abramovitch R. A.; Jeyaraman R.. Nitrenium Ions. In Azides and Nitrenes: Reactivity and Utility; Scriven E. F. V., Ed. Academic Press: Orlando, Fl, 1984; pp 297–357. [Google Scholar]
- Gassman P. G. Nitrenium Ions. Acc. Chem. Res. 1970, 3, 26–33. 10.1021/ar50025a004. [DOI] [Google Scholar]
- Henry-Riyad H.; Kobayashi S.; Tidwell T. T. The Search For Aliphatic Nitrenium Ions From Solvolysis Of N-2,2,6,6-Tetramethylpiperidinyl p-Nitrobenzoate. Arkivoc 2005, 266–276. 10.3998/ark.5550190.0006.622. [DOI] [Google Scholar]
- Falvey D. E.Nitrenium Ions In Reactive Intermediate Chemistry; Moss R. A., Platz M. S., Jones M. Jr., Eds.; John Wiley And Sons: Hoboken, NJ, 2004; pp 593–650. [Google Scholar]
- Guengerich F. P. N-Hydroxyarylamines. Drug Metab. Rev. 2002, 34, 607–623. 10.1081/dmr-120005663. [DOI] [PubMed] [Google Scholar]
- Ren D.; McClelland R. A. Carbocation-like reactivity patterns in X’-substituted-4-biphenylylnitrenium ions. Can. J. Chem. 1998, 76, 78–84. 10.1139/cjc-76-1-78. [DOI] [Google Scholar]
- Anderson G. B.; Yang L. L. N.; Falvey D. E. Photogenerated Arylnitrenium Ions–Photoisomerization Of The N-tert-Butyl-3-Methylanthranilium Ion And Spin-Selective Reactivity Of The Isomeric Arylnitrenium Ion. J. Am. Chem. Soc. 1993, 115, 7254–7262. [Google Scholar]
- Robbins R. J.; Falvey D. E. Photogenerated nitrenium ions: Singlet and triplet state reactions of tert-butyl-(2-acetyl-4-methyl)phenyl nitrenium ion. Tetrahedron Lett. 1994, 35, 4943–4946. 10.1016/s0040-4039(00)73288-6. [DOI] [Google Scholar]
- Fishbein J. C.; McClelland R. A. Azide ion trapping of the intermediate in the Bamberger rearrangement. Lifetime of a free nitrenium ion in aqueous solution. J. Am. Chem. Soc. 1987, 109, 2824–2825. 10.1021/ja00243a045. [DOI] [Google Scholar]
- Gassman P. G.; Campbell G. A. Chemistry of nitrenium ions. XXII. Thermal rearrangement of N-chloroanilines. Evidence for the intermediacy of of nitrenium ions. J. Am. Chem. Soc. 1972, 94, 3891–3896. 10.1021/ja00766a038. [DOI] [Google Scholar]
- Chakraborty M.; Jin K. J.; Brewer S. C.; Peng H.-L.; Platz M. S.; Novak M. Indirect And Direct Detection Of The 4-(Benzothiazol-2-Yl)Phenylnitrenium Ion From A Putative Metabolite Of A Model Anti-Tumor Drug. Org. Lett. 2009, 11, 4862–4865. 10.1021/ol901959z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Kubicki J.; Platz M. S. An Ultrafast Study Of Phenyl Azide: The Direct Observation Of Phenylnitrenium Ion. Org. Lett. 2007, 9, 3973–3976. 10.1021/ol701598m. [DOI] [PubMed] [Google Scholar]
- Xue J.; Guo Z.; Chan P. Y.; Chu L. M.; But T. Y. S.; Phillips D. L. Time-Resolved Resonance Raman Study Of The Reaction Of The 2-Fluorenylnitrenium Ion With 2-Fluorenylazide. J. Phys. Chem. A 2007, 111, 1441–1451. 10.1021/jp066699y. [DOI] [PubMed] [Google Scholar]
- Chan P. Y.; Zhu P.; Phillips D. L. Transient Resonance Raman And Density Functional Theory Study Of The 7-Bromo-2-Fluorenylnitrenium Cation. Res. Chem. Intermed. 2005, 31, 73–84. 10.1163/1568567053146913. [DOI] [Google Scholar]
- Kwok W. M.; Chan P. Y.; Phillips D. L. Direct Observation of the 2-Fluorenylnitrene and 4-Methoxyphenylnitrene Reactions with Water Using Picosecond Kerr-Gated Time-Resolved Resonance Raman Spectroscopy. J. Phys. Chem. B 2004, 108, 19068–19075. 10.1021/jp0467292. [DOI] [PubMed] [Google Scholar]
- Chan P. Y.; Ong S. Y.; Zhu P.; Zhao C.; Phillips D. L. Transient Resonance Raman and Density Functional Theory Investigation of 4-Methoxyphenylnitrenium and 4-Ethoxyphenylnitrenium Ions†. J. Phys. Chem. A 2003, 107, 8067–8074. 10.1021/jp0224261. [DOI] [Google Scholar]
- Ramlall P.; McClelland R. A. Photochemical Generation and Lifetimes In Water of p-Aryloxy- And p-Alkoxyphenylnitrenium Ions. J. Chem. Soc., Perkin Trans. 2 1999, 2, 225–232. 10.1039/a807567k. [DOI] [Google Scholar]
- McClelland R. A.; Kahley M. J.; Davidse P. A.; Hadzialic G. Acid–Base Properties of Arylnitrenium Ions. J. Am. Chem. Soc. 1996, 118, 4794–4803. 10.1021/ja954248d. [DOI] [Google Scholar]
- Anderson G. B.; Falvey D. E. Photogenerated arylnitrenium ions: absorption spectra and absolute rate constants for tert-butyl(4-halo-2-acetylphenyl)nitrenium ions measured by time-resolved laser spectroscopy. J. Am. Chem. Soc. 1993, 115, 9870–9871. 10.1021/ja00074a092. [DOI] [Google Scholar]
- Chen H.; Zheng X.; Yang P.; Cooks R. G. Reduction of nitroaromatics to arylnitrenium ions by vinyl halide. Chem. Commun. 2004, 688–689. 10.1039/b314713d. [DOI] [PubMed] [Google Scholar]
- Brown T. A.; Hosseini-Nassab N.; Chen H.; Zare R. N. Observation Of Electrochemically Generated Nitrenium Ions By Desorption Electrospray Ionization Mass Spectrometry. Chem. Sci. 2016, 7, 329–332. 10.1039/c5sc02939b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieker A.; Speiser B. Electrochemistry of anilines. 6. Reactions of electrogenerated biphenylylnitrenium ions. J. Org. Chem. 1991, 56, 4664–4671. 10.1021/jo00015a019. [DOI] [Google Scholar]
- Rieker A.; Speiser B. Electrochemistry of anilines. Part 5. Spectroscopic and electrochemical characterization of a persistent biphenylyl nitrenium ion. Tetrahedron Lett. 1990, 31, 5013–5014. 10.1016/s0040-4039(00)97792-x. [DOI] [Google Scholar]
- Novak M.; Zhang Y.. Antitumor Drugs And Nitrenium Ions. In Advances in Physical Organic Chemistry; Williams I. H., Williams N. H., Eds.; Academic Press Ltd-Elsevier Science Ltd: London, 2012; Vol. 46, pp 121–164. [Google Scholar]
- Skipper P. L.; Kim M. Y.; Sun H.-L. P.; Wogan G. N.; Tannenbaum S. R. Monocyclic Aromatic Amines As Potential Human Carcinogens: Old Is New Again. Carcinogenesis 2010, 31, 50–58. 10.1093/carcin/bgp267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisson J.; Stiborová M.; Martínek V.; Gamboa da Costa G.; Phillips D. H.; Arlt V. M. Mutagenic Potential Of Nitrenium Ions Of Nitrobenzanthrones: Correlation Between Theory And Experiment. Environ. Mol. Mutagen. 2008, 49, 659–667. 10.1002/em.20411. [DOI] [PubMed] [Google Scholar]
- Cui L.; Sun H.-L.; Wishnok J. S.; Tannenbaum S. R.; Skipper P. L. Identification of Adducts Formed by Reaction of N-Acetoxy-3,5-dimethylaniline with DNA. Chem. Res. Toxicol. 2007, 20, 1730–1736. 10.1021/tx700306c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onozato M.; Ohshima S. Analysis Of Mutagenicity Of Nitrobenzanthrones By Molecular Orbital Calculations. Polycyclic Aromat. Compd. 2006, 26, 93–101. 10.1080/10406630600642113. [DOI] [Google Scholar]
- McClelland R. A.; Ahmad A.; Dicks A. P.; Licence V. E. Spectroscopic Characterization of the Initial C8 Intermediate in the Reaction of the 2-Fluorenylnitrenium Ion with 2’-Deoxyguanosine. J. Am. Chem. Soc. 1999, 121, 3303–3310. 10.1021/ja9836702. [DOI] [Google Scholar]
- McClelland R. A.; Gadosy T. A.; Ren D. 1997 Alfred Bader Award Lecture Reactivities of arylnitrenium ions with guanine derivatives and other nucleophiles. Can. J. Chem. 1998, 76, 1327–1337. 10.1139/v98-187. [DOI] [Google Scholar]
- Boche G.; Andrews P.; Harms K.; Marsch M.; Rangappa K. S.; Schimeczek M.; Willeke C. Crystal and Electronic Structure of Stable Nitrenium Ions. A Comparison With Structurally Related Carbenes. J. Am. Chem. Soc. 1996, 118, 4925–4930. 10.1021/ja9536274. [DOI] [Google Scholar]
- McIlroy B.; Cramer C. J.; Falvey D. E. Singlet-triplet energy gaps in highly stabilized nitrenium tons: Experimental and theoretical study of 1,3-dimethylbenzotriazolium ion. Org. Lett. 2000, 2, 2451–2454. [DOI] [PubMed] [Google Scholar]
- Kikugawa Y. Application Of Stable Nitrenium Ions To Preparative Organic Chemistry. Heterocycles 2009, 78, 571–607. 10.3987/rev-08-644. [DOI] [Google Scholar]
- Tulchinsky Y.; Kozuch S.; Saha P.; Botoshansky M.; Shimon L. J. W.; Gandelman M. Cation-Cation Bonding In Nitrenium Metal Complexes. Chem. Sci. 2014, 5, 1305–1311. 10.1039/c3sc53083c. [DOI] [Google Scholar]
- Pogoreltsev A.; Tulchinsky Y.; Fridman N.; Gandelman M. Nitrogen Lewis Acids. J. Am. Chem. Soc. 2017, 139, 4062–4067. 10.1021/jacs.6b12360. [DOI] [PubMed] [Google Scholar]
- Schell F. M.; Ganguly R. N. Silver ion induced rearrangement of N-chloramines. Isolation of an ionic product in high yield. J. Org. Chem. 1980, 45, 4069–4070. 10.1021/jo01308a033. [DOI] [Google Scholar]
- Gassman P. G.; Cryberg R. L. Chemistry of nitrenium ions. IX. Discrete existence of singlet and triplet nitrenium ions. J. Am. Chem. Soc. 1969, 91, 5176–5177. 10.1021/ja01046a050. [DOI] [Google Scholar]
- Gassman P. G.; Carrasquillo A. Cyclopropylnitrenium Ions. J. Chem. Soc. D 1969, 495–496. 10.1039/c29690000495. [DOI] [Google Scholar]
- Hoffman R. V.; Buntain G. A. Carbon-To-Nitrogen Rearrangement in N-(Arylsulfonoxy)Amines As A Route To Azacyclic Compounds. J. Org. Chem. 1988, 53, 3316–3321. 10.1021/jo00249a034. [DOI] [Google Scholar]
- Hoffman R. V.; Kumar A.; Buntain G. A. Ionization of N-arylsulfonyloxy amines: the nitrenium ion question. J. Am. Chem. Soc. 1985, 107, 4731–4736. 10.1021/ja00302a022. [DOI] [Google Scholar]
- Piotrowski D. W.; Rolph M.; Wei L. Substituted azabicyclo[2.2.1]heptanes via nitrenium ion rearrangement. Tetrahedron Lett. 2012, 53, 1009–1012. 10.1016/j.tetlet.2011.12.065. [DOI] [Google Scholar]
- Wardrop D. J.; Burge M. S. Nitrenium Ion Azaspirocyclization–Spirodienone Cleavage: A New Synthetic Strategy for the Stereocontrolled Preparation of Highly Substituted Lactams and N-Hydroxy Lactams. J. Org. Chem. 2005, 70, 10271–10284. 10.1021/jo051252r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh C. W.; Aubé J. Aryl Nitrenium Ions From N-Alkyl-N-Arylamino-Diazonium Precursors: Synthesis and Reactivity. Chem. Sci. 2014, 5, 699–706. 10.1039/c3sc52805g. [DOI] [Google Scholar]
- Stokes B. J.; Richert K. J.; Driver T. G. Examination of the Mechanism of Rh2(II)-Catalyzed Carbazole Formation Using Intramolecular Competition Experiments. J. Org. Chem. 2009, 74, 6442–6451. 10.1021/jo901224k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter D. K.; Drouin A.; Lessard J.; Spino C. Photochemical Rearrangement of N-Chlorolactams: A Route to N-Heterocycles through Concerted Ring Contraction. J. Org. Chem. 2010, 75, 2610–2618. 10.1021/jo100181h. [DOI] [PubMed] [Google Scholar]
- Murai K.; Matsuura K.; Aoyama H.; Fujioka H. Oxidative Rearrangement Via In Situ Generated N-Chloroamine: Synthesis Of Fused Tetrahydroisoquinolines. Org. Lett. 2016, 18, 1314–1317. 10.1021/acs.orglett.6b00233. [DOI] [PubMed] [Google Scholar]
- Maiti S.; Mal P. Dehydrogenative Aromatic Ring Fusion For Carbazole Synthesis Via C-C/C-N Bond Formation And Alkyl Migration. Org. Lett. 2017, 19, 2454–2457. 10.1021/acs.orglett.7b01117. [DOI] [PubMed] [Google Scholar]
- Olah G. A. Stable carbocations, 189. The .sigma.-bridged 2-norbornyl cation and its significance to chemistry. Acc. Chem. Res. 1976, 9, 41–52. 10.1021/ar50098a001. [DOI] [Google Scholar]
- Olah G. A. 100 Years of Carbocations and Their Significance in Chemistry1. J. Org. Chem. 2001, 66, 5943–5957. 10.1021/jo010438x. [DOI] [PubMed] [Google Scholar]
- Scholz F.; Himmel D.; Heinemann F. W.; Schleyer P. v. R.; Meyer K.; Krossing I. Crystal Structure Determination Of The Nonclassical 2-Norbornyl Cation. Science 2013, 341, 62–64. 10.1126/science.1238849. [DOI] [PubMed] [Google Scholar]
- Moss R. A. The 2-Norbornyl Cation: A Retrospective. J. Phys.Org. Chem. 2014, 27, 374–379. 10.1002/poc.3290. [DOI] [Google Scholar]
- Harrison J. F.; Eakers C. W. Electronic structure of reactive intermediates. Nitrenium ions nitrenium(+), fluoronitrenium(+), and difluoronitrenium(+). J. Am. Chem. Soc. 1973, 95, 3467–3472. 10.1021/ja00792a004. [DOI] [Google Scholar]
- Peyerimhoff S. D.; Buenker R. J. Ab initio MRD-CI potential surfaces for the low-lying states of the NH+2 molecular ion. Chem. Phys. 1979, 42, 167–176. 10.1016/0301-0104(79)85176-9. [DOI] [Google Scholar]
- Stephens J. C.; Yamaguchi Y.; Sherrill C. D.; Schaefer H. F. X̃3B1, ã1A1, b̃1B1, and c̃1 Electronic States of NH2+. J. Phys. Chem. A 1998, 102, 3999–4006. 10.1021/jp980779n. [DOI] [Google Scholar]
- Van Huis T. J.; Leininger M. L.; Sherrill C. D.; Schaefer H. F. Full Configuration Interaction Energies, Geometries, And Quartic Force Fields Of The Nitrenium Ion. Collect. Czech. Chem. Commun. 1998, 63, 1107–1142. 10.1135/cccc19981107. [DOI] [Google Scholar]
- Stephens J. C.; Yamaguchi Y.; Schaefer H. F. III The adiabatic and vertical ionization potentials of NH2 to the three lowest-lying states of NH2+. J. Mol. Struct.: THEOCHEM 1999, 461–462, 41–53. 10.1016/s0166-1280(98)00420-5. [DOI] [Google Scholar]
- Willitsch S.; Merkt F.; Kállay M.; Gauss J. Thermochemical properties of small open-shell systems: experimental and high-level ab initio results for NH2. Mol. Phys. 2006, 104, 1457–1461. 10.1080/13895260500518551. [DOI] [Google Scholar]
- Winter A. H.; Gibson H. H.; Falvey D. E. Carbazolyl Nitrenium Ion: Electron Configuration And Antiaromaticity Assessed By Laser Flash Photolysis, Trapping Rate Constants, Product Analysis, And Computational Studies. J. Org. Chem. 2007, 72, 8186–8195. 10.1021/jo0708184. [DOI] [PubMed] [Google Scholar]
- Gonzalez C.; Restrepo-Cossio A.; Márquez M.; Wiberg K. B.; De Rosa M. Ab Initio Study of the Solvent Effects on the Singlet–Triplet Gap of Nitrenium Ions and Carbenes. J. Phys. Chem. A 1998, 102, 2732–2738. 10.1021/jp981150n. [DOI] [Google Scholar]
- Cramer C. J.; Truhlar D. G.; Falvey D. E. Singlet–Triplet Splittings and 1,2-Hydrogen Shift Barriers for Methylphenylborenide, Methylphenylcarbene, and Methylphenylnitrenium in the Gas Phase and Solution. What a Difference a Charge Makes. J. Am. Chem. Soc. 1997, 119, 12338–12342. 10.1021/ja9723390. [DOI] [Google Scholar]
- Chiapperino D.; Falvey D. E. N-Methyl-N-phenylnitrenium ion from photolysis of N-(methylphenylamino)-2,4,6-trimethylpyridinium tetrafluoroborate. J. Phys. Org. Chem. 1997, 10, 917–924. . [DOI] [Google Scholar]
- Lim M. H.; Worthington S. E.; Dulles F. J.; Cramer C. J.. Density-Functional Calculations Of Radicals And Diradicals. In Chemical Applications of Density-Functional Theory; Laird B. B., Ross R. B., Ziegler T., Eds.; American Chemical Society: Washington, 1996; Vol. 629, pp 402–422. [Google Scholar]
- Ford G. P.; Herman P. S. Energetics of the singlet and triplet states of alkylnitrenium ions: ab initio molecular orbital calculations. J. Am. Chem. Soc. 1989, 111, 3987–3996. 10.1021/ja00193a035. [DOI] [Google Scholar]
- Favley D. E.; Cramer C. J. Arylnitrenium And Alkylnitrenium Ions - Singlet-Triplet Gaps Via Abinitio And Semiempirical Methods. Tetrahedron Lett. 1992, 33, 1705–1708. 10.1016/s0040-4039(00)91711-8. [DOI] [Google Scholar]
- Cramer C. J.; Dulles F. J.; Falvey D. E. Ab Initio Characterization of Phenylnitrenium and Phenylcarbene: Remarkably Different Properties for Isoelectronic Species. J. Am. Chem. Soc. 1994, 116, 9787–9788. 10.1021/ja00100a069. [DOI] [Google Scholar]
- Falvey D. E. Nitrenium Ion Analogues Of Nonclassical Carbocations: Cyclopropylnitrenium, Allylnitrenium, And Azetidenium Ions And Mechanisms For Their Interconversion. Org. Lett. 2015, 17, 484–487. 10.1021/ol503488b. [DOI] [PubMed] [Google Scholar]
- Lee S. T.; Morokuma K. Ab initio molecular orbital studies on the structure of the nitrenium ion and its implication. J. Am. Chem. Soc. 1971, 93, 6863–6866. 10.1021/ja00754a028. [DOI] [Google Scholar]
- Gobbi A.; Frenking G. The singlet-triplet gap of the halonitrenium ions NHX+, NX2+ and the halocarbenes CHX, CX2(X = F, Cl, Br, I). J. Chem. Soc., Chem. Commun. 1993, 1162–1164. 10.1039/c39930001162. [DOI] [Google Scholar]
- Wright T. G.; Miller T. A. Calculation Of The Ionization Energies Of The Amidogen And Methyl-Substituted Amidogen Radicals: NH2, CH3NH, And CH3NCH3. J. Phys. Chem. 1996, 100, 4408–4412. 10.1021/jp9526760. [DOI] [Google Scholar]
- Mazur R. H.; White W. N.; Semenow D. A.; Lee C. C.; Silver M. S.; Roberts J. D. Small-ring Compounds. XXIII. The Nature of the Intermediates in Carbonium Ion-type Interconversion Reactions of Cyclopropylcarbinyl, Cyclobutyl and Allylcarbinyl Derivatives1a. J. Am. Chem. Soc. 1959, 81, 4390–4398. 10.1021/ja01525a073. [DOI] [Google Scholar]
- Casanova J.; Kent D. R.; Goddard W. A.; Roberts J. D. Quantum-mechanical calculations of the stabilities of fluxional isomers of C4H7+in solution. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 15–19. 10.1073/pnas.0136820100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah G. A.; Prakash Reddy V.; Prakash G. K. S. Long-Lived Cyclopropylcarbinyl Cations. Chem. Rev. 1992, 92, 69–95. 10.1021/cr00009a003. [DOI] [Google Scholar]
- Reddy V. P.; Rasul G.; Prakash G. K. S.; Olah G. A. Structural Studies of Nonclassical Cyclobutylmethyl Cations by the ab initio Method. J. Org. Chem. 2007, 72, 3076–3080. 10.1021/jo0701334. [DOI] [PubMed] [Google Scholar]
- Olah G. A.; Surya Prakash G. K.; Rasul G. Ab Initio/GIAO-CCSD(T) Study of Structures, Energies, and 13C NMR Chemical Shifts of C4H7+and C5H9+ Ions: Relative Stability and Dynamic Aspects of the Cyclopropylcarbinyl vs Bicyclobutonium Ions. J. Am. Chem. Soc. 2008, 130, 9168–9172. 10.1021/ja802445s. [DOI] [PubMed] [Google Scholar]
- Liu J.; Mandel S.; Hadad C. M.; Platz M. S. A Comparison Of Acetyl- And Methoxycarbonylnitrenes By Computational Methods And A Laser Flash Photolysis Study Of Benzoylnitrene. J. Org. Chem. 2004, 69, 8583–8593. 10.1021/jo048433y. [DOI] [PubMed] [Google Scholar]
- Guan J.; Randall K. R.; Schaefer H. F.; Li H. Formylmethylene: The Triplet Ground State And The Lowest Singlet State. J. Phys. Chem. A 2013, 117, 2152–2159. 10.1021/jp311681u. [DOI] [PubMed] [Google Scholar]
- Deng G.; Dong X.; Liu Q.; Li D.; Li H.; Sun Q.; Zeng X. The Decomposition Of Benzenesulfonyl Azide: A Matrix Isolation And Computational Study. Phys. Chem. Chem. Phys. 2017, 19, 3792–3799. 10.1039/c6cp08125h. [DOI] [PubMed] [Google Scholar]
- Gassman P. G.; Dygos D. K.; Trent J. E. Chemistry of nitrenium ions. X. Solvolysis of 1-chloroaziridines. J. Am. Chem. Soc. 1970, 92, 2084–2090. 10.1021/ja00710a048. [DOI] [Google Scholar]
- Sülzle D.; O’Bannon P. E.; Schwarz H. A Mass-Spectrometric Investigation of CxNO2 (x = 1, 2) Ions and Neutrals. Chem. Ber./Recl. 1992, 125, 279–283. 10.1002/cber.19921250144. [DOI] [Google Scholar]
- Bernhardi I.; Drews T.; Seppelt K. Isolation And Structure Of The OCNCO+ Ion. Angew. Chem., Int. Ed. 1999, 38, 2232–2233. . [DOI] [PubMed] [Google Scholar]
- Pichette S.; Aubert-Nicol S.; Lessard J.; Spino C. Photochemical and Thermal Ring-Contraction Of Cyclic Hydroxamic Acid Derivatives. J. Org. Chem. 2012, 77, 11216–11226. 10.1021/jo3023507. [DOI] [PubMed] [Google Scholar]
- Pichette S.; Winter D. K.; Lessard J.; Spino C. Converting Cycloalkanones into N-Heterocycles: Formal Synthesis of (−)-Gephyrotoxin 287C. J. Org. Chem. 2013, 78, 12532–12544. 10.1021/jo402217e. [DOI] [PubMed] [Google Scholar]
- Gassman P. G.; Hartman G. D. Chemistry of nitrenium ions. XXVII. Leaving group efficacy in the generation of nitrenium ions from hydroxylamine derivatives. J. Am. Chem. Soc. 1973, 95, 449–454. 10.1021/ja00783a023. [DOI] [Google Scholar]
- Winter A. H.; Falvey D. E.; Cramer C. J. Effect Of Meta Electron-Donating Groups On The Electronic Structure Of Substituted Phenyl Nitrenium Ions. J. Am. Chem. Soc. 2004, 126, 9661–9668. 10.1021/ja047677x. [DOI] [PubMed] [Google Scholar]
- Winter A.; Falvey D.. Stabilization Of An M-Xylyiene-Like Pi,Pi* Triplet State By Meta Substitution Of Aryl Ionic Species With Pi Donors. 230th National Meeting Of The American-Chemical-Society, 2005; p U3370.
- Qiu Y.; Fischer L. J.; Dutton A. S.; Winter A. H. Aryl Nitrenium and Oxenium Ions with Unusual High-Spin π,π* Ground States: Exploiting (Anti)Aromaticity. J. Org. Chem. 2017, 82, 13550–13556. 10.1021/acs.joc.7b02698. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Applications And Validations Of The Minnesota Density Functionals. Chem. Phys. Lett. 2011, 502, 1–13. 10.1016/j.cplett.2010.11.060. [DOI] [Google Scholar]
- Krishnan R.; Binkley J. S.; Seeger R.; Pople J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based On Solute Electron Density And On A Continuum Model Of The Solvent Defined By The Bulk Dielectric Constant And Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Ochterski J. W.; Petersson G. A.; Montgomery J. A. A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys. 1996, 104, 2598–2619. 10.1063/1.470985. [DOI] [Google Scholar]
- Winter A. H.; Falvey D. E. Vinyl Cations Substituted with β π-Donors Have Triplet Ground States. J. Am. Chem. Soc. 2010, 132, 215–222. 10.1021/ja906139m. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Foster J. P.; Weinhold F. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. 10.1021/ja00544a007. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.