

Abstract
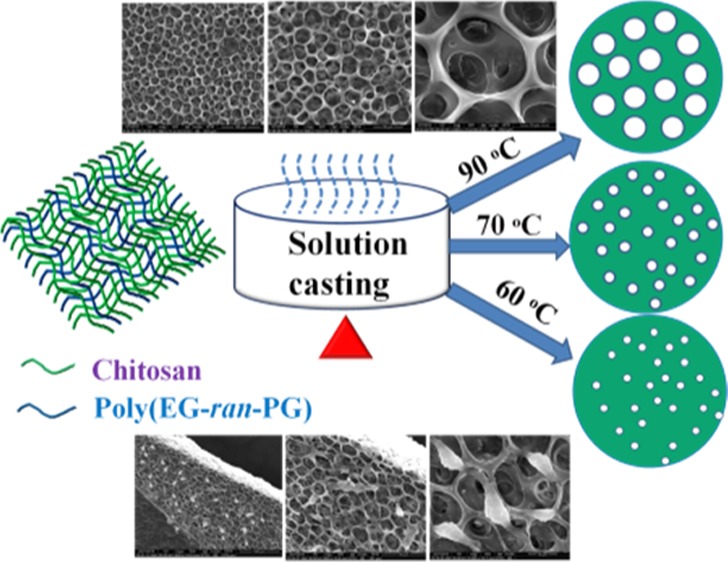
The preparation of porous films (average size variation from 1 to 32 μm) of a 1:1 blend of chitosan with poly(EG-ran-PG) by the controlled evaporation of water from a 2 wt % aqueous acetic acid solution is reported. Interestingly, the blend exhibited porosity that could be tailored from 1 to 32 μm with the temperature of preparation of the blend film. The powder X-ray diffraction, Fourier transform infrared, and differential scanning calorimetry analyses of the films suggested the formation of partially miscible blends. Temperature-induced phase separation of the blend appears to be the mechanism of pore formation. The tensile strength, cytotoxicity, and biocompatibility of the blend films for the growth of mesenchymal stem cells were assessed vis-a-vis chitosan. The 1:1 blend film was observed to lack cytotoxicity and was also viable for the growth of mesenchymal stem cells. The tensile properties of the 1:1 blend were superior to those of the chitosan film. The simple preparation of porous, nontoxic, and biocompatible films could find use as a scaffold in the growth of tissue, and especially bone tissue, in wound dressing, and in filtration if a better control over pore size is achieved.
Introduction
Among the polymeric materials, two natural polymers, namely cellulose and chitin, are of prime importance, being synthesized in nature to the extent of 1012 and 1011 tons per annum, respectively. Chitin is a linear random copolymer consisting almost exclusively β-(1,4)-2-acetamido-2-deoxy-β-d-glycopyranose repeat units and a very small extent of β-(1,4)-2-amino-2-deoxy-β-d-glycopyranose repeat units. It is present in crustaceans (as one of the components of the exoskeleton), insects, algae, fungi, and yeast.1−3 Chitosan is obtained by the deacetylation of chitin (by hot alkaline hydrolysis; the extent of deacetylation must be 50 or more mol %).1−3 Chitosan is biocompatible, biodegradable, nontoxic, hemostatic, antibacterial, and antifungal. For these reasons, chitosan and its blends find wide applications in agriculture, wastewater treatment, food packaging, beverage industries, and as a possible pharmaceutical excipient;4−13 and are ideally suited for the fabrication of biocompatible materials for tissue engineering/scaffolding,14,15 antibacterial activity,16 wound healing/care,17,18 although other biodegradable synthetic polymers such as poly(vinyl alcohol) (PVA) in combination with sodium alginate19 as well as poly-d,l-lactic acid–poly(ε-caprolactone) blends20 have also been used.
The processing of chitosan, in a manner similar to commodity plastics, has not been accomplished to date. The major limitation of chitosan is that it does not soften on heating and decomposes before melting. It is also insoluble in common organic solvents, but dissolves in acidic solutions (pH ≤ 6.5). In applications demanding specific properties such as antibacterial activity, biocompatibility, etc., chitosan is used in small quantities. Chitosan powder is used as an antibacterial additive to synthetic plastics, such as poly(lactic acid) (PLA), PVA, poly(ethylene), and nylon, for use in food packaging materials. For example, nylon-6 is endowed with antibacterial activity upon blending with chitosan.21
The development of chitosan/synthetic polymer-based blends by solution mixing and casting has been in focus over the last two decades, as the chitosan films prepared from acidic solutions are brittle.22,23 Because of the poor solubility of chitosan in common organic solvents and the requirement for specific modifications conferring aqueous solubility, films are cast from the aqueous acidic solution of chitosan mixed with the desired synthetic polymer.24−27 However, the chitosan films formed in this manner turn out to be rigid and brittle, and therefore reinforcing fillers were used to enhance the mechanical properties.27,28 The addition of plasticizers such as glycerol and sorbitol, although enables the processability of the films, invariably results in lower tensile strength and an increase in the elongation at break.29 This problem can be addressed to a certain extent through blending after a suitable modification of the structure of chitosan or through blending unmodified chitosan with other compatible polymers. Synthetic polymers can be tailored with suitable mechanical properties and are easily processed. However, the well-known problem with them is the lack of biodegradability within a reasonable period after the product’s service lifetime. Therefore, blending with synthetic polymers that can biodegrade adds value.
In the above context, the formation of miscible blends of chitosan and poly(ethylene glycol) (PEG) through intermolecular hydrogen bonds facilitated between the hydroxyl groups in chitosan and PEG (terminal) has been reported.30 It was reported that chitosan/PEG fiber could function as a potentially useful drug delivery system. Thus, chitosan and PEG, with salicylic acid of different concentrations, were spun into fibers. The results of the controlled release tests showed that the amount of salicylic acid released increased with an increase in the proportion of PEG present in the fiber. The chitosan/PEG fibers were sensitive to pH and ionic strength, with the release rate being accelerated at lower pH.31 Freeze-dried chitosan/PEG semi-interpenetrating polymer network prepared by cross-linking with glyoxal was shown to be a pH-sensitive matrix from which localized delivery of antibiotics (amoxicillin and metronidazole) in the acidic environment of the gastric fluid and simulated intestinal fluid was accomplished.32 The evaluation of a chitosan/PEG paste (CPP) hydrated suitably with phosphate-buffered saline (PBS) as a local antibiotic delivery device was reported.33 This work concluded that CPP was an injectable, bioadhesive, biodegradable, and biocompatible material, with the potential to allow variable antibiotic loading, and functioned as an active, local antibiotic released to prevent bacterial contamination, as illustrated with Staphylococcus aureus. However, it may be noted that a blend of chitosan and PEG has been shown to phase-separate, with time, partly driven by the tendency of PEG to crystallize.34 This has been overcome in the case of PLA by blending it with poly(EG-ran-PG) copolymers as plasticizers, with the PEG repeat units enabling compatibilization.35
P(EG-ran-PG) is a synthetic biocompatible and biodegradable polymer (the biodegradability of the random copolymer of EG and PG with the trade name TERGITOL L Series, Dow, has been established) that is water-soluble with a flexible backbone. It was expected that the blending of this polymer, which would be in the rubbery phase at room temperature (being much higher than the glass transition temperature and melting temperature of the crystalline domain), with chitosan may enhance the mechanical property of the chitosan film. Further blending with biocompatible polymers such as the random copolymer is required in biomedical applications such as hemostasis. Blends with a biocompatible polymer such as PEG results in phase separation arising out of crystallization, and therefore blending with P(EG-ran-PG) that has no tendency to crystallize at room temperature was explored. The requirements of biocompatibility, biodegradability, water solubility/solubility in aqueous acetic acid, poor tendency to crystallize, so that it does not phase-separate from the blend were reasons for selecting P(EG-ran-PG). The molecular weight of the random copolymer was chosen such that there were no apparent issues associated with the formation of a miscible blend, which is normally the case with high-molecular-weight polymers.
Porous blends of chitosan with a synthetic, water-soluble, random copolymer such as poly(EG-ran-PG) can find unique applications in biology and biomedical fields such as hemostasis, wound dressing, drug delivery, scaffolds for tissue regeneration, artificial organs, filtration of fluids (purification of water, separating organelles), and so forth. The objective of this work was to prepare mechanically stronger and biocompatible films of chitosan by blending with poly(EG-ran-PG), a flexible, biocompatible, and biodegradable polymer. Unexpectedly, the formation of films with regular pores, the size of which could be controlled by the temperature of film preparation, was observed. The detailed results are discussed in this paper.
Results and Discussion
The chitosan films (thickness of 0.2 mm) formed by the process reported here were translucent, and when folded failed by brittle fracture. Upon suspending a small portion of the film in aqueous acetic acid (5%), the film was swollen and disintegrated to bigger pieces over 168 h, but did not dissolve, suggesting the formation of dehydrated chitosan.36,37 The blend of chitosan with the random copolymer resulted in polymer films of thickness about 0.2 mm. The blend films (1:0.5, 1:1, 1:1.5) were semitransparent and failed by brittle fracture upon folding. The 1:2 blend film showed a rough top surface with big pin holes (fewer in number) and upon folding failed by brittle fracture. All the blend films did not dissolve or swell in aqueous 5% acetic acid over 168 h. This may be due to the formation of dehydrated chitosan or a miscible blend (partial or otherwise) and most probably not due to the formation of an immiscible blend or the presence of the random copolymer on the surface, as the random copolymer is soluble in 2% aqueous acetic acid.
The powder X-ray diffraction (PXRD) patterns of chitosan (Ch) and the blends with the random copolymer [Ch/poly(EG-ran-PG)] are presented in Figure 1. The X-ray diffraction (XRD) pattern of the chitosan film indicates the presence of broad peaks at 2θ = 10° and 2θ = 21.5° corresponding to the reflections from the [020] and [110] planes of chitosan. The main peak corresponding to the random copolymer was observed at 2θ = 21.2 [with two other peaks at 2θ = 7.8 and 41.4 of relatively much lower intensity; see Supporting Information Figure S1]. The peak around 10° is attributed to the hydrated crystalline structure of chitosan (even the well-dried samples of chitosan contain bound water to the extent of ∼5%). In comparison to the chitosan flakes, the 2θ value associated with the [110] plane in a chitosan film formed from aqueous acetic acid is shifted by ∼1.5° on the higher side and therefore lower d-spacing, possibly due to the formation of dehydrated chitosan. In this context, the formation of dehydrated crystals of chitosan from the salts of acids such as formic acid, acetic acid, and so forth, and also under prolonged heating in water, has been reported.38,39
Figure 1.
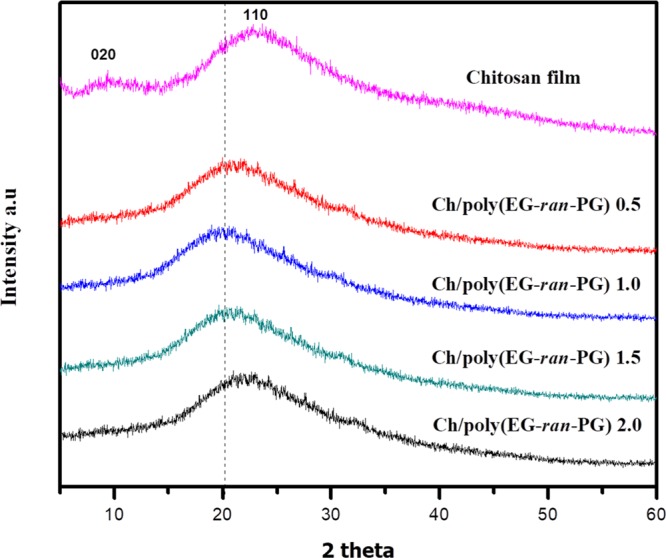
PXRD patterns of the chitosan and Ch/poly(EG-ran-PG) copolymer blends of different compositions.
Upon blending chitosan with the poly(EG-ran-PG) copolymer, the intensity of the [020] plane reduces with the concomitant shift of the diffraction from the [110] plane to a lower 2θ value. The observation of the peak from the [110] plane indicates the formation of the crystalline regions of chitosan upon heating in the presence of the random copolymer, as observed in the case of chitosan control. The decrease followed by the disappearance of the reflections from the [020] plane could arise from deacetylation, which could have taken place in the process of preparing films in the presence of acetic acid at ∼100 °C.40 The 2θ value for the [110] plane in the case of the 1:1 and 1:1.5 blends is shifted to a value (19.9°) lower than the maximum observed for the random copolymer. This suggests that the hydrated chitosan crystals may have formed, partially, facilitated by the random copolymer. Similar results have been discussed earlier for the plasticization of chitosan by glycerol29 as well as a mixture of lactic acid–glycerol–water.41 The space between the antiparallel chains in the sheets (0.447 nm) of chitosan is not likely to be accessible to the random copolymer, which is expected to have a much higher hydrodynamic diameter, whereas it is not the case for water as well as acetic acid. The reason for the facilitation of hydrated chitosan by the random copolymer requires further investigation. The crystallinity index values of the blends are presented in Table S1. This suggests the reduction in the crystallinity of chitosan upon increasing the extent of poly(EG-ran-PG). As the random copolymer is in the rubbery phase at room temperature, it follows that the amorphous regions of chitosan may facilitate the formation of a partially miscible blend with the random copolymer. As pointed out earlier, all the blend films including that of chitosan were insoluble in water–acetic acid over a period of 168 h. For comparison, and as examples of incompatible blends, the PXRD patterns of the blends of chitosan with PLA, a semicrystalline polymer, PVA, an amorphous polymer, and PEG are presented in Supporting Information Figure S2.
The structure of the Ch/poly(EG-ran-PG) blend films was investigated by Fourier transform infrared (FTIR) spectroscopy. The IR spectra of chitosan, poly(EG-ran-PG) copolymer, and the 1:1 blend are presented in Figure 2, whereas the same for the other blends are presented in Supporting Information Figure S3. The FTIR spectra of chitosan powder (data not shown) as received are characterized by three prominent peaks at 1660 (amide I, C=O stretching from the acetyl group), 1570 (amide II predominantly from N–H stretching), 1425 (amide III), 1380 (C–H bending), and 1018 cm–1 (skeletal stretching associated with the pyranose form of glucosamine repeat units). The FTIR spectra of the chitosan film (Figure 2) formed by the evaporation of a solution of chitosan powder in 2% aqueous acetic acid solution at 90 °C show characteristic peaks at 1730 (shoulder; carbonyl from acetate possibly formed by the reaction between the OH groups in chitosan and acetic acid upon drying at 90 °C; not observed in the films dried under vacuum at room temperature), 1606 (might be due to the shift of amide I to lower frequency because of the presence of a large quantity of water as assessed by thermogravimetric analysis (TGA) as well as by differential scanning calorimetry (DSC) in the first cycle of heating), 1532 (amide II, shifted by 40 cm–1 to lower frequency, in comparison to chitosan powder, possibly because of the formation of ammonium cation), 1444 (C–H bending), 1415 (carboxylate C–O, symmetric), 1259, 1018, and 800 cm–1. The shifting of the amide bands in the film form is reported to be dependent on the pH of the polymer solution from which the film is prepared,42 and hence it was not surprising that a significant shift in the amide II band was observed. Poly(EG-ran-PG) is identified by the characteristic IR peaks at 2870 (symmetric C–H stretch), 1456 (C–H bending), 1349, 1294 (C–O stretch from the random copolymer), 1248 (C–O stretch from the random copolymer), and 1089 cm–1 (C–O–C stretching; most intense). All the characteristic peaks of chitosan and poly(EG-ran-PG) are observed in the blend. The film of the 1:1 blend exhibited characteristic IR absorption peaks at 2865 (random copolymer), 1723 (possibly the acetate carbonyl from chitosan), 1630 (amide I), 1556 (amide II), 1378 (C–H bending chitosan), 1250 (random copolymer), and 1089 (random copolymer) cm–1. The shifting of the amide I band from 1660 cm–1 (chitosan powder) to 1630 cm–1 (chitosan–random copolymer blend) and the amide II band from 1570 to 1556 cm–1 suggested the presence of weak interaction between the polymers and possibly the formation of partially miscible blends. Some of the other characteristic peaks of chitosan and the random copolymer are also observed to shift on blending, suggesting the formation of partially miscible blends.43
Figure 2.
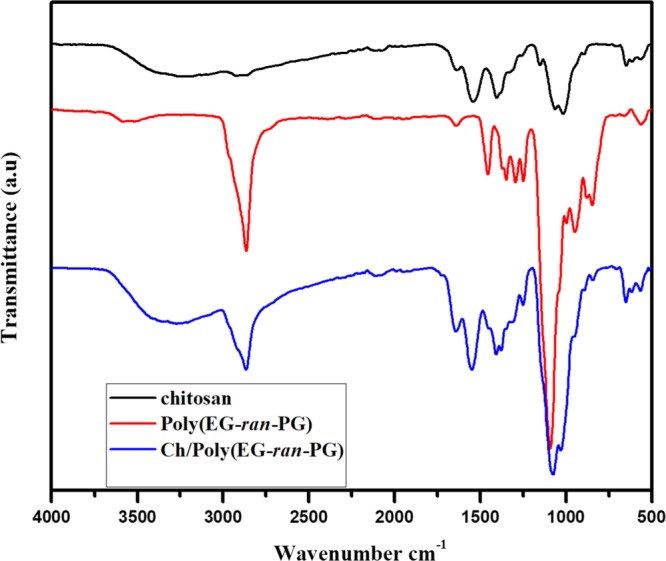
FTIR spectra of chitosan, poly(EG-ran-PG) copolymer, and 1:1 Ch/poly(EG-ran-PG) copolymer blend.
The thermal transitions and the associated enthalpy changes with the 1:1 blend of Ch/poly(EG-ran-PG), as measured by DSC, are presented in Figure 3, whereas the corresponding data for the other blends are presented in Supporting Information Figure S4. The results are summarized in Table 1. Upon the addition of chitosan, the glass transition temperature of the copolymer is reduced, suggesting that there could be a partial mixing between the two polymers. The melting point as well as the heat of fusion per gram of poly(EG-ran-PG) is also reduced upon blending, suggesting partial mixing. The temperature at which the random copolymer crystallizes and the heat of crystallization changes is significant, suggesting partial blending consistent with the trends reported for the PLA–poly(EG-ran-PG) blends.35 It could be concluded that the blending led to lowering of Tg, Tm, ΔHf, and ΔHc, suggesting partial miscibility and disruption of the crystallization of the random copolymer by chitosan.
Figure 3.
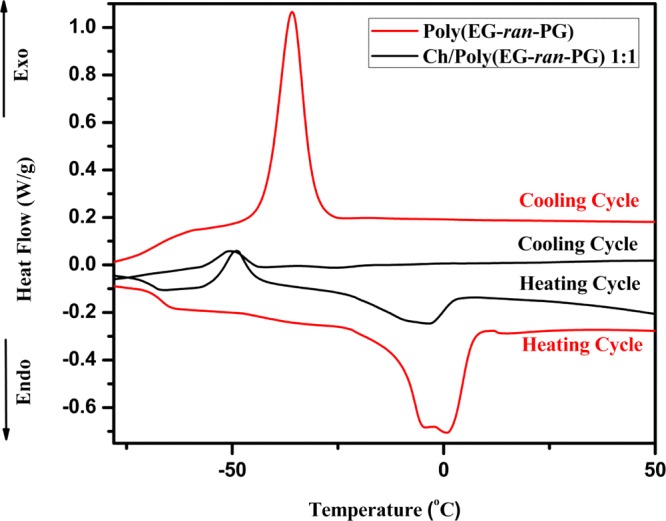
DSC analysis of poly(EG-ran-PG) (bottom-most: heating, and topmost: cooling) and Ch/poly(EG-ran-PG) 1:1 copolymer blend (second from bottom: heating, and second from top: cooling) in the second cycle of heating and cooling.
Table 1. Thermal Properties of the Blends as Assessed by DSCa.
| sample | Tg (°C) (heating cycle) | Tm (°C) | ΔH (J/g) | Tc (°C) | ΔH (J/g) |
|---|---|---|---|---|---|
| poly(EG-ran-PG) | –67.2 ± 0.7 | –0.8 ± 1.6, –16.3 ± 1.6 | 40.1 ± 0.4 | –36.3 ± 0.4 | 40.9 |
| Ch/poly(EG-ran-PG) 1:1.5 | –68.4 | –7.6 | 10.1 | –47.3 | 9.8 |
| Ch/poly(EG-ran-PG) 1:1 | –69 | –4.8 | 8.8 | –46.1 | 6.5 |
| Ch/poly(EG-ran-PG) 1:0.5 | –71 | –4.9 | 15.1 | –48.1 | 12.1 |
Tg data for poly(EG-ran-PG) is the average of two measurements in the heating cycle.
The results from the TGA and DTGA of the chitosan film, poly(EG-ran-PG) random copolymer, and the 1:1 blend of the two polymers are presented in Figure 4. It is clear from this figure that the blend shows the characteristic degradation peaks associated with the chitosan film as well as the random copolymer, thus confirming the presence of chitosan as well as poly(EG-ran-PG). The TGA and DTGA of the Ch/poly(EG-ran-PG) blend films and chitosan films prepared at room temperature and at 90 °C after Soxhlet extraction (12 h) in methanol are presented in Supporting Information Figure S5. This showed beyond any doubt that the copolymer could not be removed from the blend when the preparation was carried out at 90 °C, whereas it could be easily extracted from the blend film prepared at room temperature. These data indicate that at 90 °C, chitosan and the random copolymer form a partially miscible blend. Similar results were obtained for the other blends, but the details are not presented here.
Figure 4.

TGA (a) and DTGA (b) of chitosan film, poly(EG-ran-PG), and the Ch/poly(EG-ran-PG) copolymer blend.
The formation of Ch/poly(EG-ran-PG) blends was also accompanied by unexpected changes in morphology that were unique in comparison to chitosan, which formed a uniform and continuous film. In contrast, chitosan/PEG6000 formed a phase-separated blend. The scanning electron microscopy (SEM) images of the 1:1 Ch/poly(EG-ran-PG) copolymer blend films prepared at 90 °C are shown in Figure 5. This suggested the formation of films with pores of sizes varying between 16 and 32 μm. This was also the case for the other blends (1:0.5, 1:1.5; Supporting Information Figure S6). As the formation of pores was not expected, the mechanism of formation was investigated at some depth by varying the conditions of preparation of the film, especially temperature. The blend films prepared by drying under ambient conditions for 1–2 weeks were continuous as in the case of chitosan (Supporting Information Figure S7). The blend films dried at room temperature for 3 to 4 days followed by additional drying at 40 and 60 °C for 24 h were also continuous (Supporting Information Figure S7). When the preparation of the film was carried out directly at higher temperatures, films with different pore sizes were observed: 3–4 μm (at ∼70 °C) and 1–2 μm (at 60 °C), as shown in Figure 5. The formation of pores upon drying a polymer or polymer blend is reported to proceed by three different routes: (i) temperature-induced phase separation;44−46 (ii) breath figure mechanism;47,48 and (iii) the forced drying of solvent from a waterborne nanocomposite polymer latex.49,50 The formation of a breath figure requires the presence of a low-boiling organic solvent, which preferably does not mix with water, and high humidity. As nanofillers such as silica are also not present in our system, the forced drying of a solvent under this condition is not met. From the three known mechanisms of pore formation, temperature-induced phase separation appears to be the most likely mechanism of pore formation. Thus, the mixture of chitosan, random copolymer, acetic acid, and water, which appeared to be a uniform and homogeneous mixture at room temperature, undergoes phase separation into polymer-rich and solvent-rich phases when heated above 60 °C. The kinetics of this process are faster at higher temperatures. The rate of evaporation of the solvent and the accompanying changes in the interfacial tension may then result in pores of different sizes, with the formation of bigger pores at higher temperatures.
Figure 5.

SEM images and the pore size distribution of the 1:1 blend of Ch/poly(EG-ran-PG) formed at 90 (a), 70 (b), and 60 °C (c).
The morphology of the 1:1 blend of Ch/poly(EG-ran-PG) formed at 90 °C is retained after solvent rinsing (thrice with methanolic sodium hydroxide at room temperature to remove acetic acid, if any, followed by methanol, being a solvent for the random copolymer). The SEM images of the same showing the presence of honeycomb morphology are presented in Figure 6. It may be noted that TGA (Figure S5) confirmed the presence of chitosan and the random copolymer in the blend after the solvent rinse. It should also be noted that the chitosan film prepared under the same conditions as that of the 1:1 blend shows the formation of smooth and nonporous surface at all temperatures ranging from room temperature to 90 °C (Supporting Information Figure S8). This was also the case for the 1:1 blend films of Ch/poly(EG-ran-PG) prepared at room temperature to 60 °C (Supporting Information Figure S7). The SEM images of the chitosan film and the other blends with chitosan prepared at 90 °C are presented in Supporting Information Figure S9. It is clear that chitosan forms a smooth film under different conditions employed, whereas the blends are phase-separated. The films of a 1:1 blend of Ch/poly(EG-ran-PG) formed in this manner were observed to be nontoxic and biocompatible.
Figure 6.

SEM images of the 1:1 blend of Ch/poly(EG-ran-PG) film formed at 90 °C after rinsing with sodium hydroxide in methanol at room temperature, followed by rinsing with methanol [at different magnifications, as shown at the bottom of the figures (a–c)].
The morphologies of the 1:1 blends of Ch/PEG6000 and Ch/poly(EG-ran-PG) formed by slow evaporation on a copper grid, at room temperature, as described in the experimental section, were also analyzed using transmission electron microscopy (TEM). The TEM images of the same are shown in Figure 7. This indicated the formation of an immiscible blend in the case of Ch/PEG6000 and a phase-separated and possibly partially miscible blend in the case of the 1:1 Ch/poly(EG-ran-PG) blend. Under the same conditions, chitosan formed a smooth film (Figure S10). Considering the interlocked morphology, notably decreased crystallinity, IR spectra of the blend membranes, and the DSC data of the blends prepared under the optimized conditions, it can be deduced that these blend membranes have a partially miscible character.
Figure 7.

TEM images of the 1:1 blend of chitosan/PEG6000 (a) and the 1:1 blend of Ch/poly(EG-ran-PG) (b).
Tensile Properties of the Chitosan/Poly(EG-ran-PG) Blend Films
The stress versus strain plots for the blends of chitosan with different weight percentages of poly(EG-ran-PG) are shown in Figure 8. The chitosan film formed under the conditions reported here shows a tensile strength of 24 MPa. Upon blending with an increasing proportion of the poly(EG-ran-PG) copolymer, the tensile strength, elongation at break, and modulus increase to a maximum of 39 MPa, 1.7%, and 45.8 MPa, respectively, before decreasing. It is evident from the tensile data that the blend compositions of 1:0.5 and 1:1 might be optimum for enhancing the tensile properties. However, a further increase in the weight percentage of the copolymer causes a decrease in tensile strength from 39 to 16 MPa because of the inadequate quantity of chitosan flakes required to bind to form a film, resulting in premature failure. The 1:1.5 composition probably leads to the plasticization of the chitosan matrix by the liquid copolymer (Table 2, last row), resulting in inferior tensile strength and modulus, but surprisingly the same elongation at break characteristic of chitosan. In fact, it could be observed that the 1:1.5 blend had an excess liquid random copolymer forming a shiny surface. The increase in tensile strength can be explained as a consequence of enhanced interfacial stress transfer and synergistic effect arising out of the rubbery poly(EG-ran-PG) in the chitosan matrix. It was reported in the literature that the blending of chitosan with PEG does not improve its mechanical properties remarkably.51 The blends of chitosan with PLA prepared by a process similar to that reported here (solution mixing followed by casting film and drying) resulted in a lowering of tensile strength, and the elastic modulus of chitosan decreased with the addition of PLA. In addition to the mechanical properties, thermal properties also revealed that the blends were incompatible.52
Figure 8.
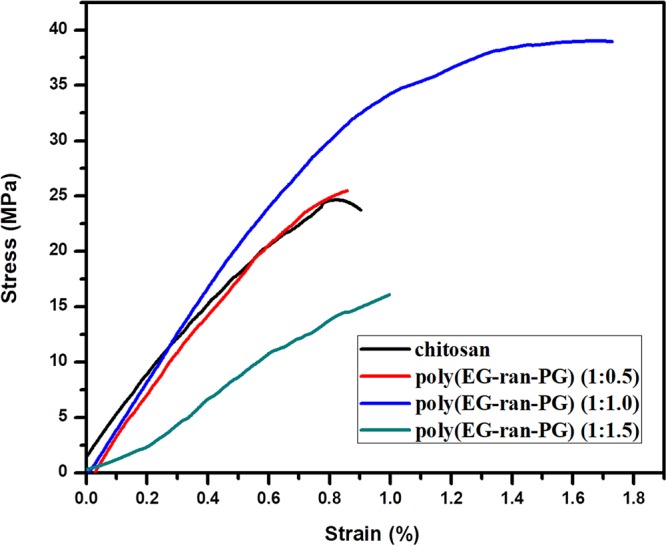
Stress vs strain plots for the Ch/poly(EG-ran-PG) blends.
Table 2. Mechanical Properties of Chitosan and the Ch/Poly(EG-ran-PG) Blend Films.
| sample code | tensile strength (MPa) | elongation at break (%) | tensile modulus (MPa) |
|---|---|---|---|
| chitosan | 24 | 0.9 | 37.8 |
| Ch/poly(EG-ran-PG) 1:0.5 | 25 | 0.9 | 43.0 |
| Ch/poly(EG-ran-PG) 1:1 | 39 | 1.7 | 45.8 |
| Ch/poly(EG-ran-PG) 1:1.5 | 16 | 1.0 | 10.4 |
Contact Angle
The contact angle of water on a surface is sensitive to the surface energy and morphology. Smaller contact angles imply more hydrophilic as well as rougher surfaces. The water contact angles of a chitosan film, the chitosan/PEG 1:1 blend, and the Ch/poly(EG-ran-PG) 1:1 blend are shown in Figure S11. From this figure, it is evident that the contact angle is reduced from 81.7 to 41.4 upon blending chitosan with PEG alone (surface free energy, 43 mJ/m2). This is due to the formation of an immiscible blend with the consequent presence of PEG segments at the air–polymer film interface. The film of the Ch/poly(EG-ran-PG) (1:1) blend, with an average porosity of 12–20 μm, exhibited a contact angle similar to that observed for the chitosan film (82.3). This, at first assessment, suggests the presence of chitosan molecules and not the random copolymer, as it is water-soluble, and would therefore lead to a low contact angle.
[3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] Assay In Vitro Cytotoxicity
The current limitations of wound dressings used for treating burn-induced septic wound injuries are that they are not porous and do not enable contact with air. The porous blends prepared in this work could act as a better microbial barrier than neat chitosan. The nontoxic nature of the blend films was ascertained by [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) assay. The relative cell viability between the mesenchymal cells grown on the Ch/poly(EG-ran-PG) scaffolds surface and on the control chitosan film substrate (without copolymer) is shown in Figure 9. This shows that after 24 h, the average cell viability for the samples Ch/PEG and Ch/poly(EG-ran-PG) (1:1) increased compared to the control (chitosan film). The cell viability values for the blends Ch/poly(EG-ran-PG) (1:0.5) and Ch/poly(EG-ran-PG) (1:1.5) were similar to that for the control, chitosan. Therefore, it can be concluded that the 1:1 blend of Ch/poly(EG-ran-PG) scaffold is nontoxic to the mesenchymal stem cell line.
Figure 9.

Results from the in vitro cytotoxicity studies of chitosan and its blends, as assessed by MTT assay.
Cell Adhesion Studies
Cell adhesion is a critical parameter when evaluating whether or not a scaffold is biocompatible and suitable for tissue regeneration. SEM was used to observe the cell morphology and the contacts between the cells and scaffolds, as shown in Figure 10. It could be inferred from these images that mesenchymal stem cells adhered strongly on the chitosan scaffolds as well as Ch/poly(EG-ran-PG) 1:1 blend films and were confluent at some areas after 48 h of cell seeding. The Ch/poly(EG-ran-PG) 1:1 blend samples not only ensure that the stem cells grow normally but also promote their proliferation, which means that the blend films were biocompatible and bioactive. One possible reason for this could be the similarity between the structures of chitosan and glycosaminoglycans. It may be noted that glycosaminoglycans constitute the main protein component of the extracellular matrixes and play an important role during the process of adhesion, proliferation, and shaping of the cell.53 The results indicate that the Ch/poly(EG-ran-PG) blends have good attachment with the mesenchymal stem cells and better biocompatibility.
Figure 10.

SEM images before (a) and after (b,c) cell adhesion on chitosan; before (d) and after (e,f) cell adhesion on the Ch/poly(EG-ran-PG) (1:1) blend; cross-sectional images (g–i) after cell adhesion on the Ch/poly(EG-ran-PG) (1:1) blend.
Conclusions
The nontoxic and biocompatible porous films of the biodegradable polymer blends of chitosan/poly(EG-ran-PG) with tailored pore sizes of the order of 1–32 μm are prepared by solvent casting from an aqueous acetic acid solution followed by drying at different temperatures. The SEM and TEM analyses suggested the formation of partially miscible blends with an organized porous morphology. The porous morphology may have arisen out of temperature-induced phase separation (followed by the extraction of the random copolymer in methanol in some cases). The 1:1 blend exhibited better tensile properties than the chitosan film. The blends were nontoxic to the mesenchymal stem cells and enabled their growth, suggesting that the method of preparation might be useful in various biomedical fields as well as in the preparation of filtration membranes.
Experimental Section
Materials
Commercial chitosan from crab shells [Mn = 51 000 Da; 80% deacetylated as determined by FTIR spectroscopy] was purchased from M/s. Matsyafed, Kochi, Kerala, India. The poly(EG-ran-PG) copolymer [Mn 12 000, liquid; propylene glycol (PG) content by 1H NMR = 22.8 mol %] was purchased from Sigma-Aldrich (St. Louis, MO, USA). PEG of molecular weights 4000 (PEG-4000; mp 58–61 °C) and 6000 (PEG-6000), PLA, and PVA were procured from Fisher Scientific Ltd., Mumbai. Laboratory-grade acetic acid was purchased from J.T. Baker, USA. Sodium hydroxide and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich, India. High-performance liquid chromatography-grade water, methanol, and ethanol were purchased from Merck, India.
Methods
Preparation of the Ch/Poly(EG-ran-PG) Blend Films
One gram of chitosan was added to 100 mL of 2% (v/v) acetic acid solution in water (0.037 mol of acetic acid and 5.444 mol of water) at 25–30 °C. The mixture was stirred under continuous agitation for 10 h (approximately) at ambient temperature. The desired quantity of the liquid poly(EG-ran-PG) copolymer (0.5 or 1 or 1.5 or 2 g, as the case may be) was added to the chitosan solutions such that the compositions of the blends of poly(EG-ran-PG):chitosan were 0.5:1.0 or 1.0:1.0 or 1.5:1.0 or 2.0:1.0 (w/w), respectively. The resulting solutions were stirred well, upon which a completely miscible solution (to the eye, no visible phase separation was observed in this process) was formed. The entire mixture was then poured onto a Petri dish, the bottom of which was covered with a Teflon-coated film (to enable smooth removal of the film after drying). The Petri dish was placed in an oven equipped with hot air circulation for 10–12 h and maintained at 90 °C for all the blends and at 50, 60, 70, and 90 °C for the 1:1 blend. The films thus obtained were peeled off and subjected to further tests.
Characterization
NMR spectroscopic measurements in solution were carried out with a Bruker Avance spectrometer (operating at 400 MHz for proton) using CDCl3 as the solvent. Gel permeation chromatography (GPC) of chitosan was performed using a Waters GPC system (two ultrahydrogel 250 SS columns of 30 cm × 7.8 mm; 0.1 N sodium nitrate in 20 mL glacial acetic acid and 1000 mL water as the mobile phase, 0.8 mL/min flow rate) equipped with a refractive index detector. Narrow molecular weight PEG standards were used for calibration. The PXRD patterns were recorded using a Bruker D8 Advance (Germany) diffractometer equipped with a Cu anode (Cu Kα source of the wavelength of 1.5406 Å) between 5 and 60° (2θ). The FTIR spectra were obtained with a Bruker Alpha model using the film samples of chitosan, chitosan/poly(EG-ran-PG) blends, and poly(EG-ran-PG). The thin films were prepared by drying 2% aqueous acetic acid solutions on a Petri dish in a hot air oven. In the case of the random copolymer, a drop of the sample was used directly for the FTIR measurements. The spectra of all the samples were recorded after baseline correction. DSC was performed using a TA Instruments Q200 modulated DSC device, equipped with a refrigerated cooling system (window of operation −90 to 300 °C). About 2.5–10 mg of the sample to be analyzed was placed in a Tzero aluminum pan and sealed with a Tzero lid for solid samples. The pan was cooled using liquid nitrogen. On the basis of the standards, the DSC data reported here are reliable above −80 °C in the heating cycle and up to −60 °C in the cooling cycle. The samples were heated and cooled at the rate of 10 °C/min. The thermal decomposition of all the materials was studied using a TA Instruments Q500 Hi-Res thermogravimetric analyzer. Around 5 mg of the sample was taken in a platinum pan and heated from 35 to 900 °C at 10 °C/min, with the sample purged by nitrogen flow at the rate of 60 mL/min. The SEM images were obtained using an FEI Quanta 400 scanning electron microscope at an acceleration voltage of 30 kV. A Cressington sputter coater (108 Auto) was used for sputtering gold onto the film surfaces before analysis. The TEM images were recorded using an FEI Tecnai T20 electron microscope at an acceleration voltage of 200 kV. The samples to be imaged were cast directly onto copper grids from solution and dried at room temperature. The intensity of the electron beam was kept very low to prevent any damage or inadvertent production of any artifacts on the sample. The tensile properties, such as tensile strength, elongation at break, and elastic modulus, of the prepared films were evaluated using an AG-1 electronic universal testing machine (UTM) controlled suitably through a computer (Shimadzu Corporation, Japan), in accordance with the ASTM Method D 882-88. Tensile testing was carried out under ambient conditions with a cross-head speed of 5 cm/min. The chitosan film required for UTM was prepared by casting a film from an aqueous solution, the details of which were described earlier. The water contact angle (sessile drop method) was measured using a Holmarc model HO-IAD-Cam-01B, equipped with a camera. Deionized (DI) water was used as the probe.
Cell Culture and Film Preparation
The mesenchymal stem cells (C3H10T1/2) were purchased from the National Centre for Cell Sciences, Pune, India. The biological reagents used for the cell culture experiments such as Dulbecco’s modified Eagle’s medium (DMEM, with 4.5 g/L glucose, 4.0 mM glutamine, and sodium pyruvate), penicillin/streptomycin (10 000 μg/mL), fetal bovine serum (FBS), 0.25% trypsin–EDTA (1×), and PBS, pH 7.4 were obtained from Gibco, India. Only 5% CO2 was used. The mesenchymal cells were plated in a flask (7 × 105 per 25 cm2), maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin (10 000 μ/mL), and maintained in a humidified incubator at 37 °C under 5% CO2. The polymer films were cut into sizes of 0.5 × 0.5 cm and 1 × 1 cm, and then the residual solvent (acetic acid) present in it, if any, was neutralized using NaOH (in methanol). The films were then sterilized using 95% ethanol for at least 30 min under a laminar flow hood. Then, they were rinsed with sterile DI water followed by 1× PBS wash. Finally, both sides of the films were exposed to UV irradiation and dried for 30 min.
In Vitro Cytotoxicity
The cytotoxicity of the chitosan film and chitosan blends prepared from the poly(EG-ran-PG) copolymers of different ratios was studied using MTT assay on mesenchymal stem cells. In brief, 10 000 cells/well in 100 μL final volumes were seeded into a 96-well plate. Once the cells were 70% confluent, the media were removed, and the wells were washed with PBS and replaced with serum-free media. The sterilized films, n = 5, for each of chitosan, Ch/PEG, and Ch/poly(EG-ran-PG) (1:0.5, 1:1 and 1:1.5) were placed gently into each well, above the cells, and the wells without the films were regarded as control. After 24 h of incubation, the wells were emptied, and 100 μL of the MTT reagent from the stock (5 mg/mL in PBS) was added to each well, such that the final concentration was 0.5 mg/mL. Then, the cells were incubated for 2 h at 37 °C in a 5% CO2 incubator. After incubation, the wells were emptied again, and 200 μL DMSO was added to each well to dissolve the insoluble purple formazan crystals. The 96-well plate was then placed on a rocker for 20 min, and then the absorbance at 570 nm was measured spectrophotometrically in an enzyme-linked immunosorbent assay reader.
Cell Adhesion
The cell morphology and their adhesion on the scaffold were observed using SEM. All the films of 1 × 1 cm (n = 3) were placed on a 24-well plate, and 50 000 cells/well in 500 μL final volume were seeded into the wells above the film. The cells were maintained at 37 °C in a 5% CO2 incubator. After 48 h, the films were removed and washed with 1× PBS (twice) to remove the unattached cells and then fixed with 2.5% glutaraldehyde for 3 h at 4 °C. The films were then dehydrated with a series of ethanol concentration (20, 40, 60, 80, and 100%) for 30 min each. Finally, the samples were dried under sterile conditions and were coated with gold to observe the cell attachment by SEM.
Acknowledgments
B.S. thanks IIT Madras for support in the form of a postdoctoral fellowship. The authors thank CLRI and Anna University for providing access to UTM, water contact angle, and elemental analyses. The support of the Department of Chemistry, IIT Madras as well as the central microscopic facilities in IIT Madras toward all the SEM and TEM imaging is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01101.
PXRD pattern of chitosan and poly(EG-ran-PG); PXRD pattern of the 1:1 blends of chitosan with PVA, PLA, PEG, and poly(EG-ran-PG); FTIR spectra of the Ch/poly(EG-ran-PG) blends; DSC analysis of the Ch/poly(EG-ran-PG) blends; TGA and DTGA of the chitosan films prepared at 90 °C and at room temperature followed by Soxhlet extraction with methanol; TGA and DTGA of 1:1 Ch/poly(EG-ran-PG) copolymer blend films prepared at 90 °C and at room temperature followed by Soxhlet extraction with methanol; SEM images of the Ch/poly(EG-ran-PG) blends; SEM images of the 1:1 blend of Ch/poly(EG-ran-PG) prepared at room temperature followed by drying at 40 and 60 °C; SEM images of the films of chitosan prepared at room temperature and 90 °C, followed by Soxhlet extraction with methanol and drying; SEM images of the 1:1 blend of Ch/poly(EG-ran-PG) prepared at room temperature and 90 °C, followed by Soxhlet extraction with methanol and drying; SEM images of the 1:1 blends Ch/PEG4000, Ch/PEG6000, and Ch/PVA prepared at 90 °C; TEM images of the chitosan film formed at 90 °C; contact angles of the films of chitosan, Ch/PEG blend, and Ch/poly(EG-ran-PG) blend; and crystallinity index values of chitosan and the Ch/poly(EG-ran-PG) blends (PDF)
The authors declare no competing financial interest.
Notes
The cell culture and film preparation, in vitro cytotoxicity, and cell adhesion studies were carried out at Anna University, MIT Campus.
Supplementary Material
References
- Muzzarelli R. A. A.Natural chelating polymers: Alginic acid, chitin, and chitosan. International Series of Monographs in Analytical Chemistry, 1st ed; Pergamon Press: United Kingdom, 1973. [Google Scholar]
- Muzzarelli R. A. A.Chitin, 1st ed.; Pergamon Press: United Kingdom, 1977. [Google Scholar]
- Pillai C. K. S.; Paul W.; Sharma C. P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. 10.1016/j.progpolymsci.2009.04.001. [DOI] [Google Scholar]
- Kumar M. N. V. R.; Muzzarelli R. A. A.; Muzzarelli C.; Sashiwa H.; Domb A. J. Chitosan chemistry and pharmaceutical perspectives. Chem. Rev. 2004, 104, 6017–6084. 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- Khor E.; Lim L. Y. Implantable applications of chitin and chitosan. Biomaterials 2003, 24, 2339–2349. 10.1016/s0142-9612(03)00026-7. [DOI] [PubMed] [Google Scholar]
- Muzzarelli R. Chitosan-based dietary foods. Carbohydr. Polym. 1996, 29, 309–316. 10.1016/s0144-8617(96)00033-1. [DOI] [Google Scholar]
- Jayakumar R.; Prabaharan M.; Nair S. V.; Tokura S.; Tamura H.; Selvamurugan N. Novel carboxymethyl derivatives of chitin and chitosan materials and their biomedical applications. Prog. Mater. Sci. 2010, 55, 675–709. 10.1016/j.pmatsci.2010.03.001. [DOI] [Google Scholar]
- Kumar M. N. V. R. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. 10.1016/s1381-5148(00)00038-9. [DOI] [Google Scholar]
- Krajewska B. Membrane-based processes performed with use of chitin/chitosan materials. Sep. Purif. Technol. 2005, 41, 305–312. 10.1016/j.seppur.2004.03.019. [DOI] [Google Scholar]
- Dash M.; Chiellini F.; Ottenbrite R. M.; Chiellini E. Chitosan-A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. 10.1016/j.progpolymsci.2011.02.001. [DOI] [Google Scholar]
- Narayanan A.; Dhamodharan R. Super water-absorbing new material from chitosan, EDTA and urea. Carbohydr. Polym. 2015, 134, 337–343. 10.1016/j.carbpol.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Narayanan A.; Kartik R.; Sangeetha E.; Dhamodharan R. Super water absorbing polymeric gel from chitosan, citric acid and urea: Synthesis and mechanism of water absorption. Carbohydr. Polym. 2018, 191, 152–160. 10.1016/j.carbpol.2018.03.028. [DOI] [PubMed] [Google Scholar]
- di Lena F. Hemostatic polymers: the concept, state of the art and perspectives. J. Mater. Chem. B 2014, 2, 3567–3577. 10.1039/c3tb21739f. [DOI] [PubMed] [Google Scholar]
- Levengood S. K. L.; Zhang M. Chitosan-based scaffolds for bone tissue engineering. J. Mater. Chem. B 2014, 2, 3161–3184. 10.1039/c4tb00027g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.; Liu X.; Wang R.; Jia F.; Dong L.; Wang Q. Emerging nanostructured materials for musculoskeletal tissue engineering. J. Mater. Chem. B 2014, 2, 6435–6461. 10.1039/c4tb00344f. [DOI] [PubMed] [Google Scholar]
- Croisier F.; Sibret P.; Dupont-Gillain C. C.; Genet M. J.; Detrembleur C.; Jérôme C. Chitosan-coated electrospun nanofibers with antibacterial activity. J. Mater. Chem. B 2015, 3, 3508–3517. 10.1039/c5tb00158g. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Shi L.; Yang M.; Zhang H.; Zhu L. Fabrication of a novel blended membrane with chitosan and silk microfibers for wound healing: characterization, in vitro and in vivo studies. J. Mater. Chem. B 2015, 3, 3634–3642. 10.1039/c5tb00226e. [DOI] [PubMed] [Google Scholar]
- Shamshina J. L.; Gurau G.; Block L. E.; Hansen L. K.; Dingee C.; Walters A.; Rogers R. D. Chitin-calcium alginate composite fibers for wound care dressings spun from ionic liquid solution. J. Mater. Chem. B 2014, 2, 3924–3936. 10.1039/c4tb00329b. [DOI] [PubMed] [Google Scholar]
- Gopishetty V.; Tokarev I.; Minko S. Biocompatible stimuli-responsive hydrogel porous membranes via phase separation of a polyvinyl alcohol and Na-alginate intermolecular complex. J. Mater. Chem. 2012, 22, 19482–19487. 10.1039/c2jm31778h. [DOI] [Google Scholar]
- Lanzetta V.; Laurienzo P.; Maglio G.; Malinconico M.; Musto P.; Schiattarella I. Development and characterization of porous membranes with ″sandwich-like″ structure based on biocompatible, immiscible polymer blends. J. Mater. Chem. 2007, 17, 4508–4520. 10.1039/b708153g. [DOI] [Google Scholar]
- Ma Y.; Zhou T.; Zhao C. Preparation of chitosan-nylon-6 blended membranes containing silver ions as antibacterial materials. Carbohydr. Res. 2008, 343, 230–237. 10.1016/j.carres.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Zivanovic S.; Li J.; Davidson P. M.; Kit K. Physical, mechanical, and antibacterial properties of chitosan/PEO blend films. Biomacromolecules 2007, 8, 1505–1510. 10.1021/bm061140p. [DOI] [PubMed] [Google Scholar]
- Kim S. Y.; Cho S. M.; Lee Y. M.; Kim S. J. Thermo- and pH-responsive behaviors of graft copolymer and blend based on chitosan andN-isopropylacrylamide. J. Appl. Polym. Sci. 2000, 78, 1381–1391. . [DOI] [Google Scholar]
- Park J. H.; Cho Y. W.; Chung H.; Kwon I. C.; Jeong S. Y. Synthesis and characterization of sugar-bearing chitosan derivatives: aqueous solubility and biodegradability. Biomacromolecules 2003, 4, 1087–1091. 10.1021/bm034094r. [DOI] [PubMed] [Google Scholar]
- Schatz C.; Viton C.; Delair T.; Pichot C.; Domard A. Typical physicochemical behaviors of chitosan in aqueous solution. Biomacromolecules 2003, 4, 641–648. 10.1021/bm025724c. [DOI] [PubMed] [Google Scholar]
- Kumar G.; Smith P. J.; Payne G. F. Enzymatic grafting of a natural product onto chitosan to confer water solubility under basic conditions. Biotechnol. Bioeng. 1999, 63, 154–165. . [DOI] [PubMed] [Google Scholar]
- Fan H.; Wang L.; Zhao K.; Li N.; Shi Z.; Ge Z.; Jin Z. Fabrication, mechanical properties, and biocompatibility of graphene-reinforced chitosan composites. Biomacromolecules 2010, 11, 2345–2351. 10.1021/bm100470q. [DOI] [PubMed] [Google Scholar]
- Suyatma N. E.; Tighzert L.; Copinet A.; Coma V. Effects of hydrophilic plasticizers on mechanical, thermal, and surface properties of chitosan films. J. Agric. Food Chem. 2005, 53, 3950–3957. 10.1021/jf048790+. [DOI] [PubMed] [Google Scholar]
- Epure V.; Griffon M.; Pollet E.; Avérous L. Structure and properties of glycerol-plasticized chitosan obtained by mechanical kneading. Carbohydr. Polym. 2011, 83, 947–952. 10.1016/j.carbpol.2010.09.003. [DOI] [Google Scholar]
- Jiang W. H.; Han S. J. Study of Interaction between Polyethylene Glycol and Chitosan by Viscosity Method. J. Polym. Sci., Part B: Polym. Phys. 1998, 36, 1275–1281. . [DOI] [Google Scholar]
- Wang Q.; Zhang N.; Hu X.; Yang J.; Du Y. Chitosan/polyethylene glycol blend fibers and their properties for drug controlled release. J. Biomed. Mater. Res., Part A 2008, 85, 881–887. 10.1002/jbm.a.31544. [DOI] [PubMed] [Google Scholar]
- Patel V. R.; Amiji M. M. Preparation and Characterization of Freeze-dried Chitosan Poly(Ethylene Oxide) Hydrogels for Site-Specific Antibiotic Delivery in the Stomach. Pharm. Res. 1996, 13, 588–593. 10.1023/a:1016054306763. [DOI] [PubMed] [Google Scholar]
- Rhodes C. S.; Alexander C. M.; Berretta J. M.; Courtney H. S.; Beenken K. E.; Smeltzer M. S.; Bumgardner J. D.; Haggard W. O.; Jennings J. A. Evaluation of a chitosan-polyethylene glycol paste as a local antibiotic delivery device. World J. Orthoped. 2017, 8, 130–141. 10.5312/wjo.v8.i2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Rogunova M.; Topolkaraev V.; Hiltner A.; Baer E. Aging of poly(lactide)/poly(ethylene glycol) blends. Part 1. Poly(lactide) with low stereoregularity. Polymer 2003, 44, 5701–5710. 10.1016/s0032-3861(03)00614-1. [DOI] [Google Scholar]
- Jia Z.; Tan J.; Han C.; Yang Y.; Dong L. Poly(ethylene glycol-co-propylene glycol) as a Macromolecular Plasticizing Agent for Polylactide: Thermomechanical Properties and Aging. J. Appl. Polym. Sci. 2009, 114, 1105–1117. 10.1002/app.30638. [DOI] [Google Scholar]
- Yui T.; Imada K.; Okuyama K.; Obata Y.; Suzuki K.; Ogawa K. Molecular and crystal structure of the anhydrous form of chitosan. Macromolecules 1994, 27, 7601–7605. 10.1021/ma00104a014. [DOI] [Google Scholar]
- Ogawa K.; Oka K.; Yui T. X-ray study of chitosan-transition metal complexes. Chem. Mater. 1993, 5, 726–728. 10.1021/cm00029a026. [DOI] [Google Scholar]
- Ogawa K.; Hirano S.; Miyanishi T.; Yui T.; Watanabe T. A new polymorph of chitosan. Macromolecules 1984, 17, 973–975. 10.1021/ma00134a076. [DOI] [Google Scholar]
- Kawada J.; Abe Y.; Yui T.; Okuyama K.; Ogawa K. Crystalline transformation of chitosan from hydrated to anhydrous polymorph via chitosan monocarboxylic acid salts. J. Carbohydr. Chem. 1999, 18, 559–571. 10.1080/07328309908544019. [DOI] [Google Scholar]
- Zhang Y.; Xue C.; Xue Y.; Gao R.; Zhang X. Determination of the degree of deacetylation of chitin and chitosan by X-ray powder diffraction. Carbohydr. Res. 2005, 340, 1914–1917. 10.1016/j.carres.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Meng Q.; Heuzey M.-C.; Carreau P. J. Hierarchical structure and physicochemical properties of plasticized chitosan. Biomacromolecules 2014, 15, 1216–1224. 10.1021/bm401792u. [DOI] [PubMed] [Google Scholar]
- Kim K. M.; Son J. H.; Kim S.-K.; Weller C. L.; Hanna M. A. Properties of Chitosan Films as a Function of pH and Solvent Type. J. Food Sci. 2006, 71, E119–E124. 10.1111/j.1365-2621.2006.tb15624.x. [DOI] [Google Scholar]
- Nunthanid J.; Puttipipatkhachorn S.; Yamamoto K.; Peck G. E. Physical Properties and Molecular Behavior of Chitosan Films. Drug Dev. Ind. Pharm. 2001, 27, 143–157. 10.1081/ddc-100000481. [DOI] [PubMed] [Google Scholar]
- La Carrubba V.; Pavia F. C.; Brucato V.; Piccarolo S. PLLA/PLA scaffolds prepared via Thermally Induced Phase Separation (TIPS): tuning of properties and biodegradability. Int. J. Material Form. 2008, 1, 619–622. 10.1007/s12289-008-0332-5. [DOI] [PubMed] [Google Scholar]
- Pavia F. C.; La Carrubba V.; Piccarolo S.; Brucato V. Polymeric scaffolds prepared via thermally induced phase separation: Tuning of structure and morphology. J. Biomed. Mater. Res., Part A 2008, 86, 459–466. 10.1002/jbm.a.31621. [DOI] [PubMed] [Google Scholar]
- Phaechamud T.; Chitrattha S. Pore formation mechanism of porous poly(DL-lactic acid) matrix membrane. Mater. Sci. Eng., C 2016, 61, 744–752. 10.1016/j.msec.2016.01.014. [DOI] [PubMed] [Google Scholar]
- Srinivasarao M.; Collings D.; Philips A.; Patel S. Three-Dimensionally Ordered Array of Air Bubbles in a Polymer Film. Science 2001, 292, 79–83. 10.1126/science.1057887. [DOI] [PubMed] [Google Scholar]
- Vivek A. V.; Babu K.; Dhamodharan R. Arborescent Polystyrene via Ambient Temperature ATRP: Toward Ordered Honeycomb Microstructured Templates. Macromolecules 2009, 42, 2300–2303. 10.1021/ma802321r. [DOI] [Google Scholar]
- You B.; Wen N.; Zhou S.; Wu L.; Zhao D. Facile Method for Fabrication of Nanocomposite Films with an Ordered Porous Surface. J. Phys. Chem. B 2008, 112, 7706–7712. 10.1021/jp802812e. [DOI] [PubMed] [Google Scholar]
- You B.; Shi L.; Wen N.; Liu X.; Wu L.; Zi J. A Facile Method for Fabrication of Ordered Porous Polymer Films. Macromolecules 2008, 41, 6624–6626. 10.1021/ma801417c. [DOI] [Google Scholar]
- Zhang M.; Li X. H.; Gong Y. D.; Zhao N. M.; Zhang X. F. Properties and biocompatibility of chitosan films modified by blending with PEG. Biomaterials 2002, 23, 2641–2648. 10.1016/s0142-9612(01)00403-3. [DOI] [PubMed] [Google Scholar]
- Suyatma N. E.; Copinet A.; Tighzert L.; Coma V. Mechanical and barrier properties of biodegradable films made from chitosan and poly (lactic acid) blends. J. Polym. Environ. 2004, 12, 1–6. 10.1023/b:jooe.0000003121.12800.4e. [DOI] [Google Scholar]
- Hong H.; Liu C.; Wu W. Preparation and characterization of chitosan/PEG/gelatin composites for tissue engineering. J. Appl. Polym. Sci. 2009, 114, 1220–1225. 10.1002/app.30619. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.