

Abstract
The effect of Zr modification on the catalytic activity of Co/SiO2 was investigated for nonoxidative propane dehydrogenation. Isolated Zr on SiO2 surface sites were prepared by organometallic synthesis using Zr(OtBu)4 as a precursor. The resulting Zr/SiO2 support was functionalized with Co2+ ions via strong electrostatic adsorption. Spectroscopic (diffuse reflectance infrared Fourier transform spectroscopy, UV–vis, electron paramagnetic resonance) and microscopic characterization (transmission electron microscopy, scanning transition electron microscopy) results are consistent with single-site cobalt that preferentially associates with the mono-dispersed Zr at a variety of loadings and Co/Zr ratios. The oxidation state of Co in the as-prepared Co/SiO2 and Co–Zr/SiO2 was both +2 with tetrahedral and octahedral geometries, respectively. In situ X-ray absorption near edge structure and extended X-ray absorption fine structure results confirmed that the oxidation state of Co remained as +2 under reaction condition for both Co/SiO2 and Co–Zr/SiO2 samples and both catalysts have tetrahedral Co2+ as the active catalyst. Despite similar Co coordination environments, the catalytic activity and selectivity was significantly improved by the Zr modification of the silica support versus Co/SiO2. This was attributed to the change in oxygen donor ability and Co–O bond strength of the ≡SiO–Zr–O sites of Co–Zr/SiO2 compared with the ≡SiO– ligands in Co/SiO2. These results show that tuning of the support SiO2 oxygen donation ability by use of an anchoring site (e.g., ≡SiO–Zr–O–) can be used to alter both rate and selectivity of propane dehydrogenation with single-site heterogeneous catalysts. These results also show some preference for Co2+ active sites to associate with ≡SiO–Zr–O– sites over ≡SiO–.
1. Introduction
On-purpose production of propylene via nonoxidative propane dehydrogenation1 (PDH) from conventional and shale gas streams is of increasing importance for the olefin and chemical industries. For this process, Pt–Sn and CrOx supported on alumina and silica catalysts are commercially available today.2−5 These catalysts have been extensively researched in the literature and the factors that affect their rate and selectivity are well-studied, even if the precise method of modification remains unclear. For example, Bariås et al. studied Pt and Pt–Sn catalytic systems on two different supports Al2O3 and SiO2 for PDH. It was found that on both supports addition of Sn promoted the catalytic activity and stability.2 Pt–Sn alloys are believed to be superior to pure Pt because of the Sn increasing Pt dispersion, decreasing propene binding energies (decreasing deep dehydrogenation and coking), and breaking up ensembles of Pt atoms that readily cleave C–C bonds.1−3,6 As might be expected, the support appears to play less of a role in metallic-phase dehydrogenation catalysts than in dehydrogenation catalysts with oxidized catalytic species. Many studies have been reported chromium-catalyzed PDH, and it is believed that isolated or low-nuclearity Cr clusters are the active catalysts for CrOx on both Al2O3 and SiO2 for dehydrogenation4,7−9 and polymerization.10−15 Copéret’s group demonstrated that Cr3+ surface sites were significantly more active than Cr2+ sites through heterolytic C–H bond activation as the rate-determining step.7 There are clearly large effects of both the support oxide and chromium oxide nuclearity.
Single-site catalysts for PDH are particularly interesting as both commercial systems (e.g., Cr and Ga) and as systems to understand the fundamental organometallic reaction mechanisms operative in this catalytic system. Over the past few years, we and others have studied PDH catalysis with a variety of ions16−19 and found that the overall catalytic rate of PDH by single-site heterogeneous catalysts is primarily governed by one of two basic rate-determining steps: heterolytic cleavage of C–H bond or β-hydride elimination of metal alkyl intermediates.7,19,20 The heterolytic cleavage of C–H bonds appears to be rate-determining for the transition-metal catalysts Co and Fe, whereas the β-hydride elimination is rate-determining for main-group ions like Zn and Ga. In addition to the overall rate, the catalytic selectivity of single-site catalysts for C–H activation over C–C bond cleavage appears to be higher for many nanoparticle catalysts. Previous work has shown that while isolated Co21,22 and Fe23 sites supported on SiO2 showed high propene selectivity during PDH under differential conditions, where background thermal cracking is low, the rates are relatively slow compared with Cr-based catalysts. In a more recent study with Y and Sc single-site catalysts and in computational study, we found that the M–O bond strength was a more important descriptor for dehydrogenation rates than the overall Lewis acidity.24 We reasoned that the catalyst-support oxide bond strength would be more tunable than the rate-determining β-hydride elimination of Zn and Ga catalysts, so we chose to focus on tuning the Lewis acidity of the support for cobalt-based PDH catalysts. For this study, we chose to prepare Zr sites on SiO2 as a test of this hypothesis that the rate of PDH could be increased while maintaining (or improving) PDH selectivity.
Bulk ZrO2 has been proved to be a good support for chromium-based dehydrogenation catalysts.25−32 De Rossi et al. reported that higher dehydrogenation activity was observed over ZrO2 compared with SiO2 as a catalyst support for chromium-catalyzed PDH.33−36 However, the low surface area of ZrO2 (<100 m2/g)28,34,35 resulted in lower overall activity than on Al2O3. For the purpose of fundamental study in this work, there are a low number of hydroxyl groups on ZrO2, likely of many different types that strongly depend on dehydration conditions (cf. Al2O3).37 For this reason, we chose to modify silica with zirconium sites to generate a more well-defined material. For silica-supported catalysts, the ligand can be viewed as ≡SiO–, though distributions of ring sizes and hydroxyl types are also well known.38−40 Replacing Si with a highly oxophilic metal species, for example Zr, will lead to ≡ZrO– which would be expected to be a weaker electron donating group than ≡SiO–. Such an effect was demonstrated during studies of metal–metal charge-transfer features by the Frei group (cross refs) that showed a decrease of the crystal field splitting energy in tetrahedral Zr–O–Co2+ sites versus Si–O–Co2+ because of the electrophilicity of the Zr. Consequently, the M–O (M is the isolated catalyst species) bond of ≡ZrO–M species should be weaker than ≡SiO–M. Zirconium sites were grafted on silica (Zr/SiO2) using Zr(OtBu)4 as a convenient starting material. Previously, the grafting of Zr though organometallic synthesis have been reported using various precursor compounds41−49 including Zr(CH2CMe3)4,50 [(ArO)Zr(CH2tBu)3],43 and Zr(NMe2)4.51 However, exploration of zirconium and other metals as anchoring sites in well-defined single-site catalysts is relatively unexplored. Zr promotion has been observed for Fischer–Tropsch catalysis52−54 and Zr on silica is a Lewis acid catalyst for converting glucose to 5-HMF (hydroxymethylfurfural) through isomerization of glucose to fructose.31,55
Herein, we report the promotion effect of zirconium addition to the Co/SiO2 catalysts for nonoxidative dehydrogenation of propane. The change in chemical properties of both Co/SiO2 and Co–Zr/SiO2 were examined by multiple characterization techniques, including under reaction conditions. Last, the effect of Zr loading to the catalytic activity of Co–Zr/SiO2 and the implications for catalyst dynamic behavior is discussed.
2. Experimental Section
2.1. Material and Methods
All manipulations of air-sensitive materials were performed with rigorous exclusion of O2 and moisture in oven-dried Schlenk-type glassware on a dual manifold Schlenk line and in N2-filled atmospheres glovebox with a high capacity recirculator (<1 ppm O2) unless otherwise noted. Solvents were sparged with N2, dried using activated alumina columns, transferred into the glovebox, and stored over 4 Å molecular sieves prior to use. Unless specified, all chemicals and other solvents were purchased and used as received from Sigma-Aldrich and Strem Chemicals. Elemental analysis (% Co, Zr) was conducted by Galbraith Laboratories, Inc. (Knoxville, TN). Brunauer–Emmett–Teller (BET) surface area and Barrett–Joyner–Halenda (BJH) pore size distribution of the samples were obtained using an accelerated surface area and porosimetry system (ASAP2020) from Micromeritics. The sample was first placed in a sample tube and held under vacuum below 5 μmHg for 30 min. The sample tube was then degassed by increasing the sample tube temperature to 130 °C for 12 h to clean the catalyst surface. After degassing, the sample tube was transferred to the analysis port where nitrogen was physiosorbed at liquid nitrogen temperature. The adsorption isotherms were collected while increasing the pressure of the tube until it reached to nitrogen saturation pressure. BET surface area was calculated on the basis of the adsorption branch of the isotherm, and the BJH pore size distribution was obtained from desorption profile of the isotherm. Transmission electron microscopy (TEM) characterization was performed by using a JEOL JEM2100F microscope equipped with a field emission gun operated at 200 keV. The solid samples were prepared by depositing a diluted sample solution (2.6-Zr/SiO2, Co/SiO2, and Co-2.6-Zr/SiO2 suspended in isopropyl alcohol reagent) on a sample holder (carbon-coated copper grid) and characterized without further treatment. The energy dispersive X-ray spectroscopy (EDX) data were collected on an Oxford Instrument X-Max 80 mm2 Silicon Drift Detector (SDD) for EDX. The scanning TEM (STEM) experiment was carried out using a FEI Talos microscope operated at 200 kV. Diffuse reflectance infrared fourier transform spectroscopy (DRIFTS) spectra were obtained using a Thermo Scientific Nicolet iS50 FT-IR spectrometer. The sample was packed inside a Praying Mantis reaction cell and sealed with a dome-shaped metal compartment including two ZnSe windows. The reaction chamber was first flushed for 30 min with He to optimize signal intensity. A sample spectrum was acquired by using a MCT/A detector at liquid nitrogen temperature where dried KBr was used as a background. DRUV–vis spectra of the samples were collected by a UV–vis–NIR spectrophotometer—Shimadzu UV-3600 Plus using a PMT (photomultiplier tube) detector. The catalyst was placed on a sample holder under atmosphere condition and the beam was aligned at the center of the sample. The sample spectrum was collected at a medium scan speed with a slit width of 3 nm in the region of 200–800 nm. To have a stable baseline, polytetrafluoroethylene was used as a background. All of the spectra shown in here were transferred to Kubelka–Munk functions. Samples were treated with 3% H2/Ar using 30 mL/min of flow rate with increasing temperature from RT to 600 °C to obtain temperature programmed reduction (TPR) profiles. The reaction was conducted inside a quartz reactor which was centered in a heated furnace. A thermocouple was inserted into the quartz reactor to measure the actual temperature during heating. A ramp rate of 10 °C/min was used. The effluent stream of the reactor was connected to a thermal conductivity detector (TCD) to monitor hydrogen consumption due to the sample. Prior to the experiment, the 3% H2/Ar gas was first introduced to the reactor continuously to obtain a stable TCD signal. Continuous wave (CW) X-band (9.48 GHz) electron paramagnetic resonance (EPR) experiments were carried out with a Bruker ELEXSYS II E500 EPR spectrometer (Bruker Biospin, Rheinstetten, Germany), equipped with a TE102 rectangular EPR resonator (Bruker ER 4102ST). A helium gas-flow cryostat (ICE Oxford, UK) and an intelligent temperature controller (ITC503, Oxford Instruments, UK) were used for measurements at cryogenic temperatures (T = 6 K). Data processing was done using Xepr (Bruker BioSpin, Rheinstetten) and MATLAB 7.11.2 (the MathWorks, Inc., Natick) environment.
2.2. Catalyst Preparation
Zr(OtBu)4 (2.24 g, 5.7 mmol) was mixed with 2.36 mL anhydrous hexanes to prepare a 4.6 mL stock solution (1.2 M) in a 50 mL round-bottom flask in a N2 glovebox. The Zr(OtBu)4 hexanes solution was added dropwise onto 4.6 g silica (dried at 200 °C under vacuum below 5 mTorr) by incipient wetness impregnation. The resulting solid was dried at room temperature under N2 protection and then was transferred onto the Schlenk line without exposure to air. The material was heated to 100 °C for 20 min under flowing nitrogen, and then the treatment temperature was increased to 200 °C and maintained at 200 °C under vacuum for 1 h, and then the solid was cooled down to room temperature. All synthetic operations were performed under air-free conditions up to this point. After exposure of the resulting material to air at room temperature, 4.92 g of the final product 2.6-Zr/SiO2 was obtained. Then, the same synthesis method was applied to synthesize 1.3-Zr/SiO2 (molar ratio of ≡SiOH/Zr = 1:0.5) and 0.26-Zr/SiO2 (≡SiOH/Zr = 1:0.1).
For Co–Zr/SiO2 catalysts, strong electrostatic adsorption (SEA)56 was applied as the synthetic method for preparing low and high loading isolated Co2+ on SiO2 and Zr/SiO2: 2 g of SiO2 (Davisil 646, 35–60 mesh, 300 m2/g and 1.1 cm3/g, Aldrich) or Zr/SiO2 were suspended in approximately 20 mL of deionized (DI) water. The pH of the solution was adjusted to 11 by using concentrated ammonium hydroxide (28.0–30.0% NH3 basis, Aldrich). In a separate flask, 0.3 g of Co(NH3)6Cl3 was dissolved in 10 mL of DI water, and the pH was adjusted to 11 with NH4OH. The basic Co3+ solution was rapidly added to the silica and stirred for 10 min at room temperature. The solid was settled for 5 min and the solution decanted. The resulting wet powder was vacuum filtered and rinsed with DI water several times. Then, the solid was dried in air at 125 °C for 2 h. Subsequently, the catalyst was calcined at 550 °C for 3 h with ramping (5 °C/min). Total cobalt loading for Co/SiO2 by elemental analysis were 1.5 wt %. Total cobalt and zirconium loadings for Co–Zr/SiO2 by elemental analysis were 1.5 and 6.0 wt %, respectively.
2.3. X-ray Absorption Near Edge Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) Measurements
Co K-edge (7709.0 eV) X-ray absorption spectroscopy (XAS) was measured in transmission mode conducted on the bending magnet beamline of the Materials Research Collaborative Access Team (MRCAT, 10-BM) at the Advanced Photon Source (APS) at Argonne National Laboratory. Ionization chambers were optimized for the maximum current with linear response (ca. 1010 photons detected per second) using 20% He in N2 (15% absorption) in the incident X-ray detector and 25% Ar in N2 (70% absorption) in the 175 transmission X-ray detector. The energy was calibrated with a cobalt foil (7709.0 eV). Third, the X-ray detector was placed in the beam path beyond the transmission detector to allow acquisition of a reference spectrum concurrent with each sample measurement. The catalyst was pressed as a 4 mm self-supporting wafer and placed in a stainless steel holder. The data were collected for as-prepared, under reaction conditions at 550 °C (3.5% propane in argon), and cooled down to ambient temperature after reaction but without exposure to air. Further XAS data analysis and fitting was processed by using WINXAS 3.2 software. The Co coordination environments were achieved by fitting of EXAFS data in R-space to the first nearest neighbor shell after a Fourier transform (k2-weighted).
2.4. Catalytic Activity Testing
Catalyst performance testing was conducted in a vertical, quartz tube reactor. Gas flow was controlled using mass flow controllers, and product distribution was determined via online gas chromatography (J&W Scientific) equipped with a 50 m GS-Alumina capillary column and a flame-ionization detector to which H2 (99.999%, Airgas USA) and air (<2 ppm H2O, Airgas) were also supplied. In a typical run, approximately 0.5 g of accurately weighed catalyst was supported on quartz wool with an internal thermocouple placed at the top of the catalyst bed. Initially, the catalyst was purged with He (99.999%, Airgas USA) that had been further purified with an oxygen trap at 40 mL/min at room temperature. The temperature was then increased to the reaction temperature of 550 °C and was allowed to stabilize for 2 h before introduction of reactant gases. For PDH, the reaction mixture comprised 3.0% propane in Ar at 20 mL/min.
3. Results and Discussion
3.1. Synthesis of Zr/SiO2, Co/SiO2 and Co–Zr/SiO2
Zirconium-modified silica was prepared via protonation of Zr(OtBu)4 by silica hydroxyls at room temperature followed by heating to 200 °C to condense any remaining hydroxyls and thermally cleave tert-butoxide groups. A similar synthesis of ZrO2 thin films by atomic layer deposition (ALD) surface chemistry has been reported by Kukli’s group.57 In our material, we utilized a solution version of an initial nucleation for an ALD reaction by dosing our precursor in solution but with the correct volume to fully saturate the SiO2 support mesoporous with Zr(OtBu)4 solution. An equimolar ratio of Zr(OtBu)4 to isolated hydroxyls (≡SiOH/Zr = 1:1) was used on the basis of the reported OH density after drying at 200 °C (2.5 OH/nm–2).58 The resulting infrared spectrum and transmission electron micrographs support the uniformity of Zr sites and lack of large crystals of ZrOx and are discussed more fully below. Our approach is also similar to the one used by Wilson et al.31,55 to prepare layers of overcoated zirconia on silica by multiple cycles of solution treatment of Zr(OiPr)4 in anhydrous organic solvent followed by hydrolysis treatment with H2O. Single-site cobalt 2+ was then grafted onto the low-zirconium density Zr/SiO2 via SEA using Co(NH3)6Cl3 concentrated ammonium hydroxide, where positively charged Co(NH3)63+ form cation–anion pairs56 with the deprotonated surface silica hydroxyls to form an orangish material. When it is heated in air to 300 °C, the Co reduced to Co2+ and the color of the material upon air exposure is pink.
3.2. Surface Area, Pore Size and Particle Size
The BET surface area and BJH pore size distribution were acquired for the bare SiO2, zirconium-modified SiO2 (Zr/SiO2), Co/SiO2 and Co–Zr/SiO2 (plot shown Figure S1). The surface area of commercial SiO2 is unchanged by the grafting of Zr to make Zr/SiO2 (324 m2/g vs 323 m2/g). Thus, the Zr(OtBu)4 precursor reacts without producing pore blockage or pore constriction. However, the grafting conditions employed for Co addition resulted method resulted in a loss of surface area of ∼50 and ∼30 m2/g for Co/SiO2 and Co–Zr/SiO2, respectively. This was found to be due to the basic conditions employed during SEA grafting (pH 11), confirmed by a control experiment that omitted cobalt from the synthetic protocol. Despite the small reduction of surface area upon cobalt grafting, the surface areas and pore size distributions are similar and within 300 ± 30 m2/g (±10%) range.
Conventional TEM (Figure S2) and high-angle annular dark-field–STEM were also utilized to characterize the dispersion and configuration of Co and Zr species on the SiO2 support for both as-prepared and post-catalysis Co–Zr/SiO2 samples shown in Figure 1a,b. Highly dispersed Co and Zr are observed on the SiO2 support with similar Z-contrast and no aggregation was found post-catalysis, nor carbon nanotubes or other coke deposits. The STEM–EDX elemental mapping result shown in Figure 1 shows that the images of Co, Zr, and Si are completely laid on each other, suggesting that Co sites could be bonded to the Zr or Si sites. The corresponding EDX line scan measured along the sample indicates that there is a relative uniform distribution of Co and Zr atomic sites along with the SiO2 support.
Figure 1.

STEM (top) images of (a) as-prepared and (b) post-catalysis Co–Zr/SiO2 samples and (bottom) the corresponding EDS line scan measured along the as prepared Co–Zr/SiO2.
3.3. DRIFTS and DRUV–Vis Spectroscopy
Comparison of DRIFTS (Figure 2a) shows that in contrast to Co/SiO2, the zirconium-modified Zr/SiO2 and Co–Zr/SiO2 do not have isolated OH peaks on the surface by the absence of the peak at 3780 cm–1. A small amount of tert-butanol may have remained coordinated during the synthesis of Zr/SiO2; however, the aqueous cobalt grafting step removes it, as expected, and no C–H ligands are observed to remain on the pre-catalysis Co–Zr/SiO2 catalyst. DRIFTS (Figure 2b) shows that after dehydration at 550 °C, the isolated OH peak appears on the Zr/SiO2 and Co–Zr/SiO2 surface at 3738 cm–1 which is same as Co/SiO2. No features were resolved that could be ascribed to Zr–OH features on the as-prepared or materials dehydrated at 550 °C.
Figure 2.

DRIFTS and DRUV–vis spectra of (a) Zr/SiO2, Co/SiO2, and Co–Zr/SiO2 in air, (b) Zr/SiO2, Co/SiO2, and Co–Zr/SiO2 dehydration at 550 °C, and (c) Zr/SiO2, Co/SiO2, and Co–Zr/SiO2 in air.
Visible inspection of Co/SiO2 and Co–Zr/SiO2 powders post-synthesis shows a striking difference in color, with the Co/SiO2 displaying a vivid and characteristic “cobalt blue” while the Co–Zr/SiO2 material has a pinkish hue due to water saturation. More careful inspection of the materials using DRUV–vis spectroscopy shows the familiar peaks of the ligand field absorption for isolated tetrahedral Co(II) of the monometallic Co/SiO2 samples are shown in red in Figure 2c. The UV–vis spectrum exhibits three absorption peaks (530, 595, and 635 nm), which can be explicitly referred to the 4A2(F)/4T1(P) transition of Co(II) ions in tetrahedral geometry.59,60 The blue shift of the spectrum is similar to that reported for Co–Zr/SiO2 prepared via a different grafting procedure. Additionally, a sharp increase in absorption band in the UV region at 200 nm was observed. This is assigned to a low-energy charge transfer between the oxygen ligands and central Co(II) ion in tetrahedral symmetry. The Co–Zr/SiO2 spectrum in green in Figure 2c shows significantly higher absorption compared with Co/SiO2 and a small peak at ∼300 nm. The increase in intensity is likely attributed to a different geometry of the cobalt environment (tetrahedral to octahedral) which was further confirmed by EXAFS analysis (vide infra). In contrast, CoO nanoparticles display a large, broad peak from 400 to 600 nm,61,62 again consistent that both of the Co sites are well-dispersed. Zr4+ cannot be examined due to weak absorption at λ > 200 nm.63,64 The results from DRUV–vis analysis clearly show that the electronic and chemical structure between the as-prepared Co/SiO2 and Co–Zr/SiO2 are fairly different. However, the familiar pink of Co(OH2)62+ ions on the Co–Zr/SiO2 catalyst disappears upon heating, replaced by the characteristic blue of tetrahedral Co2+, which is discussed further below.
EPR is potentially a more sensitive technique for identifying CoOx clusters than DRIFTS, STEM, or XAS (vide infra). A selected set of EPR spectra recorded under identical experimental conditions at 6 K are shown in Figure 3. The EPR signals of the three samples are essentially identical. The only difference is in the intensity of the signal, which is due to the concentration of the Co ions in the sample. The main signal is observed at geff ≈ 5 together with a broad signal around g ≈ 2. A substantially narrower signal close to the free electron g-value of 2.0023 was observed in Co–Zr/SiO2 post-catalysis. This latter signal is too narrow to be attributed to the cobalt. Most likely this signal is due to trace amounts of radicals/defects often found in powders. These impurities may also be the result of Zr reduction under reaction conditions; however, it is not clear at this time.65 Our materials have a low coverage of Zr on the silica and absence of large particles of ZrO2 (vide supra), and isolated Zr3+ sites on silica would not be expected to be stable to atmospheric oxygen and moisture. Previous studies have focused on bulk and doped ZrO2 EPR, rather than submonolayer coverages as in these materials, and we do not have evidence for reduction of Zr in our materials. Our analysis of the possible cobalt EPR-active species is as follows: we have confirmed that the cobalt in our samples is Co(II) via XAS analysis and Co(III) complexes should be EPR-silent under our experimental conditions. While Co(IV) has a low-spin state S = 1/2 and gives distinct EPR spectra around g ≈ 2, this type of signals was not observed in our experiment.66 On the basis of the findings by Britt and co-workers,66 the EPR signals observed here belong to Co(II) high spin state (S = 3/2), probably in tetrahedral symmetry or axially distorted tetrahedral symmetry,67 very similar to our previously reported Co/SiO2 catalysts.21 Four-coordinated square planar Co(II) complexes as well as square planar Co(II) complexes with one or two axial ligands usually demonstrate low-spin state (S = 1/2) and give EPR signals different from those observed here.68−200,70 However, as an alternative explanation, according to Peters and co-workers,67 it could also be a dimer or multimer of Co(II); as stated by Britt and co-workers:66 “The feature at geff ≈ 5 is reminiscent of EPR spectra measured for many Co(II)-containing compounds, including Co3O4, Co3(PO4)2.” However, no appreciable amounts of Co(III) were detected via XAS, as would be necessary for Co3O4 or cobalt phosphate in these materials. Thus EPR was able to demonstrate the absence of CoO but not other potential clusters with different oxidation states. However, bulk XAS analysis was able to exclude the presence of substantial amounts of Co3+ and thus the presence of Co3O4 clusters, too.
Figure 3.
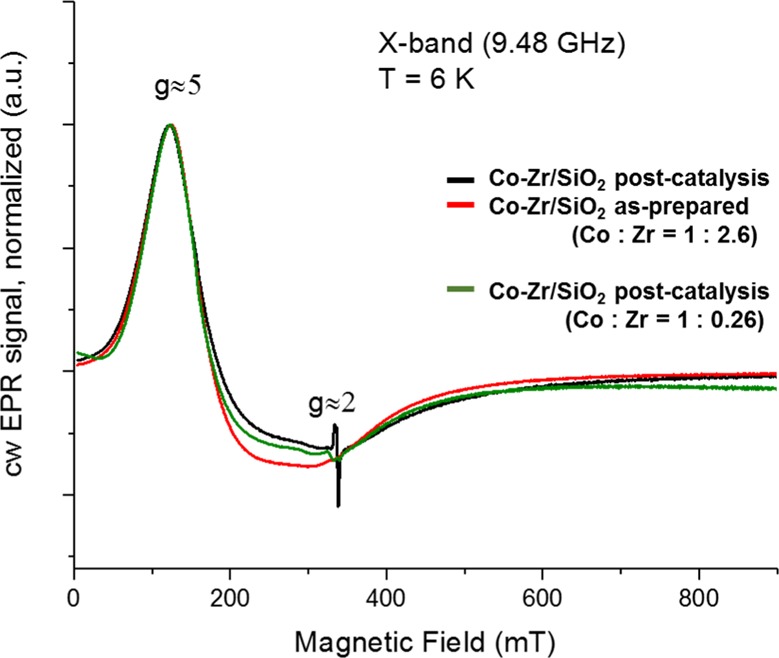
CW X-band EPR spectra obtained from various powder sample of Co/SiO2 recorded at 6 K.
3.4. XANES and EXAFS
In situ XANES and EXAFS spectra of Co/SiO2 and Co–Zr/SiO2 samples for three different conditions were collected: (1) as-prepared, (2) in situ during dehydrogenation of propane at 550 °C, and (3) post-catalysis at room temperature without exposure to air. Figure 4 shows the XANES spectra of the samples collected under these conditions. The pre-edge energy of Co in all six cases including both Co/SiO2 and Co–Zr/SiO2 were found to be 7.7092 keV which is consistent with the reported Co2+ reference compounds. This evidently indicates that the oxidation state of cobalt of Co/SiO2 and Co–Zr/SiO2 catalyst remains as +2 during PDH at 550 °C. Consistent with this observation, no consumption of hydrogen was detected from RT to 600 °C during a TPR experiment.
Figure 4.
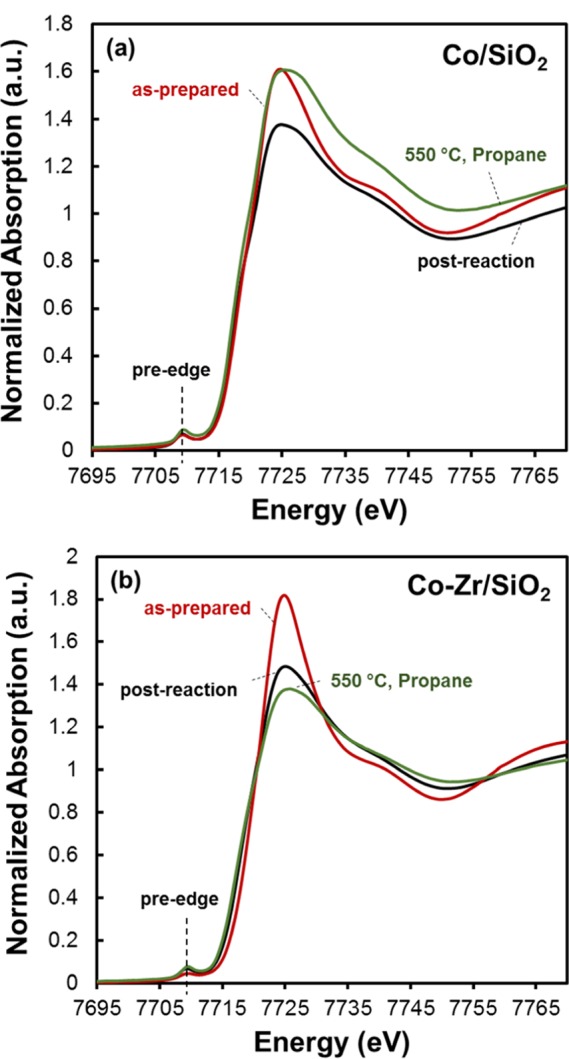
Co K-edge XANES spectra of (a) Co/SiO2 and (b) Co–Zr/SiO2 samples (1) as-prepared, (2) dehydrogenation reaction condition at 550 °C, and (3) cooled down to RT after catalysis.
Though the cobalt oxidation of state does not change upon heating, the coordination number does change for Co–Zr/SiO2 upon heating, consistent with the color changes and other previously discussed observations. Figure 5 compares the Co K-edge EXAFS spectra between Co/SiO2 and Co–Zr/SiO2. The first shell peak intensity decreased upon heating as-prepared Co–Zr/SiO2 and fitting of the first shell Co–O peak in EXAFS spectra resulted in coordination number of 6 for as-prepared Co–Zr/SiO2 that decreases to less than 4 on average upon heating (Table 1).
Figure 5.
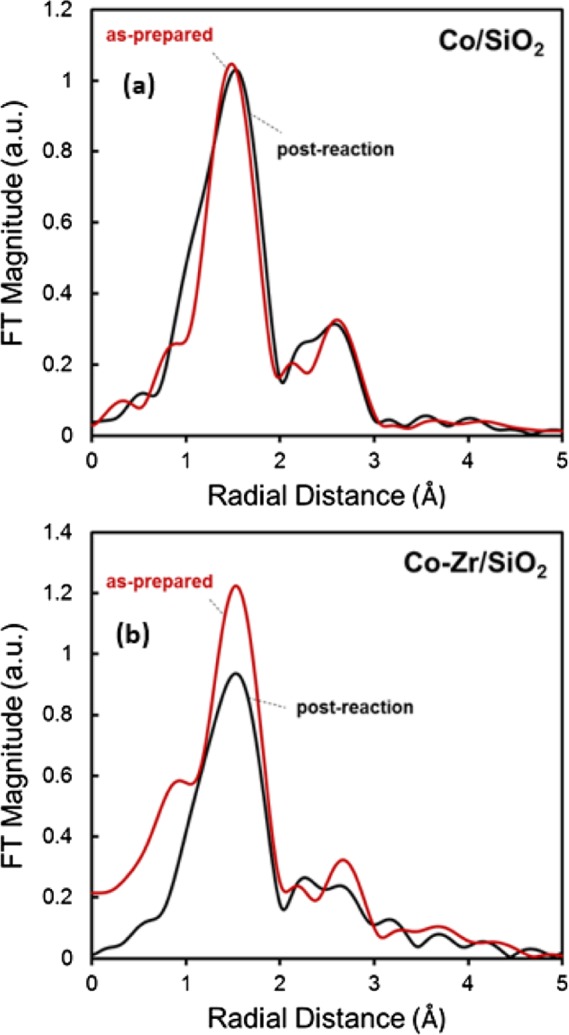
Co K-edge EXAFS spectra of (a) Co/SiO2 and (b) Co–Zr/SiO2 samples (1) as-prepared and (2) cooled down to RT after catalysis.
Table 1. XANES Pre-Edge Energies and EXAFS Fitting Data of Co/SiO2 and Co–Zr/SiO2.
| sample | pre-edge energy (keV) | oxidation state | CN | R (Å) | σ2 | E0 (eV) | S02 |
|---|---|---|---|---|---|---|---|
| Co/SiO2 as-prepared | 7.7092 | +2 | 3.7 (±0.5) | 1.98 (±0.01) | 0.005 | –8.5 (±2.0) | 0.81 |
| Co/SiO2 post-reaction | 7.7092 | +2 | 4.0 (±0.8) | 1.97 (±0.02) | 0.008 | –2.0 (±3.2) | 0.82 |
| Co–Zr/SiO2 as-prepared | 7.7092 | +2 | 5.7 (±0.7) | 2.05 (±0.01) | 0.009 | –7.9 (±1.7) | 0.88 |
| Co–Zr/SiO2 post-reaction | 7.7092 | +2 | 4.1 (±0.6) | 1.99 (±0.02) | 0.009 | –4.0 (±2.2) | 0.86 |
Heating Co–Zr/SiO2 dissociates water with increasing temperate and under reaction conditions (550 °C), the coordination number cobalt drops down to 4.1. Notably, no change in average coordination number was observed for Co/SiO2. Addition of water on the post-reaction catalyst at RT changes the color of Co–Zr/SiO2 sample back to pink (octahedral) within about half an hour, showing that the cobalt surface hydration is reversible and that Co sites remain isolated after PDH catalysis.
The fitted Co–O bond distances are different but not significantly so. The Co–O distances in Co–Zr/SiO2 (as-prepared = 2.05 ± 0.01 Å, post-reaction = 1.99 ± 0.02 Å) was found to be slightly longer than that of Co/SiO2 (as-prepared = 1.98 ± 0.01 Å, post-reaction = 1.97 ± 0.02 Å). However, these distances are notably longer than the reported Co–O bond of Co3O4 nanoparticles supported on SiO2 reduced at 553 K of 1.91 Å.71 Thus, while the average Co–O bond distances when supported on SiO2 and Zr/SiO2 are not significantly different, the fact that the cobalt of Co–Zr/SiO2 is hydrated/dehydrated readily is also consistent with a weaker cobalt–support interaction than Co/SiO2, which does not hydrate, and is consistent with the red shift in the UV–vis spectrum of Co–Zr/SiO2.
3.5. Catalytic PDH
The catalytic activity of Co–Zr/SiO2 and Co/SiO2 were compared under similar reaction conditions (at 550 °C, propane flow rate is 20 mL/min), with conversion levels controlled to below 10% in remain in the kinetically-controlled regime. The turnover frequency (TOF) value obtained over Co–Zr/SiO2 catalyst was found to be 10.0 h–1 (±10%) at 550 °C, which is three times faster than that of Co/SiO2. A control experiment under the same reaction conditions using the bare Zr/SiO2 support shows negligible catalytic activity for PDH and thus there is no contribution from Zr/SiO2 to the catalytic activity of the Co–Zr/SiO2 catalyst. This is in contrast to reports of ZrO2 and doped MZrO2 (M = La etc.) materials, where coordinatively unsaturated ZCUS are proposed as active participants.32 We do not have evidence that single-site zirconium on silica has similar catalytic behavior under these conditions. Also of note, O2– ion mobility varies widely with doping levels in doped MOx materials.72−74 In addition to having a higher PDH rate than Co/SiO2, Co–Zr/SiO2 also shows higher propene selectivity at 550 °C of ∼97% versus the Co/SiO2 catalyst of 93%. It should be noted that in most heterogeneous catalyst systems, the selectivity decreases as the conversion increases. However, in this case, both selectivity and rate for Co–Zr/SiO2 are higher than the zirconium-free catalyst. The catalysts are stable on-stream for at least 20 h and negligible coke formation (>99% carbon balance during the run). Finally, the reversible hydration of the cobalt led us to test whether long-term storage under ambient conditions would lead to different catalyst behavior. We found that after storage in air for 1 month, samples of Co–Zr/SiO2 catalysts have no detectible change in catalytic behavior when tested. These results, together with the STEM images of the post-catalysis Co–Zr/SiO2 catalysts (Figure 1) provide clear evidence that no nanoparticles are forming under reaction conditions, in contrast to cobalt on silica catalysts reported by Coperet.22
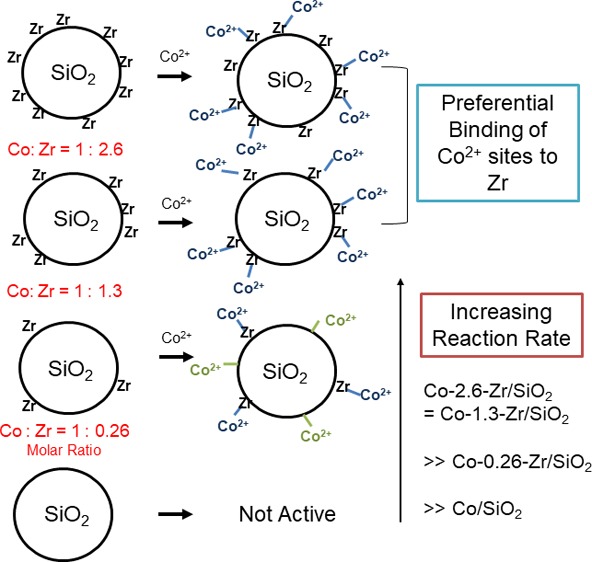
The different cobalt hydration behavior and catalytic function were obtained for Co–Zr/SiO2 containing excess zirconium sites with ratio of zirconium to cobalt of ca. 2.6:1; however, we lowered the ratio of Zr to Co for two reasons: (1) to see if there was a preference for cobalt to reside near a zirconium site on the surface of the silica and (2) to see if there is differing activity with lower density of zirconia. Thus, we found that at a 1:1.3 ratio of Co and Zr the PDH rate and selectivity were unchanged from Co–Zr/SiO2 (2.6:1 ratio) within error. Further lowering of the amount of zirconium to substoichiometric 1:0.26 resulted in an observed PDH rate that is approximately the weighted average of the Co/SiO2 and Co–Zr/SiO2 rates, also consistent with cobalt sites being directly promoted by the Zr (Figure 6). The selectivity of the PDH was found to be ca. 97% for all Co–Zr/SiO2 species (Figure 7). It is reasonable to expect that a labile octahedral Co2+ ion would be able to traverse the surface prior to dehydration based upon our previous observation of the dispersal of CoO clusters, so we infer that these results to show some thermodynamic preference for Co–O–Zr≡ linkages, though further study is clearly needed and in progress.
Figure 6.

Comparison of PDH reactivity performed at 550 °C, propane (3% in Ar) flow rate is 20 mL/min, using Co/SiO2 and Co–Zr/SiO2 with increasing Zr loading (numbers indicate molar ratio of Co to Zr).
Figure 7.
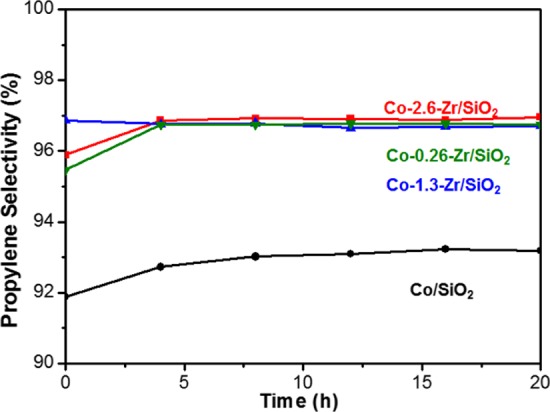
Comparison of PDH selectivity performed at 550 °C, propane (3% in Ar) flow rate is 20 mL/min, using Co/SiO2 and Co–Zr/SiO2 with increasing Zr loading (numbers indicate molar ratio of Co to Zr).
Our proposed structures for the distribution of cobalt sites in the varying Co–Zr/SiO2 catalysts are summarized in Figure 8. The surface Co2+ comprises two types of sites, Co atoms adjacent to the ≡Zr–OH and Co sites that are not adjacent to the ≡Zr–OH (neglecting siloxane ring size effects). When excess or stoichiometric zirconium sites are present, octahedral cobalt 2+ ions are preferentially associated with them. They are hydrolytically labile and hydrate under air but dehydrate to tetrahedral Co2+ under reaction conditions. Because of the highly dispersed zirconium, it is also possible to assume that Zr might deposit randomly on the catalyst surface. However, in this case, the substoichiometric zirconium loadings would not be expected to be populated by adjacent cobalt sites as a weighted average, as inferred from the TOF of the working catalysts. Furthermore, the observed weighted average TOF is also not consistent with more than one cobalt associating with a zirconium site. Thus, we hypothesize that this system has a thermodynamic preference for a cobalt to be adjacent to a surface zirconium, if one exists, and that these sites are both faster for PDH as well as more selective.
Figure 8.
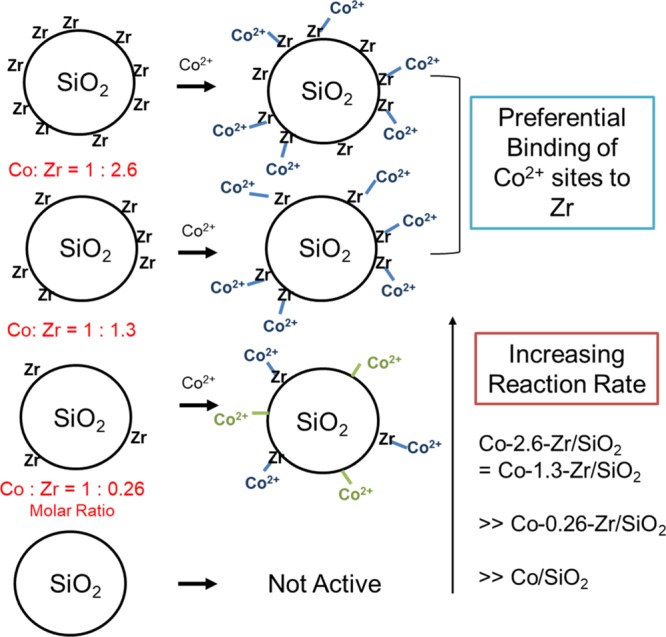
Cartoon representation of the ratio of Zr–Co in catalyst materials and Co–Zr/SiO2 catalyst site distribution. The Co–X lines (SiO2 or Zr) represent Co–O–X bonds.
In addition to the location of Co relative to Zr on the SiO2 surface, some discussion of the electronic impact of the electrophilic Zr is needed. Our Co–Zr/SiO2 catalyst shows similar UV–vis shifts as previous work by Macnaughtan, Soo, and Frei60 due to electron-withdrawing Zr decreasing the π donation of O ligand into transition-metal Co and a widening of the d-orbital splitting. This lowers the Co–O bond strength and results in a larger splitting energy between e and t2 orbitals of Co–Zr/SiO2 than Co/SiO2 (Figure 9). Altering the π-donating ability of a ligand, a common strategy for tuning rate and selectivity in homogeneous catalysis, is less understood for single-site ionic catalysts such as these. The increase in rate due to the electron-withdrawing effects of Zr is consistent with our previous calculations,40 that shows that decreasing the catalyst–oxygen bond strength leads to an increase in PDH rate. Although the impact on PDH rate in this specific case was a modest increase, the increase in catalytic selectivity to propene (ca. 92–97%) was significant. This observation shows that relatively small perturbations in catalyst–support bond strengths can result in significant changes in not only C–H heterolytic cleavage, the rate-determining step of dehydrogenation but also the catalytic selectivity of the reaction. The origin of the change in selectivity is not understood at this time and is the subject of ongoing work.
Figure 9.
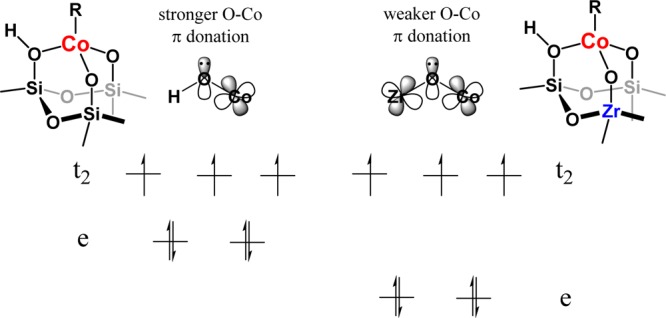
Diagram of the Co(II) splitting energy between (left) Co/SiO2 and (right) Co–Zr/SiO2. Adapted from ref (60).
4. Conclusion
Higher turnover frequencies were observed for the Zr-promoted cobalt catalysts with significant increase in propane conversion and propene selectivity compared with the Co/SiO2 catalyst. The relationship between Zr loading and catalyst selectivity is consistent with the formation of enhanced active sites at the interface between the Co metal and the Zr promoter atom. As Zr loading increases, the active sites adjacent to the promoter increases. Ultimately, the fraction of active sites that are promoted approaches unity, suggesting that this occurs near a Zr/Co atomic ratio of 1, and no further improvements to the product selectivity result from higher Zr loading. This suggests the Lewis acidity of the promoter is the relevant descriptor for metal oxide promotion effects. The improved dehydrogenation activity was attributed to the ease of heterolytic cleavage of the C–H bond over a weaker Co–O bond of ≡Zr–O–Co. The positive effects of having weaker M–O bond for facial heterolytic cleavage leads to a promising strategy for rational catalyst design.
Acknowledgments
This research was supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Biosciences and Geosciences under contract DE-AC02-06CH11357. Use of the Advanced Photon Source (APS) and the Center for Nanoscale Materials (CNM) at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences, under contract DE-AC02-06CH11357. MRCAT operations are supported by the Department of Energy and the MRCAT member institutions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00862.
BET surface areas and BJH pore size distributions, TEM images and analysis, data processing and analysis of EXAFS measurements, and dehydrogenation with Co/ZrO2 results (PDF)
Author Present Address
∥ Korea Institute of Science and Technology (H.S.).
Author Present Address
⊥ Phillips 66 (B.H.).
The authors declare no competing financial interest.
Supplementary Material
References
- Sattler J. J. H. B.; Ruiz-Martinez J.; Santillan-Jimenez E.; Weckhuysen B. M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chem. Rev. 2014, 114, 10613–10653. 10.1021/cr5002436. [DOI] [PubMed] [Google Scholar]
- Bariås O. A.; Holmen A.; Blekkan E. A. Propane Dehydrogenation over Supported Pt and Pt-Sn Catalysts: Catalyst Preparation, Characterization, and Activity Measurements. J. Catal. 1996, 158, 1–12. 10.1006/jcat.1996.0001. [DOI] [Google Scholar]
- Stagg S. M.; Querini C. A.; Alvarez W. E.; Resasco D. E. Isobutane Dehydrogenation on Pt-Sn/SiO2 Catalysts: Effect of Preparation Variables and Regeneration Treatments. J. Catal. 1997, 168, 75–94. 10.1006/jcat.1997.1617. [DOI] [Google Scholar]
- Hakuli A.; Kytökivi A.; Krause A. O. I. Dehydrogenation of i-butane on CrOx/Al2O3 catalysts prepared by ALE and impregnation techniques. Appl. Catal., A 2000, 190, 219–232. 10.1016/s0926-860x(99)00310-5. [DOI] [Google Scholar]
- Michorczyk P.; Pietrzyk P.; Ogonowski J. Preparation and characterization of SBA-1-supported chromium oxide catalysts for CO2 assisted dehydrogenation of propane. Microporous Mesoporous Mater. 2012, 161, 56–66. 10.1016/j.micromeso.2012.05.011. [DOI] [Google Scholar]
- Feng J.; Zhang M.; Yang Y. Dehydrogenation of Propane on Pt or PtSn Catalysts with Al2O3 or SBA-15 Support. Chin. J. Chem. Eng. 2014, 22, 1232–1236. 10.1016/j.cjche.2013.07.001. [DOI] [Google Scholar]
- Conley M. P.; Delley M. F.; Núñez-Zarur F.; Comas-Vives A.; Copéret C. Heterolytic Activation of C-H Bonds on CrIII-O Surface Sites Is a Key Step in Catalytic Polymerization of Ethylene and Dehydrogenation of Propane. Inorg. Chem. 2015, 54, 5065–5078. 10.1021/ic502696n. [DOI] [PubMed] [Google Scholar]
- Delley M. F.; Silaghi M.-C.; Nuñez-Zarur F.; Kovtunov K. V.; Salnikov O. G.; Estes D. P.; Koptyug I. V.; Comas-Vives A.; Copéret C. X-H Bond Activation on Cr(III),O Sites (X = R, H): Key Steps in Dehydrogenation and Hydrogenation Processes. Organometallics 2017, 36, 234–244. 10.1021/acs.organomet.6b00744. [DOI] [Google Scholar]
- Coperet C. C–H Bond Activation and Organometallic Intermediates on Isolated Metal Centers on Oxide Surfaces. Chem. Rev. 2010, 110, 656–680. 10.1021/cr900122p. [DOI] [PubMed] [Google Scholar]
- Ajjou J. A. N.; Scott S. L.; Paquet V. Synthesis and Characterization of Silica-Stabilized Chromium(IV) Alkylidene Complexes. J. Am. Chem. Soc. 1998, 120, 415–416. 10.1021/ja973177a. [DOI] [Google Scholar]
- Ajjou J. A. N.; Scott S. L. A Kinetic Study of Ethylene and 1-Hexene Homo- and Copolymerization Catalyzed by a Silica-Supported Cr(IV) Complex: Evidence for Propagation by a Migratory Insertion Mechanism. J. Am. Chem. Soc. 2000, 122, 8968–8976. 10.1021/ja001693m. [DOI] [Google Scholar]
- Brown C.; Krzystek J.; Achey R.; Lita A.; Fu R.; Meulenberg R. W.; Polinski M.; Peek N.; Wang Y.; van de Burgt L. J.; Profeta S.; Stiegman A. E.; Scott S. L. Mechanism of Initiation in the Phillips Ethylene Polymerization Catalyst: Redox Processes Leading to the Active Site. ACS Catal. 2015, 5, 5574–5583. 10.1021/acscatal.5b00927. [DOI] [Google Scholar]
- Brown C.; Lita A.; Tao Y.; Peek N.; Crosswhite M.; Mileham M.; Krzystek J.; Achey R.; Fu R.; Bindra J. K.; Polinski M.; Wang Y.; van de Burgt L. J.; Jeffcoat D.; Profeta S.; Stiegman A. E.; Scott S. L. Mechanism of Initiation in the Phillips Ethylene Polymerization Catalyst: Ethylene Activation by Cr(II) and the Structure of the Resulting Active Site. ACS Catal. 2017, 7, 7442–7455. 10.1021/acscatal.7b02677. [DOI] [Google Scholar]
- Fong A.; Peters B.; Scott S. L. One-Electron-Redox Activation of the Reduced Phillips Polymerization Catalyst, via Alkylchromium(IV) Homolysis: A Computational Assessment. ACS Catal. 2016, 6, 6073–6085. 10.1021/acscatal.6b01728. [DOI] [Google Scholar]
- Fong A.; Yuan Y.; Ivry S. L.; Scott S. L.; Peters B. Computational Kinetic Discrimination of Ethylene Polymerization Mechanisms for the Phillips (Cr/SiO2) Catalyst. ACS Catal. 2015, 5, 3360–3374. 10.1021/acscatal.5b00016. [DOI] [Google Scholar]
- Thomas J. M.; Raja R.; Lewis D. W. Single-site Heterogeneous Catalysts. Angew. Chem., Int. Ed. 2005, 44, 6456–6482. 10.1002/anie.200462473. [DOI] [PubMed] [Google Scholar]
- Nozaki C.; Lugmair C. G.; Bell A. T.; Tilley T. D. Synthesis, Characterization, and Catalytic Performance of Single-Site Iron(III) Centers on the Surface of SBA-15 Silica. J. Am. Chem. Soc. 2002, 124, 13194–13203. 10.1021/ja020388t. [DOI] [PubMed] [Google Scholar]
- Yun D.; Baek J.; Choi Y.; Kim W.; Lee H. J.; Yi J. Promotional Effect of Ni on a CrOx Catalyst Supported on Silica in the Oxidative Dehydrogenation of Propane with CO2. ChemCatChem 2012, 4, 1952–1959. 10.1002/cctc.201200397. [DOI] [Google Scholar]
- Schweitzer N. M.; Hu B.; Das U.; Kim H.; Greeley J.; Curtiss L. A.; Stair P. C.; Miller J. T.; Hock A. S. Propylene Hydrogenation and Propane Dehydrogenation by a Single-Site Zn2+ on Silica Catalyst. ACS Catal. 2014, 4, 1091–1098. 10.1021/cs401116p. [DOI] [Google Scholar]
- Chen K.; Bell A. T.; Iglesia E. Kinetics and Mechanism of Oxidative Dehydrogenation of Propane on Vanadium, Molybdenum, and Tungsten Oxides. J. Phys. Chem. B 2000, 104, 1292–1299. 10.1021/jp9933875. [DOI] [Google Scholar]
- Hu B.; Getsoian A.; Schweitzer N. M.; Das U.; Kim H.; Niklas J.; Poluektov O.; Curtiss L. A.; Stair P. C.; Miller J. T.; Hock A. S. Selective propane dehydrogenation with single-site CoII on SiO2 by a non-redox mechanism. J. Catal. 2015, 322, 24–37. 10.1016/j.jcat.2014.10.018. [DOI] [Google Scholar]
- Estes D. P.; Siddiqi G.; Allouche F.; Kovtunov K. V.; Safonova O. V.; Trigub A. L.; Koptyug I. V.; Copéret C. C-H Activation on Co,O Sites: Isolated Surface Sites versus Molecular Analogs. J. Am. Chem. Soc. 2016, 138, 14987–14997. 10.1021/jacs.6b08705. [DOI] [PubMed] [Google Scholar]
- Hu B.; Schweitzer N. M.; Zhang G.; Kraft S. J.; Childers D. J.; Lanci M. P.; Miller J. T.; Hock A. S. Isolated FeII on Silica As a Selective Propane Dehydrogenation Catalyst. ACS Catal. 2015, 5, 3494–3503. 10.1021/acscatal.5b00248. [DOI] [Google Scholar]
- Camacho-Bunquin J.; Aich P.; Ferrandon M.; Getsoian A.; Das U.; Dogan F.; Curtiss L. A.; Miller J. T.; Marshall C. L.; Hock A. S.; Stair P. C. Single-site Zinc on Silica Catalysts for Propylene Hydrogenation and Propane Dehydrogenation: Synthesis and Reactivity Evaluation Using an Integrated Atomic Layer Deposition-catalysis Instrument. J. Catal. 2017, 345, 170–182. 10.1016/j.jcat.2016.10.017. [DOI] [Google Scholar]
- Brückner A.; Radnik J.; Hoang D.-L.; Lieske H. In Situ Investigation of Active Sites in Zirconia-supported Chromium Oxide Catalysts During the Aromatization of n-octane. Catal. Lett. 1999, 60, 183–189. 10.1023/a:1019067309929. [DOI] [Google Scholar]
- Cimino A.; Cordischi D.; De Rossi S.; Ferraris G.; Gazzoli D.; Indovina V.; Minelli G.; Occhiuzzi M.; Valigi M. Studies on chromia/zirconia catalysts I. Preparation and characterization of the system. J. Catal. 1991, 127, 744–760. 10.1016/0021-9517(91)90196-b. [DOI] [Google Scholar]
- Khodakov A.; Yang J.; Su S.; Iglesia E.; Bell A. Structure and properties of vanadium oxide-zirconia catalysts for propane oxidative dehydrogenation. J. Catal. 1998, 177, 343–351. 10.1006/jcat.1998.2143. [DOI] [Google Scholar]
- Cutrufello M. G.; De Rossi S.; Ferino I.; Monaci R.; Rombi E.; Solinas V. Preparation, characterisation and activity of chromia–zirconia catalysts for propane dehydrogenation. Thermochim. Acta 2005, 434, 62–68. 10.1016/j.tca.2005.01.017. [DOI] [Google Scholar]
- Xu B.; Zheng B.; Hua W.; Yue Y.; Gao Z. Support effect in dehydrogenation of propane in the presence of CO2 over supported gallium oxide catalysts. J. Catal. 2006, 239, 470–477. 10.1016/j.jcat.2006.02.017. [DOI] [Google Scholar]
- Jiménez-López A.; Rodríguez-Castellón E.; Maireles-Torres P.; Díaz L.; Mérida-Robles J. Chromium Oxide Supported on Zirconium- and Lanthanum-doped Mesoporous Silica for Oxidative Dehydrogenation of Propane. Appl. Catal., A 2001, 218, 295–306. 10.1016/s0926-860x(01)00656-1. [DOI] [Google Scholar]
- Osatiashtiani A.; Lee A. F.; Granollers M.; Brown D. R.; Olivi L.; Morales G.; Melero J. A.; Wilson K. Hydrothermally Stable, Conformal, Sulfated Zirconia Monolayer Catalysts for Glucose Conversion to 5-HMF. ACS Catal. 2015, 5, 4345–4352. 10.1021/acscatal.5b00965. [DOI] [Google Scholar]
- Otroshchenko T. P.; Rodemerck U.; Linke D.; Kondratenko E. V. Synergy effect between Zr and Cr active sites in binary CrZrOx or supported CrOx /LaZrOx : Consequences for catalyst activity, selectivity and durability in non-oxidative propane dehydrogenation. J. Catal. 2017, 356, 197–205. 10.1016/j.jcat.2017.10.012. [DOI] [Google Scholar]
- De Rossi S.; Ferraris G.; Fremiotti S.; Garrone E.; Ghiotti G.; Campa M. C.; Indovina V. Propane Dehydrogenation on Chromia/Silica and Chromia/Alumina Catalysts. J. Catal. 1994, 148, 36–46. 10.1006/jcat.1994.1183. [DOI] [Google Scholar]
- De Rossi S.; Ferraris G.; Fremiotti S.; Cimino A.; Indovina V. Propane Dehydrogenation on Chromia/Zirconia Catalysts. Appl. Catal., A 1992, 81, 113–132. 10.1016/0926-860x(92)80264-d. [DOI] [Google Scholar]
- De Rossi S.; Ferraris G.; Fremiotti S.; Indovina V.; Cimino A. Isobutane Dehydrogenation on Chromia/Zirconia Catalysts. Appl. Catal., A 1993, 106, 125–141. 10.1016/0926-860x(93)80160-r. [DOI] [Google Scholar]
- De Rossi S.; Pia Casaletto M.; Ferraris G.; Cimino A.; Minelli G. Chromia/Zirconia Catalysts with Cr Content Exceeding the Monolayer. A comparison with Chromia/Alumina and Chromia/Silica for Isobutane Dehydrogenation. Appl. Catal., A 1998, 167, 257–270. 10.1016/s0926-860x(97)00315-3. [DOI] [Google Scholar]
- Wischert R.; Laurent P.; Copéret C.; Delbecq F.; Sautet P. γ-Alumina: The Essential and Unexpected Role of Water for the Structure, Stability, and Reactivity of “Defect” Sites. J. Am. Chem. Soc. 2012, 134, 14430–14449. 10.1021/ja3042383. [DOI] [PubMed] [Google Scholar]
- Zhuravlev L. T. Concentration of Hydroxyl Groups on the Surface of Amorphous Silicas. Langmuir 1987, 3, 316–318. 10.1021/la00075a004. [DOI] [Google Scholar]
- Zhuravlev L. T. The Surface Chemistry of Amorphous Silica. Zhuravlev model. Colloids Surf., A 2000, 173, 1–38. 10.1016/s0927-7757(00)00556-2. [DOI] [Google Scholar]
- Das U.; Zhang G.; Hu B.; Hock A. S.; Redfern P. C.; Miller J. T.; Curtiss L. A. Effect of Siloxane Ring Strain and Cation Charge Density on the Formation of Coordinately Unsaturated Metal Sites on Silica: Insights from Density Functional Theory (DFT) Studies. ACS Catal. 2015, 5, 7177–7185. 10.1021/acscatal.5b01699. [DOI] [Google Scholar]
- Copéret C.; Comas-Vives A.; Conley M. P.; Estes D. P.; Fedorov A.; Mougel V.; Nagae H.; Núñez-Zarur F.; Zhizhko P. A. Surface Organometallic and Coordination Chemistry toward Single-Site Heterogeneous Catalysts: Strategies, Methods, Structures, and Activities. Chem. Rev. 2016, 116, 323–421. 10.1021/acs.chemrev.5b00373. [DOI] [PubMed] [Google Scholar]
- El Eter M.; Hamzaoui B.; Abou-Hamad E.; Pelletier J. D. A.; Basset J.-M. Well-defined Azazirconacyclopropane Complexes Supported on Silica Structurally Determined by 2D NMR Comparative Elucidation. Chem. Commun. 2013, 49, 4616–4618. 10.1039/c3cc40385h. [DOI] [PubMed] [Google Scholar]
- Guillemot G.; Thieuleux C.; Copéret C.; Soulivong D.; Spitzmesser S.; Basset J.-M. Modifying the Reactivity in the Homologation of Propane by Introducing Aryloxide Ligands on a Silica Supported Zirconium Alkyl System. Dalton Trans. 2007, 4589–4593. 10.1039/b708883c. [DOI] [PubMed] [Google Scholar]
- Calucci L.; Forte C.; Pampaloni G.; Pinzino C.; Renili F. Chemical Implantation of Group 4 Cations on Silica via Cyclopentadienyl- and N,N-dialkylcarbamato Derivatives. Inorg. Chim. Acta 2010, 363, 33–40. 10.1016/j.ica.2009.10.004. [DOI] [Google Scholar]
- Jezequel M.; Dufaud V.; Ruiz-Garcia M. J.; Carrillo-Hermosilla F.; Neugebauer U.; Niccolai G. P.; Lefebvre F.; Bayard F.; Corker J.; Fiddy S.; et al. Supported Metallocene Catalysts by Surface Organometallic Chemistry. Synthesis, Characterization, and Reactivity in Ethylene Polymerization of Oxide-Supported Mono- and Biscyclopentadienyl Zirconium Alkyl Complexes: Establishment of Structure/Reactivity Relationships. J. Am. Chem. Soc. 2001, 123, 3520–3540. 10.1021/ja000682q. [DOI] [PubMed] [Google Scholar]
- Popoff N.; Gauvin R. M.; De Mallmann A.; Taoufik M. On the Fate of Silica-Supported Half-Metallocene Cations: Elucidating a Catalyst’s Deactivation Pathways. Organometallics 2012, 31, 4763–4768. 10.1021/om3003224. [DOI] [Google Scholar]
- Millot N.; Soignier S.; Santini C. C.; Baudouin A.; Basset J.-M. Synthesis, Characterization, and Activity in Ethylene Polymerization of Silica Supported Cationic Cyclopentadienyl Zirconium Complexes. J. Am. Chem. Soc. 2006, 128, 9361–9370. 10.1021/ja060420+. [DOI] [PubMed] [Google Scholar]
- Salinier V.; Niccolai G. P.; Dufaud V.; Basset J.-M. Silica-Supported Zirconium Complexes and their Polyoligosilsesquioxane Analogues in the Transesterification of Acrylates: Part 2. Activity, Recycling and Regeneration. Adv. Synth. Catal. 2009, 351, 2168–2177. 10.1002/adsc.200900274. [DOI] [Google Scholar]
- Thornburg N. E.; Thompson A. B.; Notestein J. M. Periodic Trends in Highly Dispersed Groups IV and V Supported Metal Oxide Catalysts for Alkene Epoxidation with H2O2. ACS Catal. 2015, 5, 5077–5088. 10.1021/acscatal.5b01105. [DOI] [Google Scholar]
- Alladin T.; Beaudoin M. C.; Scott S. L. Thermolysis of Silica-Supported Bis(neopentyl) Complexes of Titanium and Zirconium. Inorg. Chim. Acta 2003, 345, 292–298. 10.1016/s0020-1693(02)01272-0. [DOI] [Google Scholar]
- Popoff N.; Espinas J.; Pelletier J.; Macqueron B.; Szeto K. C.; Boyron O.; Boisson C.; Del Rosal I.; Maron L.; De Mallmann A.; et al. Small Changes Have Consequences: Lessons from Tetrabenzyltitanium and -zirconium Surface Organometallic Chemistry. Chem.—Eur. J. 2013, 19, 964–973. 10.1002/chem.201202737. [DOI] [PubMed] [Google Scholar]
- Feller A.; Claeys M.; van Steen E. Cobalt Cluster Effects in Zirconium Promoted Co/SiO2 Fischer–Tropsch Catalysts. J. Catal. 1999, 185, 120–130. 10.1006/jcat.1999.2497. [DOI] [Google Scholar]
- Ali S.; Chen B.; Goodwin J. G. Zr Promotion of Co/SiO2 for Fischer-Tropsch Synthesis. J. Catal. 1995, 157, 35–41. 10.1006/jcat.1995.1265. [DOI] [Google Scholar]
- Zhang H.; Ma H.; Zhang H.; Ying W.; Fang D. Effects of Zr and K Promoters on Precipitated Iron-Based Catalysts for Fischer-Tropsch Synthesis. Catal. Lett. 2011, 142, 131–137. 10.1007/s10562-011-0739-3. [DOI] [Google Scholar]
- Morales G.; Osatiashtiani A.; Hernández B.; Iglesias J.; Melero J. A.; Paniagua M.; Brown D. R.; Granollers M.; Lee A. F.; Wilson K. Conformal Sulfated Zirconia Monolayer Catalysts for the One-pot Synthesis of Ethyl Levulinate from Glucose. Chem. Commun. 2014, 50, 11742–11745. 10.1039/c4cc04594g. [DOI] [PubMed] [Google Scholar]
- Dsouza L.; Jiao L.; Regalbuto J.; Miller J.; Kropf A. Preparation of Silica- and Carbon-Supported Cobalt by Electrostatic Adsorption of Co(III) Hexaammines. J. Catal. 2007, 248, 165–174. 10.1016/j.jcat.2007.03.010. [DOI] [Google Scholar]
- Kukli K.; Ritala M.; Leskelä M. Low-Temperature Deposition of Zirconium Oxide-Based Nanocrystalline Films by Alternate Supply of Zr[OC(CH3)3]4 and H2O. Chem. Vap. Deposition 2000, 6, 297–302. . [DOI] [Google Scholar]
- Rascón F.; Wischert R.; Copéret C. Molecular Nature of Support Effects in Single-Site Heterogeneous Catalysts: Silica vs Alumina. Chem. Sci. 2011, 2, 1449. 10.1039/c1sc00073j. [DOI] [Google Scholar]
- Kim W.; Yuan G.; McClure B. A.; Frei H. Light Induced Carbon Dioxide Reduction by Water at Binuclear ZrOCoII Unit Coupled to Ir Oxide Nanocluster Catalyst. J. Am. Chem. Soc. 2014, 136, 11034–11042. 10.1021/ja504753g. [DOI] [PubMed] [Google Scholar]
- Macnaughtan M. L.; Soo H. S.; Frei H. Binuclear ZrOCo Metal-to-Metal Charge-Transfer Unit in Mesoporous Silica for Light-Driven CO2 Reduction to CO and Formate. J. Phys. Chem. C 2014, 118, 7874–7885. 10.1021/jp5014994. [DOI] [Google Scholar]
- Di Carlo G.; Liotta L. F.; Pantaleo G.; Venezia A. M.; Deganello G. Alumina and Alumina-Baria Supported Cobalt Catalysts for DeNO x : Influence of the Support and Cobalt Content on the Catalytic Performance. Top. Catal. 2009, 52, 1826–1831. 10.1007/s11244-009-9360-8. [DOI] [Google Scholar]
- Lu A.; Chen Y.; Zeng D.; Li M.; Xie Q.; Zhang X.; Peng D.-L. Shape-related Optical and Catalytic Properties of Wurtzite-type CoO Nanoplates and Nanorods. Nanotechnology 2014, 25, 035707. 10.1088/0957-4484/25/3/035707. [DOI] [PubMed] [Google Scholar]
- Emeline A. V.; Kuzmin G. N.; Basov L. L.; Serpone N. Photoactivity and photoselectivity of a dielectric metal-oxide photocatalyst (ZrO2) probed by the photoinduced reduction of oxygen and oxidation of hydrogen. J. Photochem. Photobiol., A 2005, 174, 214–221. 10.1016/j.jphotochem.2005.03.011. [DOI] [Google Scholar]
- Fernández López E.; Sánchez Escribano V.; Panizza M.; Carnasciali M. M.; Busca G. Vibrational and Electronic Spectroscopic Properties of Zirconia Powders. J. Mater. Chem. 2001, 11, 1891–1897. 10.1039/b100909p. [DOI] [Google Scholar]
- Gionco C.; Paganini M. C.; Giamello E.; Burgess R.; Di Valentin C.; Pacchioni G. Paramagnetic Defects in Polycrystalline Zirconia: An EPR and DFT Study. Chem. Mater. 2013, 25, 2243–2253. 10.1021/cm400728j. [DOI] [Google Scholar]
- McAlpin J. G.; Surendranath Y.; Dincǎ M.; Stich T. A.; Stoian S. A.; Casey W. H.; Nocera D. G.; Britt R. D. EPR Evidence for Co(IV) Species Produced During Water Oxidation at Neutral pH. J. Am. Chem. Soc. 2010, 132, 6882–6883. 10.1021/ja1013344. [DOI] [PubMed] [Google Scholar]
- Jenkins D. M.; Di Bilio A. J.; Allen M. J.; Betley T. A.; Peters J. C. Elucidation of a Low Spin Cobalt(II) System in a Distorted Tetrahedral Geometry. J. Am. Chem. Soc. 2002, 124, 15336–15350. 10.1021/ja026433e. [DOI] [PubMed] [Google Scholar]
- Malatesta V.; McGarvey B. R. Single Crystal Electron Spin Resonance Of Low Spin Co(II) And Of Cu(II) Schiff Base Complexes. Can. J. Chem. 1975, 53, 3791–3800. 10.1139/v75-548. [DOI] [Google Scholar]
- Niklas J.; Mulfort K. L.; Rakhimov R. R.; Mardis K. L.; Tiede D. M.; Poluektov O. G. The Hydrogen Catalyst Cobaloxime – a Multifrequency EPR and DFT Study of Cobaloxime’s Electronic Structure. J. Phys. Chem. B 2012, 116, 2943–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarvey B. R. Theory of the Spin Hamiltonian Parameters for Low Spin Cobalt(II) Complexes. Can. J. Chem. 1975, 53, 2498–2511. 10.1139/v75-355. [DOI] [Google Scholar]
- Nishida Y.; Kida S. Splitting of d-orbitals in square planar complexes of copper(II), nickel(II) and cobalt(II). Coord. Chem. Rev. 1979, 27, 275–298. 10.1016/s0010-8545(00)82069-x. [DOI] [Google Scholar]
- Ernst B.; Bensaddik A.; Hilaire L.; Chaumette P.; Kiennemann A. Study on a Cobalt Silica Catalyst during Reduction and Fischer-Tropsch Reaction: In situ EXAFS Compared to XPS and XRD. Catal. Today 1998, 39, 329–341. 10.1016/s0920-5861(97)00124-7. [DOI] [Google Scholar]
- Gilardi E.; Gregori G.; Wang Y.; Sigle W.; van Aken P. A.; Maier J. Interface Effects on the Ion Transport of Epitaxial Y2Zr2O7 Films. ACS Appl. Mater. Interfaces 2017, 9, 27257–27265. 10.1021/acsami.7b00773. [DOI] [PubMed] [Google Scholar]
- Levy M.; Fouletier J. Influence of Dopant Concentration on the Electronic Conductivity of Nonstoichiometric Yttria-Doped Ceria. Solid State Ionics 1984, 12, 467–472. 10.1016/0167-2738(84)90179-6. [DOI] [Google Scholar]
- Kimpton J.; Randle T. H.; Drennan J. Investigation of electrical conductivity as a function of dopant-ion radius in the systems Zr0.75Ce0.08M0.17O1.92 (M=Nd, Sm, Gd, Dy, Ho, Y, Er, Yb, Sc). Solid State Ionics 2002, 149, 89–98. 10.1016/s0167-2738(02)00136-4. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.