

Abstract
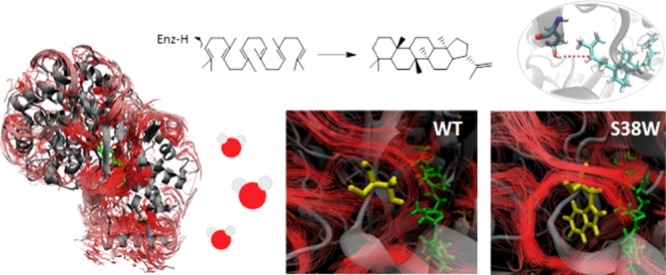
Squalene–hopene cyclase catalyzes the cyclization of squalene to hopanoids. A previous study has identified a network of tunnels in the protein, where water molecules have been indicated to move. Blocking these tunnels by site-directed mutagenesis was found to change the activation entropy of the catalytic reaction from positive to negative with a concomitant lowering of the activation enthalpy. As a consequence, some variants are faster and others are slower than the wild type (wt) in vitro under optimal reaction conditions for the wt. In this study, molecular dynamics (MD) simulations have been performed for the wt and the variants to investigate how the mutations affect the protein structure and the water flow in the enzyme, hypothetically influencing the activation parameters. Interestingly, the tunnel-obstructing variants are associated with an increased flow of water in the active site, particularly close to the catalytic residue Asp376. MD simulations with the substrate present in the active site indicate that the distance for the rate-determining proton transfer between Asp376 and the substrate is longer in the tunnel-obstructing protein variants than in the wt. On the basis of the previous experimental results and the current MD results, we propose that the tunnel-obstructing variants, at least partly, could operate by a different catalytic mechanism, where the proton transfer may have contributions from a Grotthuss-like mechanism.
Introduction
Terpenes comprise the most diversified class of natural products of high importance for life.1 The key to the myriad of observed multicyclic products is a terpene cyclase-catalyzed carbocationic polycyclization cascade of a prefolded linear polyisoprene substrate that is chaperoned in essentially one reactive conformation. The stereospecific terpene cyclization in the hydrophobic active site is initiated by either phosphate release (class I mechanism) or protonation of the terminal isoprene or oxirane (class II mechanism), which yields a carbocation that propagates in the molecule upon ring closure through carbon–carbon bond formation.2 The final carbocation is quenched by either deprotonation or water addition depending on the availability of a suitable base and the spatial organization of water in the active site.3
The cyclization step and subsequent product modification by tailoring enzymes determine the bioactivities of terpenes that range from mediating cell membrane rigidity, defense, to signaling.4,5 Triterpenes constitute one important class of terpenes with applications as medicines and fine chemical synthons.6 The potent antibacterial, anticancer, and antiviral bioactivities displayed by some cyclic terpenes,6−8 as well as their high synthetic potential as new biofuels,9,10 have spurred increasing interest from the biotechnological and chemical industries.
Stereospecific carbon–carbon bond formation is of high importance in synthetic organic chemistry and biocatalysis.11,12 Terpene cyclases are of high synthetic interest13−15 as they enable stereospecific polycyclizations that are challenging to achieve by synthetic chemistry.6 Therefore, an enhanced understanding of the reaction mechanism displayed by these fascinating enzymes would be beneficial for chemical and biotechnological industries. Squalene–hopene cyclase (SHC) has served as a model system to enhance our understanding of triterpene biosynthetic machineries. SHC converts the linear molecule squalene to the pentacyclic triterpenes hopene (Figure 1) and hopanol.
Figure 1.

Conversion from the linear squalene (A) to a prefolded geometry (B) in the active site of SHC in the polycyclization reaction to hopene (C).
SHC is a monotopic enzyme16 with a hydrophobic channel toward the membrane helix that allows for acquisition of the hydrophobic substrate. The membrane-binding region of the enzyme is highly lipophilic, and it is submerged in the nonpolar region of the membrane (Figure 2).
Figure 2.

Structure and membrane association of squalene–hopene cyclase from Alicyclobacillus acidocaldarius (PDB entry 1UMP(17)). (A) The prefolded squalene (green, stick) is shown in the active site. The membrane-binding helix is shown in red. (B) The protein inserted into a membrane patch, the substrate is located in the hydrophobic channel (green, stick).
The substrate is guided into the prefolded conformation for ring closure when entering the active site, and the shape of the active site stabilizes the substrate in a prefolded arrangement for the polycyclization to take place. The cyclization cascade is then initiated by protonation of a terminal isoprene double bond from the catalytic residue Asp376 (Figure 1), which yields a carbocation that is stabilized from premature quenching by hydrophobic side chains in the active site.14 The pKa of Asp376 is modulated by a hydrogen bond to the protonated His451 and by the close vicinity to the negatively charged Asp374:Asp377 pair.18 The latter has also been postulated to stabilize the protonated substrate.19
In a study performed by Schwab et al.,20 a higher mobility of the membrane-binding region was identified when a ligand was bound to the active site. However, there was no significant change of the narrowest part of the hydrophobic channel, either in fluctuations or the absolute structure.
The polycyclization reaction is highly exergonic and the formation of hopene theoretically releases about 48 kcal/mol.20 It has been suggested that this energy release affects the structure of the protein in such a way that the hydrophobic channel is expanded enough to accommodate the bulky product.17 Others have advocated that energy is released and dissipated much faster than the timescale of the necessary conformational changes.20,21
A previous study by Syrén et al. detected a network of tunnels leading from the active site to the surface of the protein through an in silico CAVER22 analysis.23 It was suggested that expulsion of water molecules from the active site to the bulk could compensate for the entropic cost of prefolding the substrate in one reactive conformation.23 The identified highest ranked tunnels (from CAVER) were obstructed with rational in silico protein design by introducing bulky amino acids at selected positions. In vitro experimental kinetic characterization showed that the variants display significantly altered temperature dependencies of catalysis and entropies and enthalpies of activation compared to those of the wild type (wt), suggesting the importance of unobstructed water flow for the performed catalytic reaction (Table 1). All variants, except for the control mutation, showed decreased activation enthalpy (ΔH‡) and activation entropy (ΔS‡) compared to those of the wt. However, the exact mechanism by which these mutations affect the cyclization reaction remained unknown.
Table 1. Kinetic Experimental Data from Syrén et al.23 Displaying Differences in Entropic (TΔS‡) and Enthalpic (ΔH‡) Contributions to the Activation Free Energy (ΔG‡) between the Different Protein Variantsa.
| protein variant | wt | S38W | S168F | S168W | F605W |
|---|---|---|---|---|---|
| TΔS‡ (kcal/mol) | 16 | –41 | –16 | –13 | –7.1 |
| ΔH‡ (kcal/mol) | 31 | –23 | –0.75 | 4.4 | 7.1 |
| ΔG‡ (kcal/mol) | 15 | 18 | 15 | 17 | 14 |
| (kcat/KM)H/(kcat/KM)D | 2.0 | 1.9 | 1.6 | 1.0 | 2.1 |
Data is given for apparent kcat/KM at 328 K and pH 6. Deuterium kinetic isotope effects (DKIEs) [(kcat/KM)H/(kcat/KM)D] are taken from the Supporting Information of ref (23).
The impact of water within enzyme structures has been shown to have important functions in the catalytic activities of enzymes.24,25 It has been found that water molecules can improve the steric fit of ligand binding, mediate hydrogen bonds between enzyme and active site, and affect interaction energies.24 Product release can be facilitated by refilling the active site by a flow of water molecules from the bulk solution upon product release.26 It has previously been suggested that a network of water molecules within tunnels that connect the active site to the bulk water on the outside of the protein might be a general feature of all enzymes to enable protonation, as well as rehydrating and dehydrating the active site during product release and substrate binding.27 In a study by Schiebel et al.,27 mutations were introduced in water channels in saFAbI (Staphylococcus aureus enoyl-acyl carrier protein reductase) and were found to affect the catalytic efficiency of the enzyme. These mutations were suggested to hinder the flow of water out from the active site. It was additionally argued that because the mutations are at a distance from the active site, it is more likely to be global processes that are disturbed than local processes in the active site.
The aim of the current study was to further investigate the previously suggested water tunnels in SHC23 to study the movements of water molecules between the active site and the surface of SHC in more detail, and to analyze how the introduction of bulky amino acids, predicted to block the water tunnels, affect the flow of water molecules in the active site. Therefore, we investigated the dynamics of the tunnels and water flow in the active site during sub-microsecond MD simulations of the wt as well as different protein variants.
The tunnel-blocking variants were found to have an increased flow of water around the catalytic residue Asp376 in the active site compared to that in the wt. The higher water flow is associated with a larger distance for the proton transfer between Asp376 and the substrate for the rate-determining proton transfer step of the catalytic reaction. However, there is no correlation between the protonation distance and the activation free energy, and we propose that the designed protein variants function through a different mechanism to that of the wt; one that at least partly could be dependent on Grotthuss-like proton transfer.
Results and Discussion
The stability of the protein structure without a membrane has previously been established in a simulation by Schwab et al.20 However, a recent study of membrane protein has indicated that both membrane and protein need to be considered explicitly to reproduce the hydration dynamics of the system.28 Therefore, the membrane was included in our model to account for potential contributions from water associated to the membrane. The protein structures (both 1UMP and 2SQC) were found to be stable for the full-length simulation at 330 K in water (see SI for root-mean square deviation (RMSD) plots) with and without the membrane patch.
Visual inspection of the protein structures superposed on top of the wt shows that the protein structures stay intact, and do not change significantly over the course of the simulations, for any of the protein variants. An analysis of the RMSDs was performed on averaged structures from the trajectories. The RMSD values for the backbone of the mutated protein variants compared to that of the wt were S38W: 0.510 Å; S168F: 0.385 Å; S168W: 1.270 Å; F605W: 0.405 Å; and V440F: 0.509 Å. The mutations do not introduce any major visible changes in the protein three-dimensional structure, as is depicted in Figure 5.
Figure 5.

Streamline analysis showing the flow of water (red lines) between the bulk solution and the active site. (A) Streamlines through the whole protein with the substrate in the active site. (B) Streamlines visualized through a clipping plane through the empty protein cut in half (protein not included for visualization).
Protein Flexibility
An analysis of the B-factors for each amino acid residue in the protein detected only small variations in the flexibility between the different variants. However, some parts of the enzyme are more flexible than others. Long distance effects of introduced mutations on the mobility of protein loops have previously been discussed.29
The membrane-binding region (residues 216–234) was found to display high B-factors and thus seems to be flexible in comparison to other parts of the enzyme (Figure 4). This is in agreement with a study by Wendt et al.,30 where the membrane-binding helix was described as the most mobile region, based on the variations of the B-factors from the crystal structure. There were some differences in the membrane-binding helix between the different protein variants (Figures 3 and 4). For one of the variants, F605W, the helix is slightly twisted compared to those of the other variants (Figure 3). Interestingly, this variant was the fastest at all studied temperatures among the variants analyzed by Syrén et al. (Table 1). The rate increase of F605W is probably connected to other factors, but, at least, the conformational change does not seem to have a negative impact on the catalytic conversion. In all variants, the N- and C-terminal regions show high flexibility, as could be expected for terminal regions of the protein chain.
Figure 4.

Investigated variants show small variations in B-factors. B-factors from MD simulations are given for the wt (red) and protein variants (blue). For reference, wt data is plotted in each subplot. One highly mobile region of the protein is constituted by the residues around the membrane-binding region (residues 216–234). The analysis shown was performed on the last 100 ns of the 150 ns trajectory.
Figure 3.

(A) Wt (blue) superposed onto the mutated protein variants S38W (yellow), S168F (orange), S168W (red), F605W (cyan), and V440F (purple). The structures used for generation of the figure are averaged structures from the trajectories. Squalene is indicated in green. The membrane patch is not visualized in the following images for clarity, but was included in all simulations. (B) A close-up of the protein backbone structure for the different protein variants in the active site. Squalene is indicated in green.
Previous studies have shown that there is a relationship between the activity of an enzyme and the rigidity of the residues in the active site. Some flexibility of the residues in the active site is necessary for the enzyme to perform its catalytic function throughout the catalytic cycle.31 An increased stiffness of residues has been associated with a decrease of activity for the barnase enzyme.32 Focusing on the flexibility of the amino acids in the DXDD motif (Asp374, Asp376, and Asp377), which is involved in the initial protonation step,33 some minor differences in B-factors can be noted (Table S2 in SI), but the changes do not follow the trends of the kinetic behavior of the experimental results. Thus, our analysis indicates that the variations in B-factors of the DXDD motif cannot explain the measured experimental differences between the mutated variants for this enzyme.
Substrate Entrance and Product Release
The timescales of substrate entrance and product release were investigated over the MD trajectories. Simulations with the substrate in the hydrophobic channel (Figure 2) show that the substrate can move through the channel during this time frame. In simulations with the substrate located in the hydrophobic channel at the start of the simulation, the substrate leaves the channel and moves into the membrane.
Wendt et al. suggested that the product leaves through the same channel as that through which the substrate enters.19 There are two mobile loops attached to the membrane, and the required widening of the channel could occur by membrane fluctuations.30 However, in this study, the product does not leave the active site during the simulation (even when extended to 180 ns). It is likely that the hydrophobic channel needs to expand to accommodate the bulky pentacyclic hopene for the transport from the active site to the membrane. To simulate the heat that is released during the reaction, MD was also run at a higher temperature (350 K) to allow for a possible energy-dependent widening of the channel. However, the product does not leave the active site during the course of the simulation at elevated temperature. It is difficult to assign the significance of this result, because the higher temperature may not reproduce the effect of the energy dissipation, and any associated conformational change, that results from the energy release from the reaction. Furthermore, the timescale of the simulation may not be sufficient for the event to take place.
Analysis of Water Tunnels
Visual inspection of the trajectories shows that no stationary water molecules exist in the active site, or in close proximity to the active site on the timescale of 0.1 μs, independently of whether the protein is empty, has the substrate in the hydrophobic channel or active site, or the product is in the active site. There is also a spontaneous exchange of water molecules between the protein interior and the bulk water.
The tunnels identified in the CAVER analysis (data not shown) clearly display a dynamic behavior, and the tunnels open and close over the time frame of these simulations. Because CAVER analysis does not reveal if there are water molecules moving or residing in these tunnels, an additional streamline analysis was performed.
Streamline Analysis
A streamline analysis performed on the SHC wt shows that water moves continuously through certain regions of the protein (Figure 5) when simulated in a water box. The most intense water flow takes place in the region in and around the active site. The streamline is a curve that is at any point tangent to the velocity (or flow) field. The streamline describes the path of a massless particle in a flow field. The flow field in our work is constructed from the direction and magnitude of the fastest water diffusion component in each volume cell. The diffusion parameters are obtained by analyzing and averaging the motions of all water molecules throughout the time frame of a simulation, as described in the SI. Thus, each streamline represents the most probable path of a water molecule in a simulation. For clarity, we show only streamlines originating in the cells with a magnitude and anisotropy of diffusion higher than the predefined threshold.
Interestingly, the streamline analyses of the (i) empty protein, (ii) protein with prefolded substrate, (iii) protonated substrate, and (vi) product show that water molecules are moving in and out of the active site during the entire modeled reaction sequence independent of active site occupation. Figure 6 shows the flow of water in the active site of the empty protein as well when it contains substrate, protonated substrate, and product. The flow of water is highest when the active site is empty, and the lowest flow is observed in the presence of the bulky product.
Figure 6.

Streamlines in (A) empty protein, (B) protein with substrate (green) in the active site, (C) protonated substrate (green), and (D) protein with product (blue) in the active site.
The rate-determining step of this reaction is the initial protonation of the substrate, as indicated by a significant deuterium kinetic isotope effect.23 Interestingly, the streamline analysis shows a difference in the flow of water between the systems with the protonated and unprotonated substrate in the active site (Figure 6).
Syrén et al.,23 on the basis of their CAVER analysis, introduced mutations to block the tunnels and thereby hypothetically hinder the flow of water to and from the active site. The temperature-dependent kinetic properties of these protein variants were determined experimentally and analyzed by means of Eyring transition state theory (Table 1). It was found that all tunnel-blocking mutations resulted in a lowering of the activation enthalpy (ΔH‡) and a lowering of the activation entropy (ΔS‡) compared to those of the wt, whereas some variants were faster and some slower than the wt at the normal reaction temperature (328 K).
To investigate the impact of the introduced mutations on the global water dynamics, a streamline analysis was initially performed with the S38W variant. Introducing a tryptophan at this tunnel lining position obstructs one of the (by CAVER) highest ranked tunnels in the protein,23 which is associated with profound experimental effects on the temperature dependence, and with that the activation parameters of catalysis (Table 1).23 Changing S38 to a tryptophan in silico forces water to move around the side chain of the bulkier tryptophan, which further impacts the motion of water molecules in the solvated active site (Figure 7).
Figure 7.
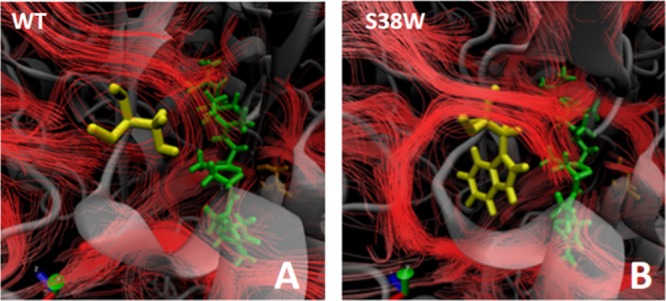
Streamline analysis on (A) the wt and (B) S38W, indicating the flow of water in the active site. The substrate (squalene) is indicated in green, and S38 (A) and S38W (B) in yellow. The flow of water is altered near the introduced mutation.
The streamline analysis was subsequently performed on all of the protein variants; Figure 8 displays the flow of water in the active site for the different protein variants. As the activation parameters (ΔH‡, ΔS‡, and ΔG‡) of the different protein variants were found to differ in the experimental study,23 we focused the analysis on the region in the active site where the rate-determining step occurs, i.e., around the catalytic residue Asp376 and the substrate, as shown in Figure 8.
Figure 8.

Streamlines around the substrate and the catalytic residue Asp376 (blue) in the wt, S38W, S168F, S168W, F605W, and V440F. The substrate (green) stays in the active site during the simulation. Each mutated residue is also indicated in each subplot. Gray: protein backbone.
The streamline analysis of the wt and variants (Figure 8) shows that there are changes in the flow of water in the active site. In particular, there is an increased flow of water between the substrate and the catalytic residue Asp376 in S38W, S168F/S168W, and F605W compared to that in the wt and V440F. This is surprising, considering that the mutations were designed to hinder the flow of water in the tunnels. The streamline analysis indeed shows that the water paths in the vicinities of the mutated residues are altered, leading to changes in active site solvation.
Analysis of Diffusion Constants in the Active Site
The streamline analysis images (Figure 8) confirm that there are more water molecules moving in the region between the catalytic residue and the substrate in the mutated protein variants (except for control mutation V440F). An investigation of the distribution of the diffusion constants in this region around the catalytic residue shows further differences between the wt and protein variants. Figure 9 shows how the distributions of diffusion coefficients change for the wt and each protein variant in a 5.5 Å region around Asp376.
Figure 9.
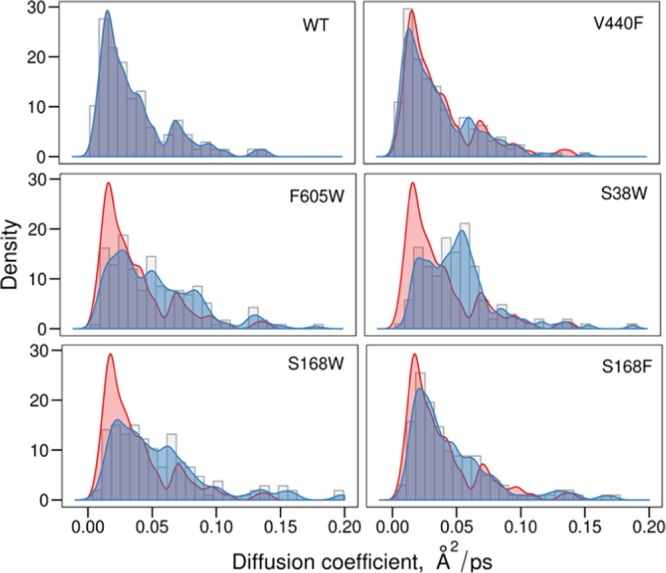
Distribution of diffusion coefficients in a 5.5 Å region around the catalytic residue for the wt and protein variants (blue). The distribution of the wt is also inserted (red) for facilitated comparison with each protein variant.
From Figure 9, it can be concluded that the wt and control mutation (V440F) have generally lower diffusion in the investigated region compared to that of the mutated protein variants F605W, S168F/W, and S38W. Although the Gibbs free energy of activation varies among the mutated variants, they all have in common that they have a much lower activation entropy than that of the wt (Table 1). The S38W variant, which has its peak for the distribution most shifted toward the higher diffusion coefficient, has the most negative activation entropy. This is also the protein variant with the highest Gibbs free energy barrier at 328 K.
The introduced mutations do not only alter the location of the water streams in the active site, but also the diffusion in the catalytic region. Looking at the median values for the diffusion coefficients in Figure 12, the trend is sustained independently of the chosen cut-off region around Asp376 for the different protein variants. There is generally a lower flow in the active site of the wt and control mutation (V440F) compared to that of the other protein variants (Figure 10). This is an interesting observation that shows how altered water access pathways by enzyme design induce a reconfigured water flow.
Figure 12.

Protonation distance for the wt and protein variants. The dotted black lines in all figures are the average value for the wt. The averages for each variant are stated in the legend of each subplot.
Figure 10.
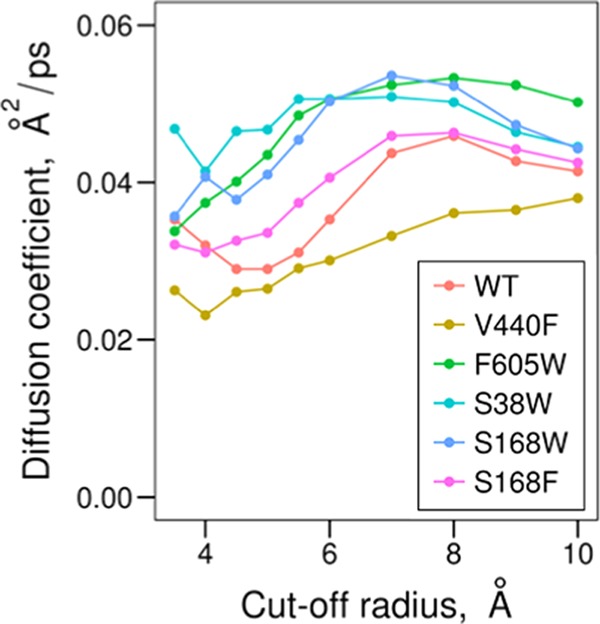
Median values for the diffusion coefficients at different cut-off radius values around the catalytic residue for the different protein variants.
Amount of Water around the Catalytic Residue and the Protonation Distance
An analysis of the number of water molecules within 5 Å of the catalytic residue Asp376 over the course of the simulation confirms that the wt and control mutation (V440F) on average have fewer water molecules surrounding the substrate than the designed variants (Figure 11). The average number of water molecules within 5 Å of Asp376 is also higher in the variants as shown in Figure 11.
Figure 11.

Number of water molecules within 5 Å of the catalytic residue Asp376 over the trajectory. The dotted black lines in all subplots are the average value for the wt. The averages for each variant are stated in the legend of each subplot.
The water molecules in the active site were hypothesized to affect the rate-determining proton transfer; if there are water molecules between the catalytic residue and the terminal isoprene group, the direct proton transfer from Asp376 to the substrate will be hindered. To quantify this effect, the distance between the acidic proton of Asp376 and the carbon on the substrate that receives the proton was compared for the different variants (Figure S2 in SI). We will refer to this distance as the “protonation distance”, although the actual distance of proton transfer is shorter by ca. 1.1 Å (the typical distance of a C–H bond).
The effect of the distance between the catalytic residue and the terminal isoprene group of the substrate on the catalytic efficiency of SHC has previously been investigated on the basis of shorter MD simulations.34,35 Hammer et al. identified a correlation between the fraction of structures with a protonation distance of 2.9 Å or shorter and the catalytic activity for different unnatural substrates.35 However, a similar correlation for the natural substrate and the different variants could not be found herein. The variations in the protonation distance along MD trajectories for the different protein variants are displayed in Figure 12.
The different plots of Figure 12 show that the protonation distance on average is shorter in the wt and the control mutation V440F than in the other variants. Furthermore, the figure clearly demonstrates the importance of a longer simulation time for this type of analysis. The first 20–30 ns are not representative of the state that the system occupies for the remaining simulation time in the majority of these trajectories (V440 changes its behavior after a longer time, ca. 80 ns). Thus, in the subsequent analysis we removed the first 50 ns and used the last 100 ns for statistics.
Table 2 indicates a qualitative correlation between the average number of water molecules around Asp376 and the average protonation distance for the different variants. Thus, it seems that on average, the larger protonation distance in the designed variants is due to intercalation of water between Asp376 and the substrate.
Table 2. Average Value of Number of Water Molecules and Average Protonation Distance between the Catalytic Residue Asp376 and the Substrate for the wt and Protein Variants, the Fraction of Structures Identified as Near Attack Conformers (NACs), and the Corresponding ΔGNACa.
| average number of water molecules around Asp376 | average protonation distance (Å) | fraction of structures (pNAC in %) with a protonation distance shorter than 3.0 (3.5) Å | ΔGNAC (kcal/mol) for a protonation distance shorter than 3.0 (3.5) Å | |
|---|---|---|---|---|
| wt | 2.9 | 4.29 | 0.04 (4.03) | 5.2 (2.2) |
| S38W | 4.0 | 4.88 | 0.01 (0.63) | 6.2 (3.4) |
| S168F | 3.2 | 5.45 | 0.00 (0.24) | >6.2 (4.0) |
| S168W | 3.1 | 5.29 | 0.00 (0.42) | >6.2 (3.7) |
| F605W | 4.0 | 5.37 | 0.00 (0.05) | >6.2 (5.1) |
| V440F | 2.6 | 3.67b | 2.21b (41.88)b | 2.6b (0.6)b |
The average values were calculated from the last 100 ns of the simulation trajectories.
On the basis of the concept of near attack conformers (NAC),36,37 the free energy of activation for the catalytic reaction has a contribution from the free energy of NAC-formation (ΔGNAC), i.e., the system has to pass a NAC before it can reach the transition state, and the free energy cost of reaching the NAC is a fraction of the total activation free energy. ΔGNAC is computed from the fraction of structures labeled as NACs (pNAC) by ΔGNAC = −RT ln(pNAC). We have previously shown38,39 that ΔGNAC can be used in enzyme design to rank the catalytic efficiencies of different protein variants for a particular reaction. Herein, we define the NAC as a structure where the distance between the acidic hydrogen of Asp376 and the accepting carbon of the substrate is within the sum of the van der Waals radii of the interacting atoms. Hammer et al. used a similar definition in their study of unnatural substrates, but they used the term transition state structure instead of NAC to denote the relevant structures.35
Studies of interactions in solid crystals have found that the mean non-bonding H–C distance of organic compounds is close to 3.0 Å, and we used this distance as an estimate of the H–C van der Waals distance and the NAC criterion in our analysis (the sum of the Bondi van der Waals radii of H and C gives 2.9 Å).40 However, as this criterion gives a very low NAC frequency for the protein variants that results in low precision, we have, for comparison, also used a slightly longer H–C distance (3.5 Å) as the NAC criterion (Table 2).
The relatively short average protonation distance and the relatively high frequency of NACs reported in Table 2 for the wt and the control mutant are consistent with a direct proton transfer between Asp376 and the substrate. On the other hand, the tunnel-blocking variants consistently have a larger average protonation distance and a much lower, in most cases negligible, NAC frequency; observations that seem to preclude a direct proton transfer mechanism in these variants. There is no overall correlation between ΔGNAC and the free energy of activation for the catalytic reaction because some of the designed variants are faster than the wt and others are slower at 328 K. On the other hand, a low NAC frequency was found to be associated with a negative activation entropy; the designed variants in all cases have a negative activation entropy whereas it is positive for the wt. One plausible explanation for these observations is that the tunnel-obstructing variants operate to some extent by a different mechanism than that of the wt. It can be noted that using the longer H–C distance as the NAC criterion, as expected, decreases ΔGNAC, but the relative difference in ΔGNAC (ΔΔGNAC) between variants shows similar variations as with the shorter H–C distance. Thus, the qualitative interpretation of the results is not sensitive to the NAC criterion.
We can speculate that the variants, in addition to direct proton transfer, can operate via a Grotthuss-like mechanism where the transfer of a proton from Asp376 to the substrate is mediated via one or more adjacent water molecules. Such a mechanism will lead to a lower activation entropy as it will require the positioning of a water molecule during the proton transfer, but it could reduce the activation enthalpy, and in principle, also the activation free energy. With the exception of S168W (where the deuterium kinetic isotope effect (DKIE) was 1.0), the observed DKIE of the wt and the variants lies in the range 1.6–2.1 (Table 1); a DKIE of this size is consistent with both a direct proton transfer or a Grotthuss-like proton transfer, and thus the DKIE does not provide a method to distinguish between the two mechanisms.41,42 In this context, it should be mentioned that a previous study reported an altered reaction mechanism by introducing mutations in a tunnel in haloalkane dehalogenase LinB.43
The introduced mutations could also affect the pKa of Asp376, and thereby indirectly affect the reaction rate. The most plausible reasons behind a pKa change are a change in the solvation of Asp376 or in the positions of the surrounding residues; in particular, the positions of His451, Asp374, and Asp377 and their protonation states are expected to be important for the pKa of Asp376. However, it should be noted that there is not likely to be a simple relationship between the pKa of Asp376 and the reaction rate. The barrier for proton transfer from Asp376 will decrease with decreasing pKa, but if the pKa is much lower than pH, only a tiny fraction of Asp376 will be protonated and the reaction rate will decrease significantly (the reaction rate is expected to reach a maximum at a pH close to the pKa).44 Furthermore, the barrier is not only dependent on the pKa but also on the distance that the proton has to be transferred, and mutations that change the pKa may simultaneously change this distance. Still, it would be very interesting to experimentally investigate how the activation parameters and the DKIE of the wt and the variants vary with the pH. Unfortunately, computations of pKa values of systems of this complexity are still affected with too great uncertainties to be directly applicable.
Water molecules have previously been recognized to play several different important roles in cell biology.45 They impact the binding of ligands to receptors and can determine ligand selectivity.46 A key role of water in the reprotonation of catalytically active amino acids has been acknowledged, and Wendt et al. proposed that the protonation of the catalytic acid in squalene–hopene cyclase from A. acidocaldarius likely occurs through a tunnel, identified in the crystal structure,30 linking the catalytic aspartate with the bulk solvent via a chain of six hydrogen-bonded water molecules. Mutations of similar proton channels that mediate solvent-assisted “Grotthuss-like” proton transfers were shown to affect the catalytic activity of other enzymes, such as the enoyl-acyl carrier protein reductase from S. aureus, where the catalytic efficacy was decreased 10000-fold in the water channel variants compared to that in the wt.27
Moreover, water molecules are prone to intercepting highly-reactive intermediates to tune product distribution, which is of high relevance for terpene cyclases that form several carbocations in their catalyzed polycyclization reactions. A structural role of water molecules in terpene cyclase active sites has been discussed in light of a sesquiterpene cyclase from Aspergillus terreus. Water molecules lining the hydrophobic active site were suggested to direct product distribution by contributing to prefolding of the substrate.47
This study has provided an enhanced understanding of active site solvation throughout the reaction sequence of SHC. It has also shown how the introduced mutations, which were designed to obstruct the water paths leading to the active site, change the flow of water in the active site. The importance of water dynamics has also been studied recently by David et al.;48 they introduced mutations in water channels that altered the dynamics of water access to the active site.
Surprisingly, the enzyme variants with obstructed water tunnels enable an increased flow of water in the active site by forcing water molecules to change path. This leads to an increase in the distance the proton has to travel in the rate-determining proton transfer step of the catalytic reaction. These variants, in contrast to the wt, all have a negative activation entropy, whereas their activation free energy varies, and for some variants, it is lower than that for the wt at 328 K.
We suggest that these observations can be explained by changes in the protonation step of the reaction mechanism in the variants, which may partially operate by a Grotthuss-like proton transfer. The SHC system seems to be very complex, and the previously identified changes in activation parameters are likely to arise from a combination of actions in the enzyme, and probably not solely from these identified changes in water flow with the associated altered protonation distance. It is possible that the introduced mutations also affect other global processes not investigated in this study. Further investigations will be necessary to fully understand the complex catalytic mechanism of SHC. In particular, we foresee that an investigation on how changes in pH affect the activation parameters and the deuterium kinetic isotope effect of the wt and variants can help elucidate variations in the mechanism.
Conclusions
Water molecules were found to move continuously between the bulk solution and the active site through specific paths in the protein; there are water molecules in the active site before the entering of the substrate and throughout the reaction. Protein variants previously designed to obstruct water paths leading to the active site by the introduction of bulky side chains were found to change the water path locally at the substitution site. Interestingly, these variants were found to be associated with an increased flow of water in the active site, particularly in the vicinity of the catalytic amino acid Asp376. Simulations with the substrate present in the active site indicate that the distance for the rate-determining proton transfer between Asp376 and the substrate is longer in the tunnel-obstructing protein variants than that in the wt and the control mutation V440F. However, the NAC analysis shows no correlation between the protonation distance and the activation free energy of the catalytic reaction at the simulation temperature. The common denominator for the tunnel-obstructing variants is that they have a negative activation entropy, whereas it is positive for the wt. On the basis of the observations of this study, we speculate that these variants could operate by a Grotthuss-like proton transfer in the rate-determining step, which is consistent with both the negative activation entropy and a larger protonation distance. Further experimental and theoretical studies will be needed to increase the mechanistic understanding of this complex enzyme.
Methods
To investigate patterns of water movement in the protein, MD simulations were conducted with AMBER49 on the bacterial SHC from A. acidocaldarius (PDB entries: 2SQC(30) and 1UMP(17)). The monomeric form (chain A) of the protein was selected for in silico analysis. The SHC structure has been reported as a dimer30 in its native form, but it has been suggested that the dimerization might be of transient nature.20
Both crystal structures 2SQC and 1UMP contain an analogue of the substrate crystallized with the protein. The analogue is either localized in the channel in (2SQC) or in the active site (1UMP). The analogue molecules were replaced by squalene and hopene molecules (Figure 1) in preparation for the MD simulations.
The monotopic membrane protein SHC was attached to an 80 Å × 80 Å palmitoyl oleoyl phosphatidylcholine (POPC) membrane, constructed in visual molecular dynamics50 (with charmm36 parameters). The binding residues (residues 216–234) were manually inserted into the membrane (Figure 2). All molecules (both POPC and associated water molecules) from the membrane that were within 3 Å of the protein were removed to avoid steric clashes.
The water molecules from the crystal structure were retained, and additional TIP3P water molecules were added by Leap51 in a box extending 10 Å around the edge of the protein–membrane complex. In total, about forty thousand water molecules were included in the simulation box.
Introduction of Mutations
Mutations were introduced manually by replacing the affected residue in each PDB-file, and processing each file with Leap, to investigate whether introduction of bulky amino acids in place of the native residue with a small side chain changes the flow of water through the previously identified tunnels23 in the protein.
Protein variants included mutations of S38, S168, and F605 to tryptophan and phenylalanine (S38W, S168F, S168W, and F605W) that block three of the previously identified tunnels23 in the enzyme. In addition, we introduced a control mutation V440F (Figure 13). V440 is considered to be neither in the active site nor in a tunnel, and the kinetic behavior of the V440F mutant is similar to that of the wt.
Figure 13.

(A) Location of introduced mutations S38 (yellow), S168 (orange), F605 (cyan), and control mutation V440 (purple). The substrate, squalene, is indicated in green. S38, S168, and F605 are considered to be in the second sphere of the active site, V440 is outside the active site. (B) A close-up of the active site and the location of the native residues and the substrate.
MD Simulations
The simulations were performed with the AMBER software using force field ff14SB52 for the proteins, lipid1453 for the membrane, and generalized Amber force field (GAFF)54 for the ligands. Parameters for the GAFF force field for the substrate and product were developed with antechamber,51 see SI for details. The simulations were around 150 ns long.
Some simulations were extended for up to 300 ns to study the processes during a longer time frame. Five parallel simulations of 20 ns length each were performed for further investigation of the sample space for the wt and the S38W mutant.
An additional simulation at higher temperature (350 K) of the protein with the product in the active site was performed to analyze whether the conformation of the hydrophobic channel would be more inclined to change to a wider diameter in order for the product to be able to be released from the active site to the membrane. A previous study20 suggested that due to the high flexibility of the channel, it is likely that it also serves as an exit channel for release of the bulky product.
Simulations were performed with (i) the empty enzyme, (ii) the substrate (squalene) in different locations of the substrate entrance channel, (iii) squalene in the active site, for each of the protein variants, (iv) protonated squalene in the active site, and (v) product in the active site. More information about which PDB structure was used for each case can be found in the SI, Table S1. Post processing of the trajectories was performed using the CPPTRAJ program55 in AmberTools.56
The stability of the protein outside its native environment of a bilayer lipid membrane was also investigated (under the same conditions as when the membrane was included, see SI for a more detailed description).
Water Analysis
The network of water tunnels in SHC was further investigated in silico with the use of different computational tools. CAVER22 was developed to identify tunnels and pores in proteins. It was originally used for investigating these features in crystal structures or other static images of macromolecules. As CAVER is not measuring water dynamics, we complemented it with a method developed by Vassiliev et al.57,58 to track the flow of water through a protein using streamline visualization of diffusion tensor fields. Previous water diffusion pathway studies have focused on proteins that have well-defined channels and a more simple geometry than SHC, such as aquaporins.59−62 A recent study also investigated water dynamics in a de novo tunnel.63 The data analysis technique applied in this study locates areas of highly anisotropic motions of water molecules from the simulation. It finds the most likely pathways of water diffusion in the protein based on the water motions detected in the MD simulations.
The streamline analysis was conducted on the 150 ns trajectories to investigate how the water molecules move in the enzyme. The water movements in the empty enzyme were compared to water movements in the enzyme with substrate, protonated substrate, and product in the active site. Water movements were also compared between the wt and the mutated protein variants. Details of streamline analysis are described in the SI.
Acknowledgments
The Swedish Research Council (VR) is acknowledged for financial support (T.B.), and Barbora Kozlikova and the CAVER team are greatly acknowledged for the time and effort to upgrade their program for these simulations.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01084.
Preparation of PDB files; detailed description of simulation procedure; RMSD plots for simulations; extended B-factor analysis for DXDD motif; details about streamline analysis method (PDF)
Author Contributions
C.G. performed simulations and analysis. The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.
C.G and T.B. received funding from Swedish Research Council (VR), grant: 621-2013-04165. S.V. received funding from Discovery grant from the National Science and Engineering Research Council of Canada. C.K. and P.-O.S. received funding from Swedish Research Council (VR), grant: 621-2013-5138.
The authors have nothing to disclose.
The authors declare no competing financial interest.
Supplementary Material
References
- Oldfield E.; Lin F. Y. Terpene Biosynthesis: Modularity Rules. Angew. Chem., Int. Ed. 2012, 51, 1124–1137. 10.1002/anie.201103110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt K. U.; Schulz G. E.; Corey E. J.; Liu D. R. Enzyme Mechanisms for Polycyclic Triterpene Formation. Angew. Chem., Int. Ed. 2000, 39, 2812–2833. . [DOI] [PubMed] [Google Scholar]
- Potter K. C.; Zi J.; Hong Y. J.; Schulte S.; Malchow B.; Tantillo D. J.; Peters R. J. Blocking Deprotonation with Retention of Aromaticity in a Plant Ent-Copalyl Diphosphate Synthase Leads to Product Rearrangement. Angew. Chem., Int. Ed. 2016, 55, 634–638. 10.1002/anie.201509060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Harris G. G.; Pemberton T. A.; Christianson D. W. Multi-Domain Terpenoid Cyclase Architecture and Prospects for Proximity in Bifunctional Catalysis. Curr. Opin. Struct. Biol. 2016, 41, 27–37. 10.1016/j.sbi.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz J. P.; Grosser D.; Bradley A. S.; Lagny T. J.; Lavrynenko O.; Broda M.; Simons K. Hopanoids as Functional Analogues of Cholesterol in Bacterial Membranes. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 11971–11976. 10.1073/pnas.1515607112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmappa R.; Geisler K.; Louveau T.; O’Maille P.; Osbourn A. Triterpene Biosynthesis in Plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. 10.1146/annurev-arplant-050312-120229. [DOI] [PubMed] [Google Scholar]
- Ajikumar P. K.; Xiao W.-H.; Tyo K. E. J.; Wang Y.; Simeon F.; Leonard E.; Mucha O.; Phon T. H.; Pfeifer B.; Stephanopoulos G. Isoprenoid Pathway Optimization for Taxol Precursor Overproduction in Escherichia coli. Science 2010, 330, 70–74. 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro D.-K.; Paradise E. M.; Ouellet M.; Fisher K. J.; Newman K. L.; Ndungu J. M.; Ho K. A.; Eachus R. A.; Ham T. S.; Kirby J.; et al. Production of the Antimalarial Drug Precursor Artemisinic Acid in Engineered Yeast. Nature 2006, 440, 940–943. 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- Jiang Z.; Kempinski C.; Bush C. J.; Nybo S. E.; Chappell J. Engineering Triterpene and Methylated Triterpene Production in Plants Provides Biochemical and Physiological Insights into Terpene Metabolism. Plant Physiol. 2016, 170, 702–716. 10.1104/pp.15.01548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta-Yahya P. P.; Zhang F.; del Cardayre S. B.; Keasling J. D. Microbial Engineering for the Production of Advanced Biofuels. Nature 2012, 488, 320–328. 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- Nestl B. M.; Hammer S. C.; Nebel B. A.; Hauer B. New Generation of Biocatalysts for Organic Synthesis. Angew. Chem., Int. Ed. 2014, 53, 3070–3095. 10.1002/anie.201302195. [DOI] [PubMed] [Google Scholar]
- Sukumaran J.; Hanefeld U. Enantioselective C-C Bond Synthesis Catalysed by Enzymes. Chem. Soc. Rev. 2005, 34, 530–542. 10.1039/b412490a. [DOI] [PubMed] [Google Scholar]
- Seitz M.; Syrén P. O.; Steiner L.; Klebensberger J.; Nestl B. M.; Hauer B. Synthesis of Heterocyclic Terpenoids by Promiscuous Squalene-Hopene Cyclases. ChemBioChem 2013, 14, 436–439. 10.1002/cbic.201300018. [DOI] [PubMed] [Google Scholar]
- Hammer S. C.; Syrén P.; Seitz M.; Nestl B. M.; Hauer B. Squalene Hopene Cyclases: Highly Promiscuous and Evolvable Catalysts for Stereoselective C–C and C–X Bond Formation. Curr. Opin. Chem. Biol. 2013, 17, 293–300. 10.1016/j.cbpa.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Yoosuf-Aly Z.; Faraldos J. A.; Miller D. J.; Allemann R. K. Chemoenzymatic Synthesis of the Alarm Pheromone (+)-Verbenone from Geranyl Diphosphate. Chem. Commun. 2012, 48, 7040. 10.1039/c2cc32883f. [DOI] [PubMed] [Google Scholar]
- Seckler B.; Poralla K. Characterization and Partial Purification of Squalene - Hopene. Biochim. Biophys. Acta 1986, 881, 356–363. 10.1016/0304-4165(86)90027-9. [DOI] [Google Scholar]
- Reinert D. J.; Balliano G.; Schulz G. E. Conversion of Squalene to the Pentacarbocyclic Hopene. Chem. Biol. 2004, 11, 121–126. 10.1016/j.chembiol.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Dang T.; Prestwich G. D. Site-Directed Mutagenesis of Squalene-Hopene Cyclase: Altered Substrate Specificity and Product Distribution. Chem. Biol. 2000, 7, 643–649. 10.1016/S1074-5521(00)00003-X. [DOI] [PubMed] [Google Scholar]
- Wendt K. U.; Poralla K.; Schulz G. E. Structure and Function of a Squalene Cyclase. Science 1997, 277, 1811–1815. 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]
- Schwab F.; van Gunsteren W. F.; Zagrovic B. Computational Study of the Mechanism and the Relative Free Energies of Binding of Anticholesteremic Inhibitors to Squalene-Hopene Cyclase. Biochemistry 2008, 47, 2945–2951. 10.1021/bi702067h. [DOI] [PubMed] [Google Scholar]
- Rajamani R.; Gao J. Balancing Kinetic and Thermodynamic Control: The Mechanism of Carbocation Cyclization by Squalene Cyclase. J. Am. Chem. Soc. 2003, 12768–12781. 10.1021/ja0371799. [DOI] [PubMed] [Google Scholar]
- Chovancova E.; Pavelka A.; Benes P.; Strnad O.; Brezovsky J.; Kozlikova B.; Gora A.; Sustr V.; Klvana M.; Medek P.; et al. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8, e1002708 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrén P.-O.; Hammer S. C.; Claasen B.; Hauer B. Entropy Is Key to the Formation of Pentacyclic Terpenoids by Enzyme- Catalyzed Polycyclization. Angew. Chem., Int. Ed. 2014, 53, 4845–4849. 10.1002/anie.201402087. [DOI] [PubMed] [Google Scholar]
- Helms V.; Wade R. C. Thermodynamics of Water Mediating Protein-Ligand Interactions in Cytochrome P450cam: A Molecular Dynamics Study. Biophys. J. 1995, 69, 810–824. 10.1016/S0006-3495(95)79955-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova M.; Klvana M.; Prokop Z.; Chaloupkova R.; Banas P.; Otyepka M.; Wade R. C.; Tsuda M.; Nagata Y.; Damborsky J. Redesigning Dehalogenase Access Tunnels as a Strategy for Degrading an Anthropogenic Substrate. Nat. Chem. Biol. 2009, 5, 727–733. 10.1038/nchembio.205. [DOI] [PubMed] [Google Scholar]
- Meyer E. Internal Water Molecules and H-Bonding in Biological Macromolecules: A Review of Structural Features with Functional Implications. Protein Sci. 1992, 1, 1543–1562. 10.1002/pro.5560011203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebel J.; Chang A.; Merget B.; Bommineni G. R.; Yu W.; Spagnuolo L. A.; Baxter M. V.; Tareilus M.; Tonge P. J.; Kisker C.; et al. An Ordered Water Channel in Staphylococcus aureus FabI: Unraveling the Mechanism of Substrate Recognition and Reduction. Biochemistry 2015, 54, 1943–1955. 10.1021/bi5014358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisette O.; Päslack C.; Barnes R.; Isas J. M.; Langen R.; Heyden M.; Han S.; Schäfer L. V. Hydration Dynamics of a Peripheral Membrane Protein. J. Am. Chem. Soc. 2016, 138, 11526–11535. 10.1021/jacs.6b07005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkovic S. J.; Hammes-Schiffer S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- Wendt K. U.; Lenhart A.; Schulz G. E. The Structure of the Membrane Protein Squalene- Hopene Cyclase at 2.0 Å Resolution. J. Mol. Biol. 1999, 286, 175–187. 10.1006/jmbi.1998.2470. [DOI] [PubMed] [Google Scholar]
- Kokkinidis M.; Glykos N. M.; Fadouloglou V. E. Protein Flexibility and Enzymatic Catalysis. Adv. Protein Chem. Struct. Biol. 2012, 87, 181. 10.1016/B978-0-12-398312-1.00007-X. [DOI] [PubMed] [Google Scholar]
- Meiering E. M.; Serrano L.; Fersht A. R. Effect of Active Site Residues in Barnase on Activity and Stability. J. Mol. Biol. 1992, 225, 585–589. 10.1016/0022-2836(92)90387-Y. [DOI] [PubMed] [Google Scholar]
- Sato T.; Hoshino T. Functional Analysis of the DXDDTA Motif in Squalene-Hopene Cyclase by Site-Directed Mutagenesis Experiments: Initiation Site of the Polycyclization Reaction and Stabilization Site of the Carbocation Intermediate of the Initially Cyclized A-Ring. Biosci. Biotechnol. Biochem. 1999, 63, 2189–2198. 10.1271/bbb.63.2189. [DOI] [PubMed] [Google Scholar]
- Hammer S. C.; Marjanovic A.; Dominicus J. M.; Nestl B. M.; Hauer B. Squalene Hopene Cyclases Are Protonases for Stereoselective Brønsted Acid Catalysis. Nat. Chem. Biol. 2015, 11, 121–126. 10.1038/nchembio.1719. [DOI] [PubMed] [Google Scholar]
- Hammer S. C.; Syrén P.-O.; Hauer B. Substrate Pre-Folding and Water Molecule Organization Matters for Terpene Cyclase Catalyzed Conversion of Unnatural Substrates. ChemistrySelect 2016, 1, 3589–3593. 10.1002/slct.201600572. [DOI] [Google Scholar]
- Bruice T. C.; Lightstone F. C. Ground State and Transition State Contributions to the Rate of Intramolecular and Enzymic Reactions. Acc. Chem. Res. 1999, 32, 127–136. 10.1021/ar960131y. [DOI] [Google Scholar]
- Hur S.; Bruice T. C. The near Attack Conformation Approach to the Study of the Chorismate to Prephenate Reaction. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12015–12020. 10.1073/pnas.1534873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder M.; Hermansson A.; Liebeschuetz J.; Brinck T. Computational Design of a Lipase for Catalysis of the Diels-Alder Reaction. J. Mol. Model. 2011, 17, 833–849. 10.1007/s00894-010-0775-8. [DOI] [PubMed] [Google Scholar]
- Linder M.; Johansson A. J.; Olsson T. S. G.; Liebeschuetz J.; Brinck T. Computational Design of a Diels-Alderase from a Thermophilic Esterase: The Importance of Dynamics. J. Comput.-Aided Mol. Des. 2012, 26, 1079–1095. 10.1007/s10822-012-9601-y. [DOI] [PubMed] [Google Scholar]
- Rowland R. S.; Taylor R. Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van Der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. 10.1021/jp953141+. [DOI] [Google Scholar]
- Hwang J. K.; Warshel A. How Important Are Quantum Mechanical Nuclear Motions in Enzyme Catalysis?. J. Am. Chem. Soc. 1996, 118, 11745–11751. 10.1021/ja962007f. [DOI] [Google Scholar]
- Marx D. Proton Transfer 200 Years after Von Grotthuss: Insights from Ab Initio Simulations. ChemPhysChem 2006, 7, 1848–1870. 10.1002/cphc.200600128. [DOI] [PubMed] [Google Scholar]
- Biedermannová L.; Prokop Z.; Gora A.; Chovancová E.; Kovács M.; Damborský J.; Wade R. C. A Single Mutation in a Tunnel to the Active Site Changes the Mechanism and Kinetics of Product Release in Haloalkane Dehalogenase LinB. J. Biol. Chem. 2012, 287, 29062–29074. 10.1074/jbc.M112.377853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland L. J.; Tomaszek T. A.; Meek T. D. Human Immunodeficiency Virus-1 Protease. 2. Use of pH Rate Studies and Solvent Kinetic Isotope Effects To Elucidate Details of Chemical Mechanism. Biochemistry 1991, 30, 8454–8463. 10.1021/bi00098a024. [DOI] [PubMed] [Google Scholar]
- Ball P. More than a Bystander. Nature 2011, 478, 467–468. 10.1038/478467a. [DOI] [PubMed] [Google Scholar]
- Snyder P. W.; Mecinovic J.; Moustakas D. T.; Thomas S. W.; Harder M.; Mack E. T.; Lockett M. R.; Héroux A.; Sherman W.; Whitesides G. M. Mechanism of the Hydrophobic Effect in the Biomolecular Recognition of Arylsulfonamides by Carbonic Anhydrase. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 17889–17894. 10.1073/pnas.1114107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Chou W. K. W.; Al-Lami N.; Faraldos J. A.; Allemann R. K.; Cane D. E.; Christianson D. W. Probing the Role of Active Site Water in the Sesquiterpene Cyclization Reaction Catalyzed by Aristolochene Synthase. Biochemistry 2016, 55, 2864–2874. 10.1021/acs.biochem.6b00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David B.; Irague R.; Jouanneau D.; Daligault F.; Czjzek M.; Sanejouand Y. H.; Tellier C. Internal Water Dynamics Control the Transglycosylation/Hydrolysis Balance in the Agarase (AgaD) of Zobellia galactanivorans. ACS Catal. 2017, 7, 3357–3367. 10.1021/acscatal.7b00348. [DOI] [Google Scholar]
- Case D. A.; Babin V.; Berryman J. T.; Betz R. M.; Cai Q.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Gohlke H.; Goetz A. W.; Gusarov S.; Homeyer N.; Janowski P.; Kaus J.; Kolossváry I.; Kovalenko A.; Lee T. S.; LeGrand S.; Luchko T.; Luo R.; Madej B.; Kollman P. A.;. et al. AMBER 14; University of California: San Francisco, 2014. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD—Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graphics Modell. 2006, 25, 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson C. J.; Madej B. D.; Skjevik Å. A.; Betz R. M.; Teigen K.; Gould I. R.; Walker R. C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Babin V.; Berryman J. T.; Betz R. M.; Cai Q.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Gohlke H.; Goetz A. W.; Gusarov S.; Homeyer N.; Janowski P.; Kaus J.; Kolossváry I.; Kovalenko A.; Lee T. S.; LeGrand S.; Luchko T.; Luo R.; Wu X.; Kollman P. A.; et al. AMBER 14 Reference Manual; University of California: San Francisco, 2014. [Google Scholar]
- Vassiliev S.; Zaraiskaya T.; Bruce D. Exploring the Energetics of Water Permeation in Photosystem II by Multiple Steered Molecular Dynamics Simulations. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1671–1678. 10.1016/j.bbabio.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Vassiliev S.; Comte P.; Mahboob A.; Bruce D. Tracking the Flow of Water through Photosystem II Using Molecular Dynamics and Streamline Tracing. Biochemistry 2010, 49, 1873–1881. 10.1021/bi901900s. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Grubmüller H. Water Permeation across Biological Membranes: Mechanism and Dynamics of Aquaporin-1 and GlpF. Science 2001, 294, 2353–2357. 10.1126/science.1062459. [DOI] [PubMed] [Google Scholar]
- De Groot B. L.; Frigato T.; Helms V.; Grubmüller H. The Mechanism of Proton Exclusion in the Aquaporin-1 Water Channel. J. Mol. Biol. 2003, 333, 279–293. 10.1016/j.jmb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Zhu F.; Tajkhorshid E.; Schulten K. Theory and Simulation of Water Permeation in Aquaporin-1. Biophys. J. 2004, 86, 50–57. 10.1016/S0006-3495(04)74082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti N.; Roux B.; Pomès R. Structural Determinants of Proton Blockage in Aquaporins. J. Mol. Biol. 2004, 343, 493–510. 10.1016/j.jmb.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Brezovsky J.; Babkova P.; Degtjarik O.; Fortova A.; Gora A.; Iermak I.; Rezacova P.; Dvorak P.; Smatanova I. K.; Prokop Z.; et al. Engineering a de Novo Transport Tunnel. ACS Catal. 2016, 6, 7597–7610. 10.1021/acscatal.6b02081. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.