

Abstract
Rationale:
The Hippo pathway plays an important role in determining organ size through regulation of cell proliferation and apoptosis. Hippo inactivation and consequent activation of Yes-associated protein (YAP), a transcription co-factor, have been proposed as a strategy to promote myocardial regeneration after myocardial infarction (MI). However, the long-term effects of Hippo deficiency on cardiac function under stress remain unknown.
Objective:
We investigated the long-term effect of Hippo deficiency upon cardiac function in the presence of pressure overload (PO).
Methods and Results:
We used mice with cardiac-specific homozygous knockout of WW45 (WW45cKO), in which activation of Mst1 and Lats2 in which Mst1 (Mammalian sterile 20-like 1) and Lats2 (large tumor suppressor kinase 2), the upstream kinases of the Hippo pathway, is effectively suppressed due to the absence of the scaffolding protein. We used male mice at 3–4 month of age in all animal experiments. We subjected WW45cKO mice to transverse aortic constriction (TAC) for up to 12 weeks. WW45cKO mice exhibited higher levels of nuclear YAP in cardiomyocytes (CMs) during PO. Unexpectedly, the progression of cardiac dysfunction induced by PO was exacerbated in WW45cKO mice, despite decreased apoptosis and activated CM cell cycle re-entry. WW45cKO mice exhibited CM sarcomere disarray and upregulation of TEAD1 target genes involved in CM de-differentiation during PO. Genetic and pharmacological inactivation of the YAP-TEAD1 pathway reduced the PO-induced cardiac dysfunction in WW45cKO mice, and attenuated CM de-differentiation. Furthermore, the YAP-TEAD1 pathway upregulated oncostatin M (OSM) and OSM receptors, which played an essential role in mediating CM de-differentiation. OSM also upregulated YAP and TEAD1 and promoted CM de-differentiation, indicating the existence of a positive feedback mechanism consisting of YAP, TEAD1 and OSM.
Conclusion:
Although activation of YAP promotes CM regeneration after cardiac injury, it induces CM de-differentiation and heart failure in the long-term in the presence of PO through activation of the YAP-TEAD1-OSM positive feedback mechanism.
Keywords: Hippo pathway, YAP, cardiac regeneration, heart failure, de-differentiation, signal transduction, pressure overload, Cell Signaling
INTRODUCTION
Heart failure is a major cause of mortality in developed countries. Despite remarkable advances in the development of medical interventions, the mortality rate of heart failure patients remains high 1. Pressure overload (PO), imposed by high blood pressure or aortic valve stenosis, induces cardiac hypertrophy to maintain wall stress and cardiac output but eventually leads to cardiac failure with increases in the death of cardiomyocytes (CMs). Since the capacity of the adult heart to compensate for CM loss through regeneration is limited, death of CMs triggers a progressive reduction in the total number of CMs and induces heart failure 2.
Among the signaling mechanisms involved in the death of CMs, the Hippo signaling pathway may play a particularly important role in the progression of heart failure 3. The Hippo signaling pathway generally serves as a major regulator of organ size, through regulation of both apoptosis and cell proliferation 4. Activation of upstream components of the Hippo pathway, including Mammalian sterile 20-like 1 (Mst1) and large tumor suppressor kinase 2 (Lats2), promotes phosphorylation and inactivation of Yes-associated protein (YAP), a transcription factor co-factor involved in cell survival and proliferation, thereby promoting cell death 5–9. We have shown previously that activation of Mst1 by myocardial stress and genetic downregulation of YAP both promote the progression of heart failure, indicating that downregulation of YAP below physiological levels is detrimental for the heart. 10–13. In contrast, inactivation of the Hippo pathway or YAP activation through genetic intervention suppresses cell apoptosis and enhances cell survival and proliferation 14, 15. In addition, it is recently reported that both constitutively active YAP and inactivation of the Hippo pathway could stimulate myocardial regeneration after adult myocardial injury, and finally protects the cardiac function 15–18. However, the physiological function of the Hippo pathway in the heart remains unknown. Although recent studies support therapeutic inhibition of the Hippo pathway during heart failure, can this concept be applied to all types of heart failure caused by distinct mechanisms without harmful consequences?
To address this question, we examined how suppression of the endogenous Hippo pathway affects the phenotype of adult hearts in the presence of a pathologically relevant stress, namely PO, using cardiac specific homozygous knockout (KO) of WW45 (WW45cKO), a scaffolding protein that induces activation of Lats by upstream kinases Mst1/2 3, 4. Although the Hippo pathway promotes apoptosis and suppresses CM proliferation/cell cycle re-entry, effects that usually negatively affect cardiac function, and despite recent reports showing the salutary effect of Hippo suppression in the heart after MI,14, 15, 18, 19 our preliminary results suggest that chronic suppression of the Hippo pathway and persistently elevated YAP facilitate the progression of heart failure in response to PO in WW45cKO. Given that stimulation of YAP through suppression of the Hippo pathway is assumed to be one of the most significant modalities for facilitating myocardial regeneration, it is important to further clarify potentially detrimental effects of YAP stimulation in the heart in the presence of PO and to elucidate the potential underlying mechanisms.
Thus, the goals of this study were to elucidate the molecular mechanism by which persistent suppression of the Hippo pathway and persistent activation of YAP in WW45cKO mice promote heart failure in response to PO and investigate whether suppression of YAP prevents progression of left ventricular (LV) dysfunction in mouse models of cardiovascular disease. Our result suggests that chronic suppression of the Hippo pathway in the presence of PO activates a gene expression profile promoting CM de-differentiation through activation of a positive feedback mechanism consisting of YAP, TEAD1 and oncostatin M (OSM), thereby causing contractile dysfunction in individual CMs.
METHODS
In order to minimize the possibility of unintentionally sharing information that can be used to re-identify private information, a subset of the data generated for this study are available at NCBI GEO and can be accessed at GSE50637 and GSE56813, and GSE92970.
Detailed methods are provided in the Online Data Supplement.
Mouse models.
Generation of mice harboring a floxed WW45/Salvador allele (C57BL/6 background) 20 and Myh6- Cre recombinase (C57BL/6 background) has been reported 21. We generated systemic TEAD1 +/− mice using homologous recombination strategies and the mice were backcrossed into a C57BL/6 background 22. Briefly, a 8954 bp fragment containing exon 3 and the alternatively spliced exon 4 (the DNA binding TEA domain) was used to insert LoxP sites into the flanking introns 23. The recombined allele in the ES cells used to generate mice inadvertently contained a duplicated exon 3. It was embryonic lethal in the homozygous mice. Downregulation of TEAD1 in the heterozygous knockout mice was confirmed at both protein and mRNA levels (Online Figure I). YAP floxed mice have been described 11, 24. We used age-matched male mice in all animal experiments to compare our results with the past studies conducted with mice with the loss of function of the Hippo pathway11. Age- and sex-matched mice without Myh6 promoter-driven Cre recombinase or targeting allele were used as littermate controls (controls). All experiments involving animals were approved by the Rutgers New Jersey Medical School’s Institutional Animal Care and Use Committee.
RESULTS
PO-induced activation of YAP was transient in control mice but was sustained in WW45cKO mice.
To evaluate the effects of PO, mice were subjected to either transverse aortic constriction (TAC) or sham operation. In the control hearts, total YAP protein increased within 1 day, reached a peak around 7 days, and gradually returned to the basal level by 4 weeks. The level of total YAP was lower than in sham-operated mice at 8 and 12 weeks of PO. Phosphorylation of YAP at Ser127, which is mediated by Lats2 and induces nuclear exit of YAP, normalized by the total amount of YAP was not significantly altered until 7 days but was significantly elevated by 8 weeks and further increased at 12 weeks (Figure 1 A, B). Immunostaining of heart sections showed that nuclear staining of YAP was increased significantly 1 week but not 4 weeks after TAC compared to in sham-operated mice (Figure 1C). Thus, although PO induces transient upregulation and nuclear accumulation of YAP through a Ser127 phosphorylation-independent mechanism, activation of YAP gradually returns to baseline around 4 weeks and further decreases thereafter, and this gradual decline in activation is accompanied by increased phosphorylation of YAP at Ser127 normalized by total YAP.
Figure 1: PO-induced YAP activation was transient in control mice but sustained in WW45cKO mice.

(A, B) Representative gel pictures and quantitative analyses of immunoblot in hearts of Ctr and WW45cKO mice after operation, at the indicated time points (n=4, each). *P<0.05, **P<0.01 by ANOVA, compared with Ctr-TAC. #P<0.05 compared with Ctr-sham. (C) Representative immunostaining and quantitative analysis of YAP (YAP, red; DAPI, blue) in Ctr and WW45cKO 1 and 4 weeks after TAC. Nuclear accumulation of YAP is indicated by arrow heads (n=4, each). p-YAP indicates phospho-YAP.
Since cardioprotective and regenerative effects of YAP have been reported in the adult heart 15, 17, we asked whether we can prevent or delay the development of heart failure by inducing sustained activation of YAP after TAC. In WW45cKO, a loss-of-function mouse model of the Hippo pathway, TAC significantly increased YAP, peaking at 7 days and remaining elevated thereafter. The level of YAP in WW45cKO mice was significantly higher than in control mice under basal conditions and 4, 8 and 12 weeks after TAC (Figure 1 A, B). The Ser127 phosphorylated YAP/total YAP ratio was significantly smaller in WW45cKO mice than in control mice 8 and 12 weeks after TAC. Immunostaining of heart sections showed that nuclear staining of YAP in CMs was significantly greater in WW45cKO than in control mice after sham operation and at 1 and 4 weeks of TAC (Figure 1C and Online Figure II).
Thus, WW45cKO mice can be used to elucidate whether sustained elevation of YAP is protective during PO.
In control mice, TAC activated Mst1, as evaluated with their phospho-MST1/2(Thr183/180)/total-MST1, and Lats2, as evaluated with their phospho-Lats2(Thr1041)/total-Lats2, with a similar time course as phosphorylation of YAP(Ser127) after PO. Activation of Mst1/2 and Lats2 and consequent phosphorylation of YAP, including at Ser127, were significantly attenuated in WW45cKO mice (Online Figure III). These results suggest that the nuclear accumulation of YAP observed after 1 week of PO cannot be sustained in control hearts because of activation of Mst1/2 and Lats2, and that endogenous WW45 plays an essential role in mediating activation of Mst1/2 and Lats2 and consequent phosphorylation of YAP at Ser127. Thus, WW45cKO mice can be used to elucidate the functional significance of chronic activation of Mst1/2 and Lats2, components of the canonical Hippo pathway, during PO.
WW45cKO mice exhibited greater CM cell cycle re-entry without organ enlargement in response to PO.
Homozygous deletion of WW45 induced marked enlargement and tumor formation in the liver 25, and homozygous KO of WW45 at the fetal stage increased heart size 26. Postnatal KO of WW45 in the heart with Myh6-Cre, however, did not significantly affect LV size (Figure 2A) or LV weight/tibial length (TL) (Figure 2B) at baseline or in the presence of PO for 4 weeks. CM cross-sectional area in WW45cKO mice, evaluated using wheat germ agglutinin staining, was similar to that in control mice at baseline, but was significantly smaller than in control mice after 4 weeks of PO (Figure 2C). Smaller CM size after stress often indicates the presence of newly generated CMs due to regeneration or proliferation 27, 28. Alternatively, activation of anti-hypertrophic signaling mechanisms may be responsible for the smaller CM size. In tissue sections and isolated CMs, the number of Ki-67-, phospho-histone H3 (pHH3)- or BrdU-positive nuclei in cardiac troponin T (TnT)-positive CMs was similar for WW45cKO and control mice at baseline but was significantly greater in WW45cKO mice 4 weeks after TAC than in control mice (Figure 2D and Online Figure IV A, B). The number of pHH3 positive CMs was also significantly greater in WW45cKO mice after TAC than in control mice when the analysis was conducted using freshly isolated CMs (Online Figure IV C). These results suggest that postnatal downregulation of WW45 and consequent upregulation of endogenous YAP stimulates CM cell cycle reentry without significantly affecting organ size in the presence of PO.
Figure 2: WW45cKO mice show severe cardiac dysfunction in response to PO, with higher level of markers of CM cycle re-entry.

(A) Representative gross morphology and longitudinal heart sections from Ctr and WW45cKO, stained with HE 4 weeks after TAC. Scale bars, 5.0 mm. (B) LV weight to TL ratio (n=7, each). (C) Representative WGA staining to assess CSA 4 weeks after operation (sham: n=6, each; TAC: n=8, each). (D) Representative immunostaining and quantitative analysis of pHH3 in the hearts of Ctr and WW45cKO mice 1 week after operation (pHH3, red; Sarc actinin, green; DAPI, blue) (n=6, each). (E) Representative echocardiographic tracings of the hearts of Ctr and WW45cKO mice 4 weeks after operation. (F) Kaplan-Meier survival curves after TAC. All results are expressed as mean ± SEM. *P<0.05, **P <0.01 by ANOVA. p-YAP indicates phosphorylated YAP.
WW45cKO mice exhibited more severe cardiac dysfunction in response to PO despite decreases in apoptosis.
Echocardiographic and hemodynamic measurements showed that WW45cKO mice have normal cardiac function at baseline. However, after 4 weeks of TAC, WW45cKO mice developed more severe heart failure. WW45cKO mice had more severe LV dysfunction, as indicated by decreases in LV fractional shortening (%FS), and a greater LV end-diastolic dimension (LVEDD) than control mice (Figure 2E, Online Figure V A and Online Table I). Hemodynamic measurements showed prominent signs of heart failure in WW45cKO mice, including a significantly higher LV end-diastolic pressure (LVEDP) and a lower pressure gradient than in control mice (Online Figure V B and Online Table II). WW45cKO mice also exhibited a significantly higher lung weight/TL, an index of lung congestion, than control mice (Online Figure V C). Consistently, WW45cKO mice exhibited significantly greater mortality in response to TAC than control mice (Figure 2F). Histological analyses of the heart showed that there was significantly more interstitial fibrosis, but not perivascular fibrosis, in WW45cKO than in control mice after 4 weeks of TAC (Online Figure V D). The number of TUNEL-positive CMs and the myocardial level of cleaved caspase 3 after 4 weeks of TAC were significantly lower in WW45cKO mice than in control mice (Online Figure V E, F), consistent with reduced activation of the upstream Hippo signaling pathway. Another important mechanism facilitating cardiac dysfunction during PO is inflammation. Myocardia infiltration of CD45 positive cells (leukocytes), CD68 positive cells (macrophages), and Ly6G positive cells (neutrophils) was not significantly different between Ctr and WW45cKO mice 1 week after PO. Infiltration of inflammatory cells was significantly but only slightly enhanced 4 weeks after PO in WW45cKO mice than in Ctr mice (Online Figure VI).
Contraction of individual CMs is decreased in WW45cKO mice in response to PO.
Since increased apoptosis was not observed in WW45cKO compared to in control mouse hearts in the presence of PO, we hypothesized that heart failure occurs due to decreases in the contractility of individual CMs. Contraction of freshly isolated CMs, evaluated using the edge detection method 29, was not significantly altered at baseline, but was significantly decreased in CMs isolated from WW45cKO mice compared to in those from control mice after PO (Online Figure VII A). Although there was no effect on myofilament Ca2+ sensitivity or the Hill coefficient (data not shown), the maximum development of force in skinned CMs was significantly reduced in WW45cKO compared to in control mice (P<0.0002) after PO (Online Figure VII B). Consistently, electron microscopic analyses showed sarcomeric disarray, especially in M-line structures, and more irregularly shaped intercalated disks in WW45cKO than in control mouse hearts after PO (Online Figure VII C).
YAP mediates the detrimental phenotype in WW45cKO mice in response to PO.
In order to investigate the role of YAP in the exacerbation of LV dysfunction in WW45cKO mice in response to PO, we generated WW45cKO-YAP heterozygous cKO (YAPhcKO) mice. Although both WW45cKO mice and YAPhcKO mice exhibited reduced cardiac function and LV dilation in the presence of PO, WW45cKO-YAPhcKO mice exhibited significantly better LV function and less LV dilation than either WW45cKO or YAPhcKO mice (Online Figure VIII A, B). WW45cKO-YAPhcKO mice exhibited significantly better survival than WW45cKO mice during PO (Online Figure VIII C). Similarly, treatment of WW45cKO mice with verteporfin, a small molecule that inhibits interaction between YAP and TEADs30, rescued LV dysfunction and dilation in response to PO (Online Figure IX A, B). These results suggest that upregulation of YAP mediates the detrimental phenotype in WW45cKO mice in the presence of PO. However, the reversal of the detrimental phenotypes in YAPhcKO mice and control mice treated with verteporfin when they were crossed with WW45cKO mice suggests that both upregulation and suppression of YAP are detrimental for the heart during PO and that maintaining YAP at appropriate levels is critical.
WW45cKO induces de-differentiation of the heart in response to PO, together with TEAD1 activation.
To elucidate the mechanism by which WW45cKO mice develop more severe cardiac dysfunction in response to PO, RNA sequencing analyses were conducted. We previously identified a set of genes whose expression changes during development are reversed in TAC.31 These genes are commonly regarded as differentiation/development markers (GO:0055007). Thus, we first tested whether WW45cKO affects changes in expression in this group of genes. As shown in Figure 3A, the difference in regulation between WW45cKO TAC mice and control TAC mice for genes that belong to GO: 0055007 was much greater than for other genes (p = 8×10−5 and 3×10−7 for genes downregulated and upregulated in embryonic development, K-S test), indicating that, under TAC, WW45cKO mice launch a stronger de-differentiation gene program than control mice. Online Table III and a heatmap (Figure 3B) of the genes that belong to GO:0055007 indicate that genes promoting CM dedifferentiation, including Bmp2, Cav3, Dll1, Fzd7, G6pdx, Pak1, Pi16, Rgs2 and Rgs4, are upregulated, whereas those promoting CM differentiation (or inhibiting dedifferentiation), including Arrb2, Efnb2, Ep300, Fdps, Gsk3b, Mef2c, Nr3c1 and Slc25a4, are downregulated in WW45cKO mice than in control mice in the presence of TAC (Figure 3B, online Table III, and Online Figure X). GO analyses also indicated that genes involved in mitochondria are downregulated, those involved in apoptosis are upregulated, whereas those involved in contractile fibers are both up- and down-regulated in WW45cKO-TAC compared to in WT-TAC (Online Table IV).
Figure 3: TEAD1 plays an essential role in mediating the exacerbation of heart failure in WW45 cKO mice in response to PO by facilitating CM de-differentiation.

(A) ECDF showing the difference in regulation between WW45cKO-TAC and Ctr-TAC for genes oppositely regulated in cardiac development and disease. Red line : genes DN in embryonic development and UP in TAC (n=438); Blue line : genes UP in embryonic development and DN in TAC (n=365); Black line : other genes. P-values are based on the K-S test comparing red or blue line genes with black line genes. (B) Heatmap showing relative expression of genes regarding cardiac differentiation. Gene set derived from the association of GO:0055007. The normalized read counts were subject to median centering before visualization. (C) TFBS analysis is based on UP or DN genes in WW45cKO vs Ctr (Arrow : TEAD1). Top five TFBS gene sets are shown with the q-values for false discovery rate control. (D) Representative gel pictures of immunoblot in the cytosolic and nuclear fractions of Ctr and WW45cKO mice 4 weeks after operation (n=4, each). (E) Representative gel pictures of immunoblot in the hearts of Ctr, WW45cKO, WW45cKO + TEAD1 +/− and TEAD1 +/− mice 4 weeks after operation. (F) Kaplan-Meier survival curves after TAC. Results are expressed as mean ± SEM. *P<0.05, **P<0.01 by ANOVA. NS indicates not significant.
Transcription factor binding site analyses of regulated genes suggested that TEAD1, a TEAD family transcription factor and known target of YAP, may play a role in mediating the changes in gene expression profile observed in WW45cKO mice or in the presence of PO (Figure 3C). Importantly, TEAD1 was upregulated in CM nuclei following PO in the heart in WW45cKO mice (Figure 3D). mRNA expression of Myh7, Acta2, and Acta1, fetal-type contractile proteins possessing TEAD1 binding sites in their promoters, was upregulated in WW45cKO hearts (Online Figure XI A). On the other hand, mRNA expression of Myh6, an adult-type contractile protein that does not possess a TEAD1 binding site, was not affected. ChIP assays showed that YAP binding to TEAD1 binding motifs located in the promoters of Acta2 and Myh7 in CMs was increased significantly in the presence of TEAD1 overexpression (Online Figure XI B), consistent with the notion that YAP binds to the TEAD1 sites in the Acta2 and Myh7 promoters through TEAD1.
Immunostaining of myocardial sections showed that cardiac TnT staining was significantly reduced, whereas the number of CMs expressing MYH7, ACTA2 or Runt-related transcription factor 1 (RUNX1), markers of CM de-differentiation, was significantly higher after 4 weeks of TAC in WW45cKO than in control mouse hearts (Online Figure XI C-E). Immunoblot analyses confirmed that downregulation of WW45 enhances upregulation of MYH7, ACTA2 and RUNX1 in response to TAC (Online Figure XI F), an effect that was attenuated by heterozygous downregulation of YAP (Online Figure VIII D-H). The level of ACTA2 was significantly greater in CMs isolated from WW45cKO mice after TAC than in those from control mice after TAC (Online Figure XI G). These results are consistent with the notion that the heart is more de-differentiated in WW45cKO mice in the presence of PO, which may be mediated by a YAP-TEAD1-dependent mechanism.
TEAD1 plays an essential role in the exacerbation of heart failure in WW45cKO mice in response to PO by facilitating de-differentiation of CMs.
In order to further elucidate the critical involvement of TEAD1 in the exacerbation of cardiac dysfunction observed in WW45cKO mice during PO, WW45cKO mice were crossed with TEAD1 +/− mice (Online Figure I A). After confirmation that TEAD1 protein and mRNA expression in the heart are significantly reduced in TEAD1 +/− mice (Online Figure I B, C), the mice were subjected to TAC for 4 weeks. As expected, heterozygous downregulation of TEAD1 inhibited the upregulation of TEAD1 during PO in WW45cKO mice (Figure 3E and Online Figure XII D). Although heterozygous downregulation of TEAD1 did not affect the cardiac phenotype at baseline or in response to TAC in the absence of WW45cKO, it abrogated the exacerbation of PO-induced cardiac dysfunction observed in WW45cKO mice and significantly reduced the mortality of WW45cKO mice during PO (Figure 3F and Online Figure XIII A, B). Heterozygous downregulation of TEAD1 normalized both decreases in CM cross-sectional area and increases in cardiac fibrosis in WW45cKO mice in response to PO (Online Figure XIII C, D). Importantly, heterozygous downregulation of TEAD1 also normalized the enhanced upregulation of MYH7, ACTA2, ACTA1 and RUNX1 protein and mRNA in WW45cKO mice during PO (Figure 3E, Online Figure XII and XIII E). Electron microscopic analyses showed that the sarcomere disarray and disturbances in the Z and M line structures observed in WW45cKO mice in response to TAC were normalized when WW45cKO mice were crossed with TEAD1 +/− mice (Online Figure XIII F). These results suggest that TEAD1 plays an essential role in the exacerbation of cardiac dysfunction and CM de-differentiation in WW45cKO mice during PO.
Oncostatin M (OSM) is a downstream target of YAP/TEAD1.
Since a previous study has suggested that OSM, a member of the interleukin-6 family of cytokines, is a major mediator of CM de-differentiation 32, we hypothesized that OSM plays an important role in mediating CM de-differentiation in WW45cKO mice during PO. Immunoblot analyses showed that both OSM and OSMR are upregulated by PO and that their levels are further increased in the presence of WW45cKO (Figure 4A). Increases in OSM staining were observed both in the cytosol and in the perinuclear areas of CMs in WW45 cKO mice after TAC (Online Figure XIV). The staining was not observed when the secondary antibody was used alone or when the primary antibody was pre-adsorbed with blocking peptide. OSM and OSMR were also increased in CMs isolated from WW45cKO mice after TAC (Figure 4B). OSM was upregulated at the level of mRNA (Figure 4C), suggesting that it is produced in the CMs rather than it is solely taken up from the extracellular space. Similarly, OSM and OSMR protein levels were increased when TEAD1 was adenovirally overexpressed in cultured neonatal rat ventricular CMs (Figure 4D). ChIP assays showed that YAP binds to a GGAATG element, the TEAD1 binding motif, in the promoter regions of the Osm and Osmr genes in CMs (Figure 4E). ChIP-sequencing analyses with anti-YAP antibody showed that YAP binds to the promoter of Osmr near the transcription start site, where there is a TEAD1 binding sequence, but not to the promoter of Actc1, in which there is no TEAD1 binding sequence, in mouse hearts subjected to PO in vivo (Figure 4F). Overexpression of TEAD1 or YAP significantly increased the activity of a reporter gene driven by the Osm promoter in CMs, but not when the TEAD1 site was mutated (Figure 4G). Conversely, short hairpin RNAs targeting YAP (sh-YAP) and TEAD1 (sh-TEAD1) significantly decreased the OSM reporter activity (Figure 4G). These results suggest that OSM and OSMR are downstream targets of the YAP-TEAD1 pathway in CMs. Consistently, immunoblot analyses showed that the enhancement of OSM and OSMR upregulation observed during PO in WW45cKO mice was attenuated by heterozygous deletion of either YAP or TEAD1 (Online Figure VIII D, I, J, Figure 3E and Online Figure XII E).
Figure 4: YAP-TEAD1 activity regulates OSM.

(A) Representative gel pictures of immunoblot in the hearts of Ctr and WW45cKO mice 4 weeks after operation (n=6, each). (B) Representative gel pictures of immunoblot in CMs isolated from adult WW45cKO mice 1 week after operation. (C) Relative mRNA expression in adult CMs isolated from the hearts 1 week after TAC (n=3, each). (D) Representative gel pictures of immunoblot in neonatal CMs transduced with adenovirus harboring LacZ or TEAD1. (E) ChIP assay of YAP binding to the Osm promoter (left) and the Osmr promoter (right) in neonatal CMs transduced with adenovirus harboring LacZ or TEAD1. PCR target regions and quantitative analyses are shown (n=4, each). (F) Representative pictures of ChIP-Seq in the Osmr promoter at the transcription start site (TSS) using YAP1, TFIIB, Pol II, or control IgG antibodies. Mice were subjected to TAC for 4 days (n=3). The fragment densities (y-axis) were aligned with the chromosomal coordinates (x-axis) using the Integrated Genome Browser (IGB). Shown are the binding sites of YAP1, TFIIB, and Pol II along the Osmr (left) and Actc1 (right) genes. The arrow defines the direction of the transcription start sites. (G) Reporter genes assays to evaluate the role of the TEAD1 binding site in the Osm promoter in neonatal CMs transduced with adenovirus harboring LacZ, TEAD1 or YAP (n=6, each). Results are expressed as mean ± SEM. *P<0.05, **P<0.01 by ANOVA. For box plots, whiskers show minima and maxima within 1.5 interquartile range. Ad-YAP indicates adenovirus harboring YAP; mutOSM, OSM promoter containing a mutated TEAD binding site; sh-TEAD1, short hairpin RNAs targeting TEAD; sh-Scr, scrambled short hairpin RNA; and sh-YAP, short hairpin RNAs targeting YAP.
OSM induced CM de-differentiation through YAP/TEAD1.
In order to investigate whether OSM is sufficient to induce cardiac dysfunction, we treated mice with OSM without stress. Treatment of mice with OSM (60 μg/kg/day ip, twice daily for 14 days) (Online Figure XV A) increased MYH7 and ACTA2, together with YAP and TEAD1, in the heart at baseline, and the upregulation was further enhanced in the presence of TAC, consistent with cardiac de-differentiation (Online Figure XV B). Interestingly, although OSM alone did not induce cardiac dysfunction without PO, OSM promoted the progression of LV dysfunction and LV dilation in response to TAC (Online Figure XV C). OSM treatment after TAC also exhibited similar results (Online Figure XVI). Taken together, these results suggest that OSM is sufficient to exacerbate PO-induced cardiac dysfunction and CM de-differentiation, thereby mimicking the effect of WW45cKO during PO. Although YAP and TEAD1 upregulated OSM and OSMRs, OSM also upregulated YAP, TEAD1, OSMRs and MYH7 in CMs isolated from adult mouse hearts (Figure 5A) and induced nuclear accumulation of YAP (Figure 5B). OSM also upregulated the total amount of the YAP-TEAD1 complex in cultured CMs in vitro (Online Figure XVII A). Luciferase assays with a reporter gene driven by 8X TEAD binding sites showed that OSM stimulates the transcriptional activity of Tead1 in a YAP-dependent manner in CMs (Figure 5C). Eventually, OSM increases transcription of the Osm gene itself through a YAP- and TEAD1-dependent mechanism in CMs (Online Figure XVII B). Functionally, OSM increased the number of BrdU-positive CMs, indicating activation of CM cell cycle re-entry, as well as YAP staining in the nuclei of BrdU-positive CMs (Online Figure XVII C). OSM also increased the number of ACTA2-positive CMs, and most ACTA2-positive CMs exhibited nuclear accumulation of YAP (Figure 5D). These results suggest that the CM cell cycle re-entry and de-differentiation induced by OSM are accompanied by upregulation of TEAD1 and activation of YAP in CMs. Indeed, in ChIP assays, OSM significantly increased binding of YAP to TEAD1 binding sequences in the Acta2 and Myh7 promoters in CMs (Online Figure XVII D). Furthermore, immunoblot analyses showed that OSM upregulated YAP and TEAD1 and that OSM-induced upregulation of MYH7, ACTA2, and OSMR is suppressed in the presence of either sh-YAP or sh-TEAD1 in CMs (Figure 5E and Online Figure XVIII). Likewise, immunostaining showed that OSM-induced upregulation of ACTA2 in CMs is attenuated in the presence of sh-TEAD1 or sh-YAP (Figure 5F). OSM-induced CM cell cycle re-entry was also attenuated in the presence of sh-TEAD1 or sh-YAP (Online Figure XVII E, F). These results suggest that OSM induces cell cycle re-entry and de-differentiation of CMs through YAP-TEAD1-dependent mechanisms. OSM inhibits p38α and Thr1041 phosphorylation of LATS2 (Figure 5E and Figure XVII G), effects that have been implicated in the upregulation of TEAD1 33 and activation of YAP 34, respectively, suggesting that OSM may stimulate YAP-TEAD1 through suppression of LATS2 and p38α. Thus, YAP and TEAD1 upregulate OSM in CMs, and OSM upregulates YAP and TEAD1, as well as OSMR through YAP and TEAD1, in CMs, suggesting that YAP, TEAD1, and OSM/OSMR form an amplification loop to enhance OSM function and promote de-differentiation of CMs.
Figure 5: OSM induces CM de-differentiation through YAP-TEAD1.
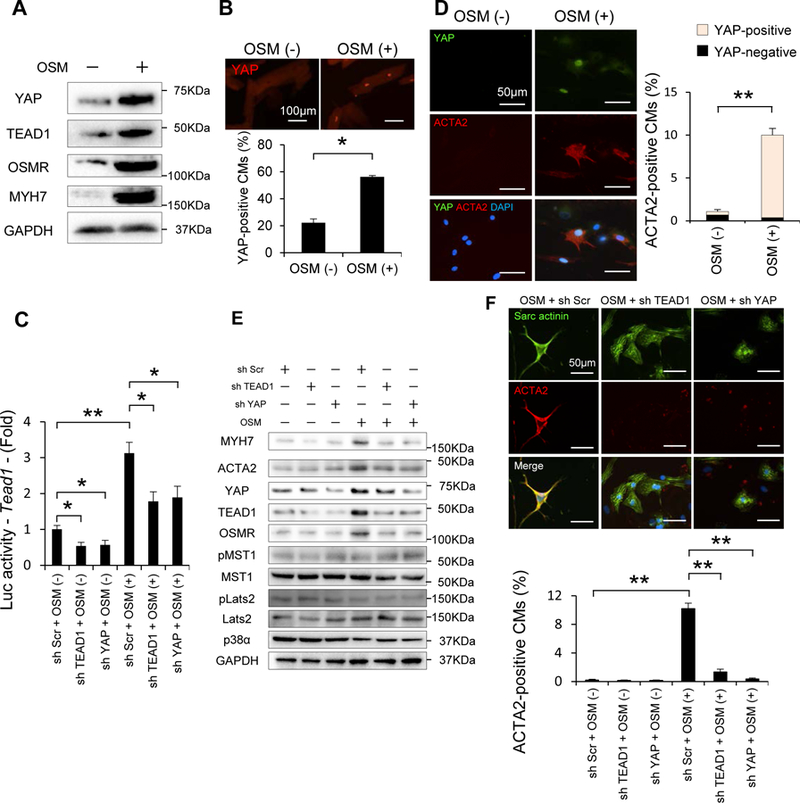
(A) Representative gel pictures of immunoblot in CMs isolated from adult mice with or without OSM treatment. (B) Representative immunostaining and quantitative analysis of YAP in CMs isolated from adult mice with or without OSM treatment (n=3, each). (C) Reporter gene assays were conducted in order to evaluate the transcriptional activity of Tead1 in response to OSM in neonatal CMs transduced with Ad-Scr, Ad-sh-TEAD1 or Ad-sh-YAP, and then transfected with 8×GTIIC-luciferase plasmid in the presence or absence of OSM (n=6, each). (D) Representative double-immunostaining and quantitative analysis of YAP and ACTA2 in neonatal CMs with or without OSM treatment (YAP, green; ACTA2, red; DAPI, blue)(n=4, each). (E) Representative gel pictures of immunoblot in neonatal CMs transduced with Ad-Scr, Ad-sh-TEAD1 or Ad-sh-YAP, in the presence or absence of OSM (n=4, each). (F) Representative double-immunostaining and quantitative analysis of Sarcomeric actinin and ACTA2 in neonatal CMs transduced with adenovirus with or without OSM treatment (Sarc actinin, green; ACTA2, red; DAPI, blue) (n=4, each). Results are expressed as mean ± SEM. *P<0.05, **P<0.01 by ANOVA.
If the detrimental phenotype in WW45cKO mice during PO depends upon this amplification loop, suppression of OSM would dramatically suppress the exacerbation of heart failure observed in WW45cKO mice. To address this hypothesis, we treated control and WW45cKO mice after PO with either control IgG or anti-OSM blocking antibody (Figure 6A). Anti-OSM blocking antibody treatment remarkably suppressed the exacerbation of LV dysfunction in response to TAC in WW45cKO mice, although the cardiac phenotype of control mice was not affected (Figure 6B–D). Furthermore, expression of MYH7, ACTA2, and RUNX1, was substantially reduced and YAP and TEAD1 were partially reduced in the presence of anti-OSM blocking antibody in WW45cKO mice during PO (Figure 6E and Online Figure XIX A-E). Anti-OSM antibody also inhibited upregulation of MYH7 and ACTA2 in CMs co-transduced with Ad-YAP and Ad-TEAD1 (Online Figure XX). These results suggest that OSM is an essential component of the YAP-TEAD1-OSM amplification loop that facilitates the progression of heart failure in response to PO in the Hippo-deficient mice. YAP may induce CM cell cycle re-entry through the Wnt and IGF pathways. However, although expression of β-catenin was increased after 4 weeks of TAC, there was no significant difference between WT and WW45cKO mice. Moreover, there were no significant changes in protein expression of IGF-1R in WT or WW45cKO mice in either the presence or absence of PO (Online Figure XIX F).
Figure 6: Figure 6 anti-OSM blocking antibody improves cardiac dysfunction in WW45cKO during PO through suppression of CM de-differentiation.
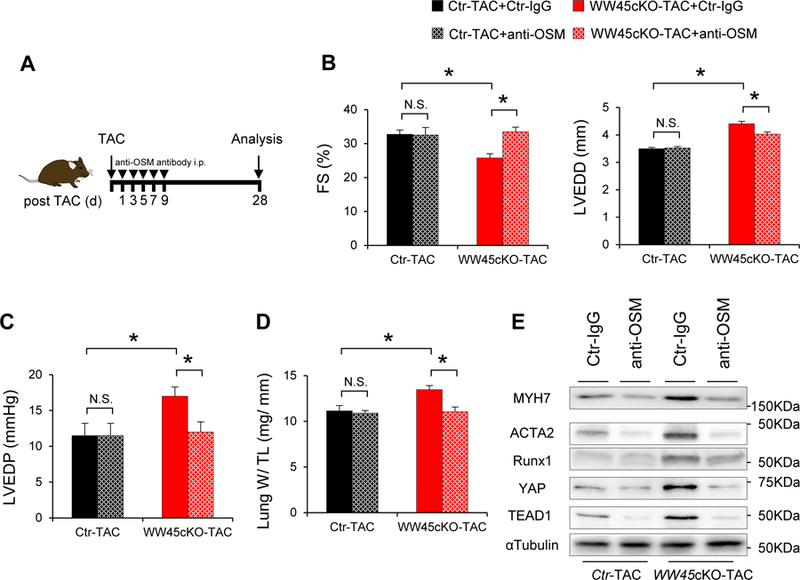
WW45cKO mice were subjected to TAC for 4 weeks in the presence of either Ctr-IgG or anti-OSM blocking antibody(anti-OSM). (B) %FS and LVEDD (n=4, each). (C) LVEDP (n=4, each). (D) Lung weight/TL (n=4, each). (E) Representative gel pictures in the hearts after TAC in the presence of either Ctr-IgG or anti-OSM. Results are expressed as mean ± SEM. *P<0.05, **P<0.01 by ANOVA.
DISCUSSION
One of the most surprising findings in this work is that chronic downregulation of Hippo and consequent activation of YAP promote cardiac dysfunction in response to PO, despite the fact that they decrease CM apoptosis and increase CM cell cycle re-entry, both of which are generally thought to be cardio-protective under conditions of stress. Our study further suggests that YAP-TEAD1-mediated mechanisms, intensified by positive feedback between YAP-TEAD1 and OSM/OSMR, decreases the contraction of individual CMs, thereby reducing LV function during PO. Previous studies have suggested that suppression of the Hippo pathway or exogenously supplied YAP effectively promotes myocardial regeneration and prevents the transition from compensation to decompensation in the post-MI heart primarily subjected to volume overload 14, 19. Thus, our results suggest that the Hippo pathway and YAP have both detrimental and salutary roles in the heart depending upon the nature of upstream stress.
OSM is a novel target of YAP.
Both RNA sequencing and ChIP sequencing analyses suggest that YAP directly stimulates transcription of genes involved in CM de-differentiation through TEAD1. This process is dramatically facilitated by a positive feedback loop consisting of OSM/OSMR, YAP and TEAD1. In the presence of persistent upregulation of YAP and PO-induced TEAD1 upregulation, OSM and OSMR are both upregulated in CMs, which in turn upregulates YAP and TEAD1, most likely through suppression of Lats2 and p38α, respectively 33, 35. Alternatively, OSM may modulate VGLL4, thereby stabilizing TEAD1 and enhancing YAP-TEAD1 interaction 36. Upregulation of OSM/OSMR and activation of TEAD1 targets are dramatically stimulated in WW45cKO mice in the presence of PO, and this effect is inhibited by attenuating any single component of the feedback loop, including YAP, TEAD1 and OSM. However, considering the fact that WW45 is a scaffolding protein, and, thus, may possess known partners, we cannot formally exclude the possibility that WW45cKO may enhance TAC-induced upregulation of OSM in part through TEAD1-indepedent mechanisms as well.
Diverse functions of YAP.
We have shown previously that downregulation of YAP below physiological levels through either heterozygous or homozygous KO exacerbates cardiac dysfunction at baseline and in response to chronic MI in mice, due to increases in CM death and a lack of compensatory cardiac hypertrophy 11. These results clearly suggest that a minimum level of YAP must be maintained and that the loss of function at sub-physiological levels is detrimental for the heart. The salutary effects of YAP are mediated in part through interaction with FoxO 16 and upregulation of either miR-206 37 or Akt 11, 38. On the other hand, the current investigation suggests that persistent elevation of YAP above baseline and concomitant upregulation of TEAD1 induce de-differentiation and cardiac dysfunction in the presence of PO. Thus, it is likely that YAP must be maintained at appropriate levels for the heart to maintain normal LV function. In addition, the in vivo effect of YAP is determined by the availability of downstream transcription factors in a given condition.
Underlying mechanisms exacerbating heart failure in WW45cKO mice in response to PO.
WW45cKO mice exhibited more severe heart failure than control mice in the presence of PO. Neither apoptosis nor fibrosis appear to explain the exacerbation of heart failure in WW45cKO mice. Since decreases in maximum force generation and sarcomere disarray were observed in individual cardiomyocytes isolated from WW45cKO mouse heart subjected to TAC, it is likely that heart failure is caused by dysfunction of individual CMs. RNA sequencing and bioinformatics analyses suggest that the chronic activation of YAP in the presence of PO facilitate gene expression pattern mimicking the fetal heart. In addition, OSM treatment, which is known to induce CM de-differentiation, induces heart failure in control hearts similar to WW45cKO mouse hearts in the presence of PO. Thus, CM de-differentiation may in part mediate CM dysfunction in the presence of PO. However, other targets of the YAP-TEAD1 pathway may also be involved in this process. For example, gene ontology analyses suggest that several groups of genes regulating cardiac function are affected in response to TAC in WW45cKO mice compared to in control hearts, although whether they are direct targets of the YAP-TEAD pathway remains to be elucidated. In addition. OSM is known to stimulate inflammation, which would in turn facilitate cardiac dysfunction in the presence of PO. Although modest increases in infiltration of inflammatory cells were observed in WW45cKO mouse hearts in the presence of PO compared to in control heart, the mechanistic involvement of inflammation in the exacerbation of heart failure remains to be elucidated.
Role of YAP during distinct forms of hemodynamic stress.
It has been shown that upregulation of YAP promotes myocardial regeneration in the postnatal heart subjected to MI 14, 15, 17, 19. In these experiments, YAP promoted proliferation of either CMs or progenitor cells, maintained the wall thickness of the infarcted areas, and improved LV function after MI 14, 15, 17. These results appear to conflict with our finding that chronic elevation of YAP is detrimental for the heart during PO, but some differences in conditions may help explain the apparent discrepancy. Perhaps the most important difference in the conditions where upregulation of YAP is salutary or different is that whether YAP is upregulated post MI or during pressure overload. Similar to the dichotomous roles of YAP, functional roles of OSM appear to be also context-dependent. OSM is cardioprotective during ischemia/reperfusion or in the post MI heart 38–41, whereas it is detrimental in some forms of cardiomyopathy, including diabetic cardiomyopathy and that induced by increased inflammation 31, 42, 44. The heart requires distinct signaling mechanisms to adapt to stress induced by volume overload, which is imposed by chronic myocardial infarction, and that by pressure overload. Further investigation is required to elucidate the underlying mechanisms to explain the differential effects of YAP and downstream signaling mechanisms, including OSM, in these conditions.
YAP-TEAD1 is a therapeutic target in some cardiovascular conditions.
YAP is generally excluded from CM nuclei in the adult heart at baseline, most likely because Mst1 and Mst2 have baseline activities. Although modest upregulation of YAP alone may not induce significant effects in the heart, simultaneous activation of TEAD1 or TEAD family transcription factors by a superimposed stress, such as high blood pressure, turns on the positive feedback loop of YAP-TEAD1-OSM, thereby inducing de-differentiation and contractile dysfunction in CMs. The consensus DNA binding sequence for TEAD family transcription factors is conserved in the OSM, OSMR, ACTA2, and MYH7 genes in humans (Online Table I). Thus, suppression of the YAP-TEAD1-OSM positive feedback loop may be effective in a subpopulation of cardiovascular patients. It is recently reported that some patients with hypertrophic cardiomyopathy exhibit high level of YAP in the heart 45 whereas patients with heart failure exhibit elevated OSM levels in plasma 46. In the future study, it is important to identify additional conditions in which YAP and TEAD1 are co-activated and test the efficacy of YAP inhibitors in these conditions. Moreover, the conditions of hemodynamic stress under which activation of the YAP pathway is detrimental should be specifically defined in humans, since TAC may not faithfully mimic the conditions of PO in patients with chronic hypertension.
Experimental limitations.
Due to limited resources and time constraints, we conducted this investigation using only male mice, as in our previous investigations of the role of the Hippo signaling pathway in the heart 5, 6,10, 11, 12, 13, 16, 37. Whether the Hippo signaling pathway functions identically in the hearts of female mice is an important question that should be addressed in the future.
In summary, our study shows that an endogenous Hippo pathway functions to maintain CM differentiation so that the heart can cope with hemodynamic overload. In conditions, where the YAP-OSM feedback loop is not opposed by the Hippo pathway, the heart undergoes failure due to de-differentiation of individual CMs.
Supplementary Material
Figure 7: A schematic representation of the current hypothesis.
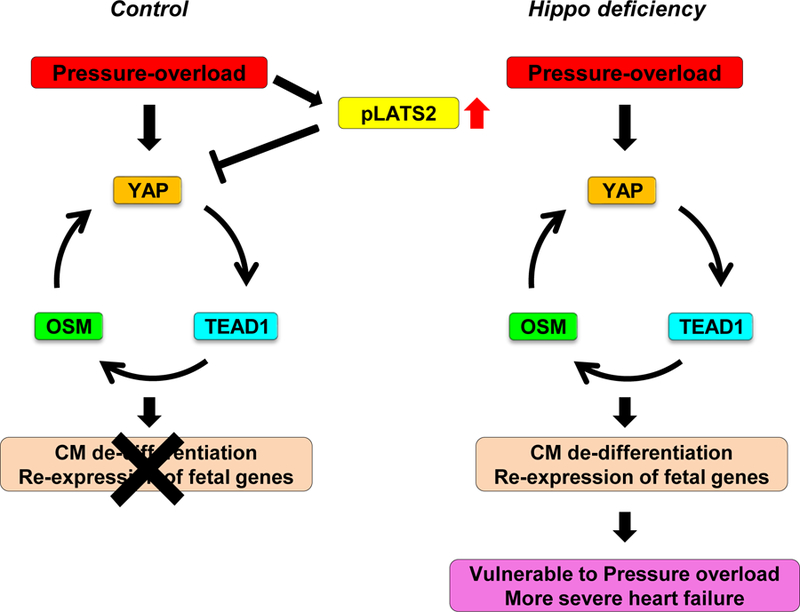
TAC suppresses the feedback loop of YAP-TEAD1-OSM through activation of the Hippo pathway and consequent suppression of YAP. In the absence of Hippo activation, TAC dramatically stimulates the feedback loop of YAP-TEAD1-OSM, thereby inducing de-differentiation of CMs and preventing the heart from maintaining cardiac output in the presence of PO.
NOVELTY AND SIGNIFICANCE.
What Is Known?
The Hippo pathway is an evolutionarily conserved signaling pathway that controls organ size by promoting apoptosis and inhibiting cell proliferation.
Mst1 and Lats2, upstream kinases of the Hippo pathway, are activated by stress in cardiomyocytes and promote heart failure.
Inhibition of the Hippo pathway and consequent activation of YAP promote myocardial regeneration after myocardial infarction.
What New Information Does This Article Contribute?
Chronic suppression of the Hippo pathway and activation of YAP are detrimental in the presence of pressure overload, despite suppression of apoptosis and promotion of cell cycle re-entry in cardiomyocytes.
Detrimental effects of YAP during pressure overload are mediated through activation of a positive feedback loop consisting of YAP, TEAD and OSM.
The Hippo pathway is activated during heart failure and promotes apoptosis of cardiomyocytes and inhibits cardiomyocyte proliferation and/or hypertrophy. Recent evidence suggests that downregulation of the Hippo pathway and/or activation of YAP, the terminal nuclear factor promoting cell survival and proliferation, is effective in inducing myocardial regeneration after myocardial infarction. This raises a possibility that either strong suppression of the Hippo pathway or stimulation of YAP may represent a promising therapeutic intervention to treat heart failure and induce cardiac regeneration. However, here we show that complete suppression of the Hippo pathway can be detrimental in the presence of pressure overload due to stimulation of a positive feedback mechanism consisting of YAP, TEAD1 and oncostatin M, which leads to a reduction in the contractility of individual cardiomyocytes. Thus, caution should be exercised when Hippo suppression and/or stimulation of YAP is considered as a modality to prevent the progression of heart failure or to stimulate myocardial regeneration, especially in the presence of pressure overload. Although excessive activation of the Hippo pathway is detrimental for the heart, excessive inhibition can also be detrimental under some conditions. We propose that the level/activity of the Hippo pathway should be maintained at appropriate levels.
ACKNOWLEDGMENTS
We thank Duojia Pan (University of Texas Southwestern Medical Center) for kindly providing mice, Junco S. Warren (University of Utah) for assisting bioinformatics analyses, and Daniela Zablocki (Rutgers New Jersey Medical School) and Christopher D. Brady (Rutgers New Jersey Medical School) for assistance with the manuscript.
SOURCES OF FUNDING
This work was supported in part by U.S. Public Health Service Grants HL130356, HL105826 and HL114749 (S.Sa.), HL071894 (I.F.), HL98802 (B.T.), and HL67724, HL91469, HL102738, HL112330 and AG23039 (J.S.), and by the Leducq Foundation Transatlantic Network of Excellence (J.S.). S.I. has been supported by a Postdoctoral Fellowship from Japan Heart Foundation / Bayer Yakuhin Research Grant Abroad and the grants-in-aid for Scientific Research (18K15876).
Nonstandard Abbreviations and Acronyms:
- cKO
cardiac-specific knockout
- CM
Cardiomyocyte
- CSA
Cross-sectional area
- Ctr
control
- ECDF
Empirical cumulative distribution function
- DN
downregulated
- FS
Fractional shortening
- GO
Gene Ontology
- Lats2
Large tumor suppressor kinase 2
- LVEDD
Left ventricular end-diastolic dimension
- LVEDP
Left ventricular end-diastolic pressure
- Mst1
Mammalian sterile 20-like 1
- OSM
oncostatin M
- pHH3
phospho-histone H3
- PO
Pressure overload
- TAC
Transverse aortic constriction
- TEAD
Transcriptional enhancer factor
- TFBS
Transcription factor binding site
- TL
Tibial length
- TnT
Troponin T
- UP
upregulated
- YAP
Yes-associated protein
- WGA
Wheat Germ Agglutinin
Footnotes
DISCLOSURES
None.
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Roger VL. Epidemiology of heart failure. Circulation research 2013;113:646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xin M, Olson EN and Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nature reviews Molecular cell biology 2013;14:529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ikeda S and Sadoshima J. Regulation of Myocardial Cell Growth and Death by the Hippo Pathway. Circulation journal : official journal of the Japanese Circulation Society 2016. [DOI] [PubMed]
- 4.Yu FX, Zhao B and Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015;163:811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, Molina CA, Yatani A, Vatner DE, Vatner SF and Sadoshima J. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. The Journal of clinical investigation 2003;111:1463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsui Y, Nakano N, Shao D, Gao S, Luo W, Hong C, Zhai P, Holle E, Yu X, Yabuta N, Tao W, Wagner T, Nojima H and Sadoshima J. Lats2 is a negative regulator of myocyte size in the heart. Circulation research 2008;103:1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Q, Li L, Zhao B and Guan KL. The hippo pathway in heart development, regeneration, and diseases. Circulation research 2015;116:1431–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piccolo S, Dupont S and Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiological reviews 2014;94:1287–312. [DOI] [PubMed] [Google Scholar]
- 9.Wackerhage H, Del Re DP, Judson RN, Sudol M and Sadoshima J. The Hippo signal transduction network in skeletal and cardiac muscle. Science signaling 2014;7:re4. [DOI] [PubMed] [Google Scholar]
- 10.Del Re DP, Matsuda T, Zhai P, Maejima Y, Jain MR, Liu T, Li H, Hsu CP and Sadoshima J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Molecular cell 2014;54:639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, Zhang N, Yabuta N, Nojima H, Pan D and Sadoshima J. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. The Journal of biological chemistry 2013;288:3977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M and Sadoshima J. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nature medicine 2013;19:1478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odashima M, Usui S, Takagi H, Hong C, Liu J, Yokota M and Sadoshima J. Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circulation research 2007;100:1344–52. [DOI] [PubMed] [Google Scholar]
- 14.Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PR, Zhou B, Chen J, Seidman JG, Wang DZ and Pu WT. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circulation research 2014;115:354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R and Olson EN. Hippo pathway effector Yap promotes cardiac regeneration. Proceedings of the National Academy of Sciences of the United States of America 2013;110:13839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao D, Zhai P, Del Re DP, Sciarretta S, Yabuta N, Nojima H, Lim DS, Pan D and Sadoshima J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nature communications 2014;5:3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL and Martin JF. Hippo signaling impedes adult heart regeneration. Development 2013;140:4683–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, Li L, Sun Z, Olson EN, Amendt BA and Martin JF. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 2016;534:119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, Segura A, Willerson JT and Martin JF. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017;550:260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JH, Kim TS, Yang TH, Koo BK, Oh SP, Lee KP, Oh HJ, Lee SH, Kong YY, Kim JM and Lim DS. A crucial role of WW45 in developing epithelial tissues in the mouse. The EMBO journal 2008;27:1231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA and Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. The Journal of clinical investigation 1997;100:169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yagi R, Kohn MJ, Karavanova I, Kaneko KJ, Vullhorst D, DePamphilis ML and Buonanno A. Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 2007;134:3827–36. [DOI] [PubMed] [Google Scholar]
- 23.Zuzarte PC, Farrance IK, Simpson PC and Wildeman AG. Tumor cell splice variants of the transcription factor TEF-1 induced by SV40 T-antigen transformation. Biochimica et biophysica acta 2000;1517:82–90. [DOI] [PubMed] [Google Scholar]
- 24.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A and Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007;130:1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS and Johnson RL. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proceedings of the National Academy of Sciences of the United States of America 2010;107:1437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL and Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011;332:458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Gao E, Vite A, Yi R, Gomez L, Goossens S, van Roy F and Radice GL. Alpha-catenins control cardiomyocyte proliferation by regulating Yap activity. Circulation research 2015;116:70–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah A, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Deberardinis R, Oz O, Lu Z, Zhang CC, Kimura W and Sadek HA. Hypoxia induces heart regeneration in adult mice. Nature 2016. [DOI] [PubMed]
- 29.Kim YK, Kim SJ, Yatani A, Huang Y, Castelli G, Vatner DE, Liu J, Zhang Q, Diaz G, Zieba R, Thaisz J, Drusco A, Croce C, Sadoshima J, Condorelli G and Vatner SF. Mechanism of enhanced cardiac function in mice with hypertrophy induced by overexpressed Akt. The Journal of biological chemistry 2003;278:47622–8. [DOI] [PubMed] [Google Scholar]
- 30.Liu R, Lee J, Kim BS, Wang Q, Buxton SK, Balasubramanyam N, Kim JJ, Dong J, Zhang A, Li S, Gupte AA, Hamilton DJ, Martin JF, Rodney GG, Coarfa C, Wehrens XH, Yechoor VK and Moulik M. Tead1 is required for maintaining adult cardiomyocyte function, and its loss results in lethal dilated cardiomyopathy JCI insight 2017; 2(17). pii: 93343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park JY, Li W, Zheng D, Zhai P, Zhao Y, Matsuda T, Vatner SF, Sadoshima J and Tian B. Comparative analysis of mRNA isoform expression in cardiac hypertrophy and development reveals multiple post-transcriptional regulatory modules. PloS one 2011;6:e22391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kubin T, Poling J, Kostin S, Gajawada P, Hein S, Rees W, Wietelmann A, Tanaka M, Lorchner H, Schimanski S, Szibor M, Warnecke H and Braun T. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell stem cell 2011;9:420–32. [DOI] [PubMed] [Google Scholar]
- 33.Ambrosino C, Iwata T, Scafoglio C, Mallardo M, Klein R and Nebreda AR. TEF-1 and C/EBPbeta are major p38alpha MAPK-regulated transcription factors in proliferating cardiomyocytes. The Biochemical journal 2006;396:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moroishi T, Hansen CG and Guan KL. The emerging roles of YAP and TAZ in cancer. Nature reviews Cancer 2015;15:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu FX, Karin M, Pan D and Guan KL. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes & development 2015;29:1271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Z, Guo H, Cao Y, Zohrabian S, Zhou P, Ma Q, VanDusen N, Guo Y, Zhang J, Stevens SM, Liang F, Quan Q, van Gorp PR, Li A, Dos Remedios C, He A, Bezzerides VJ and Pu WT. Acetylation of VGLL4 Regulates Hippo-YAP Signaling and Postnatal Cardiac Growth. Developmental cell 2016;39:466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Del Re DP, Nakano N, Sciarretta S, Zhai P, Park J, Sayed D, Shirakabe A, Matsushima S, Park Y, Tian B, Abdellatif M and Sadoshima J. miR-206 Mediates YAP-Induced Cardiac Hypertrophy and Survival. Circulation research 2015;117:891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ and Pu WT. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circulation research 2015;116:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorchner H, Poling J, Gajawada P, Hou Y, Polyakova V, Kostin S, Adrian-Segarra JM, Boettger T, Wietelmann A, Warnecke H, Richter M, Kubin T and Braun T. Myocardial healing requires Reg3beta-dependent accumulation of macrophages in the ischemic heart. Nature medicine 2015;21:353–62. [DOI] [PubMed] [Google Scholar]
- 40.Hu J, Zhang L, Zhao Z, Zhang M, Lin J, Wang J, Yu W, Man W, Li C, Zhang R, Gao E, Wang H and Sun D. OSM mitigates post-infarction cardiac remodeling and dysfunction by up-regulating autophagy through Mst1 suppression. Biochimica et biophysica acta Molecular basis of disease 2017;1863:1951–1961. [DOI] [PubMed] [Google Scholar]
- 41.Sun D, Li S, Wu H, Zhang M, Zhang X, Wei L, Qin X and Gao E. Oncostatin M (OSM) protects against cardiac ischaemia/reperfusion injury in diabetic mice by regulating apoptosis, mitochondrial biogenesis and insulin sensitivity. Journal of cellular and molecular medicine 2015;19:1296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lafontant PJ, Burns AR, Donnachie E, Haudek SB, Smith CW and Entman ML. Oncostatin M differentially regulates CXC chemokines in mouse cardiac fibroblasts. American journal of physiology Cell physiology 2006;291:C18–26. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Ma S, Zhang R, Li S, Zhu D, Han D, Li X, Li C, Yan W, Sun D, Xu B, Wang Y and Cao F. Oncostatin M-induced cardiomyocyte dedifferentiation regulates the progression of diabetic cardiomyopathy through B-Raf/Mek/Erk signaling pathway. Acta biochimica et biophysica Sinica 2016;48:257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poling J, Gajawada P, Richter M, Lorchner H, Polyakova V, Kostin S, Shin J, Boettger T, Walther T, Rees W, Wietelmann A, Warnecke H, Kubin T and Braun T. Therapeutic targeting of the oncostatin M receptor-beta prevents inflammatory heart failure. Basic research in cardiology 2014;109:396. [DOI] [PubMed] [Google Scholar]
- 45.Wang P, Mao B, Luo W, Wei B, Jiang W, Liu D, Song L, Ji G, Yang Z, Lai YQ and Yuan Z. The alteration of Hippo/YAP signaling in the development of hypertrophic cardiomyopathy. Basic research in cardiology 2014;109:435. [DOI] [PubMed] [Google Scholar]
- 46.Gruson D, Ferracin B, Ahn SA and Rousseau MF. Elevation of plasma oncostatin M in heart failure. Future cardiology 2017;13:219–227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.