

Abstract
Group B Streptococcus (GBS) is a β-hemolytic Gram-positive bacterium that colonizes the lower genital tract of approximately 18% of women globally as an asymptomatic member of the gastrointestinal and/or vaginal flora. If established in other host niches, however, GBS is highly pathogenic. During pregnancy, ascending GBS infection from the vagina to the intrauterine space is associated with preterm birth, stillbirth, and fetal injury. In addition, vertical transmission of GBS during or after birth results in life-threatening neonatal infections, including pneumonia, sepsis, and meningitis. Although the mechanisms by which GBS traffics from the lower genital tract to vulnerable host niches are not well understood, recent advances have revealed that many of the same bacterial factors that promote asymptomatic vaginal carriage also facilitate dissemination and virulence. Further, highly pathogenic GBS strains have acquired unique factors that enhance survival in invasive niches. Several host factors also exist that either subdue GBS upon vaginal colonization or alternatively permit invasive infection. This review summarizes the GBS and host factors involved in GBS’s state as both an asymptomatic colonizer and invasive pathogen. Gaining a better understanding of these mechanisms is key to overcoming the challenges associated with vaccine development and identification of novel strategies to mitigate GBS virulence.
Introduction
Group B Streptococcus (GBS or Streptococcus agalactiae) is a leading cause of neonatal infectious morbidity and mortality globally. GBS is a β-hemolytic, Gram-positive coccus that asymptomatically colonizes the lower genital and gastrointestinal tracts but is an invasive pathogen in other host niches. Fetuses and neonates are uniquely susceptible to GBS infections, which most commonly include sepsis, pneumonia, meningitis, and encephalopathy (Figure 1) [1]. Maternal colonization with GBS is associated with stillbirth and preterm birth, and thus the sequelae of prematurity in the neonate (e.g. bronchopulmonary dysplasia) [2, 3]. Rarely, GBS causes maternal sepsis [4]. In newborns, the timing of GBS bacteremia or sepsis may be early-onset (within the first week), which is thought be associated with either an in utero infection or exposure during vaginal delivery. Alternatively, late-onset GBS disease presents after the first week, but within the first three months. To date, the only clinical intervention to prevent early- and late-onset disease in neonates is the administration of intravenous antibiotics to pregnant women during birth, known as intrapartum antibiotic prophylaxis (IAP).
Figure 1. Clinical Pathways in Maternal Group B Streptococcus (GBS) Colonization.
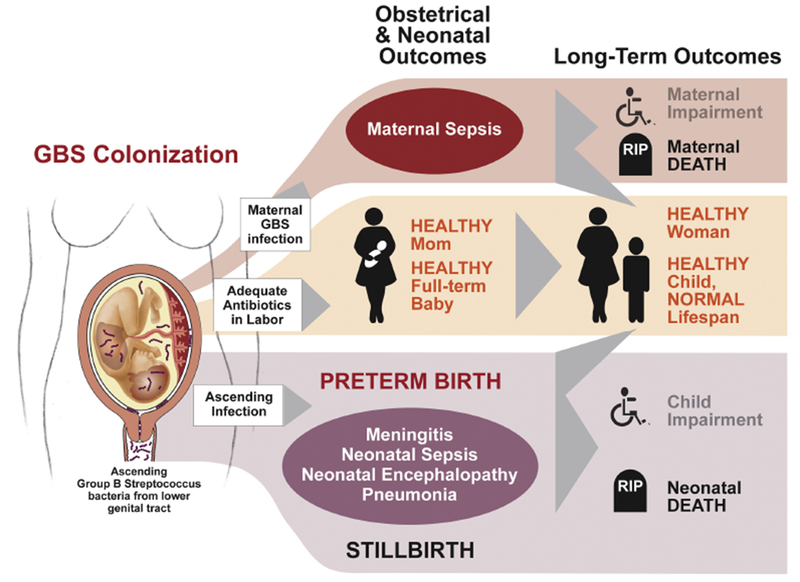
GBS is associated with several perinatal outcomes. In the case of adequate prophylaxis, most mothers and babies are healthy with normal lifespan. Virulent ascending GBS is associated with significant morbidity and mortality, some of which is not preventable with intrapartum prophylaxis. With adequate treatment of ascending infection, normal and healthy outcomes may be achieved. Figure adapted from Lawn, et al, 2017 [175].
As a colonizing microbe, GBS is detected in the gastrointestinal and genital tracts of approximately 18% of pregnant women globally; rates of colonization range from 1 in 3 pregnant women in the Caribbean to 1 in 6 women in Southern and Eastern Asia [5]. Interestingly, only a small proportion of people colonized with GBS experience invasive disease, suggesting that host-specific factors potentially play a role in determining an individual’s susceptibility to invasive disease. The most comprehensive systematic review and meta-analysis to date estimated the pooled incidence of neonatal morbidity and mortality to be 0.49 per 1000 worldwide, which includes GBS-associated preterm birth, stillbirth, and neonatal GBS infection [6]. Low- and middle-income countries experience increased rates of invasive GBS disease in neonates, and this is correlated with the lack of availability of IAP [7, 8]. In countries that have adopted screening protocols for maternal colonization and IAP, the burden of early-onset GBS disease has diminished dramatically; when data was first collected in the U.S. in 1997, the rate was 0.7 per 1,000 and in 2016 was 0.22 per 1,000 newborns [8, 9]. However, IAP appears to have little to no effect on the rate of late-onset disease in newborns or preterm birth and stillbirth, which are estimated to collectively affect 97,000 to 4 million pregnancies per year [1–3, 6, 7, 10]. Additionally, the widespread use of IAP has increased concern over antibiotic resistance in GBS. One multi-state study in the U.S.— where screening and IAP are standard of care— showed that 32% and 15% of GBS isolates were resistant to erythromycin and clindamycin, respectively [11]. In contrast, a study in South Africa— where IAP is not regularly implemented— found that only 4% of GBS isolates were resistant to erythromycin and 2% were resistant to clindamycin [12].
Overall, GBS remains a significant etiological agent of neonatal and maternal morbidity and mortality worldwide [1–3, 6, 7], and improved, rationally designed strategies to prevent invasive GBS disease are an urgent global health priority. Understanding the mechanisms that underlie GBS’s transition from asymptomatic colonizer to invasive pathogen is key to this effort. In this review, we discuss how GBS utilizes the same bacterial factors for both facets of its lifestyle, including how GBS regulates their expression using signal transduction systems. We also discuss unique factors acquired by GBS strains that are most commonly associated with disease. And finally, we review the several host processes known to either subdue GBS upon vaginal colonization or alternatively permit invasive infection.
GBS factors that promote vaginal colonization and virulence
The main host niche where GBS persists as an asymptomatic colonizer is the female rectovaginal tract. Many of the same strategies that GBS uses to colonize the vagina, such as binding host surfaces and subverting immune defenses, also promote dissemination and damage to vulnerable host niches such as newborns. Some GBS strains exhibit increased pathogenic potential due to the evolutionary acquisition of specified virulence factors that increase efficiency of dissemination, immune evasion, and tissue damage. At the cellular level, GBS tightly regulates the expression of virulence factors through signal transduction systems, which sense and respond to various host environments and subsequently enhance survival in host environments outside the lower genital tract. This section begins with a brief introduction of known signal transduction systems in GBS, and then summarizes specific factors that GBS uses to promote vaginal colonization as well as dissemination.
Signal transduction systems
In its transition from a commensal to pathogen, GBS encounters diverse host environments, which requires coordinated transcriptional control of genes involved in growth, cell wall components, stress response, metabolism, cellular processes, and virulence. To sense and respond to environmental changes, bacteria have evolved specialized signal transduction systems, such as the two-component systems (TCS). TCS are comprised of a membrane-associated sensor histidine kinase and a corresponding response regulator; the latter usually has DNA-binding abilities. Upon recognizing an external signal via the N-terminal domain, the C-terminal transmitter domain of the histidine kinase phosphorylates its cognate response regulator at a conserved active site aspartate residue, thereby causing conformational changes that alter the response regulator’s binding affinity to target DNA. Whole genomic analysis has revealed that GBS encode between 17-21 TCS [13, 14], although the function of just 7 TCS, namely CovR/S, DltR/S, RgfA/C, CiaR/H, FspR/S, LrdR/S, and SaeR/S, are described [14–24]. The most studied TCS in GBS is CovR/S, which is highly conserved in Streptococcus spp [15], In GBS, CovR/S serves as a global regulatory system, either activating or repressing the expression of over 100 genes, many of which are involved in virulence [15, 16, 25]. CovR/S regulation of several virulence genes is highly conserved among various strains of GBS (known as the “core regulon”) [26]. However, many regulatory targets of CovR/S differ among strains, suggesting that variability in pathogenic potential of GBS may be in part due to differences in sensing and responding to various host environments [26]. GBS also encodes one-component transcriptional regulators, such as MtaR, RovS, and RogB, which respond to signals from the cytosol (e.g. small molecules) or external environment via unknown sensing mechanisms [27–32]. Additionally, GBS encodes a eukaryotic-like signaling system comprising a serine/threonine kinase and its cognate phosphate Stp1, which overlaps with the CovR/S regulon [33, 34]. A membrane-bound protein known as Abx1 was also shown to mediate CovS function [32]. The bacterial-encoded factors that are under the control of many of these and other signaling systems are discussed in the sections below and summarized in Table 1 and Figure 2.
Table 1:
Summary of GBS colonization and virulence factors and their mechanisms of regulation order of their appearance in the main text
| Factor | Promotes colonization by: | Promotes invasive infection by: | Mechanism of regulation |
|---|---|---|---|
| Adhesins | |||
| Serine rich-repeat glycoproteins | Fibrinogen binding on vaginal and cervical epithelial cells (Ssr1) [38, 39] | Enhancing fibrinogen binding affinity (Ssr2) [41] Plasmin and plasminogen binding (Ssr2) [41] |
Unknown |
| HvgA | Unknown | Adherence to cells that make up the intestinal and blood brain barrier (BBB) [42] | CovR/S [42] |
| FbsA | Adherence to epithelial cells (? – not tested in vaginal model) | Adherence to epithelial cells [44] | CovR/S [16] RgfA/C [19, 20] RogB (in serotype III) [31] RovS [30] |
| FbsB | Unknown | Invading human epithelial cells [47] | CovR/S [48] RgfA/C [20] MtaR [29] |
| FbsC | Fibrinogen binding on vaginal and cervical epithelial cells [45] Promoting biofilm formation [45] |
Invasion of brain endothelial cells and crossing the BBB [46] | CovR/S [45] |
| Lmb | Binding laminin [50] (? – not tested in vaginal model) Acquiring environmental zinc [55] (? – not tested in vivo) |
Binding laminin on basement membranes of damaged cells [50, 51] Acquiring environmental zinc [55] (? – not tested in vivo) |
Unknown Some strains contain mobile genetic element that enhances expression [49] |
| C5a peptidase (ScpB) | Binding fibronectin [60–63] (? – not tested in vaginal model) Cleaving C5a [58, 59] (? – not tested in vaginal model) |
Binding fibronectin [60–63] Cleaving C5a [58, 59] |
RgfA/C (strain-specific) [19] CovR/S (strain-specific) [15] |
| Pili | Adhesion to vaginal and cervical epithelial cells [38] Resisting macrophage and neutrophil killing [74] (? – not tested in vaginal model) Resisting host antimicrobial peptides [74] (? – not tested in vaginal model) Biofilm formation [72] (? – not tested in vivo) |
Resisting macrophage and neutrophil killing [74] Disrupting barrier function of the lung and BBB [66, 70, 71, 73] Resisting host antimicrobial peptides [74] |
CovR/S (PI-1) [77] Ape1 (PI-1) [77] Rga (PI-2a) [78, 79] RogB (PI-2a) [66] Upstream hairpin structures (PI-2b, strain-specific) [80] |
| PbsP | Unknown, but up-regulated during vaginal colonization [24] | Adhering to and invading endothelial cells that make up the BBB [81, 83] | CovR/S [81] SaeR/S in the vagina [24] |
| SfbA | Invading vaginal and cervical cells [85] | Invading endothelial cells that make up the BBB [85] Invading astrocytes [86] |
Unknown |
| BibA | Binding to vaginal and cervical epithelial cells [89] Interfering with complement regulator C4bp [89] (? – not tested in vaginal model) |
Binding lung and intestinal epithelial cells [89] Interfering with complement regulator C4bp [89] |
CovR/S – dependent on pH [90] |
| Other virulence factors | |||
| Hemolytic pigment | Resisting ROS and neutrophil response [96, 98] | Disrupting barrier function of placental membranes [91, 105], lungs [106–108], and BBB [25, 109] Resisting NETs [105] Inflammasome induction [95] |
CovR/S [16] Stk1 [110] Abx1 [32] RovS [30] |
| Superoxide dismutase (SodA) | Resisting ROS [112] (? – not tested in vaginal model) | Resisting ROS [112] | RovS [30] |
| Hyaluronidase (HylB) | Dampening TLR2/4 recognition and signaling [121] | Dampening TLR2/4 recognition and signaling [121, 124] | CovR/S in strain A909 (serotype Ia) [25] |
| Sialic acid-rich capsular polysaccharide (CPS) | Interfering with complement pathway [131, 132] (? – not tested in vaginal model) Binding host Siglecs to prevent neutrophil activation [133, 134] (? – not tested in vaginal model) |
Interfering with complement pathway [131, 132] Binding host Siglecs to prevent neutrophil activation [133, 134] |
RogB (strain-specific) [31] CovR/S (strain-specific) [15, 16] |
| Cyclic di-AMP | Osmotic homeostasis [141] (? – not tested in vivo) | Osmotic homeostasis [141] (? – not tested in vivo) | CdnP [141] |
| CdnP | Unknown | Degrading cyclic di-AMP to avoid cGAS/STING recognition and signaling [142] | Unknown |
| D-alanylation of LTA | Resisting host AMPs and leukocyte killing [18, 146] (? – not tested in vaginal model) | Resisting host AMPs and leukocyte killing [18, 146] | DltR/S [17] |
Figure 2. Host and bacterial factors that contribute to GBS’s status as either an asymptomatic colonizer or invasive pathogen.
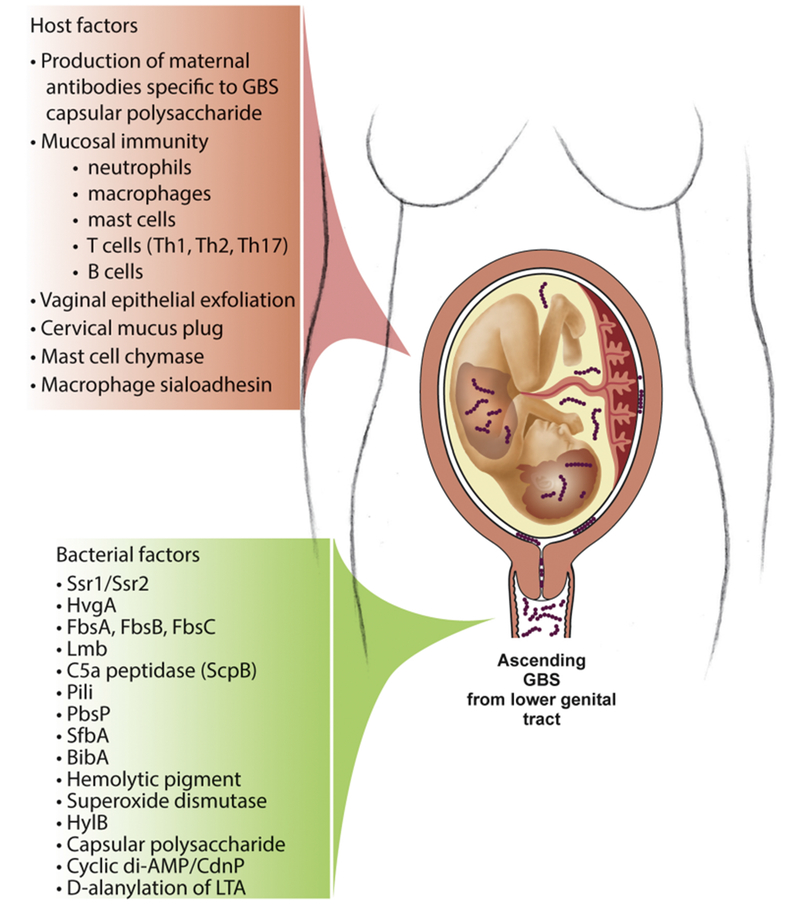
GBS typically colonizes the gastrointestinal/vaginal tract asymptomatically but is highly pathogenic in other host compartments. The host responses to GBS are multifaceted and can promote asymptomatic colonization and clearance or alternatively permit invasive infection and disease. Many of the bacterial factors that promote colonization are also involved in dissemination and tissue damage. GBS tightly regulates the expression of these factors using signal transduction systems, which sense and respond to variations in the external environment.
Adhesins
A major strategy that GBS employs to colonize the lower genital tract is adherence to epithelial cells via surface-associated adhesins. These include fibrinogen binding proteins (Fbs), the hypervirulent GBS adhesin (HvgA), laminin-binding protein (Lmb), C5a peptidase, pili, plasminogen binding surface protein (PbsP), fibronectin binding protein (SfbA), and the immunogenic bacterial adhesin (BibA) (Summarized in Table 1, Figure 2). A common ability conferred by these adhesins is GBS binding to components of the extracellular matrix (ECM). Some GBS adhesins execute additional functions, such as cellular invasion and immune evasion. Importantly, contact between GBS and host cells via adhesins is vital for establishing a niche in the gastrointestinal and vaginal mucosa and for invasion into other host compartments. In general, binding to surfaces improves GBS’s ability to cross host barriers, which is most likely why the enhanced ability to bind host cells increases GBS’s pathogenic potential [35].
Ssr1/Ssr2
Some highly virulent GBS clonal lineages have accomplished enhanced binding ability by evolving surface-associated adhesins that confer greater binding affinity to host cellular components than less pathogenic strains. Take, for example, the highly virulent clonal complex 17 (CC17) strains (capsular serotype III), which are frequently associated with neonatal meningitis. The GBS serine-rich repeat (SRR) glycoproteins are surface-associated Fbs that bind to a single tandem repeat region of human fibrinogen via a “lock, dock, and latch” mechanism [36]. Most GBS strains express Ssr1 and use it to bind to fibrinogen on vaginal and cervical epithelial cells via the “latch” domain, which promotes persistent vaginal colonization [37–40]. CC17 strains, on the other hand, produce a homolog of Ssr1, known as Ssr2 [36, 37]. In a mouse model of systemic infection, a CC17 strain lacking Ssr2 was less able to persist in the liver and brain, indicating that this adhesin promoted dissemination to peripheral organs [41]. Interestingly, Ssr2 was found to have greater binding affinity to fibrinogen compared to Ssr1 and could also bind immobilized plasmin and plasminogen, which Ssr1 could not [41]. Enhanced fibrinogen binding by Ssr2 likely provides CC17 strains with a competitive advantage in the vaginal niche while also promoting binding to epithelial and endothelial cells in invasive niches. Additionally, Ssr2-mediated plasminogen and plasmin binding could be a mechanism by which CC17 strains usurp host coagulation mechanisms to disseminate to peripheral organs, thereby making them more pathogenic than other GBS strains typically associated with asymptomatic colonization.
HvgA
Another surface-associated adhesin exclusively encoded in hyper-virulent CC17 strains is the hypervirulent GBS adhesin (HvgA), which is negatively regulated by the CovR/S TCS [42, 43]. Although the host ligand for HvgA has not been discovered, expression of this adhesin augmented adherence to intestinal epithelial cells, choroid epithelial cells, and microvascular endothelial cells which make up the blood brain barrier (BBB) [42]. Adherence to these cells via HvgA promoted intestinal and meningeal tropism, as HvgA-expressing GBS was better able to colonize and cross the intestinal barrier and BBB than a strain lacking this adhesin in orally inoculated mice [42].
FbsA, FbsB, FbsC
In addition to Ssr1/Ssr2, other fibrinogen-binding proteins encoded by GBS include FbsA, FbsB, and FbsC (also known as BsaB). FbsA and FbsC are central to GBS’s ability to adhere to human epithelial cells, which likely promotes vaginal colonization [44, 45]. In addition, FbsC mediates biofilm formation, which could facilitate vaginal colonization by binding to other commensal microbes in the gastrointestinal and/or vaginal tract [45]. Studies have also shown a role for FbsC in facilitating GBS dissemination; deletion of FbsC reduced GBS’s ability to bind to and invade human brain endothelial cells in vitro and colonize the brain in a murine model of systemic infection [46]. FbsB is also thought to be important for GBS dissemination by promoting bacterial invasion of human epithelial cells [47]. The signaling systems that regulate the expression of fbs genes are multifaceted and complex and appear to show strain dependence. For instance, FbsA expression is negatively regulated by CovR/S, RgfA/C, and RovS [16, 19, 20, 30], and in serotype III strains, positively regulated by RogB [31]. FbsB, on the other hand, is regulated by CovR/S (represses expression), RgfA/C (activates expression), and in some strains, MtaR [20, 29, 48]. Finally, FbsC is negatively regulated CovR/S [45].
Lmb
The lmb gene encodes the laminin binding protein of GBS (Lmb), which mediates binding to laminin, a major component of the basement membrane in tissues throughout the human body [49, 50]. The lmb gene is present in almost all human isolates of GBS [49], which suggests that this factor is crucial for colonization in humans. Additionally, the ability to bind laminin on the basement membrane of damaged tissues may be a key mechanism that GBS uses to disseminate in the human host. Indeed, deletion of lmb results in decreased invasion into human brain microvascular endothelial cells [51], indicating that Lmb contributes to GBS’s neurotropism. To bind laminin, the Lmb adhesin utilizes a zinc-binding pocket, which likely keeps Lmb protein in a structural confirmation conducive to laminin binding and/or enhances affinity to zinc finger motifs present in laminin [52–54]. Intriguingly, the zinc-binding pocket of Lmb also plays a role in zinc acquisition from the environment, which is important for bacterial survival and growth in various host niches [55]. Whether Lmb is under the control of a signal transduction system is unknown. However, increased Lmb surface expression and enhanced laminin-binding ability and has been reported in GBS strains harboring the mobile genetic element IS1548 upstream of the putative lmb promoter [49], indicating that some GBS strains have evolved to constitutively up-regulate Lmb.
C5a peptidase
Upstream of the lmb gene is the scpB gene, which encodes a surface-associated serine protease known as C5a peptidase or ScpB [56, 57]. C5a peptidase was first described to facilitate immune evasion by cleaving the human complement component C5a, a chemotaxin involved in neutrophil recruitment [58, 59]. Later, C5a peptidase was shown to promote GBS adherence to and invasion of human epithelial cells by binding fibronectin independently of its peptidase activity [60–63]. Although the regulation of C5a peptidase by RgfA/C and CovR/S is strain-specific [26, 64, 65], both response regulators in these TCS repress expression of the scpB gene [15, 19].
Pili
GBS encodes cell wall-anchored appendages known as pili, which protrude from the bacterial surface and are comprised of three structural subunit proteins: the pilus shaft backbone protein (PilB), the pilus tip (PilA, the pilus-associated adhesin), and the pilus base (PilC) [66–69]. Targeted mutagenesis has revealed distinct roles of the pili components in colonization and virulence. GBS strains lacking PilA were less able to adhere to vaginal and cervical epithelial cells in vitro and were out-competed by wild-type GBS in a vaginal model of colonization [38]. Furthermore, PilA knockouts were significantly impaired in their ability to adhere to human pulmonary epithelial cells and brain microvascular endothelial cells in vitro [66, 70], and in systemic mouse models, caused less neutrophil infiltration and bacterial dissemination to the brain [71]. PilB knockouts, on the other hand, were less able to form biofilms [72], transcytose monolayers of immortalized cervical epithelial cells [73], resist macrophage and neutrophil killing, and combat the effects of host antimicrobial peptides compared to wild-type strains [74]. Unsurprisingly, systemic infection with GBS lacking PilB resulted in enhanced clearance and reduced mortality [74].
The genetic loci that encode GBS pili are pilus islands-1 and -2, (PI-1 and PI-2, respectively) [67, 75, 76], and the PI-2 locus exists in two variants: PI-2a (present in the majority of GBS isolates belonging to all capsular serotypes) and PI-2b (restricted to CC17 and a few other clinical isolates) [67]. All characterized GBS strains contain at least one variant or a combination of the two pilus islands [67, 75, 76], and variation among pili likely account for differential ability to colonize and invade among GBS strains. Variation also exists in the regulatory mechanisms for the pili genetic loci. PI-1 expression is repressed by CovR/S TCS and activated by Ape1, an Arac-type regulator encoded immediately upstream of the PI-1 locus [77]. Interestingly, acidic pH activated the expression of PI-1 independently of CovR/S and Ape1 [77], suggesting that another unidentified signal transduction system is involved in PI-1 regulation. On the other hand, PI-2a is positively regulated by the transcriptional regulators RogB, which is encoded upstream of the PI-2a locus, and Rga, which binds to the PI-2a promoter region [66, 78, 79]. Intriguingly, differential expression of PI-2b among strains may be due to variations in DNA folding; a recent study by Périchon, et al found that decreased expression of PI-2b in GBS strain BM110 compared to strain A909 was due to the presence of a 43-base pair DNA hairpin structure upstream of the PI-2b operon in BM110 [80]. Further studies on the variations in regulatory mechanisms of GBS pili will improve our understanding of how GBS pili contribute to adhesion, biofilm formation, and resistance to host immune mechanisms in GBS isolates.
PbsP
PbsP is a cell wall-associated surface protein that binds to the ECM component plasminogen [81]. PbsP expression is highly conserved among GBS strains, but strains vary in the level of PbsP surface expression [81]. GBS employs PbsP to promote vaginal colonization; in a murine model, the pbsP transcript was significantly increased in the vaginal tract compared to GBS grown in culturally defined medium, and a GBS strain lacking PbsP was a deficient vaginal colonizer compared to wild-type GBS [24]. Interestingly, vaginal lavage fluid from mice induced SaerR/S-dependent up-regulation of pbsP [24], indicating that the histidine kinase SaeS senses a signal in the vaginal milieu to control the expression of pbsP through the SaeR response regulator. Expression of the pbsP locus is also repressed by CovR/S [81, 82], but direct binding of CovR to the pbsP promoter could not be proven, suggesting that this regulation may be indirect [81]. Plasminogen-binding via PbsP also promotes invasive infection, as PbsP is required for GBS to adhere to brain endothelial cells and cross the BBB [81, 83]. Additionally, PbsP confers GBS the ability to bind human vitronectin, a glycoprotein abundant in the ECM and in serum, which promotes GBS adherence to and invasion of epithelial cells [84]. Because PbsP is surface-associated, highly conserved, up-regulated in vivo, and is important for both vaginal colonization and invasion, it represents a promising target for vaccine development.
SfbA
The Streptococcal fibronectin binding protein (SfbA) is well conserved among GBS strains and promotes invasion of human vaginal and cervical cells, brain microvascular endothelial cells, and astrocytes [85, 86]. Thus, SfbA is likely involved in bacterial tropism to the gastrointestinal/vaginal tract and central nervous system. In a murine model of hematogenous GBS meningitis, a SfbA-deficient GBS strain was less able to colonize the brain, indicating that SfbA mediates GBS translocation of the BBB [85]. The mechanisms that regulate SfbA expression in GBS remain unexplored. Interestingly, hosts have evolved a strategy to counteract GBS fibronectin binding to prevent bacterial dissemination. A recent study revealed that in response to GBS infection, mast cells release a protease known as chymase, which degrades fibronectin and thus impairs bacterial adherence to fibronectin and downstream dissemination [87]. Mice lacking mast cell chymase experienced greater rates of preterm birth and bacterial dissemination when infected with wild-type GBS compared to chymase-sufficient mice. Furthermore, this effect was abrogated when chymase-deficient mice were infected with GBS lacking SfbA [87]. Together, these findings indicate that fibronectin degradation by host mast cell chymase prevents SfbA-mediated dissemination.
BibA
The GBS immunogenic bacterial adhesin, BibA, is another highly conserved, multifunctional, cell wall-tethered protein that GBS uses to promote colonization at mucosal surfaces and invasion to peripheral compartments [88, 89]. BibA is involved in adherence to human cervical, lung, and intestinal epithelial cells, and was proposed to interfere with host antimicrobial defense mechanisms by binding to the human complement regulator C4-binding protein [88, 89]. Recombinant BibA protein was shown to induce protective, opsonizing antibodies against GBS in mice [88], and high surface expression of BibA on GBS enhanced opsonophagocytic killing of GBS in vitro [90]. These findings indicate that BibA is a target of effective host immune responses to GBS and could be a potential vaccine candidate. Expression of BibA, however, is repressed by the CovR/S TCS and dependent on pH [16, 90]; BibA expression is up-regulated in neutral versus acidic environmental conditions [90]. Consequently, a vaccine targeting BibA may be more effective in protecting against GBS infection of the blood, where pH is close to neutral, versus the lower genital tract, where pH is low.
Hemolytic pigment
Hemolytic activity in GBS is due to the ornithine rhamnolipid pigment (hereafter called “hemolytic pigment” or “pigment”) [91], which is produced by the gene products of the cyl operon [92, 93]. Several genes within the cyl operon have homology to those involved in fatty acid biosynthesis, and the cylE gene, which encodes an N-acyl transferase, is necessary for pigment production [91–93]. Transcription of cyl genes is negatively regulated by the CovR/S TCS, so loss of function in this TCS results in a hyper-hemolytic/hyper-pigmented phenotype in GBS [16]. The hemolytic pigment contains a polyene chain consisting of 12 double-bonds [91, 94], and serves both as a potent cytotoxin [91, 95] and an antioxidant with reactive oxygen species (ROS)-quenching properties [96], In a murine model of vaginal colonization, a GBS strain lacking the hemolytic pigment was a deficient vaginal colonizer compared to wild-type GBS and was associated with amplified neutrophil response in the vagina [97]. However, GBS over-expressing the hemolytic pigment was more readily cleared from the murine vagina [98, 99], likely due to an inflammatory response resulting in an infiltration of neutrophils [98] and/or mast cell degranulation [99]. The finely tuned expression of the hemolytic pigment in the lower genital tract, therefore, is essential to GBS survival; GBS must achieve an optimum level of pigment production to resist ROS but also avoid inflammation and subsequent clearance.
In addition to modulating vaginal colonization, the hemolytic pigment permits GBS dissemination to host niches outside the vagina and facilitates tissue damage. In a murine model of vaginal colonization, non-hemolytic GBS was less likely to disseminate or cause fetal injury [100], suggesting the hemolytic pigment plays a role in GBS’s ability to breach the placental barrier. Indeed, hyper-hemolytic GBS strains lacking CovR were found to more readily invade amniotic epithelial cells, cause amniotic epithelial cell barrier disruption, and penetrate the human placenta compared to wild-type GBS and non-hemolytic GBS also lacking CovR (GBSΔcovRΔcylE) [91]. Furthermore, hyper-hemolytic GBS with CovR/S mutations have been isolated from women in preterm labor [91] and non-pregnant adults with invasive infections [101–104], suggesting that some GBS isolates have evolved the ability to constitutively over express the hemolytic pigment and that these are associated with human disease. In a non-human primate (NHP) study of GBS infection during pregnancy, GBS lacking CovR invaded the amniotic cavity more efficiently, resulting in uterine contractions and inflammation indicative of preterm labor [105]. Notably, even though hyper-hemolytic GBS caused an infiltration of neutrophils to the chorioamniotic (placental) membranes of NHP and the subsequent release of neutrophils extracellular traps (NETS), hyper-hemolytic GBS were able to resist killing by NETs [105]. Other inflammatory cascades initiated by the pigment during pregnancy, such as the NLRP3 inflammasome, can likewise result in fetal injury and preterm birth [95].
Neonates, particularly those born prematurely, are especially susceptible to the toxic effects of the hemolytic pigment. Many infants acquire early onset GBS by aspirating infected vaginal or amniotic fluid, resulting in colonization of the lung. Higher expression of the hemolytic pigment correlates to greater cytotoxicity of the very cells that constitute the mucosal barrier of the lung: alveolar epithelial cells [106] and capillary endothelial cells [107]. Additionally, mutant GBS strains lacking the cylE gene are less capable of adhering to, invading, and initiating inflammatory cascades in lung epithelial cells [108]. Once GBS breaches the lung’s barrier, it deploys several virulence factors (such as adhesins and invasins described elsewhere in this review) to disseminate to peripheral organs, resulting in septicemia and neurotropism. At the BBB, the hemolytic pigment facilitates bacterial penetration into the brain and promotes acute inflammatory responses in microvascular endothelial cells, thus contributing to the severity of GBS meningitis [25, 109].
Beyond pH [90], little is known about the stimuli sensed by CovS to induce or relieve expression of the cyl genes via CovR. Other strain-specific regulators have been shown to alter expression of cyl genes, such as the sensor kinase Stk1 [110, 111] and the transmembrane protein Abx1 [32], which interact with CovR and CovS, respectively. Additionally, the one-component transcriptional regulator RovS can bind to and activate genes in the cyl operon [30]. In short, the signal transduction mechanisms responsible for regulating expression of the hemolytic pigment are complex. Further studies on how the hemolytic pigment is regulated in the vagina and in vulnerable niches are critical to understanding GBS’s conversion from commensal to pathogen.
Superoxide dismutase
In addition to the hemolytic pigment, GBS resists host ROS during infection using the Mn2+-cofactored superoxide dismutase, SodA, which detoxifies superoxide radical species (O2−) [112, 113]. A GBS mutant deficient in SodA was more susceptible to killing following macrophage internalization compared to wild-type GBS, and in a systemic murine model of infection, was attenuated for virulence [112]. The transcriptional regulator RovS was shown to bind directly to the promoter region of the sodA gene, and a rovS deletion mutant exhibited over a two-fold decrease in sodA expression [30]. These data indicate that RovS is an activator of sodA, but the signals that activate or repress sodA expression through RovS are unknown and warrant further study.
HylB
The GBS hyaluronidase, also known as HylB, is an endoglycosidase that cleaves glycosaminoglycan chains, such as hyaluronic acid (HA) into disaccharides [114]. HylB activity has long been demonstrated to correlate to GBS’s invasive potential; in a 1978 study, increased expression of HylB was observed in GBS strains isolated from infected neonates [115, 116]. Similarly, in 1989, the highly virulent GBS clonal type now known as CC17 was shown to have increased expression of hyaluronidase compared to strains less often associated with invasive disease in neonates [116–118]. Even in a GBS strain that lacks the potent hemolytic pigment toxin, over-expression of HylB confers hypervirulence [119].
GBS mutants lacking HylB induce more pro-inflammatory cytokines in vitro and in vivo [120], and the precise mechanism of HylB-mediated immune suppression was elucidated in a recent study by Kolar, et al [121]. HylB’s substrate, HA, is present in almost all human tissues and is important for stability and structure of the ECM as well as immune surveillance [122, 123]. Upon sterile or microbial-induced tissue injury, HA is degraded into multimeric fragments by ROS, which act as damage-associated molecular patterns (DAMPs) that bind to pattern recognition receptors such as TLRs on innate immune cells to initiate a pro-inflammatory response. The HA dimers generated by GBS HylB inhibit TLR2 and TLR4 inflammatory pathways by blocking the direct binding of stimulatory HA fragments and other pathogen-associated molecular patterns (PAMPs) to TLR2 and TLR4 [121]. Using a non-pregnant mouse model of vaginal inoculation, Kolar, et al. established that the immune-suppressing properties of HylB facilitate vaginal colonization of GBS [121]. In pregnant mice, wild-type GBS caused increased ascending infection into the uterus, decreased uterine inflammation, and higher rates of fetal demise compared to HylB-deficient GBS [124], indicating suppression of inflammatory responses in the uterus also facilitates GBS-associated fetal injury. These findings show that like many other virulence factors, GBS uses HylB to promote both vaginal colonization and invasive disease. GBS therefore likely regulates expression of the hylB gene to modulate the immune response during colonizing and invasive states. One study found that CovR activated hylB expression in GBS strain A909 (capsular serotype la), suggesting that the CovR/S TCS may be involved in hylB gene regulation [25]. Beyond this, little evidence exists about how, when, and where GBS modulates hylB expression during colonization or invasive infection.
Sialic acid-rich capsular polysaccharide
GBS is encased by a capsular polysaccharide (CPS) rich in sialic acid and made up of one of the ten capsular serotypes: Ia, Ib, or II-IX. The CPS is assembled by the gene products of the cps operon [125–127] and is known to promote virulence; CPS-deficient GBS strains have attenuated virulence in systemic models of infection [128–130]. The sialic acid present in the CPS is a nine-carbon monosaccharide that is also widely distributed on the terminal glycan structures present on host cell surfaces. Because of this, the CPS sialic acid facilitates GBS innate immune evasion by molecular mimicry. For instance, CPS sialic acid prevents deposition of the C3b complement component on the surface of GBS and subsequent opsonophagocytosis [131, 132], likely by interfering with regulatory complement enzymes. Additionally, CPS sialic acid binds to host inhibitory Siglecs (Sialic acid-binding immunoglobulin-type lectins), which reduces neutrophil activation and inflammation of the placental membranes [133–135], dampens pro-inflammatory signaling in vivo [136], and facilitates bacterial dissemination [136]. Interestingly, diminished CPS expression enhances GBS’s ability to invade epithelial and endothelial cells in vitro [137, 138], and acapsular, hyper-hemolytic GBS more readily disseminates to peripheral organs and crosses the BBB in vivo [139]. Also, sialoadhesin present in splenic macrophages binds CPS sialic acid and promotes phagocytic uptake, thereby restricting GBS dissemination [140]. These findings indicate that up- and down-regulation of CPS confers differential advantages to GBS in certain situations during infection. RogB, a transcriptional regulator of the RofA-like protein family, suppresses expression of the first gene in the cps operon, cpsA [31], which encodes a cell-wall anchored protein involved in surface attachment of capsular polysaccharides [127]. However, the gene locus encoding RogB is absent in some GBS strains, including A909 (serotype la) and COH1 (serotype III), indicating that its regulation is strain-specific. CovR/S activation of the cps genes is also strain-specific [15, 16]. To date, our understanding of gene regulation by the cps operon is limited, and further research will lend valuable clues into the mechanisms that GBS uses to survive various host defenses.
Cyclic di-AMP and CndP
GBS synthesizes the signaling nucleotide cyclic di-AMP, which plays a critical role in maintaining osmotic homeostasis [141]. Osmotic regulation through cyclic di-AMP is likely a key strategy that GBS uses to survive osmotic stress encountered in various host niches, such as the acidic vagina or more alkaline blood. However, the cyclic di-AMP synthesized by GBS is recognized by intracellular host pattern recognition receptors c-GAS and STING, leading to the production of type 1 interferons (e.g. IFN-β) [142], which promote neutrophil recruitment and subsequent bacterial clearance [142–144]. It was recently discovered that GBS encodes a cell wall-anchored ectonucleotidase, CdnP, which degrades extracellular cyclic di-AMP [142]. Interestingly, inactivation of CdnP led to increased IFN-β and reduced bacteremia in wild-type mice in a murine model of intravenous GBS infection. This effect was not observed in mice lacking cGAS or STING, indicating that GBS expresses CdnP to dampen STING-mediated detection of cyclic di-AMP and subsequent pro-inflammatory cascades to promote invasive infection [142]. Whether GBS fine-tunes the expression of CdnP to promote cyclic di-AMP-mediated osmotic homeostasis in some host environments and immune evasion in others remains unknown.
D-alanylation of LTA
Like many Gram-positive bacteria, GBS contains a cell wall densely packed with lipoteichoic acid (LTA), a phosphate-containing polymer that contains glycosyl and D-alanine ester substituents. LTA mediates ligand binding and surface charge, and the incorporation of D-alanine to LTAs can modify these properties in bacteria. For instance, D-alanylation of LTAs decreases the net negative charge on the bacterial surface, which confers improved resistance to cationic antimicrobial compounds [145]. In GBS, D-alanylation of LTAs is regulated by the dltRSABCD operon, which encodes the DltR/S TCS and structural genes dltA-D [17]. GBS strains lacking dltA cannot produce the D-alanine-D-alanyl carrier ligase and are severely attenuated for virulence compared to wild-type GBS due to increased susceptibility to host cationic defensins and leukocyte killing [18, 146]. GBS uses the DltR/S TCS to control the level of D-alanine esters in LTAs; upon activation by DltS, DltR binds to promoter DNA to induce the expression of the dlt operon [17]. Although the specific signal sensed by DltS is unknown, up-regulation of the dlt operon is observed when amounts of D-alanine esters in LTAs decrease [17], suggesting that GBS may detect changes in their own surface charge or cell wall composition. Additionally, the presence of host cationic antimicrobial peptides may trigger activation of dlt genes by the DltR/S TCS. In fact, antimicrobial peptides have been shown to trigger TCS regulation of virulence-associated genes in Group A Streptococci [147, 148].
Host factors that mediate GBS’s transition from asymptomatic colonizer to invasive pathogen
Although some host defenses are effective at clearing GBS, others are permissive to or exploited by GBS, allowing GBS to gain a foothold in invasive niches. Thus, many host responses to GBS are promising targets for therapeutic intervention. Only a small proportion of people who are colonized with GBS experience invasive infection, indicating host-specific responses to GBS may vary by individual. More research on GBS-host interactions as well as the variations or deficiencies in the host response to GBS are critical to identifying individuals most at risk for invasive GBS disease and developing effective therapies. The known host factors involved in GBS colonization and disease are discussed below and summarized in Table 2.
Table 2:
Host factors that mediate GBS’s transition from asymptomatic colonizer to invasive pathogen in order of their appearance in the main text
| Factor | Effect on GBS colonization/virulence | Intervention |
|---|---|---|
| Maternal CPS-specific antibody production | Associated with asymptomatic vaginal colonization [149, 150] Associated with enhanced vaginal clearance [150] Associated with reduced risk of early onset disease in newborns [151] |
CPS-based conjugate vaccines [152–158] |
| Mucosal immunity | Neutrophils [97, 98, 161], macrophages [97], mast cells [99] promote clearance of GBS from the vagina Soluble inflammatory mediators promote clearance of GBS from the vagina [97, 98, 161] IL-17 and IL-17+ cells promoted clearance of persistent strain [161] Humoral immunity in vaginal mucosa promotes clearance of GBS from the vagina [162] Deficiencies may increase risk for invasive disease |
Mucosal vaccination tested using in vivo mouse models [162] |
| Vaginal epithelial exfoliation | Disrupts barrier function of vaginal epithelia, promotes ascending GBS infection [170] | None known |
| Cervical mucus plug | Physical barrier between lower genital tract and uterus Can secrete pro-inflammatory cytokines, chemokines (not tested in vivo) [171–173] Can retain antibiotic drugs that kill GBS [174] Antimicrobial compounds within CMPs insufficient to kill GBS [174] |
None known |
| Mast cell chymase | Inhibits GBS’s ability to bind fibronectin [87] | None known |
| Macrophage sialoadhesin | Binds GBS sialic acid, promotes phagocytic uptake and clearance [140] | None known |
Production of CPS-specific antibodies
The adaptive immune response to the GBS CPS plays an important role in GBS’s state as an asymptomatic colonizer or invasive pathogen. Clinical studies have suggested a correlation between naturally acquired maternal CPS-specific antibodies and asymptomatic vaginal carriage and/or clearance [149, 150], and maternal titers of CPS-specific antibodies >1μg/ml at birth were associated with a 70% reduction in the risk of early onset disease in newborns [151]. Given the CPS-dependent antibody protection observed in these studies, the CPS is a frequent target for GBS vaccine candidates, and several multivalent CPS conjugate vaccines have shown promising safety and immunogenicity outcomes in phase I and II clinical trials [152–158]. However, the diversity of GBS CPS types and CPS switching are significant challenges to the development of broadly efficacious GBS vaccines targeting the CPS [159, 160].
Mucosal immune response
Recently, in vivo models have revealed that GBS vaginal colonization invokes the recruitment of neutrophils [97, 98, 161], macrophages [97], and mast cells [99], which promote clearance. In addition, several soluble inflammatory mediators, including histamine, 1β, IL-6, IL-8, IL-23, and IL-17, have been shown to be important for decreased colonization [97, 98, 161]. The mucosal T cell response to GBS colonization is poorly defined, although one study found that cytokines involved in Th1, Th2, and Th17 differentiation pathways are important for decreased colonization [97]. Similarly, another study found that IL-17 and IL-17-producing cells in the vaginal tract promote clearance of a persistent GBS strain [161]. The mucosal humoral response to GBS is also incompletely understood, but a recent study showed that B cell-deficient mice vaginally inoculated with GBS demonstrated prolonged time to colonization clearance compared to wild-type mice [162]. This trend was recapitulated immunoglobulin-deficient mice, suggesting a role for GBS-specific antibodies in clearing GBS from the vagina [162]. Further, mice immunized intranasally with heat-killed GBS more readily cleared GBS from the vagina in a serotype-specific manner [162]. Together, these data indicate that specialized immune responses to GBS at the mucosal surface mediate vaginal clearance and thus have implications for designing effective delivery routes for vaccines and/or therapeutics. Some evidence suggests that deficiencies in mucosal immune response may render some hosts less able to combat persistent vaginal colonization and/or invasive GBS disease. For instance, a recent study found that HIV-positive women vaginally colonized with GBS experienced a 19-39% reduction in the concentration of transplacental transfer of GBS surface protein antibodies compared to HIV-negative women [163]. This could, in part, explain increased susceptibility to GBS infection in HIV-exposed uninfected infants [164].
Vaginal epithelial exfoliation
A common host defense mechanism use to clear mucosal surfaces of adherent pathogenic bacteria is epithelial exfoliation, a process in which epithelial cells lose their tight junctions and detach from the basement membrane [165]. In fact, Neisseria gonorrheae and uropathogenic E. coli have evolved specific mechanisms to prevent clearance from the lower genital tract by epithelial exfoliation [166–169]. A recent study by Vornhagen, et al showed that GBS induce exfoliation of vaginal epithelial cells by activating β-catenin signaling to induce epithelial-to-mesenchymal transition (EMT) [170]. Surprisingly, GBS-induced exfoliation of vaginal epithelial cells had no effect on vaginal colonization within the short time window of the study (3 days), but instead permitted ascending infection to the intrauterine space, resulting in fetal demise and preterm birth in pregnant mice [170]. The specific GBS factors involved in binding integrin to induce EMT and how EMT (or lack thereof) impacts long-term vaginal colonization are unknown and warrant further study.
CMP composition
During pregnancy, a dense and viscous mucoid structure known as the cervical mucus plug (CMP) forms in the cervical canal. In addition to acting as a physical barrier, CMPs can harbor pro-inflammatory cytokines as well as a variety of antimicrobial compounds, including lysozyme, lactoferrin, calprotectin, and secretory leukoprotease inhibitor 1, which vary by individual [171–173]. Although our understanding of the CM P’s role in defending against pathogenic bacteria is poor, a recent study examining 60 human CMPs showed that CMP-associated proteins activated leukocytes in whole blood, resulting in increased GBS killing [174]. However, the concentration of antimicrobial factors was insufficient to kill several strains of GBS except in those CMPs still harboring antibiotics administered during Caesarean section and prior to CMP collection [174]. These findings suggest that GBS have evolved mechanisms to resist the antibiotic properties of CMPs. Resisting the antimicrobial effects of CMPs may be a mechanism GBS employ to promote vaginal colonization during pregnancy, a host state that has unique immune characteristics. Additionally, CMP antimicrobial resistance may be a factor in permitting GBS to cross the cervix, although the mechanisms by which GBS bind to and ascend the CMP remain unknown.
Conclusion
Group B Streptococcus, or GBS, leads a double life as both an asymptomatic colonizer and a potent pathogen. Although most people who are colonized with GBS do not experience invasive disease, invasion of GBS into host niches outside of the gastrointestinal and/or vaginal mucosa causes severe damage to the host, resulting in devastating clinical outcomes in pregnant women and newborns. Even with the development of screening and IAP protocols in some countries, the global burden of GBS disease remains high. New, rationally designed strategies to prevent GBS colonization and disease are an urgent priority and require a comprehensive understanding of the dynamics of commensal versus invasive states. The factors mediating GBS’s transition from commensal to pathogen are derived both from the bacteria themselves and from the host. Many of the same strategies that GBS uses to reside in the lower genital tract also allow it to wreak havoc in invasive niches. Further, GBS tightly regulates the expression of bacterial products such as adhesins, the hemolytic pigment, and immune modulators, to best equip itself for its surroundings. While several host defenses to GBS have been identified, they vary by individual and can be inadequate to suppress GBS or are exploited by GBS for its dissemination. Despite prolific research in recent years, our understanding of GBS’s dual lifestyle as a commensal and pathogen is perforated with many knowledge gaps. More basic research revealing the molecular underpinnings of signal transduction systems, bacterial products, and the host response to GBS is essential to developing novel therapeutic interventions that keep GBS at bay.
Highlights.
Group B Streptococcus is a major cause of preterm birth and neonatal infection.
GBS produces factors that promote asymptomatic vaginal colonization and virulence.
Host defenses against GBS are multifaceted, and some can be exploited by GBS.
Knowing how GBS shifts from commensal to pathogen is key to therapeutic advances.
Acknowledgements:
We would like to thank Jan Hamanishi for help with graphic design, and Connie Hughes for administrative support. This work was supported by funding from the National Institutes of Health grants R01AI112619, R01AI33976, R01AI00989, and R21AI125907 to L.R. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest: None.
References:
- [1].Madrid L, Seale AC, Kohli-Lynch M, Edmond KM, Lawn JE, Heath PT, et al. Infant Group B Streptococcal Disease Incidence and Serotypes Worldwide: Systematic Review and Meta-analyses. Clin Infect Dis. 2017;65:S160–S72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Seale AC, Blencowe H, Bianchi-Jassir F, Embleton N, Bassat Q, Ordi J, et al. Stillbirth With Group B Streptococcus Disease Worldwide: Systematic Review and Meta-analyses. Clin Infect Dis. 2017;65:S125–S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bianchi-Jassir F, Seale AC, Kohli-Lynch M, Lawn JE, Baker CJ, Bartlett L, et al. Preterm Birth Associated With Group B Streptococcus Maternal Colonization Worldwide: Systematic Review and Meta-analyses. Clin Infect Dis. 2017;65:S133–S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hall J, Adams NH, Bartlett L, Seale AC, Lamagni T, Bianchi-Jassir F, et al. Maternal Disease With Group B Streptococcus and Serotype Distribution Worldwide: Systematic Review and Meta-analyses. Clin Infect Dis. 2017;65:S112–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Russell NJ, Seale AC, O’Driscoll M, O’Sullivan C, Bianchi-Jassir F, Gonzalez-Guarin J, et al. Maternal Colonization With Group B Streptococcus and Serotype Distribution Worldwide: Systematic Review and Meta-analyses. Clin Infect Dis. 2017;65:S100–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Seale AC, Bianchi-Jassir F, Russell NJ, Kohli-Lynch M, Tann CJ, Hall J, et al. Estimates of the Burden of Group B Streptococcal Disease Worldwide for Pregnant Women, Stillbirths, and Children. Clin Infect Dis. 2017;65:S200–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Le Doare K, O’Driscoll M, Turner K, Seedat F, Russell NJ, Seale AC, et al. Intrapartum Antibiotic Chemoprophylaxis Policies for the Prevention of Group B Streptococcal Disease Worldwide: Systematic Review. Clin Infect Dis. 2017;65:S143–S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].CDC. Active Bacterial Core Surveillance (ABCs) Report Emerging Infections Program Network group B Streptococcus, 1997 1998.
- [9].CDC. Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Group B Streptococcus, 2016 2016.
- [10].Nanduri SA, Petit S, Smelser C, Apostol M, Alden NB, Harrison LH, et al. Epidemiology of Invasive Early-Onset and Late-Onset Group B Streptococcal Disease in the United States, 2006 to 2015: Multistate Laboratory and Population-Based Surveillance. JAMA Pediatr. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, et al. Epidemiology of invasive group B streptococcal disease in the United States, 1999-2005. JAMA. 2008;299:2056–65. [DOI] [PubMed] [Google Scholar]
- [12].Africa CWJ, Kaambo E. Group B Streptococcus Serotypes in Pregnant Women From the Western Cape Region of South Africa. Front Public Health. 2018;6:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tettelin H, Masignani V, Cieslewicz MJ, Eisen JA, Peterson S, Wessels MR, et al. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc Natl Acad Sci U S A. 2002;99:12391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Faralla C, Metruccio MM, De Chiara M, Mu R, Patras KA, Muzzi A, et al. Analysis of two-component systems in group B Streptococcus shows that RgfAC and the novel FspSR modulate virulence and bacterial fitness. MBio. 2014;5:e00870–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jiang SM, Cieslewicz MJ, Kasper DL, Wessels MR. Regulation of virulence by a two-component system in group B streptococcus. Journal of bacteriology. 2005;187:1105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lamy MC, Zouine M, Fert J, Vergassola M, Couve E, Pellegrini E, et al. CovS/CovR of group B streptococcus: a two-component global regulatory system involved in virulence. Mol Microbiol. 2004;54:1250–68. [DOI] [PubMed] [Google Scholar]
- [17].Poyart C, Lamy MC, Boumaila C, Fiedler F, Trieu-Cuot P. Regulation of D-alanyl-lipoteichoic acid biosynthesis in Streptococcus agalactiae involves a novel two-component regulatory system. J Bacteriol. 2001;183:6324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Poyart C, Pellegrini E, Marceau M, Baptista M, Jaubert F, Lamy MC, et al. Attenuated virulence of Streptococcus agalactiae deficient in D-alanyl-lipoteichoic acid is due to an increased susceptibility to defensins and phagocytic cells. Mol Microbiol. 2003;49:1615–25. [DOI] [PubMed] [Google Scholar]
- [19].Spellerberg B, Rozdzinski E, Martin S, Weber-Heynemann J, Lutticken R. rgf encodes a novel two-component signal transduction system of Streptococcus agalactiae. Infect Immun. 2002;70:2434–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Al Safadi R, Mereghetti L, Salloum M, Lartigue MF, Virlogeux-Payant I, Quentin R, et al. Two-component system RgfA/C activates the fbsB gene encoding major fibrinogen-binding protein in highly virulent CC17 clone group B Streptococcus. PLoS One. 2011;6:e14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Quach D, van Sorge NM, Kristian SA, Bryan JD, Shelver DW, Doran KS. The CiaR response regulator in group B Streptococcus promotes intracellular survival and resistance to innate immune defenses. J Bacteriol. 2009;191:2023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mu R, Cutting AS, Del Rosario Y, Villarino N, Stewart L, Weston TA, et al. Identification of CiaR Regulated Genes That Promote Group B Streptococcal Virulence and Interaction with Brain Endothelial Cells. PLoS One. 2016;11:e0153891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Deng L, Mu R, Weston TA, Spencer BL, Liles R, Doran KS. Characterization of a two-component system transcriptional regulator LtdR that impacts Group B Streptococcal colonization and disease. Infect Immun. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cook LCC, Hu H, Maienschein-Cline M, Federle MJ. A Vaginal Tract Signal Detected by the Group B Streptococcus SaeRS System Elicits Transcriptomic Changes and Enhances Murine Colonization. Infect Immun. 2018;86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lembo A, Gurney MA, Burnside K, Banerjee A, de los Reyes M, Connelly JE, et al. Regulation of CovR expression in Group B Streptococcus impacts blood-brain barrier penetration. Mol Microbiol. 2010;77:431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jiang SM, Ishmael N, Dunning Hotopp J, Puliti M, Tissi L, Kumar N, et al. Variation in the group B Streptococcus CsrRS regulon and effects on pathogenicity. J Bacteriol. 2008;190:1956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ulrich LE, Koonin EV, Zhulin IB. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 2005;13:52–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shelver D, Rajagopal L, Harris TO, Rubens CE. MtaR, a regulator of methionine transport, is critical for survival of group B streptococcus in vivo. J Bacteriol. 2003;185:6592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bryan JD, Liles R, Cvek U, Trutschl M, Shelver D. Global transcriptional profiling reveals Streptococcus agalactiae genes controlled by the MtaR transcription factor. BMC Genomics. 2008;9:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Samen UM, Eikmanns BJ, Reinscheid DJ. The transcriptional regulator RovS controls the attachment of Streptococcus agalactiae to human epithelial cells and the expression of virulence genes. Infect Immun. 2006;74:5625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gutekunst H, Eikmanns BJ, Reinscheid DJ. Analysis of RogB-controlled virulence mechanisms and gene repression in Streptococcus agalactiae. Infect Immun. 2003;71:5056–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Firon A, Tazi A, Da Cunha V, Brinster S, Sauvage E, Dramsi S, et al. The Abi-domain protein Abx1 interacts with the CovS histidine kinase to control virulence gene expression in group B Streptococcus. PLoS Pathog. 2013;9:e1003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rajagopal L, Clancy A, Rubens CE. A eukaryotic type serine/threonine kinase and phosphatase in Streptococcus agalactiae reversibly phosphorylate an inorganic pyrophosphatase and affect growth, cell segregation, and virulence. J Biol Chem. 2003;278:14429–41. [DOI] [PubMed] [Google Scholar]
- [34].Rajagopal L, Vo A, Silvestroni A, Rubens CE. Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol Microbiol. 2005;56:1329–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rosenau A, Martins K, Amor S, Gannier F, Lanotte P, van der Mee-Marquet N, et al. Evaluation of the ability of Streptococcus agalactiae strains isolated from genital and neonatal specimens to bind to human fibrinogen and correlation with characteristics of the fbsA and fbsB genes. Infect Immun. 2007;75:1310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Seo HS, Minasov G, Seepersaud R, Doran KS, Dubrovska I, Shuvalova L, et al. Characterization of fibrinogen binding by glycoproteins Srr1 and Srr2 of Streptococcus agalactiae. J Biol Chem. 2013;288:35982–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Seifert KN, Adderson EE, Whiting AA, Bohnsack JF, Crowley PJ, Brady LJ. A unique serine-rich repeat protein (Srr-2) and novel surface antigen (epsilon) associated with a virulent lineage of serotype III Streptococcus agalactiae. Microbiology. 2006;152:1029–40. [DOI] [PubMed] [Google Scholar]
- [38].Sheen TR, Jimenez A, Wang NY, Banerjee A, van Sorge NM, Doran KS. Serine-rich repeat proteins and pili promote Streptococcus agalactiae colonization of the vaginal tract. J Bacteriol. 2011;193:6834–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang NY, Patras KA, Seo HS, Cavaco CK, Rosier B, Neely MN, et al. Group B streptococcal serine-rich repeat proteins promote interaction with fibrinogen and vaginal colonization. J Infect Dis. 2014;210:982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brochet M, Couve E, Zouine M, Vallaeys T, Rusniok C, Lamy MC, et al. Genomic diversity and evolution within the species Streptococcus agalactiae. Microbes Infect. 2006;8:1227–43. [DOI] [PubMed] [Google Scholar]
- [41].Six A, Bellais S, Bouaboud A, Fouet A, Gabriel C, Tazi A, et al. Srr2, a multifaceted adhesin expressed by ST-17 hypervirulent Group B Streptococcus involved in binding to both fibrinogen and plasminogen. Mol Microbiol. 2015;97:1209–22. [DOI] [PubMed] [Google Scholar]
- [42].Tazi A, Disson O, Bellais S, Bouaboud A, Dmytruk N, Dramsi S, et al. The surface protein HvgA mediates group B streptococcus hypervirulence and meningeal tropism in neonates. J Exp Med. 2010;207:2313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lamy MC, Dramsi S, Billoet A, Reglier-Poupet H, Tazi A, Raymond J, et al. Rapid detection of the “highly virulent” group B Streptococcus ST-17 clone. Microbes Infect. 2006;8:1714–22. [DOI] [PubMed] [Google Scholar]
- [44].Schubert A, Zakikhany K, Pietrocola G, Meinke A, Speziale P, Eikmanns BJ, et al. The fibrinogen receptor FbsA promotes adherence of Streptococcus agalactiae to human epithelial cells. Infect Immun. 2004;72:6197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jiang S, Wessels MR. BsaB, a novel adherence factor of group B Streptococcus. Infect Immun. 2014;82:1007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Buscetta M, Papasergi S, Firon A, Pietrocola G, Biondo C, Mancuso G, et al. FbsC, a novel fibrinogen-binding protein, promotes Streptococcus agalactiae-host cell interactions. J Biol Chem 2014;289:21003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gutekunst H, Eikmanns BJ, Reinscheid DJ. The novel fibrinogen-binding protein FbsB promotes Streptococcus agalactiae invasion into epithelial cells. Infect Immun. 2004;72:3495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Park SE, Jiang S, Wessels MR. CsrRS and environmental pH regulate group B streptococcus adherence to human epithelial cells and extracellular matrix. Infect Immun. 2012;80:3975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Al Safadi R, Amor S, Hery-Arnaud G, Spellerberg B, Lanotte P, Mereghetti L, et al. Enhanced expression of lmb gene encoding laminin-binding protein in Streptococcus agalactiae strains harboring IS1548 in scpB-lmb intergenic region. PLoS One 2010;5:e10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Spellerberg B, Rozdzinski E, Martin S, Weber-Heynemann J, Schnitzler N, Lutticken R, et al. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infect Immun. 1999;67:871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tenenbaum T, Spellerberg B, Adam R, Vogel M, Kim KS, Schroten H. Streptococcus agalactiae invasion of human brain microvascular endothelial cells is promoted by the laminin-binding protein Lmb. Microbes Infect. 2007;9:714–20. [DOI] [PubMed] [Google Scholar]
- [52].Ragunathan P, Spellerberg B, Ponnuraj K. Structure of laminin-binding adhesin (Lmb) from Streptococcus agalactiae. Acta Crystallogr D Biol Crystallogr. 2009;65:1262–9. [DOI] [PubMed] [Google Scholar]
- [53].Ragunathan P, Sridaran D, Weigel A, Shabayek S, Spellerberg B, Ponnuraj K. Metal binding is critical for the folding and function of laminin binding protein, Lmb of Streptococcus agalactiae. PLoS One. 2013;8:e67517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sridharan U, Ragunathan P, Spellerberg B, Ponnuraj K. Molecular dynamics simulation of metal free structure of Lmb, a laminin-binding adhesin of Streptococcus agalactiae: metal removal and its structural implications. J Biomol Struct Dyn. 2018:1–12. [DOI] [PubMed] [Google Scholar]
- [55].Moulin P, Patron K, Cano C, Zorgani MA, Camiade E, Borezee-Durant E, et al. The Adc/Lmb System Mediates Zinc Acquisition in Streptococcus agalactiae and Contributes to Bacterial Growth and Survival. J Bacteriol. 2016;198:3265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Franken C, Haase G, Brandt C, Weber-Heynemann J, Martin S, Lammler C, et al. Horizontal gene transfer and host specificity of beta-haemolytic streptococci: the role of a putative composite transposon containing scpB and lmb. Mol Microbiol. 2001;41:925–35. [DOI] [PubMed] [Google Scholar]
- [57].Chmouryguina I, Suvorov A, Ferrieri P, Cleary PP. Conservation of the C5a peptidase genes in group A and B streptococci. Infect Immun. 1996;64:2387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bohnsack JF, Widjaja K, Ghazizadeh S, Rubens CE, Hillyard DR, Parker CJ, et al. A role for C5 and C5a-ase in the acute neutrophil response to group B streptococcal infections. J Infect Dis. 1997;175:847–55. [DOI] [PubMed] [Google Scholar]
- [59].Cheng Q, Carlson B, Pillai S, Eby R, Edwards L, Olmsted SB, et al. Antibody against surface-bound C5a peptidase is opsonic and initiates macrophage killing of group B streptococci. Infect Immun. 2001;69:2302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cheng Q, Stafslien D, Purushothaman SS, Cleary P. The group B streptococcal C5a peptidase is both a specific protease and an invasin. Infect Immun. 2002;70:2408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Beckmann C, Waggoner JD, Harris TO, Tamura GS, Rubens CE. Identification of novel adhesins from Group B streptococci by use of phage display reveals that C5a peptidase mediates fibronectin binding. Infect Immun. 2002;70:2869–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tamura GS, Hull JR, Oberg MD, Castner DG. High-affinity interaction between fibronectin and the group B streptococcal C5a peptidase is unaffected by a naturally occurring four-amino-acid deletion that eliminates peptidase activity. Infect Immun. 2006;74:5739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hull JR, Tamura GS, Castner DG. Interactions of the streptococcal C5a peptidase with human fibronectin. Acta Biomater. 2008;4:504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Glaser P, Rusniok C, Buchrieser C, Chevalier F, Frangeul L, Msadek T, et al. Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol Microbiol. 2002;45:1499–513. [DOI] [PubMed] [Google Scholar]
- [65].Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102:13950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dramsi S, Caliot E, Bonne I, Guadagnini S, Prevost MC, Kojadinovic M, et al. Assembly and role of pili in group B streptococci. Mol Microbiol 2006;60:1401–13. [DOI] [PubMed] [Google Scholar]
- [67].Rosini R, Rinaudo CD, Soriani M, Lauer P, Mora M, Maione D, et al. Identification of novel genomic islands coding for antigenic pilus-like structures in Streptococcus agalactiae. Mol Microbiol 2006;61:126–41. [DOI] [PubMed] [Google Scholar]
- [68].Konto-Ghiorghi Y, Mairey E, Mallet A, Dumenil G, Caliot E, Trieu-Cuot P, et al. Dual role for pilus in adherence to epithelial cells and biofilm formation in Streptococcus agalactiae. PLoS Pathog. 2009;5:e1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Nobbs AH, Rosini R, Rinaudo CD, Maione D, Grandi G, Telford JL. Sortase A utilizes an ancillary protein anchor for efficient cell wall anchoring of pili in Streptococcus agalactiae. Infect Immun. 2008;76:3550–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Maisey HC, Hensler M, Nizet V, Doran KS. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J Bacteriol. 2007;189:1464–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Banerjee A, Kim BJ, Carmona EM, Cutting AS, Gurney MA, Carlos C, et al. Bacterial Pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat Commun. 2011;2:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Rosini R, Margarit I. Biofilm formation by Streptococcus agalactiae: influence of environmental conditions and implicated virulence factors. Front Cell Infect Microbiol. 2015;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pezzicoli A, Santi I, Lauer P, Rosini R, Rinaudo D, Grandi G, et al. Pilus backbone contributes to group B Streptococcus paracellular translocation through epithelial cells. J Infect Dis. 2008;198:890–8. [DOI] [PubMed] [Google Scholar]
- [74].Maisey HC, Quach D, Hensler ME, Liu GY, Gallo RL, Nizet V, et al. A group B streptococcal pilus protein promotes phagocyte resistance and systemic virulence. FASEB J. 2008;22:1715–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Springman AC, Lacher DW, Waymire EA, Wengert SL, Singh P, Zadoks RN, et al. Pilus distribution among lineages of group b streptococcus: an evolutionary and clinical perspective. BMC Microbiol. 2014;14:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Margarit I, Rinaudo CD, Galeotti CL, Maione D, Ghezzo C, Buttazzoni E, et al. Preventing bacterial infections with pilus-based vaccines: the group B streptococcus paradigm. J Infect Dis. 2009;199:108–15. [DOI] [PubMed] [Google Scholar]
- [77].Jiang S, Park SE, Yadav P, Paoletti LC, Wessels MR. Regulation and function of pilus island 1 in group B streptococcus. J Bacteriol. 2012;194:2479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Samen U, Heinz B, Boisvert H, Eikmanns BJ, Reinscheid DJ, Borges F. Rga is a regulator of adherence and pilus formation in Streptococcus agalactiae. Microbiology. 2011;157:2319–27. [DOI] [PubMed] [Google Scholar]
- [79].Dramsi S, Dubrac S, Konto-Ghiorghi Y, Da Cunha V, Couve E, Glaser P, et al. Rga, a RofA-like regulator, is the major transcriptional activator of the PI-2a pilus in Streptococcus agalactiae. Microb Drug Resist. 2012;18:286–97. [DOI] [PubMed] [Google Scholar]
- [80].Perichon B, Szili N, du Merle L, Rosinski-Chupin I, Gominet M, Bellais S, et al. Regulation of PI-2b Pilus Expression in Hypervirulent Streptococcus agalactiae ST-17 BM110. PLoS One. 2017;12:e0169840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Buscetta M, Firon A, Pietrocola G, Biondo C, Mancuso G, Midiri A, et al. PbsP, a cell wall-anchored protein that binds plasminogen to promote hematogenous dissemination of group B Streptococcus. Mol Microbiol. 2016;101:27–41. [DOI] [PubMed] [Google Scholar]
- [82].Papasergi S, Galbo R, Lanza-Cariccio V, Domina M, Signorino G, Biondo C, et al. Analysis of the Streptococcus agalactiae exoproteome. J Proteomics. 2013;89:154–64. [DOI] [PubMed] [Google Scholar]
- [83].Lentini G, Midiri A, Firon A, Galbo R, Mancuso G, Biondo C, et al. The plasminogen binding protein PbsP is required for brain invasion by hypervirulent CC17 Group B streptococci. Sci Rep 2018;8:14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].De Gaetano GV, Pietrocola G, Romeo L, Galbo R, Lentini G, Giardina M, et al. The Streptococcus agalactiae cell wall-anchored protein PbsP mediates adhesion to and invasion of epithelial cells by exploiting the host vitronectin/alphav integrin axis. Mol Microbiol. 2018;110:82–94. [DOI] [PubMed] [Google Scholar]
- [85].Mu R, Kim BJ, Paco C, Del Rosario Y, Courtney HS, Doran KS. Identification of a group B streptococcal fibronectin binding protein, SfbA, that contributes to invasion of brain endothelium and development of meningitis. Infect Immun. 2014;82:2276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Stoner TD, Weston TA, Trejo J, Doran KS. Group B streptococcal infection and activation of human astrocytes. PLoS One. 2015;10:e0128431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Gendrin C, Shubin NJ, Boldenow E, Merillat S, Clauson M, Power D, et al. Mast cell chymase decreases the severity of group B Streptococcus infections. J Allergy Clin Immunol. 2018;142:120–9 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Santi I, Maione D, Galeotti CL, Grandi G, Telford JL, Soriani M. BibA induces opsonizing antibodies conferring in vivo protection against group B Streptococcus. J Infect Dis. 2009;200:564–70. [DOI] [PubMed] [Google Scholar]
- [89].Santi I, Scarselli M, Mariani M, Pezzicoli A, Masignani V, Taddei A, et al. BibA: a novel immunogenic bacterial adhesin contributing to group B Streptococcus survival in human blood. Mol Microbiol. 2007;63:754–67. [DOI] [PubMed] [Google Scholar]
- [90].Santi I, Grifantini R, Jiang SM, Brettoni C, Grandi G, Wessels MR, et al. CsrRS regulates group B Streptococcus virulence gene expression in response to environmental pH: a new perspective on vaccine development. J Bacteriol. 2009;191:5387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Whidbey C, Harrell MI, Burnside K, Ngo L, Becraft AK, Iyer LM, et al. A hemolytic pigment of Group B Streptococcus allows bacterial penetration of human placenta. J Exp Med. 2013;210:1265–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Spellerberg B, Pohl B, Haase G, Martin S, Weber-Heynemann J, Lutticken R. Identification of genetic determinants for the hemolytic activity of Streptococcus agalactiae by ISS1 transposition. J Bacteriol 1999;181:3212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pritzlaff CA, Chang JC, Kuo SP, Tamura GS, Rubens CE, Nizet V. Genetic basis for the beta-haemolytic/cytolytic activity of group B Streptococcus. Mol Microbiol. 2001;39:236–47. [DOI] [PubMed] [Google Scholar]
- [94].Rosa-Fraile M, Rodriguez-Granger J, Haidour-Benamin A, Cuerva JM, Sampedro A. Granadaene: proposed structure of the group B Streptococcus polyenic pigment. Appl Environ Microbiol. 2006;72:6367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Whidbey C, Vornhagen J, Gendrin C, Boldenow E, Samson JM, Doering K, et al. A streptococcal lipid toxin induces membrane permeabilization and pyroptosis leading to fetal injury. EMBO Mol Med. 2015;7:488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Liu GY, Doran KS, Lawrence T, Turkson N, Puliti M, Tissi L, et al. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc Natl Acad Sci U S A. 2004;101:14491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Carey AJ, Tan CK, Mirza S, Irving-Rodgers H, Webb RI, Lam A, et al. Infection and cellular defense dynamics in a novel 17beta-estradiol murine model of chronic human group B streptococcus genital tract colonization reveal a role for hemolysin in persistence and neutrophil accumulation. J Immunol. 2014;192:1718–31. [DOI] [PubMed] [Google Scholar]
- [98].Patras KA, Wang NY, Fletcher EM, Cavaco CK, Jimenez A, Garg M, et al. Group B Streptococcus CovR regulation modulates host immune signalling pathways to promote vaginal colonization. Cell Microbiol. 2013;15:1154–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Gendrin C, Vornhagen J, Ngo L, Whidbey C, Boldenow E, Santana-Ufret V, et al. Mast cell degranulation by a hemolytic lipid toxin decreases GBS colonization and infection. Sci Adv. 2015;1:e1400225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Randis TM, Gelber SE, Hooven TA, Abellar RG, Akabas LH, Lewis EL, et al. Group B Streptococcus beta-hemolysin/cytolysin breaches maternal-fetal barriers to cause preterm birth and intrauterine fetal demise in vivo. J Infect Dis. 2014;210:265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Whidbey C, Burnside K, Martinez RM, Gendrin C, Vornhagen J, Frando A, et al. A Hyperhemolytic/Hyperpigmented Group B Streptococcus Strain with a CovR Mutation Isolated from an Adolescent Patient with Sore Throat. Clin Res Infect Dis. 2015;2. [PMC free article] [PubMed] [Google Scholar]
- [102].Sendi P, Johansson L, Dahesh S, Van-Sorge NM, Darenberg J, Norgren M, et al. Bacterial phenotype variants in group B streptococcal toxic shock syndrome. Emerg Infect Dis. 2009;15:223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lupo A, Ruppen C, Hemphill A, Spellerberg B, Sendi P. Phenotypic and molecular characterization of hyperpigmented group B Streptococci. Int J Med Microbiol. 2014;304:717–24. [DOI] [PubMed] [Google Scholar]
- [104].Almeida A, Rosinski-Chupin I, Plainvert C, Douarre PE, Borrego MJ, Poyart C, et al. Parallel Evolution of Group B Streptococcus Hypervirulent Clonal Complex 17 Unveils New Pathoadaptive Mutations. mSystems. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Boldenow E, Gendrin C, Ngo L, Bierle C, Vornhagen J, Coleman M, et al. Group B Streptococcus circumvents neutrophils and neutrophil extracellular traps during amniotic cavity invasion and preterm labor. Sci Immunol 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Nizet V, Gibson RL, Chi EY, Framson PE, Hulse M, Rubens CE. Group B streptococcal beta-hemolysin expression is associated with injury of lung epithelial cells. Infect Immun. 1996;64:3818–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Gibson RL, Nizet V, Rubens CE. Group B streptococcal beta-hemolysin promotes injury of lung microvascular endothelial cells. Pediatr Res. 1999;45:626–34. [DOI] [PubMed] [Google Scholar]
- [108].Doran KS, Chang JC, Benoit VM, Eckmann L, Nizet V. Group B streptococcal beta-hemolysin/cytolysin promotes invasion of human lung epithelial cells and the release of interleukin-8. J Infect Dis. 2002;185:196–203. [DOI] [PubMed] [Google Scholar]
- [109].Doran KS, Liu GY, Nizet V. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest. 2003;112:736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Rajagopal L, Vo A, Silvestroni A, Rubens CE. Regulation of cytotoxin expression by converging eukaryotic-type and two-component signalling mechanisms in Streptococcus agalactiae. Mol Microbiol. 2006;62:941–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lin WJ, Walthers D, Connelly JE, Burnside K, Jewell KA, Kenney LJ, et al. Threonine phosphorylation prevents promoter DNA binding of the Group B Streptococcus response regulator CovR. Mol Microbiol. 2009;71:1477–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Poyart C, Pellegrini E, Gaillot O, Boumaila C, Baptista M, Trieu-Cuot P. Contribution of Mn-cofactored superoxide dismutase (SodA) to the virulence of Streptococcus agalactiae. Infect Immun. 2001;69:5098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Bannister JV, Bannister WH, Rotilio G. Aspects of the structure, function, and applications of superoxide dismutase. CRC Crit Rev Biochem. 1987;22:111–80. [DOI] [PubMed] [Google Scholar]
- [114].Pritchard DG, Lin B, Willingham TR, Baker JR. Characterization of the group B streptococcal hyaluronate lyase. Arch Biochem Biophys. 1994;315:431–7. [DOI] [PubMed] [Google Scholar]
- [115].Milligan TW, Baker CJ, Straus DC, Mattingly SJ. Association of elevated levels of extracellular neuraminidase with clinical isolates of type III group B streptococci. Infect Immun. 1978;21:738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Pritchard DG, Lin B. Group B streptococcal neuraminidase is actually a hyaluronidase. Infect Immun. 1993;61:3234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Jones N, Bohnsack JF, Takahashi S, Oliver KA, Chan MS, Kunst F, et al. Multilocus sequence typing system for group B streptococcus. J Clin Microbiol. 2003;41:2530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Musser JM, Mattingly SJ, Quentin R, Goudeau A, Selander RK. Identification of a high-virulence clone of type III Streptococcus agalactiae (group B Streptococcus) causing invasive neonatal disease. Proc Natl Acad Sci U S A. 1989;86:4731–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Gendrin C, Vornhagen J, Armistead B, Singh P, Whidbey C, Merillat S, et al. A Nonhemolytic Group B Streptococcus Strain Exhibits Hypervirulence. J Infect Dis. 2018;217:983–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wang Z, Guo C, Xu Y, Liu G, Lu C, Liu Y. Two novel functions of hyaluronidase from Streptococcus agalactiae are enhanced intracellular survival and inhibition of proinflammatory cytokine expression. Infect Immun. 2014;82:2615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Kolar SL, Kyme P, Tseng CW, Soliman A, Kaplan A, Liang J, et al. Group B Streptococcus Evades Host Immunity by Degrading Hyaluronan. Cell Host Microbe. 2015;18:694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev. 2011;91:221–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–61. [DOI] [PubMed] [Google Scholar]
- [124].Vornhagen J, Quach P, Boldenow E, Merillat S, Whidbey C, Ngo LY, et al. Bacterial Hyaluronidase Promotes Ascending GBS Infection and Preterm Birth. MBio. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Rubens CE, Heggen LM, Haft RF, Wessels MR. Identification of cpsD, a gene essential for type III capsule expression in group B streptococci. Mol Microbiol. 1993;8:843–55. [DOI] [PubMed] [Google Scholar]
- [126].Yamamoto S, Miyake K, Koike Y, Watanabe M, Machida Y, Ohta M, et al. Molecular characterization of type-specific capsular polysaccharide biosynthesis genes of Streptococcus agalactiae type Ia. J Bacteriol. 1999;181:5176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Toniolo C, Balducci E, Romano MR, Proietti D, Ferlenghi I, Grandi G, et al. Streptococcus agalactiae capsule polymer length and attachment is determined by the proteins CpsABCD. J Biol Chem. 2015;290:9521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Rubens CE, Wessels MR, Heggen LM, Kasper DL. Transposon mutagenesis of type III group B Streptococcus: correlation of capsule expression with virulence. Proc Natl Acad Sci U S A. 1987;84:7208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Wessels MR, Rubens CE, Benedi VJ, Kasper DL. Definition of a bacterial virulence factor: sialylation of the group B streptococcal capsule. Proc Natl Acad Sci U S A. 1989;86:8983–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Martin TR, Ruzinski JT, Rubens CE, Chi EY, Wilson CB. The effect of type-specific polysaccharide capsule on the clearance of group B streptococci from the lungs of infant and adult rats. J Infect Dis. 1992;165:306–14. [DOI] [PubMed] [Google Scholar]
- [131].Takahashi S, Aoyagi Y, Adderson EE, Okuwaki Y, Bohnsack JF. Capsular sialic acid limits C5a production on type III group B streptococci. Infect Immun. 1999;67:1866–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Marques MB, Kasper DL, Pangburn MK, Wessels MR. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect Immun. 1992;60:3986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Carlin AF, Uchiyama S, Chang YC, Lewis AL, Nizet V, Varki A. Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood. 2009;113:3333–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ali SR, Fong JJ, Carlin AF, Busch TD, Linden R, Angata T, et al. Siglec-5 and Siglec-14 are polymorphic paired receptors that modulate neutrophil and amnion signaling responses to group B Streptococcus. J Exp Med. 2014;211:1231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Carlin AF, Lewis AL, Varki A, Nizet V. Group B streptococcal capsular sialic acids interact with siglecs (immunoglobulin-like lectins) on human leukocytes. J Bacteriol. 2007;189:1231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Chang YC, Olson J, Beasley FC, Tung C, Zhang J, Crocker PR, et al. Group B Streptococcus engages an inhibitory Siglec through sialic acid mimicry to blunt innate immune and inflammatory responses in vivo. PLoS Pathog. 2014;10:e1003846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Hulse ML, Smith S, Chi EY, Pham A, Rubens CE. Effect of type III group B streptococcal capsular polysaccharide on invasion of respiratory epithelial cells. Infect Immun. 1993;61:4835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Nizet V, Kim KS, Stins M, Jonas M, Chi EY, Nguyen D, et al. Invasion of brain microvascular endothelial cells by group B streptococci. Infect Immun. 1997;65:5074–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Gendrin C, Merillat S, Vornhagen J, Coleman M, Armistead B, Ngo L, et al. Diminished Capsule Exacerbates Virulence, Blood-Brain Barrier Penetration, Intracellular Persistence, and Antibiotic Evasion of Hyperhemolytic Group B Streptococci. J Infect Dis. 2018;217:1128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Chang YC, Olson J, Louie A, Crocker PR, Varki A, Nizet V. Role of macrophage sialoadhesin in host defense against the sialylated pathogen group B Streptococcus. J Mol Med (Berl) 2014;92:951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Devaux L, Sleiman D, Mazzuoli MV, Gominet M, Lanotte P, Trieu-Cuot P, et al. Cyclic di-AMP regulation of osmotic homeostasis is essential in Group B Streptococcus. PLoS Genet. 2018;14:e1007342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, et al. Group B Streptococcus Degrades Cyclic-di-AMP to Modulate STING-Dependent Type I Interferon Production. Cell Host Microbe. 2016;20:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Gratz N, Hartweger H, Matt U, Kratochvill F, Janos M, Sigel S, et al. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog 2011;7:e1001345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Parker D, Planet PJ, Soong G, Narechania A, Prince A. Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog 2014;10:e1003951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, et al. D-alanylation of teichoic acids promotes group a streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol. 2005;187:6719–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Saar-Dover R, Bitler A, Nezer R, Shmuel-Galia L, Firon A, Shimoni E, et al. D-alanylation of lipoteichoic acids confers resistance to cationic peptides in group B streptococcus by increasing the cell wall density. PLoS Pathog. 2012;8:e1002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Gryllos I, Tran-Winkler HJ, Cheng MF, Chung H, Bolcome R, 3rd, Lu W, et al. Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc Natl Acad Sci U S A. 2008;105:16755–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Froehlich BJ, Bates C, Scott JR. Streptococcus pyogenes CovRS mediates growth in iron starvation and in the presence of the human cationic antimicrobial peptide LL-37. J Bacteriol. 2009;191:673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Baker CJ, Kasper DL. Correlation of maternal antibody deficiency with susceptibility to neonatal group B streptococcal infection. N Engl J Med. 1976;294:753–6. [DOI] [PubMed] [Google Scholar]
- [150].Kwatra G, Adrian PV, Shiri T, Buchmann EJ, Cutland CL, Madhi SA. Natural acquired humoral immunity against serotype-specific group B Streptococcus rectovaginal colonization acquisition in pregnant women. Clin Microbiol Infect. 2015;21:568 e13-21. [DOI] [PubMed] [Google Scholar]
- [151].Baker CJ, Carey VJ, Rench MA, Edwards MS, Hillier SL, Kasper DL, et al. Maternal antibody at delivery protects neonates from early onset group B streptococcal disease. J Infect Dis. 2014;209:781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Baker CJ, Rench MA, Fernandez M, Paoletti LC, Kasper DL, Edwards MS. Safety and immunogenicity of a bivalent group B streptococcal conjugate vaccine for serotypes II and III. J Infect Dis. 2003;188:66–73. [DOI] [PubMed] [Google Scholar]
- [153].Baker CJ, Rench MA, McInnes P. Immunization of pregnant women with group B streptococcal type III capsular polysaccharide-tetanus toxoid conjugate vaccine. Vaccine. 2003;21:3468–72. [DOI] [PubMed] [Google Scholar]
- [154].Baker CJ, Paoletti LC, Wessels MR, Guttormsen HK, Rench MA, Hickman ME, et al. Safety and immunogenicity of capsular polysaccharide-tetanus toxoid conjugate vaccines for group B streptococcal types Ia and Ib. J Infect Dis. 1999;179:142–50. [DOI] [PubMed] [Google Scholar]
- [155].Baker CJ, Paoletti LC, Rench MA, Guttormsen HK, Edwards MS, Kasper DL. Immune response of healthy women to 2 different group B streptococcal type V capsular polysaccharide-protein conjugate vaccines. J Infect Dis. 2004;189:1103–12. [DOI] [PubMed] [Google Scholar]
- [156].GlaxoSmithKline. Safety and Immunogenicity of a Trivalent Group B Streptococcus Vaccine in Healthy Pregnant Women (NCT02046148). 2017.
- [157].Novartis. Safety and Immunogenicity of a Group B Streptococcus Vaccine in Non Pregnant and Pregnant Women 18-40 Years of Age (NCT01193920). ClinicalTrials.gov2014. [Google Scholar]
- [158].Novartis. Immune Response Induced by a Vaccine Against Group B Streptococcus and Safety in Pregnant Women and Their Offsprings (NCT01446289). ClinicalTrials.gov2014. [Google Scholar]
- [159].Bellais S, Six A, Fouet A, Longo M, Dmytruk N, Glaser P, et al. Capsular switching in group B Streptococcus CC17 hypervirulent clone: a future challenge for polysaccharide vaccine development. J Infect Dis. 2012;206:1745–52. [DOI] [PubMed] [Google Scholar]
- [160].Neemuchwala A, Teatero S, Athey TB, McGeer A, Fittipaldi N. Capsular Switching and Other Large-Scale Recombination Events in Invasive Sequence Type 1 Group B Streptococcus. Emerg Infect Dis. 2016;22:1941–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Patras KA, Rosler B, Thoman ML, Doran KS. Characterization of host immunity during persistent vaginal colonization by Group B Streptococcus. Mucosal Immunol. 2015;8:1339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Baker JA, Lewis EL, Byland LM, Bonakdar M, Randis TM, Ratner AJ. Mucosal vaccination promotes clearance of Streptococcus agalactiae vaginal colonization. Vaccine. 2017;35:1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Dzanibe S, Adrian PV, Mlacha SZK, Dangor Z, Kwatra G, Madhi SA. Reduced Transplacental Transfer of Group B Streptococcus Surface Protein Antibodies in HIV-infected Mother-Newborn Dyads. J Infect Dis. 2017;215:415–9. [DOI] [PubMed] [Google Scholar]
- [164].Slogrove AL, Goetghebuer T, Cotton MF, Singer J, Bettinger JA. Pattern of Infectious Morbidity in HIV-Exposed Uninfected Infants and Children. Front Immunol 2016;7:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Muenzner P, Bachmann V, Zimmermann W, Hentschel J, Hauck CR. Human-restricted bacterial pathogens block shedding of epithelial cells by stimulating integrin activation. Science. 2010;329:1197–201. [DOI] [PubMed] [Google Scholar]
- [167].Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, et al. Induction and evasion of host defenses by type 1 -piliated uropathogenic Escherichia coli. Science. 1998;282:1494–7. [DOI] [PubMed] [Google Scholar]
- [168].Muenzner P, Rohde M, Kneitz S, Hauck CR. CEACAM engagement by human pathogens enhances cell adhesion and counteracts bacteria-induced detachment of epithelial cells. J Cell Biol. 2005;170:825–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Nagamatsu K, Hannan TJ, Guest RL, Kostakioti M, Hadjifrangiskou M, Binkley J, et al. Dysregulation of Escherichia coli alpha-hemolysin expression alters the course of acute and persistent urinary tract infection. Proc Natl Acad Sci U S A. 2015;112:E871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Vornhagen J, Armistead B, Santana-Ufret V, Gendrin C, Merillat S, Coleman M, et al. Group B streptococcus exploits vaginal epithelial exfoliation for ascending infection. J Clin Invest. 2018;128:1985–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Becher N, Adams Waldorf K, Hein M, Uldbjerg N. The cervical mucus plug: structured review of the literature. Acta Obstet Gynecol Scand. 2009;88:502–13. [DOI] [PubMed] [Google Scholar]
- [172].Arko D, Dovnik A, Fokter N, Takac I. The role of genital pathogens in morbidity following diathermy loop excision of the transformation zone of the uterine cervix. Int J Gynaecol Obstet. 2012;117:27–9. [DOI] [PubMed] [Google Scholar]
- [173].Hein M, Valore EV, Helmig RB, Uldbjerg N, Ganz T. Antimicrobial factors in the cervical mucus plug. Am J Obstet Gynecol. 2002;187:137–44. [DOI] [PubMed] [Google Scholar]
- [174].Vornhagen J, Quach P, Santana-Ufret V, Alishetti V, Brokaw A, Armistead B, et al. Human Cervical Mucus Plugs Exhibit Insufficiencies in Antimicrobial Activity Towards Group B Streptococcus. J Infect Dis. 2018;217:1626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Lawn JE, Bianchi-Jassir F, Russell NJ, Kohli-Lynch M, Tann CJ, Hall J, et al. Group B Streptococcal Disease Worldwide for Pregnant Women, Stillbirths, and Children: Why, What, and How to Undertake Estimates? Clin Infect Dis. 2017;65:S89–S99. [DOI] [PMC free article] [PubMed] [Google Scholar]