

Abstract
Myeloid-derived suppressor cells (MDSC) expand during sepsis, suppress both innate and adaptive immunity, and promote chronic immunosuppression, which characterizes the late/chronic phase of sepsis. We previously reported that the transcription factors Stat3 and C/EBPβ synergize to induces the expression of microRNA (miR)-21 and miR-181b to promote MDSC expansion in a mouse model of polymicrobial sepsis that progresses from an early/acute proinflammatory phase to a late/chronic immunosuppressive stage. We also showed that Gr1+CD11b+ cells, the precursors of MDSCs, from mice genetically deficient in the inflammatory protein S100A9 lack miR-21 or miR-181b in late sepsis, and are not immunosuppressive. In the present study, we show that S100A9 induces miR-21 and miR-181b during the late sepsis phase. We find that S100A9 associates with and stabilizes the Stat3-C/EBPβ protein complex that activates the miRNA promoters. Reconstituting Gr1+CD11b+ cells from S100A9 knockout mice with late sepsis with S100A9 protein restores the Stat3-C/EBPβ protein complex and miRNA expressions, and switches the Gr1+CD11b+ cells into the immunosuppressive, MDSC phenotype. Importantly, we find that this process requires IL-10 mediated signaling, which induces S100A9 translocation from the cytosol to the nucleus. These results demonstrate that S100A9 promotes MDSC expansion and immunosuppression in late/chronic sepsis by inducing the expression of miR-21 and miR-181b.
Keywords: Sepsis, Immune suppression, MDSC, S100A9
Graphical abstract
Introduction
Myeloid-derived suppressor cells (MDSC) are characterized by the immature myeloid phenotype Gr1+CD11b+ in mouse and by their immunosuppressive functions, as they suppress both innate and adaptive immunity (Cuenca et al., 2011; Gabrilovich and Nagaraj, 2009; Kong et al., 2013). Generation and accumulation of MDSCs are observed under a variety of pathological conditions where inflammation or infection is a common theme (Dai et al., 2015; Ostrand-Rosenberg and Fenselau, 2018; Veglia et al., 2018). We previously reported expansion and accumulation of MDSCs in the bone marrow and spleens in mouse with polymicrobial sepsis, which progresses from an early/acute proinflammatory phase to a late/chronic stage characterized by protracted immunosuppression (Brudecki et al., 2012), and is associated with elevated mortality and morbidity in animal and humans with sepsis (Delano et al., 2007; Efron et al., 2018; Hotchkiss et al., 2013a; Patil et al., 2016). While Gr1+CD11b+ cells (precursors of MDSCs) expand and accumulate throughout sepsis, early sepsis Gr1+CD11b+ cells produce proinflammatory mediators and promote inflammation, whereas their counterparts in late sepsis are immunosuppressive (i.e., functional MDSCs) as they produce immunosuppressive mediators such as IL-10 and arginase 1 (Brudecki et al., 2012; McPeak et al., 2017a).
The mechanisms underlying MDSC expansion remain unclear, but different mediators, including inflammatory cytokines and growth factors, have been thought to drive MDSC expansion under various inflammatory conditions (Condamine and Gabrilovich, 2011; Kong et al., 2013; Ostrand-Rosenberg et al., 2018). In the context of sepsis, we showed that expression of microRNA (miR)-21 and miR-181b is induced after sepsis initiation, where they couple with the myeloid differentiation-related transcription factor NFI-A to drive Gr1+CD11b+ cell expansion in early and late sepsis (McClure et al., 2016; McClure et al., 2014). The expression of these miRNAs is induced by the synergistic effect of Stat3 and C/EBPβ, which bind to and activate the miR-21 and miR-181b promoters after sepsis initiation (McClure et al., 2017).
Recently, we reported that mice genetically deficient in the calcium-binding S100A9 protein do not generate MDSCs during the late sepsis phase; however, they still make proinflammatory Gr1+CD11b+ cells during early sepsis (Dai et al., 2017). Interestingly, expression of miR-21 and miR-181b is lost only in late sepsis Gr1+CD11b+ cells from the S100A9 deficient mice, and these cells are generated at normal/baseline levels and are not immunosuppressive (Dai et al., 2017). These studies suggested that S100A9 may play a role in the induction of miR-21 and miR-181b expressions and MDSC generation during the late, immunosuppressive phase of sepsis. How S100A9 differentially regulates miR-21 and miR-181b expressions in late/chronic sepsis is unknown.
S100A9 protein is expressed and secreted by various cell types, including phagocytes (Ehrchen et al., 2009; Foell and Roth, 2004a; Vogl et al., 2012), and is induced by many stimuli, including inflammatory mediators and bacterial products (Foell et al., 2004b; Goyette and Geczy, 2011; Vogl et al., 2007). S100A9 is constitutively expressed in immature myeloid cells, including Gr1+CD11b+ cells, but its expression decreases with differentiation and maturation (Roth et al., 1993; Vogl et al., 2012). Most studies on S100A9 have focused on its role as a proinflammatory, soluble mediator that amplifies inflammation and infection via immune cell activation and recruitment (Ehrchen et al., 2009; Foell et al., 2004; Goyette et al., 2011). Importantly, our studies indicated that S100A9 might play a role in sepsis pathogenesis at the molecular level, by promoting MDSC generation in late sepsis. S100A9 protein translocates from the cytosol to the nucleus in MDSCs during late/chronic sepsis only (Dai et al., 2017). In the present study, we investigated the molecular mechanism by which S100A9 induces miR-21 and miR-181 expressions, and MDSC generation during murine chronic sepsis. We find that nuclear S100A9 acts as a transcription cofactor to facilitate miR-21 and miR-181b induction by Stat3 and C/EBP β during the late phase of sepsis. The results support that molecular targeting of S100A9 to inhibit MDSC expansion and sepsis-associated immunosuppression might improve sepsis survival.
Materials and Methods
Mice
The C57BL/6N S100a9 knockout mice used for this study have been described previously (Dai et al., 2017). Homozygous (−/−) mice were bred and housed in a pathogen-free facility in the Division of Laboratory Animal Resources. Wild-type C57BL/6N mice were purchased from Jackson Laboratory (Bar Harbor, ME) and used as controls. Male mice, 8–10 weeks old were used in this study and were acclimated to the new environment for a week before surgery. All experiments followed the National Institutes of Health guidelines the East Tennessee State University Animal Care and Use Committee approved the study.
Sepsis
Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) as described (Brudecki et al., 2012). A midline abdominal incision was made and the cecum was ligated distal to the ileocecal valve, and punctured twice with a 23-gauge needle. A small amount of feces into the abdominal cavity. The abdominal wall and skin were sutured in layers with 3–0 silk. Shamoperated mice were treated identically except that the cecum was neither ligated nor punctured. Mice received (i.p.) 1 ml lactated Ringers plus 5% dextrose for fluid resuscitation. To establish intra-abdominal infection and approximate the clinical condition of early human sepsis where there is a delay between the onset of sepsis and the delivery of therapy (Mazuski et al., 2002), mice were subcutaneously administered antibiotic (Imipenem; 25 mg/kg body weight) or an equivalent volume of 0.9% saline at 8 and 16 hr after CLP. These levels of injury and manipulation create prolonged infections with high mortality (~60–70%) during the late/chronic phase (Brudecki et al., 2012). We followed survival 28 days. We euthanized mice moribund within 5 days after CLP (early sepsis) and after that (Brudecki et al., 2012). We euthanized a corresponding number of healthy appearing mice from the control group to provide control samples.
Gr1+CD11b+ cell isolation
Bone marrow Gr1+CD11b+ cells were isolated using magnetic beads according to the manufacturer’s protocol (Miltenyi Biotech, Auburn, CA). Briefly, the bone marrow was flushed out of the femurs with RPMI-1640 medium (without serum) under aseptic conditions. A single cell suspension was made by pipetting up and down and filtering through a 70-μm nylon strainer, followed by incubation with erythrocyte lysis buffer and washing. The cell suspension was subjected to a positive selection of the Gr1+ cells by incubating with biotin-coupled mouse antiGr1 antibody (Clone RB6–8C5; eBioscience, San Diego, CA) for 15 min at 4°C. Cells were then incubated with anti-biotin magnetic beads for 20 min at 4°C and subsequently passed over an MS column. The cell purity was more than 90% as determined by flow cytometry.
Gr1+CD11b+ cells were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine (all from Hyclone Laboratories, Logan, UT), and 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) at 37°C and 5% CO2. In some experiments, cells were stimulated for 6 hr with recombinant murine IL-10.
S100A9 expression construct and transfection
Full-length mouse S100a9 cDNA was cloned in pEZ-M02 expression vector downstream of the CMV promoter. A pReceiver-M02 vector served as a negative control.
For Gr1+CD11b+ cell, S100A9 plasmid DNA or empty vector was suspended in HiPerFect reagent at a 0.5 μg/ml final concentration (Qiagen, Valencia, CA). Cells were transfected using the Gene Pulser MXCell system (Bio-Rad, Hercules, CA) and then stimulated with or without recombinant murine IL-6 or IL-10 (PeproTech; Rocky Hill, NJ).
Preparation of protein extracts
Whole cell extract was prepared by cell lysis in 1× RIPA buffer containing 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholic acid, and 1 mM EDTA (Millipore, Temecula, CA) plus 1× protease inhibitor cocktail. After 30 min on ice, cell lysate was cleared by centrifugation for 5 min at 4°C and 14,000 rpm. Protein concentrations were determined by Bradford assay (Bio-Rad), and aliquots were kept at −20°C.
Cytoplasmic and nuclear proteins were prepared using the NE-PER nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL) per the manufacturer’s instructions. Immediately after harvesting, cells were washed in PBS and resuspended in CER1 lysis buffer with protease inhibitor cocktail and incubated on ice for 1 min. CER2 buffer was added, and the incubation continued for 5 min. Supernatants (cytoplasmic proteins) were removed by centrifugation for 5 min at 4°C and 14,000 rpm. The nuclear pellets were resuspended in NER lysis buffer with protease inhibitor cocktail and incubated for 40 min on ice with occasional vortexing. The nuclear proteins were recovered by centrifugation for 10 min at 4°C and 14,000 rpm.
Co-immunoprecipitation
Immunoprecipitation (IP) of Stat3 or S100A9 proteins was performed to assess protein complex formations in Gr1+CD11b+ cells during sepsis. Briefly, whole-cell extracts were pre-cleared by incubation with pre-blocked protein G-agarose beads for one h at 4°C. The beads were pre-blocked by incubation for one h with 100 μg/ml of BSA. Next, The beads were washed with buffer C (250 mM sucrose, 10 mM Tris-HCl [pH 7.5], 25 mM KCl, 5 mM MgCl2, 2 mM DTT, 30 U/ml RNase inhibitor, and 1× protease inhibitor cocktail). Cell extract was centifuged at 2,000 rpm for 5 min, and supernatant (900 μl) was added to 100 μl of the pre-blocked beads coated with 10 μl antibody against pStat3 (phospho tyrosine705), S100A9, or IgG control antibody. After rotating overnight at 4°C, the beads were centrifuged and washed three times with buffer C. Aliquots of bound protein complexes were analyzed by western blotting as described below.
Western blots
Equal amounts of protein extracts or co-immunoprecipitated protein complexes were mixed with 5× Laemmeli sample buffer, separated by SDS-10% polyacrylamide gel (Bio-Rad, Hercules, CA) and transferred to nitrocellulose membranes (Thermo Fisher Scientific, Waltham, MA). Membranes were blocked with 5% milk in Tris-buffered saline/Tween-20 for 1 hr at room temperature, and then probed overnight at 4°C with the following primary antibodies: anti-pStat3 tyrosine 705 (sc-8059), anti-C/EBPβ (sc-7962) (Santa Cruz Biotechnology, Santa Cruz, CA), or anti-Rabbit S100A9 (ab75478; Abcam, Cambridge, MA). After washing, blots were incubated with the appropriate HRP-conjugated secondary antibody for 2 hr at room temperature. Proteins were detected with the enhanced chemiluminescence detection system (Thermo Fisher Scientific, Waltham, MA). The protein bands were visualized using the ChemiDoc XRS System (Bio-Rad), and the images were captured with the Image Lab Software V3.0. Membranes were stripped and reprobed with β-actin or nucleoporin antibody (Santa Cruz Biotechnology) as a loading control.
Chromatin immunoprecipitation (ChIP)
ChIP was performed to assess in vivo DNA-protein interactions at the miR-21 and miR-181b promoters using ChIP-IT Express Enzymatic Shearing kit according to the manufacturer’s instructions (Active Motif, Carlsbad, CA). Briefly, Gr1+CD11b+ cells were harvested and fixed in 1% formaldehyde in minimal culture medium for 10 min at room temperature, to cross-link protein-DNA complexes. After washing, cells were lysed in 1× lysis buffer containing protease inhibitor cocktail. The cell lysate was cleared by centrifugation at 5,000 rpm for 10 min at 4°C. The pelleted nuclei were then resuspended in digestion buffer and incubated with the enzymatic shearing cocktail at 37°C for 10 min. The sheared chromatin solution was recovered by centrifugation at 15,000 rpm for 10 min at 4°C. Ten microliters of the chromatin solution were reserved as an “input” sample. The remaining chromatin solution was immunoprecipitated overnight at 4°C with protein G magnetic beads and 3 μg of antibody specific to pStat3, C/EBPβ (Santa Cruz Biotechnology), S100A9 (Abcam, Cambridge, MA), or isotype control antibody (Santa Cruz Biotechnology). The chromatin/antibody complexes were washed three times in ChIP buffer and then eluted by incubation for 15 min in 50 μl elution buffer. Next, the DNA-protein cross-links were reversed by incubation with 50 μl of reverse cross-linking buffer. The supernatant containing the DNA was then incubated, along with the “input” DNA samples, at 95°C for 15 min. After treatment with proteinase K for 1 h at 37°C, the reaction was stopped, and the resulting DNA was extracted and stored at −20°C.
Quantitative real-time PCR was used to measure the enrichment of miR-21 and miR-181b promoter sequences in the ChIPed DNA using primer and fluorescently labeled internal probe sequences (Integrated DNA Technologies, Coralville, IA) specific to the miR-21 and miR-181b promoters as described previously (McClure et al., 2017).
miRNA measurement
Quantitative real-time qPCR was used to determine levels of miR-21 and miR-181b in Gr1+CD11b+ cells. miRNA-enriched RNA was isolated and measured using miScript SYBR Green PCR kit with miScript Primer Assays specific to miR-21 and miR-181b according to the manufacturer’s protocol (Qiagen). The relative expression of each miRNA was calculated using the 2−ΔΔCt cycle threshold method after normalization to the endogenous U6 RNA as an internal control.
CD4+ T cells suppression assay
To determine the suppressive effects of Gr1+CD11b+ cells on T cell functions, we tested Gr1+CD11b+ CD4+ T cells for proliferation and IFNγ production. Splenocytes from naive wildtype were used to purify CD4+ T cells by positive selection using biotinylated anti-CD4 magnetic beads (Myltenyi). Purified cells were labeled with carboxyfluorescein diacetate, succinimidyl ester (CFSE) dye using the Vybrant CFDA SE Cell Tracer Kit (Invitrogen Molecular Probes, Eugene, OR). Cells were incubated for 10 min at room temperature with 10 μM CFSE dye. Labeled cells were co-cultured (1:1 ratio) with Gr1+CD11b+ cells. We induced T cell proliferation by the stimulation with an anti-CD3 antibody plus an anti-CD28 antibody (1 μg/ml/each). After three days, cells CD4+ T cell proliferation was determined by stepwise dilution of CFSE dye in dividing, CD3-gated CD4+ T cells using flow cytometry. Culture supernatants were collected for the IFN-γ measurement by ELISA.
Statistical analysis
Data were analyzed with Microsoft Excel, V3.0., and expressed as mean ± s.d. Differences between 2 groups were determined by use of unpaired student’s t-test. One-way analysis of variance was used to analyze the data with more than two groups. Statistical significance is reported for p-values < 0.05.
Results
S100A9 knockout disrupts pStat3 and C/EBPβ bindings at miR-21 and miR-181b promoters
Expression of miR-21 and miR-181b is induced in Gr1+CD11b+ cells after sepsis initiation via pStat3 and C/EBPβ mediated activation of their promoters during the early and late sepsis phases (McClure et al., 2017). We recently discovered that genetic ablation of S100A9 prevents expression of miR-21 and miR-181b only during the late sepsis phase, despite elevated levels of pStat3 and C/EBPβ (Dai et al., 2017).
To determine the differential effect of S100A9 protein on the miRNA expression, we compared pStat3 and C/EBPβ occupancy at the miRNA promoters in early and late sepsis Gr1+CD11b+ cells from wild-type and S100A9 knockout mice using chromatin immunoprecipitation assay. As shown in Fig. 1, both pStat3 and C/EBPβ proteins were detected at the miR-21 promoter in early sepsis Gr1+CD11b+ cells from the wild-type and knockout mice. In late sepsis, however, binding of pStat3 and C/EBPβ occurred in Gr1+CD11b+ cells from wildtype mice, but was abolished in cells from the S100A9 knockout mice. Similar binding patterns of pStat3 and C/EBPβ were detected at the miR-181b promoter (Supplemental Fig. 1). These results suggest that S100A9 supports the expression of miR-21 and miR-181b in late sepsis only.
Figure 1: Lack of S100A9 expression prevents assembly of the pStat3-C/EBP β protein complex at the miR-21 promoter in Gr1+CD11b+ cells in late sepsis.

Detection of transcription factor binding at the miRNA promoters. Gr1+CD11b+ cells were isolated from the bone marrow of sham and septic mice (n = 3–4 mice per group). Formaldehyde-fixed cells were lysed, and the pelleted nuclei were digested with chromatin shearing enzymatic cocktail. The chromatin was immunoprecipitated with antibodies specific to pStat3 (Tyr705), C/EBP β, or IgG isotype control antibody. Next, chromatin cross-links were reversed to recover the protein-bound DNA. The purified DNA was amplified by quantitative real-time PCR to measure the level of enrichment of miR-21 and miR-181b promoter DNAs in chromatin, using promoter-specific primer/probe sets. PCR reactions were performed in triplicate. Samples values were normalized to the “input” DNA (DNA isolated before immunoprecipitation) and are presented as fold enrichment relative to the IgG-immunoprecipitated samples (set at 1-fold). Data are mean ± s.d. (n = 3–4 mice per group). *p < 0.05 vs. sham; **p < 0.05 vs. early sepsis. The results are representative of three independent experiments. KO, knockout.
S100A9 is required for assembly of the pStat3-C/EBPβ protein complex
PStat3 and C/EBPβ bind at the miR-21 and miR-181b promoters in Gr1+CD11b+ cells as one protein complex (McClure et al., 2017). Western blotting confirmed high levels of S100A9 along with pStat3 and C/EBPβ in early and late sepsis cells (Fig. 2A). To determine whether S100A9 is required for the pStat3-C/EBPβ complex assembly, we performed co-immunoprecipitation using Stat3 and S100A9 antibodies. Western blotting of Stat3 immunoprecipitates detected pStat3-C/EBPβ protein complex in early and late sepsis Gr1+CD11b+ cells from the wild-type mice, whereas S100A9 immunoprecipitation showed that S100A9 associated with pStat3-C/EBPβ complex in late sepsis cells only (Fig. 2B and C). Notably, lack of S100A9 protein expression resulted in inhibition of the pStat3-C/EBPβ complex formation in late sepsis Gr1+CD11b+ cells only (Fig. 2D). These results demonstrate that S100A9 promotes formation of pStat3-C/EBPβ protein complex in Gr1+CD11b+ cells in late sepsis, but has no effect during early sepsis response.
Figure 2: The S100A9 forms a protein complex with pStat3 and C/EBPβ proteins in late sepsis.

Gr1+CD11b+ cells were isolated from the bone marrow of sham and septic mice. (A) Levels of pStat3, C/EBPβ, and S100A9 proteins were determined by western blot. (B-D) Coimmunoprecipitation analysis of pStat3(Tyr705), C/EBPβ and S100A9 protein interactions. Gr1+CD11b+ cell lysates were prepared and immunoprecipitated with pStat3, S100A9, or IgG isotype control antibody using protein G-agarose beads. The immunoprecipitated protein complexes were resolved on denaturing polyacrylamide gel and then immunoblotted with specific antibody against Stat3, pStat3, C/EBPβ, or S100A9. To obtain enough cell lysates for immunoprecipitation, cells were pooled from 2–3 mice per group. The results are representative of three independent experiments. KO, knockout.
Reconstituting late sepsis Gr1+CD11b+ cells from the S100A9 knockout mice with S100A9 restores the pStat3-C/EBPβ protein complex binding at the miRNA promoters.
We investigated whether S100A9 is sufficient to induce the pStat3-C/EBPβ complex assembly and binding at the miR-21 and miR-181b promoters. To do this, we introduced S100A9 expression plasmid into Gr1+CD11b+ cells from the S100A9 knockout mice with late sepsis, where formation of the pStat3-C/EBPβ protein complex is disrupted (Fig. 2D). The cells were stimulated with the immunosuppressive IL-10 cytokine because our previous studies showed that IL-10 induced Stat3 phosphorylation, as well as S100A9 translocation to the nucleus (Bah et al., 2018). IL-10 is poduced by many myeloid cells during inflammation and infection (Hotchkiss et al., 2013b; Shubin et al., 2011). Notably, IL-10 production is increased as sepsis progresses to the immunosuppressive stage (Bah et al., 2018), and late sepsis MDSCs produce high levels of IL-10 upon ex vivo stimulation with bacterial LPS (McPeak et al., 2017a). Western blotting confirmed S100A9 expression after the transfection with the S100A9 plasmid (Fig. 3A). S100A9 immunoprecipitation assay showed that S100A9 formed a protein complex with pStat3 and C/EBPβ in the presence of IL-10, and these results were similar to what we observed in late sepsis Gr1+CD11b+ cells from wild-type mice (Fig. 3B). Importantly, PCR analysis of the immunoprecipitated chromatin revealed simultaneous bindings of pStat3, C/EBPβ and S100A9 at the miR-21 promoter (Fig. 3C) and the miR-181b promoter (data not shown). These results indicate that pStat3-C/EBPβ protein complex assembly and binding to the miR-21 and miR-181b promoters in Gr1+CD11b+ cells during late sepsis requires S100A9.
Figure 3: Ectopic expression of S100A9 in late sepsis Gr1+CD11b+ cells from the S100A9 knockout mice restores the pStat3-C/EBPβ protein complex formation and binding at the miRNA promoter.

Gr1+CD11b+ cells were isolated from the bone marrow of S100A9 knockout mice during late sepsis response. Cells were transfected with S100A9 expression plasmid or control vector for 36 hr and then stimulated with 10 ng/ml of recombinant murine IL-10 for 6 hr. Cells from WT mice were used as a control. (A) Cell lysates were prepared, and S100A9 protein expression was determined by western blot. (B) Cell lysates were immunoprecipitated with an anti-S110A9 antibody, and proteins were detected by western blot. To obtain enough cell lysates for immunoprecipitation, cells were pooled from 2–3 mice per group. (C) Analysis of pStat3 (tyrosine 705), C/EBP β and S100A9 binding at the miRNA promoter by chromatin immunoprecipitation (ChIP). ChIP DNA was extracted and amplified by quantitative real-time PCR to measure the level of miR-21 promoter DNA enrichment in chromatin. Samples values were normalized to the “input” DNA (DNA isolated before immunoprecipitation) and are presented as fold enrichment relative to the IgG-immunoprecipitated samples (set at 1-fold). Data are mean ± s.d. (n = 3–4 mice per group). *p < 0.05 vs. KO + S100A9. The results are representative of three independent experiments. KO, knockout.
Reconstituting late sepsis Gr1+CD11b+ cells from the S100A9 knockout mice with S100A9 activates expression of miR-21 and miR-181b, and induces NFI-A
Expression of miR-21 (Fig. 4A) and miR-181b (Supplemental Fig. 2) decreases in the S100A9 knockout mice in late sepsis. We next examined whether reconstituting late sepsis Gr1+CD11b+ cells from the S100A9 knockout mice with S100A9 can restore the miRNA expressions, concurrently with the pStat3-C/EBPβ complex formation and binding at their promoters (Fig. 3). Transfection with S100A9 plasmid restored expression of miR-21 (Fig. 4B) and miR-181b (Supplemental Fig. 2) in the presence of IL-10.
Figure 4: Ectopic expression of S100A9 in late sepsis Gr1+CD11b+ cells from the S100A9 knockout mice restores miR-21 expression.

Gr1+CD11b+ cells were isolated from the bone marrow of sham and septic mice. (A) miR-21 expression. miRNA-enriched RNA was isolated, and levels of miR-21 were determined by quantitative real-time PCR using miR-21 specific primers. (B) Gr1+CD11b+ cells, isolated from the S100A9 knockout mice during late sepsis response (days 6–28 after sepsis induction), were transfected with S100A9 expression plasmid or control vector for 36 hr and then stimulated with 10 ng/ml of recombinant murine IL-10 for 6 hr. Expression of miR-21 was determined as in A. Sample values were normalized to U6 RNA as an internal control and are presented relative to values (set at 1-fold) from sham wild-type mice (A) or vector alone (B). (C) Overexpression of miR-21 and miR-181b does not restore Gr1+CD11b+ cell suppressive function. Late sepsis Gr1+CD11b+ cells from the S100A9 knockout mice were transfected with control precursors or a mixture of miR-21 and miR-181b precursors for 36 hr. The cells, along with S100A9-transfected cells, were washed and stimulated with 1 μg/ml of gramnegative bacterial LPS for 12 hr. Levels of IL-10 in the culture supernatants were determined by ELISA. Data are means ± s.d. (n = 4–6 mice per group). *p < 0.05 vs. sham; **p < 0.05 vs. wildtype; ♯p < 0.05 vs. S100A9; §p < 0.05. The results are representative of three independent experiments. KO, knockout.
Because S100A9 transfection induced miR-21 and miR-181b expressions in MDSCs from the S100A9 knockout mice, and because S100A9 also is required for the production of immunosuppressive MDSCs (Dai et al., 2017), we investigated whether overexpression of miR-21 and miR-181b in these cells can restore their immunosuppressive function. MDSCs were transfected with control or miR-21 and miR-181b precursors, and stimulated with bacterial LPS, which induces IL-10 expression in MDSCs (McPeak et al., 2017b). Overexpression of miR-21 and miR-181b could not induce IL-10, whereas LPS stimulation in S100A9-trasfected cells significantly induced IL-10 (Fig. 4C). These results show that S100A9, in addition to inducing the miRNA expressions, also play a role in promoting MDSC immunosuppressive function.
S100A9 protein resides in the cytosol in early sepsis Gr1+CD11b+ cells, which are not immunosuppressive (Brudecki et al., 2012), but moves to the nucleus during late sepsis (Fig. 5A). We have shown that IL-10 signaling shuttles S100A9 protein from the cytosol to the nucleus in naive Gr1+CD11b+ cells from wild-type mice (Bah et al., 2018). Here, we observed in late sepsis Gr1+CD11b+ cells from S100A9 knockout mice that, concurrent with the activation of miR-21 and miR-181b expressions (Fig. 4), IL-10 induced translocation of the ectopically expressed S100A9 protein to the nucleus (Fig. 5B).
Figure 5: Nuclear S100A9 protein promotes NFI-A expression in Gr1+CD11b+ cells during late sepsis.

Gr1+CD11b+ cells were isolated from the bone marrow of sham and septic mice. S100A9 or NFI-A protein levels were determined by western blot. (A) Levels of S100A9 protein in the nucleus and cytosol of Gr1+CD11b+ cells from wild-type mice. (B) Levels of S100A9 proteins in the nucleus and cytosol of Gr1+CD11b+ cells from S100A9 knockout mice. The cells were isolated during the late sepsis phase (days 6–28 after sepsis induction) and transfected with S100A9 expression plasmid for 36 hr and then stimulated with 10 ng/ml of recombinant murine IL-10 for 6 hr. Cells were pooled from 2 mice per group. (C) Levels of NFI-A protein in Gr1+CD11b+ cells from wild-type mice. (D) Levels of NFI-A proteins in Gr1+CD11b+ cells from S100A9 knockout mice. The cells were isolated during the late sepsis phase and transfected and treated as in A. Cells from sham and sepsis mice, and without transfection were used as controls. Cells were pooled from 2 mice per group. The results are representative of three independent experiments. KO, knockout.
Activation of miR-21 and miR-181b expressions during sepsis leads to induction of NFIA, and subsequently promotes Gr1+CD11b+ cell expansion (McClure et al., 2016). Similar to the miRNA expression pattern (Fig. 4), NFI-A increased in early, but not late, sepsis in Gr1+CD11b+ cells in S100A9 knockout mice (Fig. 5C and D). Introducing S100A9 plasmid into these cells restored NFI-A expression in the presence of IL-10 (Fig. 5D). Collectively, the results presented above show that S100A9 induces expression of miR-21 and miR-181b in late sepsis Gr1+CD11b+ cells, leading to NFI-A expression and that IL-10 signaling mediated these effects by shuttling S100A9 from the cytosol to the nucleus.
S100A9 promotes an immunosuppressive phenotype in Gr1+CD11b+ cells
Gr1+CD11b+ cells generated during late sepsis are immunosuppressive (i.e., MDSCs), as they can suppress T cell proliferation and activation (McPeak et al., 2017a). We have shown that S100A9 knockout mice still generate Gr1+CD11b+ cells at baseline levels during late sepsis, and that these cells are not immunosuppressive (Dai et al., 2017), i.e., like normal myeloid precursors that are produced under normal conditions. We examined whether introducing S100A9 into Gr1+CD11b+ cells from S100A9 knockout mice with late sepsis can promote an immunosuppressive phenotype, concurrently with the induction of miRNA expressions (Fig. 4B and Supplemental Fig. 1B). Gr1+CD11b+ cells from sham and late septic wild-type mice were used as positive and negative controls, respectively. As shown in Fig. 6, Gr1+CD11b+ cells from wild-type, control mice significantly suppressed T cell proliferation and activation (IFNγ production). Late sepsis Gr1+CD11b+ cells derived from S100A9 knockout mice and transfected with S100A9 plasmid could not suppress T cells, whereas S100A9 plasmid transfection with IL-10 stimulation led to significant suppression of T cell proliferation and IFNγ production (Fig. 6). These results suggest that S100A9 promotes an immunosuppressive phenotype in Gr1+CD11b+ cells during late sepsis and that IL-10 signaling mediates these effects.
Figure 6: Reconstituting late sepsis Gr1+CD11b+ cells from the S100A9 knockout mice with S100A9 restores their immunosuppressive functions.

Gr1+CD11b+ cells were isolated from the bone marrow of S100A9 knockout mice during the late sepsis phase. The cells were transfected with S100A9 expression plasmid for 36 hr and then stimulated with 10 ng/ml of recombinant murine IL-10 for 6. (A) Effect of Gr1+CD11b+ cells on CD4+ T cell proliferation. Spleen CD4+ T cells were isolated from normal (naive) mice by positive selection and labeled with the fluorescent dye CFSE for 10 min at room temperature. Gr1+ CD11b+ cells were then cocultured (1:1 ratio) with T CD4+ cells. The culture was stimulated with anti-CD3 plus anti-CD28 antibodies (1 μ g/ml/each). After three days, CD4+ T cell proliferation was determined by the step-wise dilution of CFSE dye in dividing CD4+ T cells by flow cytometry. Representative dot plots (upper panels) of CFSE positive T cells gated on CD4 are shown. Percentages of cell proliferation (lower) were calculated as follow: % cell proliferation = 100 × (count from T cell + Gr1+CD11b+ cell culture/count from T cell culture). *p < 0.001 vs. sham or early sepsis; **p < 0.001 vs. late sepsis WT. (B) ELISA determined levels of IFNγ in the culture supernatants. Cells from sham and sepsis wild-type mice, and without transfection were used as controls. Cells were pooled from 2–3 mice per group. Data are means ± s.d. (n = 3–4 mice per group). *p < 0.05 vs. sham wild-type; **p < 0.05 vs. late sepsis + vector or late sepsis + S100A9. The results are representative of three independent experiments. KO, knockout.
Discussion
The initial/acute phase of sepsis, if not resolved early to restore immune homeostasis, can progress to a late/chronic stage with protracted immunosuppression (Hotchkiss et al., 2013b; Patil et al., 2016) and continued mortality and morbidity (Delano and Ward, 2016; Hotchkiss et al., 2013a). Increases in functional Gr1+CD11b+ cells accumulate within a few days of sepsis onset, but then switch their phenotypes to become MDSCs later (Brudecki et al., 2012). We reported that nuclear S100A9 accumulation in Gr1+CD11b+ cells parallels increases in miR-21 and miR-181b levels during late sepsis in wild-type mice, but not in S100A9 deficient mice (Dai et al., 2017). The present study’s new discovery is that S100A9 mediates the expression of miR-21 and miR-181b during late sepsis concomitant with increasing MDSCs repressor function. These findings suggest that S100A9 is a significant regulator of MDSC expansion during late/chronic sepsis in mice.
Activation of miR-21 and miR-181b expressions is a dynamic process. In the steady-state, transcription factor C/EBPα, which supports normal myeloid cell development and differentiation (Hirai et al., 2006), binds the miR-21 and miR-181b promoters (McClure et al., 2014; McClure et al., 2017). After sepsis initiation, C/EBPα is replaced by C/EBPβ, which forms a complex with pStat3 (McClure et al., 2017). Fig. 7 depicts the dynamic binding process of pStat3 and C/EBPβ. The protein complex then activates the expression of miR-21 and miR-181b. The current study shows that pStat3 and C/EBPβ do not bind miR-21 and miR-181b promoters in Gr1+CD11b+ cells during late sepsis in the S100A9 knockout mice (Dai et al., 2017). Thus, these findings indicate that S100A9 protein plays an essential role in miR-21 and miR-181b expression induction during late sepsis. In support of this, we showed that ectopic expression of S100A9 in these cells restored pStat3 and C/EBPβ bindings at the miRNA promoters and induced their expression.
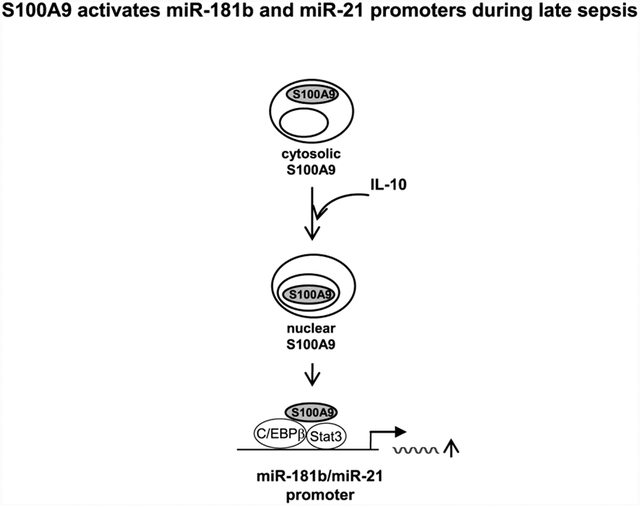
Figure 7: A proposed model for regulation of miR-21 and miR-181b promoters by S100A9 in Gr1+CD11b+ cells during late sepsis response.

During late sepsis, S100A9 protein promotes assembly of pStat3 and C/EBP β into one protein complex and induces its binding at the miR-181b and miR-21 promoters in Gr1+ CD11b+ cells, leading to activation of the miRNA expressions and subsequent upregulation of NFI-A. This process is mediated by IL-10 signaling, which promotes S100A9 protein translocation from the cytosol to the nucleus (Bah et al., 2018). In the absence of S100A9 (S100A9 KO), pStat3 and C/EBP β cannot bind/activate the miRNA promoters. Not shown is the regulatory node downstream of NFI-A that leads to Gr1+CD11b+ cell expansion and suppressive functions.
The mechanism by which S100A9 contributes to the pStat3-C/EBPβ protein complex binding at the miR-21 and miR-181b promoters only during late sepsis remains unclear. In early sepsis Gr1+CD11b+ cells, S100A9 resides in the cytosol and does not affect miR-21 and miR-181b promoters, because they are expressed in the S100A9 knockout mice (Dai et al., 2017). The current study shows that nuclear S100A9 may stabilize pStat3-C/EBPβ complex binding at the miRNA promoters. Moreover, we found that IL-10, which induces S100A9 nuclear translocation during late sepsis, supports Stat3 phosphorylation (Bah et al., 2018). Nuclear S100A9 acting as a coregulator might modify the chromatin structure around the miRNA promoters and facilitate pStat3-C/EBPβ complex binding. In this regard, we previously reported that HMGB1 protein, which like S100A9 functions as a proinflammatory mediator when secreted by immune cells (Andersson et al., 2000; Park et al., 2006), modifies chromatin at promoters of proinflammatory genes in endotoxin-tolerant monocytes (El Gazzar et al., 2009).
MiR-21 and miR-181b couple to promotes NFI-A expression, which propagates its effects downstream in the path leading to Gr1+CD11b+ cell expansion in sepsis (McClure et al., 2016; McClure et al., 2014). This regulatory mechanism operates during the early and late sepsis phases in wild-type mice. However, Gr1+CD11b+ cells that expand during early sepsis are not immunosuppressive and can differentiate and mature ex vivo with growth factors (Brudecki et al., 2012; McPeak et al., 2017a). In contrast, late sepsis Gr1+CD11b+ cells, in which S100A9 resides in the nucleus (Dai et al., 2017), are immunosuppressive (Brudecki et al., 2012). On the other hand, the lack of Gr1+CD11b+ cell expansion and immunosuppressive functions in the S100A9 knockout mice during late sepsis suggests that S100A9 not only is vital for Gr1+CD11b+ cell expansion, but also for switching them into the immunosuppressive, MDSC phenotype.
S100 proteins released from leukocytes can act as proinflammatory mediators and acute phase proteins (Ehrchen et al., 2009; Foell et al., 2004a; Foell et al., 2004b; Vogl et al., 2012). S100A9 protein released at the site of inflammation can serve as an “alarmin” by binding to and activating toll-like receptor 4 (TLR4) on innate immunity cells, which increases phagocyte activation and recruitment (Foell et al., 2004a; Hiratsuka et al., 2006; Vogl et al., 2007). A previous study reported that S100A9 promotes MDSC expansion in tumor-bearing mice by inhibiting myeloid progenitor differentiation via increasing production of reactive oxygen species (Cheng et al., 2008). In our polymicrobial sepsis model, S100A9 protein is expressed throughout sepsis but its secretion markedly decreases concurrently with its nuclear accumulation in Gr1+CD11b+ cells during the late/immunosuppressive phase (Dai et al., 2017). Together, the new data from this study support the notion that S100A9 is a multifunctional protein and promote or repress inflammation, depending on the context (Foell et al., 2004b; Ikemoto et al., 2007; Vogl et al., 2012; Vogl et al., 2007). Our novel findings that S100A9 contribute to sepsis phenotype shift from proinflammatory to immunosuppressive at the molecular level are significant. Post-sepsis immunosuppression attenuates both innate and adaptive immune responses (Delano et al., 2016; Hotchkiss et al., 2013b), which leads to the acquisition of secondary, opportunistic infections, and thus elevates mortality (Otto et al., 2011; Torgersen et al., 2009).
Our findings that genetically targeting S100A9 in the mouse can inhibit MDSC expansion during late sepsis could be biologically significant. There are no molecular-based treatments for sepsis. Limiting MDSC-mediated support of immune suppression or chronic sepsis by targeting S100A9 might restore immune competency and improve survival during the post-sepsis chronic illness syndrome.
Supplementary Material
Highlights.
A mechanism for S100A9-mediated induction of miR-21 and miR-181b in sepsis is proposed.
S100A9 contributes to the activation of miR-21 and miR-181b promoters only during the late phase of sepsis.
S100A9 is sufficient to switch normal myeloid precursors into immunosuppressive MDSCs.
Acknowledgments
This work was supported by National Institutes of Health Grants R01GM103887 (to ME) and C06RR0306551 (to College of Med.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflicts of interest.
REFERENCES
- 1.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ, 2000. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med 192, 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bah I, Kumbhare A, Nguyen L, McCall CE, El Gazzar G, 2018. IL-10 induces an immune repressor pathway in sepsis by promoting S100A9 nuclear localization and MDSC development. Cell Immunol 332, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brudecki L, Ferguson DA, McCall CE, El Gazzar M, 2012. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect. Immun 80, 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brudecki L, Ferguson DA, Yin D, Lesage GD, McCall CE, El Gazzar M, 2012. Hematopoietic stem-progenitor cells restore immunoreactivity and improve survival in late sepsis. Infect. Immun 80, 602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI, 2008. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med 205, 2235–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Condamine T, Gabrilovich DI, 2011. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 32, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, LaFace DM, Heyworth PG, Efron PA, Moldawer LL, 2011. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol. Med 17, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai J, El Gazzar M, Li GY, Moorman JP, Yao ZQ, 2015. Myeloid-derived suppressor cells: paradoxical roles in infection and immunity. J. Innate. Immun 7, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai J, Kumbhare A, Youssef D, McCall CE, El Gazzar M, 2017. Intracellular S100A9 Promotes Myeloid-Derived Suppressor Cells during Late Sepsis. Front Immunol 8, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL, 2007. MyD88-dependent expansion of an immature GR-1(+) CD11b (+) population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med 204, 1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delano MJ, Ward PA, 2016. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J. Clin. Invest 126, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Efron PA, Mohr AM, Bihorac A, Horiguchi H, Hollen MK, Segal MS, Baker HV, Leeuwenburgh C, Moldawer LL, Moore FA, Brakenridge SC, 2018. Persistent inflammation, immunosuppression, and catabolism and the development of chronic critical illness after surgery. Surgery 164, 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehrchen JM, Sunderkotter C, Foell D, Vogl T, Roth J, 2009. The endogenous Tolllike receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol 86, 557–566. [DOI] [PubMed] [Google Scholar]
- 14.El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE, 2009. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol. Cell Biol 29, 1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foell D, Frosch M, Sorg C, Roth J, 2004b. Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin. Chim. Acta 344, 37–51. [DOI] [PubMed] [Google Scholar]
- 16.Foell D, Roth J, 2004a. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum 50, 3762–3771. [DOI] [PubMed] [Google Scholar]
- 17.Gabrilovich DI, Nagaraj S, 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goyette J, Geczy CL, 2011. Inflammation-associated S100 proteins: new mechanisms that regulate function. Amino. Acids 41, 821–842. [DOI] [PubMed] [Google Scholar]
- 19.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, Tenen DG, 2006. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat. Immunol 7, 732–739. [DOI] [PubMed] [Google Scholar]
- 20.Hiratsuka S, Watanabe A, Aburatani H, Maru Y, 2006. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol 8, 1369–1375. [DOI] [PubMed] [Google Scholar]
- 21.Hotchkiss RS, Monneret G, Payen D, 2013a. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis 13, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hotchkiss RS, Monneret G, Payen D, 2013b. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol 13, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikemoto M, Murayama H, Itoh H, Totani M, Fujita M, 2007. Intrinsic function of S100A8/A9 complex as an anti-inflammatory protein in liver injury induced by lipopolysaccharide in rats. Clin. Chim. Acta 376, 197–204. [DOI] [PubMed] [Google Scholar]
- 24.Kong YY, Fuchsberger M, Xiang SD, Apostolopoulos V, Plebanski M, 2013. Myeloid derived suppressor cells and their role in diseases. Curr. Med. Chem 20, 1437–1444. [DOI] [PubMed] [Google Scholar]
- 25.Mazuski JE, Sawyer RG, Nathens AB, DiPiro JT, Schein M, Kudsk KA, Yowler C, 2002. The Surgical Infection Society guidelines on antimicrobial therapy for intraabdominal infections: an executive summary. Surg. Infect. (Larchmt.) 3, 161–173. [DOI] [PubMed] [Google Scholar]
- 26.McClure C, Ali E, Youssef D, Yao ZQ, McCall CE, El Gazzar M, 2016. NFI-A disrupts myeloid cell differentiation and maturation in septic mice. J. Leukoc. Biol 99, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McClure C, Brudecki L, Ferguson DA, Yao ZQ, Moorman JP, McCall CE, El Gazzar M, 2014. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect. Immun 82, 3816–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McClure C, McPeak MB, Youssef D, Yao ZQ, McCall CE, El Gazzar M, 2017. Stat3 and C/EBPbeta synergize to induce miR-21 and miR-181b expression during sepsis. Immunol. Cell Biol 95, 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McPeak MB, Youssef D, Williams DA, Pritchett C, Yao ZQ, McCall CE, El Gazzar M, 2017a. Myeloid Cell-Specific Knockout of NFI-A Improves Sepsis Survival. Infect. Immun 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McPeak MB, Youssef D, Williams DA, Pritchett CL, Yao ZQ, McCall CE, El Gazzar M, 2017b. Frontline Science: Myeloid cell-specific deletion of Cebpb decreases sepsis-induced immunosuppression in mice. J. Leukoc. Biol 102, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostrand-Rosenberg S, Fenselau C, 2018. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol 200, 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, Bauer M, Riedemann NC, 2011. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care 15, R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E, 2006. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol Cell Physiol 290, C917–C924. [DOI] [PubMed] [Google Scholar]
- 34.Patil NK, Bohannon JK, Sherwood ER, 2016. Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol. Res 111, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth J, Goebeler M, van den Bos C, Sorg C, 1993. Expression of calcium-binding proteins MRP8 and MRP14 is associated with distinct monocytic differentiation pathways in HL-60 cells. Biochem. Biophys. Res. Commun 191, 565–570. [DOI] [PubMed] [Google Scholar]
- 36.Shubin NJ, Monaghan SF, Ayala A, 2011. Anti-inflammatory mechanisms of sepsis. Contrib. Microbiol 17, 108–124. [DOI] [PubMed] [Google Scholar]
- 37.Torgersen C, Moser P, Luckner G, Mayr V, Jochberger S, Hasibeder WR, Dunser MW, 2009. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth. Analg 108, 1841–1847. [DOI] [PubMed] [Google Scholar]
- 38.Veglia F, Perego M, Gabrilovich D, 2018. Myeloid-derived suppressor cells coming of age. Nat. Immunol 19, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogl T, Gharibyan AL, Morozova-Roche LA, 2012. Pro-inflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexes. Int. J. Mol. Sci 13, 2893–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J, 2007. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med 13, 1042–1049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.