

Abstract
Root system architecture is crucial for wheat adaptation to drought stress, but phenotyping for root traits in breeding programmes is difficult and time-consuming owing to the belowground characteristics of the system. Identifying quantitative trait loci (QTLs) and linked molecular markers and using marker-assisted selection is an efficient way to increase selection efficiency and boost genetic gains in breeding programmes. Hundreds of QTLs have been identified for different root traits in the last few years. In the current study, consensus QTL regions were identified through QTL meta-analysis. First, a consensus map comprising 7352 markers was constructed. For the meta-analysis, 754 QTLs were retrieved from the literature and 634 of them were projected onto the consensus map. Meta-analysis grouped 557 QTLs in 94 consensus QTL regions, or meta-QTLs (MQTLs), and 18 QTLs remained as singletons. The recently published genome sequence of wheat was used to search for gene models within the MQTL peaks. As a result, gene models for 68 of the 94 Root_MQTLs were found, 35 of them related to root architecture and/or drought stress response. This work will facilitate QTL cloning and pyramiding to develop new cultivars with specific root architecture for coping with environmental constraints.
Subject terms: Agricultural genetics, Plant breeding
Introduction
Wheat is the most widely cultivated crop in the world, providing humans with about 18% of their daily intake of calories and 20% of their protein (http://faostat.fao.org/). Because of its wide adaptability, wheat is grown in a wide range of environments, many of them in dryland regions, where soil water deficit is the main limiting factor for crop growth, and production depends on stored soil moisture. Moreover, as a consequence of human-induced climate change, warmer temperatures and lower and more erratic water availability affecting the major producing areas are expected in the next few decades1.
Breeding for adaptation to drought-prone environments is challenging because of its complexity and that of the plant mechanisms involved in drought tolerance2. Among the mechanisms adopted by plants, the root system architecture plays an important role in crop performance under low water input conditions3. Modelling studies conducted in Australia have shown that direct selection for deeper, more effective roots could considerably increase the capture of water and nitrogen from soil, thus resulting in a wheat yield improvement under rainfed conditions4,5. However, because of the inherent difficulties in measuring and assessing it, root system architecture has not been considered a selection trait in the breeding process6. Discovering quantitative trait loci (QTLs) controlling root architecture and identifying molecular markers linked to them are an essential tool for exploiting root traits in breeding programmes through marker-assisted selection.
A number of QTL studies for a wide range of root traits have been carried out in the last few years, and some of them are reviewed in Table 1. These studies identified hundreds of QTLs in different mapping populations with different types of markers. In order to identify consensus QTL regions in the genome, Goffinet and Gerber7 developed QTL meta-analysis. This method allows results of independent QTL studies to be integrated in a consensus or reference map. The power of QTL meta-analysis lies in identifying regions of the genome that are most frequently involved in trait variation and narrowing down the QTL supporting intervals, thus facilitating the identification of candidate genes for positional cloning. In selecting meta QTLs (MQTLs) to be used for breeding purposes, three criteria must be met8: (1) a small supporting interval, (2) clustering of a high number of initial QTLs, and (3) a high effect of the phenotypic variance explained by the initial QTLs.
Table 1.
Summary of QTL studies included in the meta-analysis.
| Reference | Cross | Type | Size | Traitsa | N QTLs | Projected QTLs |
|---|---|---|---|---|---|---|
| An et al.71 | Hanxuan 10 × Lumai 14 | DH | 120 | RDW | 4 | 3 |
| Ayalew et al.32 | W7984 × Opata 85 | RIL | 104 | RDW, TRL | 13 | 13 |
| Bai et al.72 | Avalon × Cadenza | DH | 199 | RDW, RSA, RSR, RV, SAL, SASA, SAVol, SLL, SLSA, SLVol, TRL | 32 | 32 |
| Ballesteros et al.73 | USG3209 × Jaypee | RIL | 130 | TRB, TRL | 7 | 6 |
| Botwright Acuña et al.74 | Cranbrook × Halberd | DH | 161 | AWSDW, BWSDW, RDW | 21 | 18 |
| Christopher et al.29 | SeriM82 × Hartog | DH | 184 | RGA, TRN | 11 | 4 |
| Czyczyło-Mysza et al.75 | Chinese spring × SQ1 | DH | 90 | RDW, TRL | 18 | 17 |
| Ehdaie et al.76 | Iran #49 × Yecora Rojo | RIL | 168 | DRW, LRL, RPR, RSR,SRW, TRL | 12 | 6 |
| Guo et al.77 | Chuan 35050 × Shannong 483 | RIL | 131 | ARN, RDW, RKC, RKUE, RNC, RPC, RPUE | 13 | 13 |
| Hamada et al.78 | U24 × Ayahikari | DH | 103 | DRR, RAH, RER, SRN | 7 | 7 |
| Horn et al.79 | Spark × Rialto | DH | 119 | RHL | 3 | 3 |
| Horn et al.79 | Charger × Badger | DH | 95 | RHL | 1 | 1 |
| Ibrahim et al.80 | Devon × Syn084 | BC | 177 | ARD, CRS,FRK, RSA, RV, TIP, TRL | 32 | 17 |
| Iehisa et al.81 | Ldn/KU-2159 × Ldn/IG126387 | F2 | 100 | RRGI, TRL | 3 | 3 |
| Iehisa et al.82 | Chinese spring × Hope5A | F2 | 110 | RRGI, TRL | 2 | 2 |
| Kabir et al.83 | Nongda 338 × Jingdong 6 | DH | 216 | MRL, RSA, RV, TIP, TRL | 39 | 26 |
| Kabir et al.83 | Nongda 331 × Zang 1817 | RIL | 217 | MRL, RSA, RV, TIP, TRL | 14 | 9 |
| Kadam et al.84 | WL711 × C306 | RIL | 206 | MaxRL, RBB30, RBU30, TRB | 5 | 5 |
| bKubo et al.21 | Jennah Khetifa × Cham1 | RIL | 110 | RDW, RP | 2 | 2 |
| Landjeva et al.85 | Chinese Spring × Synthetic 6× | RIL | 85 | RSR | 5 | 5 |
| Li et al.86 | Rio Blanco × IDO444 | RIL | 159 | LRL, TRL, TRN | 15 | 14 |
| Liu et al.87 | Hanxuan 10 × Lumai 14 | DH | 150 | MaxRL, PRA, SRA, SRN, TRL | 46 | 42 |
| bMaccaferri et al.22 | Colosseo × Lloyd | RIL | 176 | ARL, LRN, PRD, PRL, RDW, RGA, RSR, RT6, RV, SRA, TRD, TRL, TRN | 41 | 41 |
| bMaccaferri et al.22 | Meridiano × Claudio | RIL | 181 | ARL, LRN, PRD, PRL, PRS, PRV, RDW, RGA, RT6, RV, SRA, TRD, TRL, TRN | 67 | 66 |
| bPetrarulo et al.23 | Creso × Pedroso | RIL | 123 | RV, SRA, TIP, TRL | 57 | 57 |
| Ren et al.88 | Xiaoyan 54 × Jing 411 | RIL | 142 | LatRL, MaxRL, RDW | 35 | 31 |
| Ren et al.89 | Xiaoyan 54 × Jing 411 | RIL | 142 | MaxRL, RDW | 17 | 15 |
| Sun et al.90 | Chuan 35050 × Shannong 483 | RIL | 131 | REW, RFW, RSR | 31 | 26 |
| Xie et al.91 | Forno × Oberkulmer | RIL | 226 | MasRD, MaxRW, SRL, SRN | 38 | 25 |
| Yu and Chen92 | W7984 × Opata85 | RIL | 112 | RDW | 8 | 8 |
| Zhang et al.93 | Weimai 8 xLuohan 2 | RIL | 229 | LRL, RDW, RFW, RSR, TRN | 38 | 27 |
| Zhang et al.93 | Weimai 8 × Yannong 19 | RIL | 302 | LRL, RDW, RFW, RSR, TRN | 17 | 8 |
| Zhang et al.94 | Weimai 8 × Jimai 20 | RIL | 172 | LRL, RDW, RSR, TRN | 17 | 15 |
| Zhang et al.94 | Weimai 8 × Luohan 2 | RIL | 179 | LRL, RDW, RSR, TRN | 20 | 17 |
| Zhang et al.95 | Weimai 8 × Yannong 19 | RIL | 175 | LRL, RDW, RSR, TRN | 19 | 15 |
| Zhao et al.95 | Huapei 3 × Yumai 57 | DH | 168 | ACT, ARD, CCR, KCR, MCR, NCR, RDW, RSR, RV, SRA, TRL | 44 | 35 |
aAbbreviation list for the traits is shown in Supplementary Material 2.
bStudies carried out in durum wheat.
QTL meta-analysis has been performed in the last few years in wheat for traits such as grain yield9,10, crop phenology10,11, disease resistance8,12–14, plant height15, grain-related traits16,17 and sprouting tolerance and dormancy18. QTL meta-analysis for root-related traits was performed previously by Darzi-Ramandi et al.6, who only considered 53 QTLs from chromosome groups 2 and 3 for the analysis, and by Iannucci et al.19, who performed the analysis in durum wheat using 100 QTLs retrieved from the literature together with 17 QTLs discovered in their study.
The present study reports the results of the largest QTL meta-analysis conducted for root traits in wheat. QTL projection was carried out on a consensus map also developed in this study by integrating three maps used as a reference composed mainly of simple sequence repeat (SSR) and DArT markers (Composite-2004, available at http://wheat.pw.usda.gov; SSR-200420; and Integrated-201313). The main objective of the study was to produce a repository of root QTL information to define consensus regions controlling root architecture in wheat.
Results
Consensus map
The Wheat_Consensus_2018 map included 7352 markers (Supplementary Material 1) after combining the Composite_2004, SSR_200420 and Integrated_201313 maps. The total length of the consensus map was 4994.0 cM, with an average chromosome length of 237.8 cM and a range of 155.6 cM (5D) to 347.5 cM (7A). The average number of markers per chromosome was 350, chromosome 3B carrying the highest number of markers (580) and chromosome 4D the lowest (178). The overall marker density was 1.5 markers/cM, ranging from 3.1 markers/cM on chromosome 3B to 0.8 markers/cM on chromosome 7D. Genome A covered a distance of 1810.2 cM with 2497 markers, genome B covered a distance of 1789.1 cM with 3230 markers, showing the highest marker density (1.8 markers/cM), and genome D covered a distance of 1394.7 cM with 1625 markers.
The three maps shared 8% of the markers (566), whereas Composite_2004 had 3531 unique markers (48%), SSR_2004 81 had markers (1%) and Integrated_2013 had 2351 markers (32%). Composite_2004 and SSR_2004 shared 71 markers (1%), Composite_2004 and Integrated_2013 shared 235 markers (3%) and SSR_2004 and Integrated_2013 517 shared 517 markers (7%) (Fig. 1).
Figure 1.
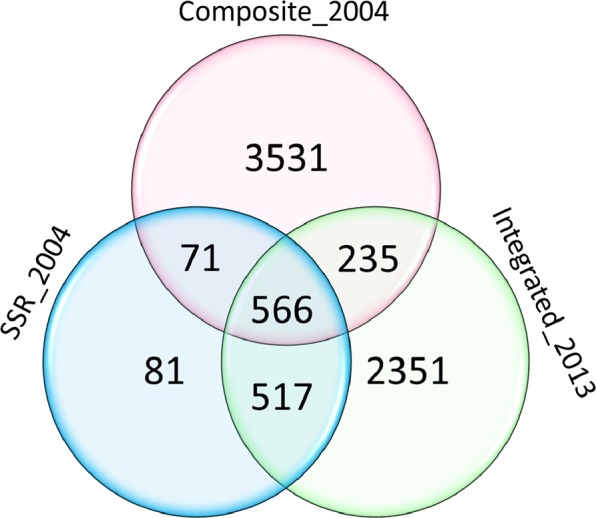
Venn diagram showing the number of unique and common markers among the three maps used to construct the consensus map.
QTL distribution and projection
Thirty studies published from 2006 to 2017 based on bi-parental populations reporting 754 QTLs for root traits were collected (Table 1, Supplementary Material 2). The studies covered 31 different populations with 56 lines as parents. QTLs were distributed throughout the whole genome. The number of QTLs per chromosome ranged from 7 on chromosome 3D to 63 on chromosome 2B, with an average of 36 QTLs per chromosome. Of the QTLs, 39% were identified in genome A, 42% in genome B and 19% in genome D (Fig. 2a). Three studies21–23 were carried out in durum wheat, where only genomes A and B are present, reporting a total of 167 QTLS. The most reported QTLs corresponded to traits involved in root length (30%), followed by QTLs involved in root number and weight (19% and 14% respectively) (Fig. 2b). Supporting intervals (SI) ranged from 0 to 75.1 cM, with an average of 14.8 cM. Approximately half of the collected QTLs (47%) had an SI lower than 10 cM, and 79% had an SI lower than 20 cM (Fig. 2c). The proportion of phenotypic variance explained (PVE) by single QTLs followed an L-shaped distribution, with most of the QTLs showing a PVE lower than 0.20 (96%) (Fig. 2d). PVE ranged from 0.01 to 0.76, with an average of 0.1.
Figure 2.

Traits estimated from the collected QTL studies. Number of QTLs per (a) chromosome, (b) trait, (c) supporting interval and (d) phenotypic variance explained (PVE).
A total of 634 out of the 754 collected QTLs were projected onto the Wheat_Consensus_2018 map. One hundred and twenty-one QTLs were not projected because (1) they lacked common markers between initial and consensus maps, and (2) the QTL showed a low PVE, causing a large SI (>50 cM). The projection onto the consensus map showed a clustering of QTLs in the centromeric and pericentromeric regions (Figs 3 and 4).
Figure 3.
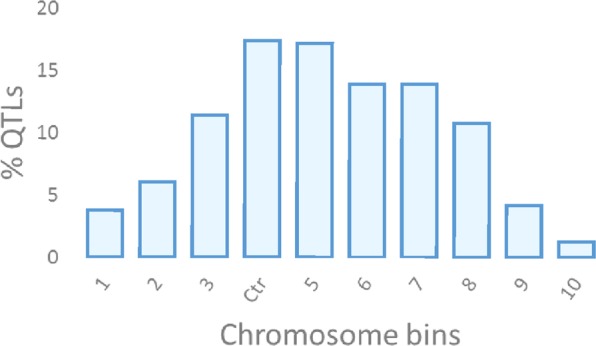
Representation of the QTL coverage along a consensus chromosome Ctr: centromere (bin 4). Each bin represents 10% of the whole chromosome.
Figure 4.

Concentric circles representing the root QTLome. (A) Wheat_Consensus_2018 map. (B) Frequency of QTLs computed as QTL-overview index. (C) MQTL positions with an SI of 95%. (D) Heatmap representing the number of QTLs for 25 cM bins (number of QTLs increasing from light to dark).
Figure 3 represents a consensus chromosome among the 21 wheat chromosomes divided into 10 bins. Each bin represents 10% of the total length of the consensus chromosome. Centromeric (4) and pericentromeric bins (3 and 5) included almost half of the QTLs (46%), decreasing towards telomeres. Central regions of chromosome arms, bin 2 (short arm) and bins 6 to 8 (long arm), carried 45% of the QTLs and, finally, the telomeric regions, bin 1 (short arm) and bins 9 and 10 (long arm), contained only 9% of the QTLs.
To detect the genome regions most frequently involved in the genetic control of root traits, the QTL-overview index24 was calculated for each cM of the consensus map (Fig. 4, Supplementary Material 3). A total of 150 overview peaks were obtained, of which 126 exceeded the average value of the statistic for each chromosome and represent hot spots for root QTLs. Additionally, 41 of the 126 peaks exceeded a high value threshold calculated as five times the average value of the overview index for each chromosome (Supplementary Material 3). These 41 peaks corresponded to 35 of the 94 MQTL (37%) and included 292 QTLs (52%).
QTL meta-analysis
From 634 QTLs projected onto the Wheat_Consensus_2018 map 557, were grouped into 94 meta-QTLs (MQTLs), whereas 18 remained as single QTLs (sQTLs) not overlapping with MQTLs (Table 2, Fig. 4). Fifty-nine QTLs were not assigned to any MQTL because of their large SI overlapping with different MQTLs or because the predicted QTL peaks were not included within any MQTL SI. However, they were not considered as sQTLs as their SI overlapped with MQTLs.
Table 2.
Summary of the meta-QTLs for root traits including the gene model based on the wheat genome sequence annotation for the closest marker and its protein description.
| MQTL | Chr | Position (cM) | SI (95%) | N QTLs | N studies | Traits | Closest marker | Gene modela | Description |
|---|---|---|---|---|---|---|---|---|---|
| Root_MQTL_1 | 1A | 48.0 | 4.5 | 5 | 2 | RDW, PRV,LRL, RV | wPt-1862_1A | TraesCS1A01G038700 | NBS-LRR like resistance protein |
| Root_MQTL_2 | 66.3 | 2.9 | 16 | 3 | TRL, RHL, SRA, TIP, LatRL, RV, MaxRL | Xgwm33_1A | … | … | |
| Root_MQTL_3 | 89.2 | 3.2 | 5 | 3 | RDW, RFW, CCR, TRL | Xwmc286_1A | TraesCS1A01G069600 | Zinc finger CCCH domain-containing protein | |
| Root_MQTL_4 | 99.2 | 8.3 | 2 | 2 | RDW, TRL | Xgwm135_1A | TraesCS1A01G228000 | Phosphatase 2C family protein | |
| Root_MQTL_5 | 106.2 | 2.7 | 7 | 5 | RAH, NCR, RSR, LRL, RV, RDW, TRL | Xpsr1327_1A | TraesCS1A01G431000 | F-box family protein | |
| Root_MQTL_6 | 121.3 | 7.1 | 2 | 1 | TRL | XksuE3_1A | … | … | |
| Root_MQTL_7 | 1B | 27.2 | 3.5 | 12 | 6 | RV, RSR, TRL, BWSDW, TIP, SRA, AWSDW, RDW | Xwmc619_1B | TraesCS1B01G086300 | Glycosyl hydrolase family protein |
| Root_MQTL_8 | 51.1 | 2.8 | 15 | 4 | ACT, TRL, SRA, ARN, RPUE, RPC, RNC, RSR, RKC, RKUE, RV, TRN | Xwmc500.2_1B | … | … | |
| Root_MQTL_9 | 73.8 | 4.8 | 4 | 3 | RDW, TRL, TIP, MaxRL | Xpsr949_1B | TraesCS1B01G144600 | Hexosyltransferase | |
| Root_MQTL_10 | 84.2 | 3.5 | 5 | 4 | TRL, ARL, LRL, RDW, RV | Xbarc240_1B | TraesCS1B01G214900 | Peroxidase | |
| Root_MQTL_11 | 99.3 | 7.9 | 2 | 1 | LRL | Xwmc85a_1B | TraesCS1B01G312100 | Glycosyltransferase | |
| Root_MQTL_12 | 147.4 | 10.6 | 2 | 2 | DRR, TRL | wPt-8832_1B | TraesCS1B01G440200 | Root Primordium Defective 1 | |
| Root_MQTL_13 | 187.1 | 1.1 | 3 | 2 | TRB, RSR | wPt-6142_1B | … | … | |
| Root_MQTL_14 | 1D | 50.0 | 6.4 | 3 | 3 | AWSDW, SRA, RFW | P32/M59-2_1D | TraesCS1D01G040100 | Poly [ADP-ribose] polymerase |
| Root_MQTL_15 | 87.7 | 3.6 | 7 | 3 | RDW, RSR, BWSDW | Xwmc590_1D | TraesCS1D01G238300 | Signal peptidase I | |
| Root_MQTL_16 | 100.8 | 4.5 | 2 | 2 | FRK, RFW | Xbarc62_1D | TraesCS1D01G451200 | Early flowering 3 | |
| Root_MQTL_17 | 2A | 47.0 | 3.5 | 11 | 5 | TRN, LRN, LRL, TRL, TIP, RDW, RSA | Xbarc1138_2A | TraesCS2A01G004600 | C2H2-like zinc finger protein |
| Root_MQTL_18 | 91.7 | 2.2 | 14 | 4 | MaxRW, RSR, RV, TRL, RPR, SRA, TIP, SRW, RT6 | Xwmc296_2A | TraesCS2A01G134400 | Plasma-membrane choline transporter family protein | |
| Root_MQTL_19 | 99.6 | 1.9 | 8 | 6 | TRL, RHL, RV, SRN,MRL, TRN | Xbarc309_2A | TraesCS2A01G160000 | Pentatricopeptide repeat-containing protein | |
| Root_MQTL_20 | 144.4 | 9.2 | 3 | 2 | LRN, RV, RDW | Xcfd168_2A | … | … | |
| Root_MQTL_21 | 2B | 1.2 | 3.3 | 2 | 1 | TRN | Lr16_2B | … | … |
| Root_MQTL_22 | 22.3 | 1.0 | 14 | 5 | RAH, MaxRL, PRL, RSR, TRL, LRL, RFW, PRE, LatRL | wPt-8737_2B | TraesCS2B01G029100 | Glycosyltransferase | |
| Root_MQTL_23 | 30.0 | 6.4 | 4 | 1 | TRL, SRA, PRL, ARL | Xcdo456_2B | TraesCS2B01G040500 | Glycosyltransferase | |
| Root_MQTL_24 | 97.5 | 5.8 | 3 | 2 | TRL, SRN | Xwmc770_2B | |||
| Root_MQTL_25 | 116.5 | 1.9 | 18 | 7 | RDW, RGA, BWSDW, PRA, SRA, TRL, TRN | Xbarc55_2B | TraesCS2B01G159800 | Terpene synthase | |
| Root_MQTL_26 | 154.3 | 0.5 | 17 | 8 | RGA, RHL, RFW, RDW, RT6, ARL, PRL, CRS, PRD, TRN, SRA | wPt-6522_2B | TraesCS2B01G436400 | Oleosin | |
| Root_MQTL_27 | 2D | 29.2 | 12.4 | 5 | 1 | SASA, RSA, RDW, SAVol, RV | Xgwm296_2D | TraesCS2D01G100800 | NAC domain protein, |
| Root_MQTL_28 | 49.2 | 5.0 | 4 | 4 | MaxRL, RSR, TRB, TRL | Xcfd255_2D | TraesCS2D01G118300 | E3 ubiquitin-protein ligase | |
| Root_MQTL_29 | 75.2 | 7.8 | 2 | 2 | RDW, RSR | Xwmc18_2D | TraesCS2D01G185600 | Dihydroflavonol-4-reductase | |
| Root_MQTL_30 | 100.5 | 2.8 | 7 | 3 | RSR, TRN, RDW | Xbarc228_2D | TraesCS2D01G464800 | Multidrug resistance protein ABC transporter family protein | |
| Root_MQTL_31 | 3A | 37.7 | 4.6 | 6 | 3 | TRD, RDW, PRD | Xbcd1428_3A | TraesCS3A01G025200 | E3 ubiquitin-protein ligase |
| Root_MQTL_32 | 72.3 | 2.9 | 7 | 4 | TRL, MaxRD, TRD, RSA, SRL, TIP | Xgwm2_3A | TraesCS3A01G197700 | Serine/threonine-protein phosphatase | |
| Root_MQTL_33 | 90.7 | 5.5 | 7 | 2 | RSA, TRL, SLL, SLVol, SLSA, TIP | Xwmc695_3A | … | … | |
| Root_MQTL_34 | 99.2 | 5.7 | 6 | 3 | TIP, SRA, TRN, PRE, RV, TRL | Xwmc264_3A | TraesCS3A01G376500 | E3 ubiquitin-protein ligase SINA-like 10 | |
| Root_MQTL_35 | 113.8 | 4.5 | 5 | 3 | RDW, RV, TRL, SRA | wPt-4725_3A | TraesCS3A01G477800 | glutamyl-tRNA amidotransferase | |
| Root_MQTL_36 | 146.7 | 3.4 | 2 | 2 | RDW, RSR | wPt-1864_3A | … | … | |
| Root_MQTL_37 | 169.6 | 1.3 | 4 | 3 | LRL, TRN, RT6, TRL | wPt-9422_3A | TraesCS3A01G512900 | Regulator of chromosome condensation (RCC1) family with FYVE zinc finger domain | |
| Root_MQTL_38 | 3B | 35.3 | 4.3 | 11 | 4 | RSR, RDW, TRD, ARD, LRL, PRS, RV | Xwmc597_3B | … | … |
| Root_MQTL_39 | 68.3 | 3.6 | 5 | 4 | MaxRD, TRL, RSR | wPt-9170_3B | TraesCS3B01G306100 | transmembrane protein (DUF616) | |
| Root_MQTL_40 | 82.6 | 2.4 | 11 | 2 | SRN, TRL, SRA, PRA | Xwmc527a_3B | TraesCS3B01G334000 | F-box family protein | |
| Root_MQTL_41 | 92.4 | 6.3 | 2 | 1 | TRL, MRL | Xwmc787_3B | TraesCS3B01G463600 | F-box domain containing protein | |
| Root_MQTL_42 | 109.7 | 3.7 | 8 | 5 | AWSDW, BWSDW, RGA, TRD, RSR, RDW, TRN | wPt-10071_3B | TraesCS3B01G504900 | B3 domain-containing protein | |
| Root_MQTL_43 | 158.0 | 5.9 | 3 | 3 | LRN, RDW, TRN | Xfba167b_3B | TraesCS3B01G586400 | Late embryogenesis abundant protein | |
| Root_MQTL_44 | 4A | 40.7 | 5.1 | 8 | 5 | RV, TIP, TRL, PRA, SRA, LRL | Xwmc89_4A | TraesCS4A01G216800 | Late embryogenesis abundant hydroxyproline-rich glycoprotein |
| Root_MQTL_45 | 94.5 | 2.7 | 3 | 1 | SRN, MaxRD, TRL | Xwmc513_4A | TraesCS4A01G306600 | Germin-like protein | |
| Root_MQTL_46 | 130.6 | 3.1 | 5 | 3 | MaxRW, TRL, SRN, RDW, RNC | Xpsr115_4A | … | … | |
| Root_MQTL_47 | 136.2 | 2.3 | 3 | 2 | RPC, RKC, RSR | XS25M49(350)_4A | … | … | |
| Root_MQTL_48 | 160.2 | 3.7 | 4 | 3 | RFW, KCR, RDW | XgbxG557_4A | TraesCS4A01G372100 | ABC transporter family protein | |
| Root_MQTL_49 | 187.6 | 5.0 | 6 | 4 | TRN, BWSDW, RDW, LRL | wPt-5543_4A | TraesCS4A01G431700 | Late embryogenesis abundant hydroxyproline-rich glycoprotein | |
| Root_MQTL_50 | 229.5 | 2.1 | 5 | 2 | TRN, RDW, RSR, CCR, KCR | P31/M53-4_4A | … | … | |
| Root_MQTL_51 | 4B | 114.8 | 2.3 | 5 | 2 | TRL, LRL, LRN | Xcdo795_4B | … | … |
| Root_MQTL_52 | 133.0 | 2.1 | 18 | 9 | TIP, RDW, MaxRL, LRL, RBU30, ACT, TRB, RSR, RV, ARN, RBB30, TRL | QPhs.ocs-4B.1_4B | TraesCS4B01G136500 | Root Hair Defective 3 homolog | |
| Root_MQTL_53 | 153.4 | 2.5 | 16 | 4 | RV, TRL, LRL, SRA, RT6, PRA, PRS, PRL, RT6, RDW, TRN, RGA, LRN | Xwmc413_4B | TraesCS4B01G347600 | Peroxidase | |
| Root_MQTL_54 | 4D | 16.3 | 3.9 | 12 | 2 | SAL, TIP, RSA, SASA, RDW, RSR, RSV, RV, TRL, MRL | Xgpw2180_4D | … | … |
| Root_MQTL_55 | 5A | 48.5 | 3.9 | 4 | 1 | MaxRD, TRL, MaxRW, SRN | Xwmc51_5A | TraesCS5A01G012600 | ERD (Early-responsive to dehydration stress) |
| Root_MQTL_56 | 84.3 | 8.1 | 4 | 3 | MRL, MaxRL, TRL, SRN | Xcfd17b_5A | TraesCS5A01G055000 | Root Hair Defective 3 homolog | |
| Root_MQTL_57 | 103.9 | 3.4 | 7 | 5 | SAVol, TRN, AWSDW, TRN, SRN, RT6, PRL | Xbarc180_5A | TraesCS5A01G163400 | Cinnamoyl-CoA reductase | |
| Root_MQTL_58 | 123.6 | 3.0 | 4 | 3 | RRGI, TRL, MaxRL, RSA | Xcdo1090A_5A | … | … | |
| Root_MQTL_59 | 141.7 | 1.5 | 6 | 4 | MRL, RDW, TRL, RSR, LRN | Xbarc40_5A | TraesCS5A01G228300 | E3 ubiquitin-protein ligase | |
| Root_MQTL_60 | 5B | 80.6 | 8.1 | 3 | 2 | TRN, ARL, RGA | Xgdm146_5B | … | … |
| Root_MQTL_61 | 122.4 | 3.2 | 16 | 7 | TRL, SLVol, RV, SLL, RDW, SRA, MaxRL, SLSA, MRL | Xbarc109_5B | TraesCS5B01G127200 | Pentatricopeptide repeat-containing protein | |
| Root_MQTL_62 | 211.9 | 0.7 | 11 | 6 | RV, PARA, TRL, ARD, MaxRD, TRN, LRL | Xbarc232_5B | TraesCS5B01G447800 | Cytosine-specific methyltransferase | |
| Root_MQTL_63 | 5D | 35.7 | 8.0 | 3 | 2 | MaxRL | Xcfd78_5D | TraesCS5D01G096700 | Ribosomal RNA apurinic site specific lyase |
| Root_MQTL_64 | 47.2 | 8.1 | 2 | 2 | TRL, RER | Xwmc805_5D | TraesCS5D01G176000 | Late embryogenesis abundant hydroxyproline-rich glycoprotein | |
| Root_MQTL_65 | 73.2 | 5.1 | 7 | 3 | TRL, RSR, RDW | XgbxG083_5D | TraesCS5D01G342500 | ALWAYS EARLY 2 | |
| Root_MQTL_66 | 121.6 | 2.3 | 3 | 3 | RV, CCR, RDW | Xcfd86_5D | TraesCS5D01G504400 | E3 ubiquitin-protein ligase | |
| Root_MQTL_67 | 6A | 35.2 | 8.6 | 3 | 3 | RGA, RDW | Xgwm334_6A | … | … |
| Root_MQTL_68 | 55.9 | 4.9 | 5 | 3 | TRN, NCR, RDW | wPt-7565_6A | TraesCS6A01G023100 | Acid beta-fructofuranosidase | |
| Root_MQTL_69 | 100.1 | 4.0 | 14 | 5 | TRL, RFW, RHL, SRA, RV, RT6 | Xbarc171_6A | TraesCS6A01G210900 | Root meristem growth factor 1 | |
| Root_MQTL_70 | 110.0 | 10.4 | 3 | 3 | TIP, MRL | Xbarc107_6A | TraesCS6A01G269500 | Protein BREVIS RADIX | |
| Root_MQTL_71 | 128.4 | 19.7 | 3 | 1 | TRL, SAL, SLVol | Xcdo1373_6A | … | … | |
| Root_MQTL_72 | 157.5 | 2.8 | 4 | 3 | RDW, TRL LatRL, TIP | Xfba111b_6A | … | … | |
| Root_MQTL_73 | 180.7 | 8.9 | 4 | 3 | PRV, RDW, TRL, SRA, ARL | Xgwm169_6A | TraesCS6A01G393200 | Heavy metal transport / detoxification superfamily | |
| Root_MQTL_74 | 195.7 | 0.9 | 6 | 2 | TRL, PRL, RP, PRS, RGA | Xcdo836_6A | TraesCS6A01G410100 | Embryogenesis transmembrane protein-like | |
| Root_MQTL_75 | 6B | 66.7 | 9.3 | 2 | 2 | LRN, TRL | Gli-B2_6B | TraesCS6B01G054600 | F-box family protein |
| Root_MQTL_76 | 97.7 | 12.8 | 2 | 1 | TIP, LatRL | TC85307_6B | TraesCS6B01G075800 | SAUR-like auxin-responsive protein family, putative | |
| Root_MQTL_77 | 141.8 | 4.8 | 5 | 3 | TRN, ARL, RT6, RDW | Xbarc198_6B | TraesCS6B01G212200 | Pentatricopeptide repeat-containing protein | |
| Root_MQTL_78 | 153.8 | 7.4 | 3 | 1 | PRS, TRD, PRL | Yr36_6B | … | … | |
| Root_MQTL_79 | 170.4 | 5.8 | 4 | 2 | TRN, MaxRL, PRL | Xfba67b_6B | TraesCS6B01G356400 | E3 ubiquitin-protein ligase COP1 | |
| Root_MQTL_80 | 193.8 | 1.4 | 4 | 3 | TIP, TRL, RDW | P32/M54-2_6B | TraesCS6B01G398300 | Embryogenesis transmembrane protein-like | |
| Root_MQTL_81 | 6D | 42.9 | 7.5 | 2 | 2 | RSA, TRL | XksuG48_6D | … | … |
| Root_MQTL_82 | 108.7 | 12.1 | 2 | 2 | TRL | P43/M62-1_6D | TraesCS6D01G245400 | Protein BREVIS RADIX | |
| Root_MQTL_83 | 7A | 82.5 | 6.1 | 2 | 2 | ARN, RSR | Xgbx3480a_7A | … | … |
| Root_MQTL_84 | 135.0 | 6.1 | 3 | 1 | PRV, SRA, PRD | Xgwm900_7A | TraesCS7A01G154600 | Gibberellin receptor GID1A | |
| Root_MQTL_85 | 159.9 | 6.2 | 3 | 2 | RDW, RSR | Xbarc195_7A | TraesCS7A01G428400 | Peroxidase | |
| Root_MQTL_86 | 175.2 | 7.6 | 4 | 3 | RGA, SRN, RDW | Xwmc9_7A | … | … | |
| Root_MQTL_87 | 206.4 | 7.9 | 3 | 2 | RDW, TRN | Xcfa2257_7A | TraesCS7A01G481200 | F-box protein | |
| Root_MQTL_88 | 7B | 51.0 | 5.2 | 2 | 2 | LRL, TRD | wPt-2278_7B | TraesCS7B01G184000 | 1-phosphatidylinositol-3-phosphate 5-kinase-like |
| Root_MQTL_89 | 95.7 | 2.6 | 15 | 8 | LRL, RT6, TRN, TRL, PRL, MaxRL | wPt-7925_7B | TraesCS7B01G134800 | Root hair defective 3 GTP-binding protein (RHD3) | |
| Root_MQTL_90 | 160.5 | 2.4 | 2 | 2 | ARD, MaxRL | wPt-5280_7B | TraesCS7B01G429700 | Glycosyltransferase | |
| Root_MQTL_91 | 173.3 | 2.0 | 8 | 3 | ARD, RSR, TRL, KCR, MCR, SRN, MaxRW | Psy-B1_7B | TraesCS7B01G482000 | Phytoene synthase | |
| Root_MQTL_92 | 196.1 | 1.2 | 8 | 3 | SRA, ARL, PRL, TRL, TRN, PRS, LRL | Xwmc273_7B | TraesCS7B01G472200 | NAC domain-containing protein | |
| Root_MQTL_93 | 7D | 102.9 | 4.8 | 3 | 1 | TRL, SRA, PRA | Stb5_7D | … | … |
| Root_MQTL_94 | 176.8 | 8.8 | 2 | 2 | RDW | D1.1_ctg10053_7D | … | … |
a …: closest or flanking markers not mapped on the genome sequence.
The number of MQTLs per chromosome ranged from 1 on chromosome 4D to 8 on chromosome 6A. The QTLs on chromosome 3D did not cluster into MQTLs. The number of clustered QTLs per MQTL ranged from 2 on several chromosomes to 18 on chromosomes 2B (Root_MQTL25) and 4B (Root_MQTL52). Fifty-two MQTLs (55%) were derived from the clustering of QTLs from three or more different studies, thus involving different mapping populations. These MQTLs were more likely to be stable across different environments. The number of traits involved per MQTL ranged from 1 to 13 in Root_MQTL53. The SI of the MQTLs ranged from 0.47 to 19.68 cM with an average of 4.96 cM, showing a significant reduction from the initial QTLs ranging from 0.00 to 75.10 cM with an average of 14.80 cM.
Based on the criteria defined by Löffler et al.8 to consider a MQTL for marker assisted breeding, i.e: small supporting intervals of the MQTLs, high number of initial QTLs and high PVE of the initial QTLs, eight root MQTLs, the “Breeding MQTLs”, were selected (Table 3). Firstly, seventeen MQTLs including more than 10 QTLs with SI lower than 5 cM were identified. Subsequently those with a PVE mean of the initial QTLs of 0.10 or higher were chosen.
Table 3.
Breeding MQTLs.
| MQTL | Chr | Position (cM) | CI (95%) | Marker interval | N QTLs | Mean PVE | Trait category |
|---|---|---|---|---|---|---|---|
| Root_MQTL_2 | 1A | 66.3 | 2.9 | Xcfd15 - Xwmc104 | 16 | 0.11 | Angle, Length, Number, Volume |
| Root_MQTL_8 | 1B | 51.1 | 2.8 | Xwmc500 - XKsu136 | 15 | 0.11 | Nutrient Concentration, Activity, Angle, Length, Number, Volume, Root Shoot Ratio |
| Root_MQTL_18 | 2A | 91.7 | 2.2 | Xwmc32 - Xgwm339 | 14 | 0.13 | Activity, Angle, Biomass, Length, Number, Root Shoot Ratio, Volume, Width |
| Root_MQTL_22 | 2B | 22.3 | 1.0 | wPt-3459 - Xwmc382 | 14 | 0.27 | Angle, Length, Root Shoot Ratio, Weight |
| Root_MQTL_25 | 2B | 116.5 | 1.9 | Xcfa2043 - Xwmc272 | 18 | 0.10 | Angle, Area, Length, Number, Weight |
| Root_MQTL_40 | 3B | 82.6 | 2.4 | Xcdo1164 - Xfbb110 | 11 | 0.12 | Angle, Area, Length, Number |
| Root_MQTL_52 | 4B | 133.0 | 2.1 | Xgwm710 - Xgwm368 | 18 | 0.10 | Activity, Angle, Biomass, Length, Number, Root Shoot Ratio, Volume, Weight |
| Root_MQTL_69 | 6A | 100.1 | 4.0 | Xbarc146 - Xcdo29 | 14 | 0.10 | Angle, Length, Number, Volume, Weight |
Gene models
Identification of gene models using the Gbrowse tool available at https://wheat-urgi.versailles.inra.fr/Seq-Repository/Assemblies was successful for 68 of the 94 MQTL peaks reported in Table 2. Most of the gene models corresponded only to one peak. However, E3 ubiquitin ligases were found in 6 peaks, F-box domain in 5 peaks, glycosyltransferases and late embryogenesis abundant (LEA) hydroxyproline-rich glycoprotein in 4 peaks, root hair defective (RHD) 3 protein, peroxidases and pentatricopeptide repeat-containing protein in 3 peaks, and finally, ABC transporter proteins, NAC domains, embryogenesis transmembrane proteins, zinc finger domains and brevis radix proteins in 2 peaks.
Discussion
Increasing wheat productivity is considered one of the major challenges to cope for wheat producers worldwide due to the need to ensure sufficient food supply for a growing world population in the current global climate change scenario.
Breeding for drought adaptation is particularly challenging because of the complexness of the target environments and the stress-adaptive mechanisms that plants use to diminish the negative effects of water deficit2. These mechanisms allow the plant to skip, evade or accept the negative effects of drought, and therefore have an important effect on final yield25–27.
Roots anchor the plant to the soil and take up water and nutrients, thus interacting with the water available in the soil and affecting biomass production28. The role of root architecture in the response to drought stress has been reported by several authors3,29–32. The wide morphological plasticity of root system to the different soil conditions, allow plants to adapt better, particularly under drought conditions. It has been demonstrated that wheat roots reduce their growth during periods of water scarcity while increasing the water uptake rates33 and extracting water stored in deep soil layers34. It has been suggested that a deep root system with an appropriate distribution of root density along the soil profile would confer some yield advantage on wheat grown in rainfed systems, where grain filling relies on deep soil water35. Therefore, identifying and introgressing favourable alleles controlling different root traits are desirable approaches for breeding programmes. The use of molecular markers accounting for a significant amount of the variability for relevant root traits, such as those analysed in the current study, may accelerate genetic gain by improving the efficiency of selection in segregating populations and conferring breeding programmes a great advantage in terms of selection for adaptation to drought.
In the last ten years, numerous studies identifying QTLs controlling root traits have been published, each of them using different traits, genetic backgrounds, mapping populations and/or environmental conditions. In order to reduce redundancies and to find consensus genomic regions harbouring the most robust and reliable QTLs among the mapping populations, in the current study QTL meta-analysis was performed.
The first step in the QTL meta-analysis is the projection of QTLs onto a reference or consensus map integrating a high number of molecular markers. However, not all of the QTLs reported for root traits were included in the analysis based on (1) the lack common markers between initial and consensus maps, and (2) some of the QTLs showed a low PVE, causing a large SI (>50 cM). Other studies reporting root QTLs were not considered (and not included in the reference list) because the map position of QTLs or the genetic maps were not reported in the papers. Although GBS and SNPs are being common used makers in the last years, most of the maps published before 2017 do not include other types of markers36–39, thus they are not useful for map comparison with previously developed maps. Or when SSRs and SNPs are present as in Iannucci et al.19 and Maccaferri et al.22 the studies are based on durum wheat where genome D is not represented. Additionally the use of the reference sequence of the wheat genome40 has the inconvenience that not all of SSR markers reported in this study are mapped and the different physical:genetic distance rate depending on chromosome regions, as previously reported41–43, could lead to a wrong determination of supporting intervals, increasing the complexity to identify common QTL regions. The majority of QTL studies reported in this work were based on mapping populations genotyped with SSR or DArT markers. Therefore, to include most of the QTLs, a consensus map integrating different types of markers (SSR, DArT, restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), expressed sequence tag (EST), sequence-tagged sites (STS)) was constructed by merging three reference maps: (1) Wheat Composite 2004 (http://wheat.pw.usda.gov), (2) the consensus SSR map developed by Somers et al.20, and (3) the integrated map developed by Marone et al.13.
The usefulness of QTL meta-analysis is to integrate the QTL information previously published to define consensus genomic regions for a given trait and narrow down the confidence intervals of QTLs to tackle map-based cloning strategies more efficiently. However, QTL meta-analysis is highly dependent on the individual QTL mapping studies, their SI and projection quality7. For this reason, we projected only QTLs that had all the information required for QTL projection following an homothetic approach24 and meta-analysis using the BioMercator v4.2 software, such as LOD score, PVE, peak position, SI and flanking markers.
Using the Wheat_Consensus_2018 map, we were able to project 84% of the retrieved QTLs, most of them reporting a low PVE (96% lower than 0.2) and supporting the implication of many loci in the trait variation, each of them with a small effect.
QTL meta-analysis revealed the presence of 112 genomic regions harbouring root QTLs. Of these 94 represented consensus QTLs (MQTLs) and 18 remained as singletons (sQTL). Results of meta-analysis were supported by the QTL-overview density index24, which was computed to picture the regions involved in the genetic control of root-related traits. According to the QTL-overview index, 126 peaks exceeded the average value of the statistic and represented hotspots for root QTLs. Seventy-eight of the 94 MQTLs (83%) were located within overview peaks and 7 (7%) were located between two overview peaks. The remaining 10% did not correspond to overview peaks. Of the 18 sQTLs, 14 (78%) corresponded to overview peaks exceeding the average value and the remaining 4 (22%) were located within non-significant overview peaks. In addition, 37% of the MQTL corresponded to high-value overview peaks grouping 52% of the projected QTLs. The number of QTLs in the wheat genome was reduced by 82% or 85%, respectively, when all of the reported genome regions were considered, or only MQTLs. The average SI of MQTLs was reduced three-fold, increasing the mapping precision.
Previous studies reporting MQTLs for root traits have been carried out by Darzi-Ramandi6 and Iannucci19. From an initial database comprising 243 QTLs reported from 12 different experiments, Darzi-Ramandi6 used for meta-analysis only 53 QTLs located on chromosome groups 2 and 3, as they carried most of the QTLs for root traits. After meta-analysis, these authors grouped the QTLs into 8 MQTLs. Based on the position of flanking SSR markers, common regions for MQTLs were found between their results and those of the present study. On chromosome 2A, M-QTL16 was located in a similar position to Root_MQTL_18. On chromosome 2B, M-QTL2 to M-QTL5 corresponded to the positions for Root_MQTL_22, Root_MQTL_23, Root_sQTL_3 and Root_MQTL_24, respectively. On chromosome 3A, M-QTL6 was positioned between Root_MQTL_32 and Root_MQTL_33, and M-QTL7 in a common region with Root_MQTL_35. Finally, on chromosome 3B, M-QTL8 corresponded to the position of Root_MQTL_39. Iannucci19 performed a QTL meta-analysis in durum wheat; from 12 previously published QTL studies comprising 100 QTLs, and 17 QTLs identified in their study, the authors found 34 MQTLs and 29 QTLs on 8 out of the 14 durum wheat chromosomes. Common MQTL regions with our study were found for the 8 chromosomes reported by these authors19 based on common flanking markers between Wheat_Consensus_2018 and the map reported by Maccaferri22 integrating SNP, SSR and DArT markers. Chromosomes 1B, 2A, 4B, 5B, 6A and 7B shared one MQTL among studies. Chromosome 3A harboured the highest number of common MQTL positions (5), and MQTL7-11 shared flanking markers with Root_MQTL_32-36. Finally, on chromosome 6B, 2 MQTLs were mapped in similar regions (MQTL30 – Root_MQTL_77 and MQTL31 – Root_MQTL_80).
The annotation of the wheat genome sequence40 allowed us to identify gene models near the MQTL peaks with implications in a wide range of biological functions. Among them, those involved in root development or response to abiotic stress may be of special interest for breeding.
Six MQTL peaks corresponded to gene models with similarity to E3 ubiquitin protein ligases located on chromosomes 2D, 3A, 5A, 5D and 6B. These proteins play a role in the signal pathway of abiotic stresses tolerance, such as dehydration. Moreover, they are also involved in the regulation of plant development44. Additionally, five peaks corresponded to F-box domains that are the protein subunit of E3 ubiquitin ligases involved in the responses to abiotic stresses45. The overexpression of TaFBA1, a wheat F-box gene, improved the heat tolerance of transgenic tobacco46. These transgenic plants showed longer roots than wild type plants. Four MQTL peaks were located near gene models for LEA hydroxyproline-rich glycoproteins (chromosomes 3B, 4A and 5D). These proteins are involved in the response to abiotic stresses and they accumulated during the late embryo development. Although they are mainly expressed in seeds, they have also been found in roots during the whole cycle of development47.
Gene models for three MQTL peaks on chromosomes 2A, 5B and 6B corresponded to proteins containing pentatricopeptide repeats. These repeats have been found in the protein TaMRRP (TaMOR-related protein), which interacts with the wheat transcription factor gene TaMOR (MORE ROOT)48. The overexpression of this gene in Arabidopsis led to an increase in the number of lateral roots, and rice produced more crown roots, thus enhancing grain yield48. Based on the expression pattern, the authors suggested that TaMOR is involved in root initiation48. In our study, peroxidases were identified for three MQTL peaks (chromosomes 1B, 4B, 7A). The role of extracellular peroxidases in the production of superoxide in root cells as a part of the inducible defence response against abiotic and biotic stresses has been reported49.
Root hair defective 3 (RHD3) proteins were identified for three MQTL peaks on chromosomes 4B, 5A and 7B. Root hairs increase the surface area of the roots, providing major water and nutrient uptakes. In mature Arabidopsis plants, the rhd3 mutation produces a reduction in plant ize50. In wheat, Shan et al.51 found that under salt stress the expression of RHD3 was inhibited, stopping root growth.
Zinc finger proteins play important roles in several plant processes from growth regulation and development, signalling and responses to abiotic stresses. Two zinc finger domains were found for two MQTL peaks on chromosomes 1A and 2A. In wheat, the overexpression of the zinc finger protein TaZFP34 in roots resulted in an increased root-to-shoot ratio, reducing shoot growth but maintaining root elongation52. The expression of TaZFP34 in roots was upregulated during plant adaptation to drying soils caused by high salinity and dehydration.
On chromosomes 2D and 4A, ABC transporter proteins were located within the peaks for the Root_MQTL30 and 48. In Arabidopsis, a new ABC transporter controlling root development, AtMRP5, was identified53. A T-DNA insertion in this gene produced mutant plants with a decrease in root growth but an increase in the formation of lateral roots. The authors concluded that AtMRP5 acts as an auxin conjugate transporter or that the mutants are defective for ion uptake, leading to changes in auxin concentration.
NAC domain-containing proteins are also present within MQTL positions in chromosomes 2D and 7B. Transcription factors of this kind have been described in many developmental processes and stress responses. In Arabidopsis NAC1 is induced by auxins, promoting the development of lateral roots54, and the expression of AtNAC2 is induced in response to salt stress, leading to an increase in the development of lateral roots55.
Brevis radix (BRX) acts as a key regulator of cell proliferation and elongation in the roots. In this work, two MQTLs were found on chromosomes 6A and 6D with gene models corresponding to this type of protein. In Arabidopsis, brx-2, an abscisic acid-hypersensitive mutant in root growth was isolated, providing evidence of the role of BRX-2 as a modulator of abscisic acid sensitivity in roots56.
The gene model TraesCS6B01G075800 was identified in the Root_MQTL76 coding for a small auxin-upregulated RNA (SAUR)-like auxin-responsive protein. According to Guo et al.57, these genes are regulated by auxin and environmental factors. The authors identified in wheat the gene TaSAUR75, which was downregulated in roots under salt stress conditions. Its overexpression in Arabidopsis increased salt and drought tolerance and the plants showed increased root length.
Heavy metals are essential for plant growth, but high concentrations of them result in growth inhibition and toxicity. A heavy metal transport/detoxification gene model (TraesCS6A01G393200) was found for the Root_MQTL73 peak. As the organ in contact with the soil metals, roots play an important role in adsorption and detoxification. These mechanisms are reviewed in Hall58.
The role of root primordium defective 1 (RPD1) has been described in Arabidopsis59. This gene is involved in maintaining active cell proliferation in the root primordium. When rpd1 mutants were used, initially cell proliferation led to root primordia formation, although it stopped at an early stage59. The authors concluded that rpd1 could not maintain the highly active cell division during the first stages of the root primordium. In our study, the gene model TraesCS1B01G440200 located within the peak position of Root_MQTL12 on chromosome 1B corresponded to an RPD1 protein.
The gene model TraesCS7A01G154600 found in the peak of Root_QTL84 codes for a gibberellin receptor GID1A. Its interest lies in the fact that gibberellins are essential hormones regulating growth and development in plants.
Among other genes involved in root development although not found within MQTL peaks are expansin genes. The overexpression of TaEXPB23, a wheat expansin gene, improved drought tolerance stimulating the growth of the root system in tobacco without affecting other developmental processes60. VERNALIZATION1 (Vrn1), a key regulator of flowering time in cereals, was also involved in root architecture in wheat and barley61. QTLs for root traits were also detected in barley in the vicinity of Vrn162,63.
The current study is the largest QTL meta-analysis for root traits carried out in wheat, although it only includes a fraction of all the published QTL information. Integrating the recently developed maps based on high-throughput genotyping with SNP arrays with the earlier maps will allow the regions involved in trait variation to be identified more precisely in order to tackle QTL cloning approaches successfully. Additionally, the integration of genome wide association studies will improve our understanding of the molecular bases of the quantitative traits involved in root architecture.
By integrating maps for meta-analysis, we were able to detect consensus QTL regions where robust SSR markers flanking MQTLs can be identified in the consensus map and used to transfer relevant QTLs by marker-assisted selection (MAS). The identification of several root traits within the same MQTL region will help breeders pyramiding different genes to model the architecture of new cultivars in response to the changing environment. From a breeding point of view, Löffler et al.8 defined the criteria that must be taken into consideration for MQTL selection to be used to accelerate breeding programs: (1) MQTLs should have small supporting intervals, (2) the MQTL should include a high number of initial QTLs and (3) the initial QTLs should be characterized by high PVE. Based on these criteria in this work we identified the most promising MQTL to be used for root improvement in wheat. Additionally, the use of common SSR markers in this study would allow to any laboratory or breeding company to implement MAS in their programs. In this respect, breeders have to take into consideration the quantitative inheritance of these traits, thus more than one MQTL must be selected for crop improvement.
The genome sequence of wheat (IWGSC 2018) published recently in open access will be an excellent tool for research and for the breeding community. As reported in this work, the identification of putative candidate genes for the traits of interest will accelerate the breeding process through finely directed research of specific gene models.
Materials and Methods
Consensus map construction
In order to include the maximum number of studies for QTL projection, a consensus map, Wheat_Consensus_2018, was constructed by integrating three wheat maps using the MergeMap software64 available at http://www.mergemap.org (Supplementary Material 1). MergeMap takes into account the individual maps to calculate the consensus marker order in each linkage group (LG). Initially, each LG is converted to directed acyclic graphs (DAGs)65 that are then merged into a consensus graph on the basis of their shared vertices. MergeMap tries to resolve conflicts by deleting a minimum set of marker occurrences. Finally, each consensus DAG was linearized using a mean distance approximation. Equal weight was given to all genetic maps (weight = 1.0).
The selected maps were (1) the Wheat_Composite_2004 map composed of 4403 markers, including SSR, RFLP and AFLP, available at http://wheat.pw.usda.gov (Composite_2004); (2) the consensus SSR map developed by Somers et al.20 composed of 1235 SSR markers (SSR_2004); and (3) the integrated map developed by Marone et al.13 composed of 3669 markers, including DArT, SSR, EST, STS and RFLP (Integrated_2013).
The root QTL database
The literature was retrieved using the keywords ‘wheat root QTL’ from the Web of Science server (http://apps.webofknowledge.com). The QTL database was created from 30 studies published from 2006 to 2017, three of them carried out in durum wheat. A summary of the QTL studies is reported in Table 1. Although some of the papers reported QTLs for different traits, we took into account only those involving root traits. A total of 64 traits were considered and grouped into 15 main categories (Supplementary Material 2).
The QTL database reported information on type of cross, parents used, number of progenies, name of QTL, trait, LOD score, proportion of PVE by each QTL, QTL position on the original linkage map, SI and flanking markers.
QTL projection
To compare the QTL positions detected in the studies, the original QTL data were projected onto the Wheat_Consensus_2018 map developed in the present work. Flanking markers for each QTL were located on the consensus map and projected on it following a homothetic approach proposed by Chardon et al.24. When SI was not reported in the original studies, the distance between flanking markers was taken as the SI. When the QTL peak position was projected, the SI on the consensus map was estimated for a confidence interval of 95% using the empirical formula proposed by Guo et al.66.
The frequency of identification of a root QTL for every cM position on the Wheat_Consensus_2018 map was estimated using the QTL-overview index24. To highlight regions with significant peaks, the average value of the statistic and a threshold for high values, calculated as five times the average value, were plotted. The statistic increases in a region with (1) the number of QTLs from different experiments, (2) the proximity between QTL positions and (3) the precision of the individual QTL position estimation.
QTL meta-analysis
QTL meta-analysis was conducted using BioMercator v.4.267,68, available at https://urgi.versailles.inra.fr/Tools/BioMercator-V4, for each chromosome. Two different approaches were used according to the number of QTLs per chromosome. The approach of Goffinet and Gerber7 was used when the number of QTLs per chromosome was 10 or lower. According to these authors, for n individual QTLs, BioMercator tests the most likely assumption between 1, 2, 3, 4 and n underlying QTLs. Decision rules are based on an Akaike information criterion (AIC), and the one with the lowest AIC value is considered the best fit. However, when the number of QTLs per chromosome was higher than 10, the approach of Veyrieras et al.69 was used. In this case meta-analysis determines the best QTL model based on model choice criteria from AIC, AICc, AIC3, a bayesian information criterion and average weight of evidence. The best QTL model was selected when values of the model selection criteria were the lowest in at least three of the five models. Consensus QTLs from the optimum model were regarded as meta-QTLs (MQTLs).
Graphical representation
The QTL-overview index and MQTLs, together with a heatmap of the number of projected QTLs per 25 cM bin, were represented graphically using ClicO FS70.
Gene annotation
Gene annotation corresponding to the MQTL peaks was performed using the gene models for high-confidence genes reported for the wheat genome sequence40, available at https://wheat-urgi.versailles.inra.fr/Seq-Repository/Assemblies. When the closest marker was not located in the sequence, flanking markers were used to project the peak position in the physical sequence.
Supplementary information
Acknowledgements
This study was funded by the projects AGL2015-65351-R of the Spanish Ministry of Economy and Competitiveness and RTA2015-00072-C03 of the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), and the CERCA Programme/Generalitat de Catalunya. JMS was hired by the INIA-CCAA programme funded by the INIA and the Generalitat de Catalunya. The authors thank Conxita Royo for her critical review of the manuscript.
Author Contributions
J.M.S. and F.A. obtained funding. J.M.S. performed the analyses. J.M.S. and F.A. conceived the manuscript. J.M.S. wrote the manuscript. J.M.S. and F.A. edited and reviewed the manuscript. J.M.S. and F.A. read and approved the final manuscript.
Data Availability
Data generated or analysed during this study are included in this published article (and its Supplementary Information files). Datasets are also available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-47038-2.
References
- 1.Pereira A. Plant abiotic stress challenges from the changing environment. Front Plant Sci. 2016;7:1123. doi: 10.3389/fpls.2016.01123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reynolds MP, Mujeeb-Kazi A, Sawkins M. Prospects for utilising plant-adaptive mechanisms to improve wheat and other crops in drought- and salinity-prone environments. Annals of Applied Biology. 2005;146:239–259. doi: 10.1111/j.1744-7348.2005.040058.x. [DOI] [Google Scholar]
- 3.Paez-García A, et al. Root traits and phenotyping strategies for plant improvement. Plants. 2015;4:334–355. doi: 10.3390/plants4020334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manschadi AM, Christopher J, Devoil P, Hammer GL. The role of root architectural traits in adaptation of wheat to water-limited environments. Functional Plant Biology. 2006;33:823–837. doi: 10.1071/fp06055. [DOI] [PubMed] [Google Scholar]
- 5.Lilley JM, Kirkegaard JA. Benefits of increased soil exploration by wheat roots. Field Crops Research. 2011;122:118–130. doi: 10.1016/j.fcr.2011.03.010. [DOI] [Google Scholar]
- 6.Darzi-Ramandi H, et al. Detection of consensus genomic regions associated with root architecture of bread wheat on groups 2 and 3 chromosomes using QTL meta-analysis. Australian Journal of Crop Science. 2017;11:777–785. doi: 10.21475/ajcs.17.11.07.pne312. [DOI] [Google Scholar]
- 7.Goffinet B, Gerber S. Quantitative trait loci: a meta-analysis. Genetics. 2000;155:463–473. doi: 10.1093/genetics/155.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Löffler M, Schoen CC, Miedaner T. Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Mol Breed. 2009;23:473–488. doi: 10.1007/s11032-008-9250-y. [DOI] [Google Scholar]
- 9.Zhang LY, et al. Genomic distribution of quantitative trait loci for yield and yield-related traits in common wheat. Journal of Integrative Plant Biology. 2010;52:996–1007. doi: 10.1111/j.1744-7909.2010.00967.x. [DOI] [PubMed] [Google Scholar]
- 10.Soriano JM, Malosetti M, Roselló M, Sorrells ME, Royo C. Dissecting the old Mediterranean durum wheat genetic architecture for phenology, biomass and yield formation by association mapping and QTL meta-analysis. PLoS One. 2017;12:e0178290. doi: 10.1371/journal.pone.0178290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanocq E, Laperche A, Jaminon O, Laine AL, Le Gouis J. Most significant genome regions involved in the control of earliness traits in bread wheat, as revealed by QTL meta-analysis. Theor Appl Genet. 2007;114:569–584. doi: 10.1007/s00122-006-0459-z. [DOI] [PubMed] [Google Scholar]
- 12.Goudemand E, et al. Association mapping and meta-analysis: two complementary approaches for the detection of reliable Septoria tritici blotch quantitative resistance in bread wheat (Triticum aestivum L.) Mol Breed. 2013;32:563–584. doi: 10.1007/s11032-013-9890-4. [DOI] [Google Scholar]
- 13.Marone D, et al. Genetic basis of qualitative and quantitative resistance to powdery mildew in wheat: from consensus regions to candidate genes. BMC Genomics. 2013;14:562. doi: 10.1186/1471-2164-14-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soriano JM, Royo C. Dissecting the genetic architecture of leaf rust resistance in wheat by QTL meta-analysis. Phytopathology. 2015;105:1585–1593. doi: 10.1094/PHYTO-05-15-0130-R. [DOI] [PubMed] [Google Scholar]
- 15.Griffiths S, et al. Meta-QTL analysis of the genetic control of crop height in elite European winter wheat germplasm. Mol Breed. 2012;29:159–171. doi: 10.1007/s11032-010-9534-x. [DOI] [PubMed] [Google Scholar]
- 16.Quraishi UM, et al. Combined metagenomics analyses unravel candidate genes for the grain dietary fiber content in bread wheat (Triticum aestivum L.) Funct Integr Genom. 2011;11:71–83. doi: 10.1007/s10142-010-0183-2. [DOI] [PubMed] [Google Scholar]
- 17.Tyagi S, Balyan HS, Gupta PK. Interval mapping and meta-QTL analysis of grain traits in common wheat (Triticum aestivum L.) Euphytica. 2015;201:367–380. doi: 10.1007/s10681-014-1217-y. [DOI] [Google Scholar]
- 18.Tyagi S, Gupta PK. Meta-analysis of QTLs involved in pre-harvest sprouting tolerance and dormancy in bread wheat. Triticeae Genomics Genet. 2012;3:9–24. doi: 10.5376/tgg.2012.03.0002. [DOI] [Google Scholar]
- 19.Iannucci Anna, Marone Daniela, Russo Maria Anna, De Vita Pasquale, Miullo Vito, Ferragonio Pina, Blanco Antonio, Gadaleta Agata, Mastrangelo Anna Maria. Mapping QTL for Root and Shoot Morphological Traits in a Durum Wheat × T. dicoccum Segregating Population at Seedling Stage. International Journal of Genomics. 2017;2017:1–17. doi: 10.1155/2017/6876393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Somers DJ, Isaac P, Edwards K. A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.) Theor Appl Genet. 2004;109:1105–1114. doi: 10.1007/s00122-004-1740-7. [DOI] [PubMed] [Google Scholar]
- 21.Kubo K, et al. Quantitative trait loci for soil-penetrating ability of roots in durum wheat. Plant Breed. 2007;126:375–378. doi: 10.1111/j.1439-0523.2007.01368.x. [DOI] [Google Scholar]
- 22.Maccaferri M, et al. Prioritizing quantitative trait loci for root system architecture in tetraploid wheat. Journal of Experimental Botany. 2016;67:1161–78. doi: 10.1093/jxb/erw039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrarulo M, et al. Genetic analysis of root morphological traits in wheat. Molecular Genetics and Genomics. 2015;290:785–806. doi: 10.1007/s00438-014-0957-7. [DOI] [PubMed] [Google Scholar]
- 24.Chardon F, et al. Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics. 2004;168:2169–2185. doi: 10.1534/genetics.104.032375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blum Abraham. Plant Breeding for Stress Environments. 2018. Plant Breeding and Yield Stability; pp. 15–42. [Google Scholar]
- 26.Passioura JB. Environmental biology and crop improvement. Functional Plant Biology. 2002;29:537–546. doi: 10.1071/FP02020. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds M, Tuberosa R. Translational research impacting on crop productivity in drought-prone environments. Current Opinion in Plant Biology. 2008;11:171–179. doi: 10.1016/j.pbi.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Subira J, et al. Changes in durum wheat root and aerial biomass caused by the introduction of the Rht-B1b dwarfing allele and their effects on yield formation. Plant and Soil. 2016;403:291–304. doi: 10.1007/s11104-015-2781-1. [DOI] [Google Scholar]
- 29.Christopher J, et al. QTL for root angle and number in a population developed from bread wheats (Triticum aestivum) with contrasting adaptation to water-limited environments. Theor Appl Genet. 2013;126:1563–1574. doi: 10.1007/s00122-013-2074-0. [DOI] [PubMed] [Google Scholar]
- 30.Chesnokov YV, Goncharova EA, Sitnikov N, Kocherina NV, Lohwasser U. Börne, r A. Mapping QTL for water regime in spring bread wheat. Russian Journal of Plant Physiology. 2014;61:834–841. doi: 10.1134/S1021443714060041. [DOI] [Google Scholar]
- 31.Maccaferri M, et al. A high-density, SNP-based consensus map of tetraploid wheat as a bridge to integrate durum and bread wheat genomics and breeding. Plant Biotechnol J. 2015;13:648–63. doi: 10.1111/pbi.12288. [DOI] [PubMed] [Google Scholar]
- 32.Ayalew H, Liu H, Yan G. Identification and validation of root length QTLs for water stress resistance in hexaploid wheat (Titicum aestivum L.) Euphytica. 2017;213:1–11. doi: 10.1007/s10681-017-1914-4. [DOI] [Google Scholar]
- 33.Asseng S, Ritchie JT, Smucker AJM, Robertson MJ. Root growth and water uptake during water deficit and recovering in wheat. Plant Soil. 1998;201:265–273. doi: 10.1023/A:1004317523264. [DOI] [Google Scholar]
- 34.Narayanan S, Mohan A, Gill KS, Prasad PVV. Variability of root traits in spring wheat germplasm. PLoS One. 2014;9(6):e100317. doi: 10.1371/journal.pone.0100317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wasson AP, et al. Traits and selection strategies to improve root systems and water uptake in water-limited wheat crops. J Exp. Bo.t. 2012;63:3485–3498. doi: 10.1093/jxb/ers111. [DOI] [PubMed] [Google Scholar]
- 36.Cavanagh CR, et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl. Acad. Sci. USA. 2013;110:8057–8062. doi: 10.1073/pnas.1217133110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S, et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 SNP array. Plant Biotechnol. J. 2014;12:787–796. doi: 10.1111/pbi.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delhaize E, Rathjen TM, Cavanagh CR. The genetics of rhizosheath size in a multiparent mapping population of wheat. Journal of Experimental Botany. 2015;66:4527–4536. doi: 10.1093/jxb/erv223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu F, et al. Identification of major quantitative trait loci for root diameter in synthetic hexaploid wheat under phosphorus-deficient conditions. Journal of Applied Genetics. 2017;58:437–447. doi: 10.1007/s13353-017-0406-5. [DOI] [PubMed] [Google Scholar]
- 40.IWGSC (International Wheat Genome Sequencing Consortium) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361:eaar7191. doi: 10.1126/science.aar7191. [DOI] [PubMed] [Google Scholar]
- 41.Akhunov E. D. The Organization and Rate of Evolution of Wheat Genomes Are Correlated With Recombination Rates Along Chromosome Arms. Genome Research. 2003;13(5):753–763. doi: 10.1101/gr.808603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goyal A, et al. Physical molecular maps of wheat chromosomes. Theor Appl Genet. 2005;5:260–263. doi: 10.1007/s10142-005-0146-1. [DOI] [PubMed] [Google Scholar]
- 43.Mester, D., Ronin, Y., Schnable, P., Aluru, S. & Korol, A. Fast and Accurate Construction of Ultra-Dense Consensus Genetic Maps Using Evolution Strategy Optimization PLoS One, 10, e0122485, https://doi.org/journal.pone.0122485 (2015). [DOI] [PMC free article] [PubMed]
- 44.Guerra D, Cattivelli L, Mazzucotelli E. The E3 ubiquitin ligase WVIP2 highlights the versatility of protein ubiquitination. Plant Signaling & Behavior. 2012;7:1155–1157. doi: 10.4161/psb/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hua Z, Vierstra RD. The cullin-RING ubiquitin-protein ligases. Annu. Rev. Plant Biol. 2011;62:299–334. doi: 10.1146/annurev-arplant-042809-112256. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, et al. Wheat F-Box protein gene TaFBA1 is involved in plant tolerance to heat stress. Frontiers in Plant Science. 2018;9:521. doi: 10.3389/fpls.2018.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao J, Lan T. Functional characterization of the late embryogenesis abundant (LEA) protein gene family from Pinus tabuliformis (Pinaceae) in Escherichia coli. Scientific Reports. 2016;6:19467. doi: 10.1038/srep19467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B, et al. Overexpression of wheat gene TaMOR improves root system architecture and grain yield in Oryza sativa. J. Exp. Bot. 2016;67:4155–67. doi: 10.1093/jxb/erw193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minibayeva FV, Gordon LK, Kolesnikov OP, Chasov AV. Role of extracellular peroxidase in the superoxide production by wheat root cells. Protoplasma. 2001;217:125–128. doi: 10.1007/BF01289421. [DOI] [PubMed] [Google Scholar]
- 50.Wang H, Lockwood SK, Hoeltzel MF, Schiefelbein JW. The Root Hair Defective3 gene encodes an evolutionarily conserved protein with GTP-binding motifs and is required for regulated cell enlargement in Arabidopsis. Genes Dev. 1997;11:799–811. doi: 10.1101/gad.11.6.799. [DOI] [PubMed] [Google Scholar]
- 51.Shan L, Zhao SY, Xia GM. Cloning of the Full-length cDNA of the Wheat Involved in Salt Stress: Root Hair Defective 3 Gene (RHD3) Journal of Integrative Plant Biology. 2005;47:881–891. doi: 10.1111/j.1744-7909.2005.00090.x. [DOI] [Google Scholar]
- 52.Chang H, et al. Abiotic stress upregulated TaZFP34 represses the expression of type-B response regulator and SHY2 genes and enhances root to shoot ratio in wheat. Plant Science. 2016;252:88–102. doi: 10.1016/j.plantsci.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 53.Gaedeke N, et al. The Arabidopsis thaliana ABC transporter AtMRP5 controls root development and stomata movement. The EMBO Journal. 2001;20:1875–1887. doi: 10.1093/emboj/20.8.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie Q, Frugis G, Colgan D, Chua NH. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes &. Development. 2000;14:3024–3036. doi: 10.1101/Gad.852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He XJ, et al. AtNAC2, a transcription factor downstream of ethylene and auxin signalling pathways, is involved in salt stress response and lateral root development. Plant J. 2005;44:903–916. doi: 10.1111/j.1365-313X.2005.02575.x. [DOI] [PubMed] [Google Scholar]
- 56.Rodrigues A, et al. The short-rooted phenotype of the brevis radix mutant partly reflects root abscisic acid hypersensitivity. Plant Physiology. 2009;149:1917–1928. doi: 10.1104/pp.108.133819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo Y, et al. Function of the auxin-responsive gene TaSAUR75 under salt and drought stress. The Crop Journal. 2018;6:181–190. doi: 10.1016/j.cj.2017.08.005. [DOI] [Google Scholar]
- 58.Hall JL. Cellular mechanisms for heavy metals detoxification and tolerance. Journal of Experimental Botany. 2002;366:1–11. doi: 10.1093/jexbot/53.366.1. [DOI] [PubMed] [Google Scholar]
- 59.Konishi M, Sugiyama M. A novel plant-specific family gene, Root Primordium Defective 1, is required for the maintenance of active cell proliferation. Plant Physiology. 2006;140:591–602. doi: 10.1104/pp.105.074724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li AX, et al. Root-specific expression of wheat expansin gene TaEXPB23 enhances root growth and water stress tolerance in tobacco. Environmental and Experimental Botany. 2015;110:73–84. doi: 10.1016/j.envexpbot.2014.10.002. [DOI] [Google Scholar]
- 61.Volss-Fels KP, et al. Vernalization1 modulates root system architecture in wheat and barley. Molecular Plant. 2018;11:226–229. doi: 10.1016/j.molp.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 62.Arifuzzaman M, et al. Detection and validation of novel QTL for shoot and root traits in barley (Hordeum vulgare L.) Mol Breed. 2014;34:1373–1387. doi: 10.1007/s11032-014-0122-3. [DOI] [Google Scholar]
- 63.Arifuzzaman M, et al. Genetic mapping reveals broader role of Vnr-H3 gene in root and shoot development beyond heading in barley. PLoS One. 2016;11:e0158718. doi: 10.1371/journal.pone.0158718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Y, Close TJ, Lonardi S. Accurate construction of consensus genetic maps via integer linear programming. IEEE/ACM Tran. Computational Biol. Bioinformatics. 2011;8:381–394. doi: 10.1109/TCBB.2010.35. [DOI] [PubMed] [Google Scholar]
- 65.Yap IV, et al. A graph-theoretic approach to comparing and integrating genetic, physical and sequence-based maps. Genetics. 2003;165(2235–22):47. doi: 10.1093/genetics/165.4.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo B, et al. QTLs associated with resistance to soybean cyst nematode in soybean: meta-analysis of QTL locations. Crop Science. 2006;46:595–602. doi: 10.2135/cropsci2005.04-0036-2. [DOI] [Google Scholar]
- 67.Arcade A, et al. BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics. 2004;20:2324–2326. doi: 10.1093/bioinformatics/bth230. [DOI] [PubMed] [Google Scholar]
- 68.Sosnowski O, Charcosset A, Joets J. BioMercator V3: an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics. 2012;28:2082–2083. doi: 10.1093/bioinformatics/bts313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veyrieras JB, Goffinet B, Charcosset A. MetaQTL: a package of new computational methods for the meta-analysis of QTL mapping experiments. BMC Bioinformatics. 2007;8:49. doi: 10.1186/1471-2105-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheong WH, Tan YC, Yap SJ, Ng KP. ClicO FS: an interactive web-based service of Circos. Bioinformatics. 2015;31:3685–3687. doi: 10.1093/bioinformatics/btv433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.An D, et al. Mapping QTLs for nitrogen uptake in relation to the early growth of wheat (Triticum aestivum L.) Plant and Soil. 2006;284:73–84. doi: 10.1007/s11104-006-0030-3. [DOI] [Google Scholar]
- 72.Bai C, Liang Y, Hawkesford MJ. Identification of QTLs associated with seedling root traits and their correlation with plant height in wheat. J. Exp. Bot. 2013;64:1745–53. doi: 10.1093/jxb/ert041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ballesteros DC, et al. Tolerance of wheat to vegetative stage soil waterlogging is conditioned by both constitutive and adaptive QTL. Euphytica. 2015;201:329–343. doi: 10.1007/s10681-014-1184-3. [DOI] [Google Scholar]
- 74.Botwright Acuña TL, Rebetzke GJ, He X, Maynol E, Wade LJ. Mapping quantitative trait loci associated with root penetration ability of wheat in contrasting environments. Molecular Breeding. 2014;34:631–642. doi: 10.1007/s11032-014-0063-x. [DOI] [Google Scholar]
- 75.Czyczyło-Mysza I, et al. QTL mapping for germination of seeds obtained from previous wheat generation under drought. Cent. Eu. J. Biol. 2014;9:374–382. doi: 10.2478/s11535-013-0273-y. [DOI] [Google Scholar]
- 76.Ehdaie B, Mohammadi SA, Nouraein M, Bektas H, Waines JG. QTLs for root traits at mid-tillering and for root and shoot traits at maturity in a RIL population of spring bread wheat grown under well-watered conditions. Euphytica. 2016;211:17–38. doi: 10.1007/s10681-016-1670-x. [DOI] [Google Scholar]
- 77.Guo Y, et al. QTL mapping for seedling traits in wheat grown under varying concentrations of N, P and K nutrients. Theor. Appl. Genet. 2012;124:851–865. doi: 10.1007/s00122-011-1749-7. [DOI] [PubMed] [Google Scholar]
- 78.Hamada A, et al. Novel QTLs for growth angle of seminal roots in wheat (Triticum aestivum L.) Plant Soil. 2012;354:395. doi: 10.1007/s11104-011-1075-5. [DOI] [Google Scholar]
- 79.Horn R, Wingen LU, Snape JW, Dolan L. Mapping of quantitative trait loci for root hair length in wheat identifies loci that co-locate with loci for yield components. J Exp Bot. 2016;67:4535–4543. doi: 10.1093/jxb/erw228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ibrahim SE, Schubert A, Pillen K, Léon J. QTL analysis of drought tolerance for seedling root morphological traits in an advanced backcross population of spring wheat. International Journal of AgriScience. 2012;2:619–629. [Google Scholar]
- 81.Iehisa JC, et al. Identification of quantitative trait loci for abscisic acid responsiveness in the D-genome of hexaploid wheat. J Plant Physiol. 2014;171:830–841. doi: 10.1016/j.jplph.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 82.Iehisa JC, Matsuura T, Mori IC, Takumi S. Identification of quantitative trait locus for abscisic acid responsiveness on chromosome 5A and association with dehydration tolerance in common wheat seedlings. J Plant Physiol. 2014;171:25–34. doi: 10.1016/j.jplph.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 83.Kabir MR, et al. Mapping QTLs associated with root traits using two different populations in wheat (Triticum aestivum L.) Euphytica. 2015;206:175–190. doi: 10.1007/s10681-015-1495-z. [DOI] [Google Scholar]
- 84.Kadam S, et al. Genomic associations for drought tolerance on the short arm of wheat chromosome 4B. Funct Integr Genomics. 2012;12:447–464. doi: 10.1007/s10142-012-0276-1. [DOI] [PubMed] [Google Scholar]
- 85.Landjeva S, Lohwasser U, Börner A. Genetic mapping within the wheat D genome reveals QTL for germination, seed vigour and longevity, and early seedling growth. Euphytica. 2010;171:129–143. doi: 10.1007/s10681-009-0016-3. [DOI] [Google Scholar]
- 86.Li P, et al. Quantitative trait loci analysis for the effect of Rht-B1 dwarfing gene on coleoptile length and seedling root length and number of bread wheat. Crop Sci. 2011;51:2561–2568. doi: 10.2135/cropsci2011.03.0116. [DOI] [Google Scholar]
- 87.Liu X, Li R, Chang X, Jing R. Mapping QTLs for seedling root traits in a doubled haploid wheat population under different water regimes. Euphytica. 2013;189:51–66. doi: 10.1007/s10681-012-0690-4. [DOI] [Google Scholar]
- 88.Ren, et al. Major quantitative trait loci for seminal root morphology of wheat seedlings. Molecular Breeding. 2012;30:139–148. doi: 10.1007/s11032-011-9605-7. [DOI] [Google Scholar]
- 89.Ren, et al. Characterization of QTLs for root traits of wheat grown under different nitrogen and phosphorus supply levels. Front. Plant Sci. 2017;8:2096. doi: 10.3389/fpls.2017.02096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun J, et al. QTL mapping for seedling traits under different nitrogen forms in wheat. Euphytica. 2013;191:317–331. doi: 10.1007/s10681-012-0834-6. [DOI] [Google Scholar]
- 91.Xie Q, Fernando KMC, Mayes S, Sparkes DL. Identifying seedling root architectural traits associated with yield and yield components in wheat. Ann Bot. 2017;119:1115–1129. doi: 10.1093/aob/mcx001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu M, Chen GY. Conditional QTL mapping for waterlogging tolerance in two RILs populations of wheat. SpringerPlus. 2013;2:245. doi: 10.1186/2193-1801-2-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang H, et al. Conditional and unconditional QTL mapping of drought-tolerance-related traits of wheat seedling using two related RIL populations. J Genet. 2013;92:213–31. doi: 10.1007/s12041-013-0253-z. [DOI] [PubMed] [Google Scholar]
- 94.Zhang H, Cui F, Wang H. Detection of quantitative trait loci (QTLs) for seedling traits and drought tolerance in wheat using three related recombinant inbred line (RIL) populations. Euphytica. 2014;196:313–330. doi: 10.1007/s10681-013-1035-7. [DOI] [Google Scholar]
- 95.Zhao Y, et al. Mapping QTLs for potassium-deficiency tolerance at the seedling stage in wheat (Triticum aestivum L.) Euphytica. 2014;198:185–198. doi: 10.1007/s10681-014-1091-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated or analysed during this study are included in this published article (and its Supplementary Information files). Datasets are also available from the corresponding author on reasonable request.