

Abstract
Cancer stem cells play a critical role in disease initiation and insensitivity to chemotherapy in numerous hematologic malignancies and some solid tumors, and these stem cells need to be eradicated to achieve a cure. Key to successful targeting of cancer stem cells is to identify and functionally test critical target genes and to fully understand their associated molecular network in these stem cells. Human chronic myeloid leukemia (CML) is well accepted as one of the typical types of hematopoietic malignancies that are derived from leukemia stem cells (LSCs), serving as an excellent model disease for understanding the biology of LSCs and developing effective, selective, and curative strategies through targeting LSCs. Here, we discuss LSCs in CML with a focus on identification of unique biological features of these stem cells to emphasize the feasibility and significance of specific targeting of LSCs while sparing normal stem cell counterparts in leukemia therapy. stem cells translational medicine 2019;8:768&774
Keywords: Cancer stem cells, BCR‐ABL, Chronic myeloid leukemia, Bone marrow, Leukemia
Significance Statement.
Molecular mechanisms by which leukemia stem cells (LSCs) survive and self‐renew are poorly understood, and an effective anti‐LSC therapeutic strategy for chronic myeloid leukemia is yet to be developed. In discussing the establishment of anti‐LSC methods in the present study, much attention has been paid to the identification of fundamental biological differences between LSCs and normal hematopoietic stem cells (HSCs) with a goal of eradicating LSCs specifically to avoid or minimize unwanted cytotoxic side effects on normal HSCs. The authors hope to provide convincing arguments to emphasize that it is feasible to specifically target LSCs while sparing normal HSCs.
Introduction
Cancer stem cells are believed to be associated with cancer initiation and insensitivity to chemotherapy in numerous hematologic malignancies and some solid tumors involving the breast, brain, pancreas, colon, lung, and prostate, and need to be eradicated for achieving a cure 1, 2, 3, 4, 5, 6, 7, 8, 9. Although the cancer stem cell theory cannot be used to explain the pathological features of all types of cancers, it has become clear that some major forms of human hematopoietic malignancies such as chronic myeloid leukemia (CML) and acute myeloid leukemia (AML) are derived from leukemia stem cells (LSCs) that are responsible for leukemia initiation, progression, and relapse 10. To develop effective and curative anti‐stem‐cell strategies, CML and AML are good model diseases for understanding the molecular biology of LSCs, and a key initial step is to identify and functionally test critical target genes and the molecular pathways they communicate with in LSCs. In this article, we intend to focus on CML because we have more direct evidence showing the biology of LSCs and their insensitivity to tyrosine kinase inhibitors (TKIs), the first‐line treatments for CML patients.
LSCs are leukemia‐initiating cells with the capacity to self‐renew, differentiate, and remain in a state of quiescence 1, 2. In CML, a myeloproliferative disease that originates from an abnormal hematopoietic stem cell (HSC) harboring the Philadelphia chromosome (Ph+) 11, functional LSCs in mice reside in a cell population that does not express cell lineage markers but express both c‐Kit and Sca‐1 (Lin−c‐Kit+Sca‐1+, LSK) 12, recapitulating the cell surface markers expressed on normal HSCs. LSCs in human CML also reside in the HSC population 13, displaying phenotypically Lin−CD34+CD38−CD90+ with some specific surface markers such as interleukin‐1 receptor accessory protein (IL1RAP) and CD26 14, 15.
At a molecular level, gene expression profiling studies using leukemia mice and human patient samples have shown some dramatic changes in gene expression of LSCs 16. These findings help to lay a foundation for characterizing LSCs for the treatment of hematopoietic malignancies. However, a challenging question still remains: are there fundamental differences between LSCs and their normal stem cell counterparts at a molecular level? In other words, can we specifically target LSCs while sparing normal stem cells when treating leukemias? To answer this question, we need to identify and test key target molecules/genes that are solely or more specifically required for survival and proliferation by LSCs in CML. Although eradication of LSCs in the treatment of CML patients is yet to be achieved, we believe that for therapeutic benefit, it is critical to identify unique biological features of LSCs for developing effective strategies aiming to kill LSCs while protecting normal HSCs with a hope of curing CML. In this article, we will pay much attention to discussing the potential strategies for targeting LSCs more specifically.
Biological Features of LSCs
With self‐renewal and multipotency at the hub of what defines a LSC (Fig. 1), the major focus of current and future research should be on studying the biology of LSCs with a goal of fully understanding the underlying molecular and cellular processes.
Figure 1.
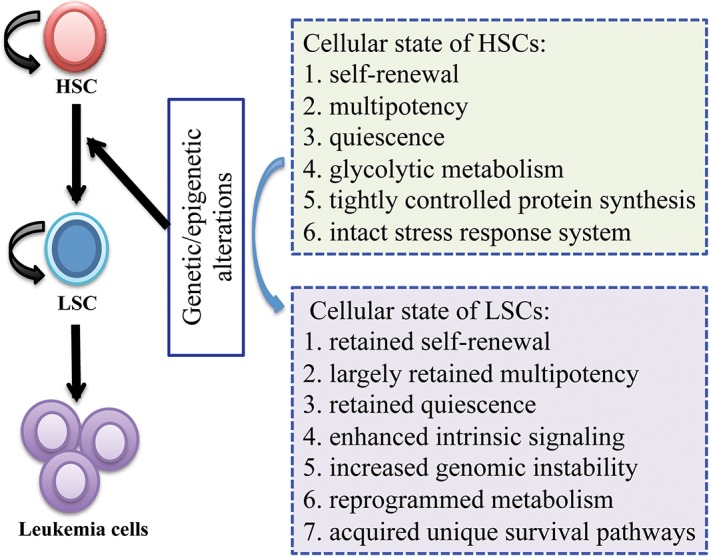
Biological properties of normal HSCs and LSCs. Normal HSCs have three major biological features or stem cell properties: self‐renewal, multipotency, and quiescence. After acquiring genetic lesions, HSCs undergo cellular transformation to become LSCs that retain the major stem cell properties of HSCs with enhanced signaling activities and also acquire some unique biological features. These biological features define the cellular states of HSCs and LSCs, and provide opportunities to develop strategies for specifically targeting LSCs while sparing normal HSCs. Abbreviations: HSC, hematopoietic stem cell; LSC, leukemia stem cell.
Leukemia Stem Cells Display Unique Cellular State
The developmental processes and biological characteristics of normal HSCs have been extensively investigated in the past decades. It is commonly accepted that normal HSCs are largely in a state of quiescence with autophagy‐dependent, glycolytic, and tightly controlled levels of protein synthesis 17, 18, 19, 20. Leukemogenesis occurs because of the serial genetic and epigenetic alterations that transform normal HSC/progenitor cell into LSCs 21, 22. This transformation changes the steady cellular state of normal HSCs. Using CML as an example, the molecular evolution of CML LSCs initiates from the formation of reciprocal translocation between chromosomes 9 and 22, leading to generation of the BCR‐ABL oncogene in a HSC and subsequent expansion of myeloid progenitors 23. As a result, kinase activity of BCR‐ABL tyrosine kinase is constitutively activated, causing uncontrolled activation of some growth‐related signaling pathways such as Wnt/β‐catenin 24, hedgehog 25, JAK/STAT 26, Hif1a 27, 28, TGFβ‐FOXO 29, etc. These intrinsic genetic and signaling changes increase the abilities of CML LSCs in self‐renewal, resistance to apoptosis, and genomic instability 30. However, these pathways also play important roles in normal development, and when searching for potential therapeutic targets in LSCs, we suggest that we should pay more attention to the genes that are more specifically required for survival, self‐renewal, and proliferation of LSCs.
Besides inheriting common stem cell characteristics, LSCs also have some unique functional changes, as exemplified by LSCs that undergo reprogrammed cellular metabolism, a hallmark of cancers 31. Fatty acid metabolism enzyme stearoyl‐CoA desaturase (Scd1) is an endoplasmic reticulum enzyme in a family of Δ9‐fatty acid desaturase isoforms and catalyzes the biosynthesis of monounsaturated fatty acids from saturated fatty acids, which are the most abundant fatty acids present in mammalian organisms 32. Fatty acid synthesis has been found to be associated with tumorigenesis and tumor progression 33. However, we found that Scd1 is downregulated in LSCs and plays a tumor‐suppressive role in LSCs with no notable inhibitory effect on normal HSCs 34, suggesting a cell‐content‐dependent role of fatty acid in cancer. In addition, BCAT1, a cytosolic aminotransferase for branched‐chain amino acids is aberrantly activated and functionally required for AML LSCs 35. It also plays an essential role in the progression of CML chronic phase to blast crisis through induction of cell differentiation arrest 36. Furthermore, a metabolic analysis on both stem‐cell‐enriched (CD34+ and CD34+CD38−) and differentiated cells (CD34−) derived from CML patients reveals that CML LSCs rely on upregulated oxidative metabolism for their survival 37. Compared with differentiated CML cells, LSCs show an increase in glycerol‐3‐phosphate, carnitine, acylcarnitine derivatives, and a decrease in free fatty acid such as oleic and stearic acids. Another example for the functional changes in LSCs is that Alox5, a lipid‐metabolic gene encoding the arachidonate 5‐lipoxygenase, is required for survival of CML LSCs and essential for CML development 38.
Heterogeneity of LSCs
Cellular heterogeneity is one of well‐recognized characteristics of both normal HSCs and LSCs. With respect to the clonal heterogeneity of differentiation and self‐renewal properties in normal HSCs, two distinct subtypes of HSCs (lymphoid‐deficient and lymphoid‐myeloid‐balanced) have been identified and distinguished by assessing the contributions of individual HSCs to the circulating cell lineages in serial transplantation experiments 39, 40. Also, the post‐transplant clonal analysis of HSC expansion suggests that both HSC subtypes display an extensive but variable self‐renewal activity with occasional interconversion 40. Similarly, heterogeneity of LSCs has been recognized. Using the SCL‐tTA/BCR‐ABL mouse model of CML, a recent study reveals that long‐term repopulation and leukemia‐initiating capacity of LSCs after transplantation is restricted to BCR‐ABL‐expressing long‐term HSCs (LT‐HSCs) with remarkable heterogeneity 41. This heterogeneity of BCR‐ABL‐expressing LT‐HSCs is determined based on comparing the global gene expression between leukemic and nonleukemic LT‐HSCs by RNA sequencing. A higher level of MPL expression is found in some leukemic LT‐HSCs with enhanced JAK/STAT signaling and cell proliferation in response to stimulation of the thrombopopoietin (TPO) receptor MPL 41. In contrast, BCR‐ABL‐expressing LT‐HSCs with low MPL expression show a reduced response to TPO‐induced JAK/STAT signaling and decreased leukemogenic potential, suggesting that this subtype of LSCs may be insensitive to inhibition by JAK inhibitors. Therefore, this study identifies MPL expression levels as a key determinant of heterogeneous leukemia‐initiating capacity of LSCs in CML 41. Importantly, the heterogeneity of LSCs is thought to contribute to leukemia initiation, progression, and relapse. It has been reported that residual BCR‐ABL+ stem cells persist in some CML patients who have maintained long‐term remission, and after discontinuing the treatment with a TKI, molecular relapse occurs in a significant number of CML patients 12, 13, 42, 43, 44, 45. The discrepancies in leukemogenic potential between MPL‐high and MPL‐low LSCs could be explained by the heterogeneity of CML LSCs, which likely reflects uniqueness of LSCs determined by the intrinsic molecular machinery or extrinsic microenvironment.
Insensitivity of Leukemia Stem Cells to Drug Therapy and Possible Mechanisms
BCR‐ABL TKIs including imatinib mesylate (Gleevec, Novartis) are highly effective in controlling chronic phase CML, but they fail to eradicate leukemia‐initiating cells or LSCs in CML mice 12 and patients 13, 46, 47. Clinically, a complete and sustained molecular remission (undetectable levels of BCR‐ABL transcripts) is difficult to attain even after a complete cytogenetic remission achieved through imatinib treatment 48, 49, 50, 51, suggesting that imatinib and probably other BCR‐ABL kinase inhibitors can effectively kill highly proliferating leukemia cells but are incapable of eradicating LSCs for cure. An anti‐LSC strategy other than the use of a TKI alone needs to be developed to eradicate LSCs, and the success of this approach relies on uncovering the underlying mechanisms by which LSCs survive drug therapy (Fig. 2).
Figure 2.
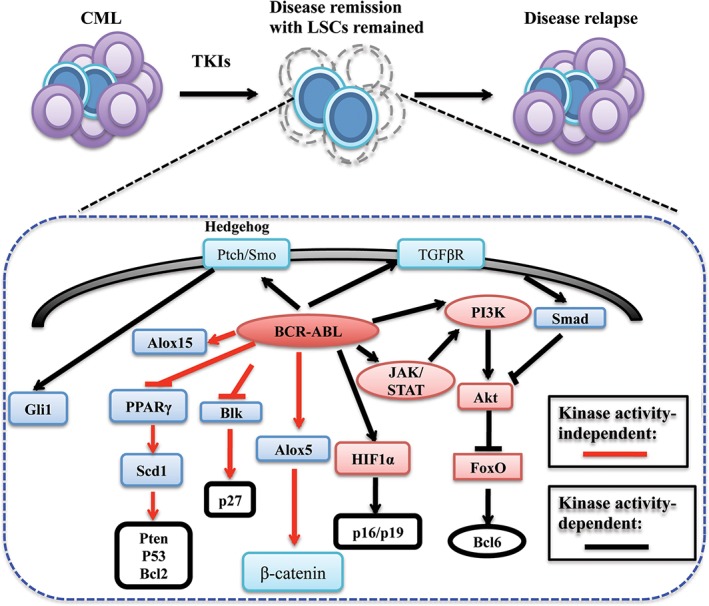
BCR‐ABL kinase activity‐dependent and kinase activity‐independent pathways. TKIs are effective in controlling chronic phase CML, resulting in clinical remission in the majority of CML patients. However, LSCs are insensitive to inhibition by TKIs, as their survival is not dependent on BCR‐ABL kinase activity. Besides altering signaling pathways in LSCs through its kinase activity, BCR‐ABL activates or inhibits some survival‐ or growth‐related pathways in a kinase‐activity‐independent manner. In other words, BCR‐ABL kinase activity‐independent pathways are not completely shut down by TKIs and must be targeted to inhibit or kill LSCs. In addition, some of these BCR‐ABL kinase activity‐independent pathways, including Alox5, Blk, and Scd1, are uniquely required by LSCs for survival and proliferation, serving as potential targets for eradicating LSCs. Abbreviations: Blk, B lymphocyte kinase; CML, chronic myeloid leukemia; LSC, leukemia stem cell; Scd1, stearoyl‐CoA desaturase 1; TKI, tyrosine kinase inhibitor.
LSCs Are Insensitive to Inhibition by TKIs
TKIs have become first‐line drugs in treating CML, and the majority of patients achieve a complete hematological response 52, 53, 54, 55. However, the fact that clinical relapse occurs in a significant number of CML patients once treatment is interrupted 56 indicates that CML LSCs are insensitive to drug therapy. In support of this idea, cells from CML patients in chronic phase were labeled with carboxy‐fluorescein diacetate succinimidyl diester to track cell division, and imatinib treatment caused eradication of almost all dividing CD34+ cells, but the nonproliferating quiescent cells remained 13. In addition, BCR‐ABL+CD34+ cells persisted in CML patients who achieved complete cytogenetic response with imatinib treatment 57. Furthermore, although treatment with TKIs dramatically prolonged the survival of CML mice, the mice eventually died of this disease 12, indicating the failure of TKIs to completely eradicate leukemia cells. The incomplete therapeutic response of CML cells to TKI inhibition in mice is related to the inability of imatinib to eradicate LSCs 58. Together, these studies indicate that CML LSCs are insensitive to TKI treatment, prompting us to provide a mechanistic explanation for TKI resistance of LSCs. It should be pointed out that the TKI resistance of LSCs we discuss here is not relevant to TKI‐resistant BCR‐ABL kinase domain mutations.
LSC Survival Is Not Dependent on BCR‐ABL Kinase Activity
The failure of TKIs to completely eradicate CML LSCs suggests that survival of these LSCs is not dependent on BCR‐ABL kinase activity. We provided a biochemical evidence showing that dasatinib, a second‐generation TKI, inhibits BCR‐ABL phosphorylation in BCR‐ABL‐expressing HSCs but fails to kill these stem cells 12, suggesting that LSCs likely use BCR‐ABL kinase activity‐independent pathways for survival. Similarly, BCR‐ABL kinase activity is inhibited by TKIs in CD34+CD38+ and CD34+CD38− cell populations from newly diagnosed CML patients, and phospho‐CRKL, which is stimulated by BCR‐ABL kinase activity, is reduced upon inhibition of BCR‐ABL kinase activity as detected by immunoblots of sorted quiescent (Ki67−) and cycling (Ki67+) cells 46. Additionally, in human CML CD34+ cells cultured in serum‐free media and treated with dasatinib, phospho‐CRKL is completely inhibited by dasatinib, but the abilities of proliferation and self‐renewal of the cells are retained 44. These results demonstrate that the insensitivity of CML LSCs to inhibition by TKIs is not due to the inability of TKIs to inhibit BCR‐ABL kinase activity in LSCs. It is likely that BCR‐ABL also activates other signaling pathways in a kinase activity‐independent manner, and it will be critical to identify and test these pathways in survival regulation of LSCs.
BCR‐ABL Kinase Activity‐Independent Pathways in LSCs
As described above, compared with proliferative leukemia cells, CML LSCs are much less sensitive to inhibition by TKIs even in the absence of BCR‐ABL kinase domain mutations that cause TKI resistance. We believe that when its kinase activity is suppressed by TKIs, BCR‐ABL can still activate some pathways that render CML LSCs insensitive to TKI inhibition. As a result, the cells continue to survive, whereas BCR‐ABL kinase activity is inhibited by TKIs, indicating that this TKI‐insensitive pathway activated by BCR‐ABL must be targeted to lead to eradication of LSCs. This idea is supported by the essential role of Alox5 in survival regulation of CML LSCs 38. We show that Alox5 is upregulated by BCR‐ABL and essential for CML development, but this upregulation is not reduced by TKI treatment. These results provide a mechanistic explanation for why CML LSCs is insensitive to inhibition of BCR‐ABL kinase activity by TKIs even in the absence of BCR‐ABL kinase domain mutations. Thus, Alox5 represents a unique pathway that cannot be shut down upon kinase inhibition by TKIs in BCR‐ABL signaling and plays a critical role in mediating TKI resistance in LSCs. Another example is that B lymphocyte kinase is significantly downregulated by BCR‐ABL in a kinase activity‐independent manner, and this pathway plays a tumor‐suppressive role in regulating the survival of CML LSCs 59. Again, the abovementioned intrinsic mechanism provides one explanation for the insensitivity of LSCs to TKIs. It should be mentioned that some studies also suggest that TKI resistance of LSCs is related to receiving extrinsic signals from bone marrow niche with which LSCs interact 60. Further research in this area will be beneficial for developing new strategies for eradicating LSCs.
Strategies for Targeting LSCs
It is obvious that one of the best strategies for inhibiting LSCs is to target the key genes that are required for survival regulation of LSCs but not normal HSCs. It may also be acceptable that as a potential anti‐LSC target, a candidate gene is required more by LSCs than by normal HSCs, providing a therapeutic window for inhibiting LSCs more specifically. In other words, the unique biological features of LSCs provide better opportunities for specifically targeting LSCs while sparing normal stem cell counterparts.
Targeting Critical Molecular Pathways of LSCs
In CML, some genes have been shown to be involved in survival regulation of LSCs, including Wnt/β‐catenin 24, 58, Hedgehog 25, Bim‐1 61, 62, p53 63, p16INK4a 64, p19ARF 65, Pten 66, PML 67, PP2A 68, TGF‐β/FOXO 29, Musashi 69, Alox5 38, SIRT1 70, Alox15 71, and Hif1a 27. However, only some of these studies emphasize specific targeting of LSCs, although it is hoped that the target genes required for both LSCs and normal HSCs would only produce tolerable side effects after normal HSCs are inhibited to a certain degree. In fact, several chemical inhibitors against these targets have been developed and studied. For example, pharmacological blockade of Hedgehog signaling by clinical‐grade SMO inhibitors (such as GDC‐0449 and LDE225) 25, 72, 73, 74, inhibition of the TGFβ‐FoxO pathway by Ly364947 29, inactivation of BCL6 by the retro‐inverso BCL6 peptide inhibitor RI‐BPI 75, and suppression of autophagy by pharmacological inhibitors 76 have been shown to inhibit CML development by inhibiting LSCs. Inhibition of the HIF1α pathway by echinomycin is also effective in suppressing LSCs 27, 77. It will be important to further evaluate these inhibitors for their clinical benefit in treating leukemia patients.
We have been focusing on identification of target genes uniquely or more specifically required for cellular functions by LSCs but not normal HSCs. In fact, we have identified Alox5 as a key gene that regulates the function of LSCs but not normal HSCs, because Alox5 deficiency or inhibition of function of this gene impairs survival and self‐renewal of LSCs and prevents the initiation of BCR‐ABL‐induced CML with no significant inhibitory effect on normal HSC function 38. Additionally, Scd1 plays a tumor‐suppressive role specifically in LSCs, and we and others have tested and shown the inhibitory or apoptotic effect of PPARγ agonists on CML LSCs 34, 78. Mechanistically, LSC apoptosis induced by the PPARγ agonist rosiglitazone is associated with an increased expression of Scd1, Pten, and p53 34. Furthermore, deficiency of Alox15 and inhibition of Alox15 function lead to remarkable inhibition of LSCs with much less effect on normal HSCs in CML mice 71. Finally, it has been recently shown that simultaneous targeting of P53 (by blocking its degradation) and c‐MYC (by suppressing its transcription) has more dramatic inhibitory effect on CD34+ cells from CML patients than on normal CD34+ cells 79. Taken together, these results support our belief that it is realistic and approachable to identify and target critical molecular pathways that play an essential role more specifically in LSCs. In other words, it is possible to develop new therapeutic strategies aiming to specifically eradicate LSCs while sparing normal HSCs.
Targeting Epigenetic Properties of LSCs
Besides acquiring genetic lesions, LSCs also undergo epigenetic changes. Targeting of epigenetic regulators has recently shown to be effective in eliminating CML LSCs. EZH2, the catalytic subunit of polycomb repressive complex 2, is overexpressed in CML LSCs 80, 81, which is associated with extensive reprogramming of H3K27me3 targets in the cells. Genetic inactivation of EZH2 in conventional conditional knockout mice and through CRISPR/Cas9‐mediated gene editing reduces survival of LSCs and prolongs survival of CML mice 80. An EZH2‐specific inhibitor promotes apoptosis of LSCs from CML patients without impairing normal HSCs, which is more predominant when the combined treatment with an EZH2 inhibitor and a TKI is used 81. These findings suggest a promising epigenetic‐based therapeutic strategy for more specifically targeting LSCs.
Targeting LSCs Using Antibodies Against Cell Surface Antigens
Although cell surface markers expressed on CML LSCs and normal HSCs are similar, the levels of expression for some markers are much higher in LSCs than in HSCs, providing an opportunity for preferentially targeting LSCs using antibodies. For example, a gene‐expression profiling study in CML CD34+ cells and cord blood CD34+ cells transduced with retroviral BCR‐ABL showed that expression of IL1RAP is upregulated in the cells 82. In this study, normal (Ph−) and leukemic (Ph+) cells within the CML CD34+CD38− cell compartment were distinguished by fluorescence in situ hybridization, and the results showed that the CML CD34+CD38−IL1RAP+ cells were Ph+, whereas CML CD34+CD38−IL1RAP− cells were almost exclusively Ph−. Furthermore, a long‐term culture‐initiating cell assays showed that Ph+ and Ph− candidate CML stem cells could be prospectively separated based on IL1RAP expression, and an anti‐IL1RAP antibody could be used as a target on CML CD34+CD38− cells to induce antibody‐dependent cell‐mediated cytotoxicity. Another example is CD33 that was found to have a much higher expression in CD34+CD38−CD123+ cells from CML patients than in normal CD34+CD38− stem cells 83. Interestingly, colony formation and long‐term culture‐initiating cell assays showed that the CD33‐targeting drug germtuzumab/ozogamicin produced growth inhibition of leukemic progenitor cells. These studies support a strong scientific premise for targeting CML LSCs using antibodies against cell surface antigens. Other examples include expression of cell surface molecules that are linked to signaling pathways in LSCs. In particular, CD25, a STAT5‐dependent cell surface marker, regulates the growth of CML LSCs, which is associated with the PI3K/mTOR pathway 84, 85. It is hopeful that CD25 could be a legitimate target for eradicating CML LSCs.
Conclusion
A full understanding of biology of LSCs allows exploiting the critical differences between LSCs and normal HSCs at a molecular level. This approach will subsequently lead to identification of unique biological features of LSCs for developing effective therapeutic strategies aiming to target LSCs specifically while sparing normal HSCs. Although there are still some difficult hurdles to cross, we believe that we are much closer to applying anti‐LSC strategies for achieving durable disease remission or even a cure. However, the reality is that an effective anti‐LSC therapy is yet to be developed, implying difficult challenges we are facing. Based on the recent scientific advances made in the LSC field, it is hopeful that we begin to understand how LSCs use unique molecular pathways to maintain their abilities of survival and self‐renewal, which will lead to future clinical trails for testing new anti‐LSC strategies.
Author Contributions
H.Z., S.L.: manuscript writing, final revision and approval of the manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
H.Z. is supported by grants from the National Key Research and Development Program of China (2017YFA0505600), the National Natural Science Foundation of China (81722003 and 81870124), the Wuhan Science and Technology Program for Application and Basic Research Project (2018060401011325), and the Hubei Provincial Natural Science Foundation for Creative Research Group (2018CFA018). S.L. is supported by National Institutes of Health (R01CA176179, R01CA222590, and R21CA209298).
Contributor Information
Haojian Zhang, Email: haojian_zhang@whu.edu.cn.
Shaoguang Li, Email: shaoguang.li@umassmed.edu.
References
- 1. Dick JE. Acute myeloid leukemia stem cells. Ann N Y Acad Sci 2005;1044:1–5. [DOI] [PubMed] [Google Scholar]
- 2. Wang JC, Dick JE. Cancer stem cells: Lessons from leukemia. Trends Cell Biol 2005;15:494–501. [DOI] [PubMed] [Google Scholar]
- 3. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–737. [DOI] [PubMed] [Google Scholar]
- 4. Al‐Hajj M, Wicha MS, Benito‐Hernandez A et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh SK, Clarke ID, Terasaki M et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821–5828. [PubMed] [Google Scholar]
- 6. Ricci‐Vitiani L, Lombardi DG, Pilozzi E et al. Identification and expansion of human colon‐cancer‐initiating cells. Nature 2007;445:111–115. [DOI] [PubMed] [Google Scholar]
- 7. Li C, Heidt DG, Dalerba P et al. Identification of pancreatic cancer stem cells. Cancer Res 2007;67:1030–1037. [DOI] [PubMed] [Google Scholar]
- 8. Kim CF, Jackson EL, Woolfenden AE et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005;121:823–835. [DOI] [PubMed] [Google Scholar]
- 9. Goldstein AS, Huang J, Guo C et al. Identification of a cell of origin for human prostate cancer. Science 2010;329:568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer‐stem‐cell research. Nat Rev Cancer 2005;5:311–321. [DOI] [PubMed] [Google Scholar]
- 11. Wong S, Witte ON. The BCR‐ABL story: Bench to bedside and back. Annu Rev Immunol 2004;22:247–306. [DOI] [PubMed] [Google Scholar]
- 12. Hu Y, Swerdlow S, Duffy TM et al. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci USA 2006;103:16870–16875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Graham SM, Jorgensen HG, Allan E et al. Primitive, quiescent, Philadelphia‐positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002;99:319–325. [DOI] [PubMed] [Google Scholar]
- 14. Wisniewski D, Affer M, Willshire J et al. Further phenotypic characterization of the primitive lineage‐ CD34+CD38‐CD90+CD45RA‐ hematopoietic stem cell/progenitor cell sub‐population isolated from cord blood, mobilized peripheral blood and patients with chronic myelogenous leukemia. Blood Cancer J 2011;1:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herrmann H, Sadovnik I, Cerny‐Reiterer S et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014;123:3951–3962. [DOI] [PubMed] [Google Scholar]
- 16. Radich JP, Dai H, Mao M et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA 2006;103:2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cabezas‐Wallscheid N, Buettner F, Sommerkamp P et al. Vitamin A‐retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell 2017;169:807–823 e819. [DOI] [PubMed] [Google Scholar]
- 18. Ho TT, Warr MR, Adelman ER et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017;543:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takubo K, Nagamatsu G, Kobayashi CI et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013;12:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Signer RA, Magee JA, Salic A et al. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014;509:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018;22:157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li S, Garrett‐Bakelman FE, Chung SS et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 2016;22:792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer 2008;8:341–350. [DOI] [PubMed] [Google Scholar]
- 24. Zhao C, Blum J, Chen A et al. Loss of beta‐catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007;12:528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao C, Chen A, Jamieson CH et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009;458:776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR‐Abl‐expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol 1997;159:4720–4728. [PubMed] [Google Scholar]
- 27. Zhang H, Li H, Xi HS et al. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012;119:2595–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheloni G, Tanturli M, Tusa I et al. Targeting chronic myeloid leukemia stem cells with the hypoxia‐inducible factor inhibitor acriflavine. Blood 2017;130:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Naka K, Hoshii T, Muraguchi T et al. TGF‐beta‐FOXO signalling maintains leukaemia‐initiating cells in chronic myeloid leukaemia. Nature 2010;463:676–680. [DOI] [PubMed] [Google Scholar]
- 30. Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017;129:1595–1606. [DOI] [PubMed] [Google Scholar]
- 31. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 32. Scaglia N, Igal RA. Stearoyl‐CoA desaturase is involved in the control of proliferation, anchorage‐independent growth, and survival in human transformed cells. J Biol Chem 2005;280:25339–25349. [DOI] [PubMed] [Google Scholar]
- 33. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016;16:732–749. [DOI] [PubMed] [Google Scholar]
- 34. Zhang H, Li H, Ho N et al. Scd1 plays a tumor‐suppressive role in survival of leukemia stem cells and the development of chronic myeloid leukemia. Mol Cell Biol 2012;32:1776–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raffel S, Falcone M, Kneisel N et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut‐like DNA hypermethylation. Nature 2017;551:384–388. [DOI] [PubMed] [Google Scholar]
- 36. Hattori A, Tsunoda M, Konuma T et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017;545:500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuntz EM, Baquero P, Michie AM et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy‐resistant chronic myeloid leukemia stem cells. Nat Med 2017;23:1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen Y, Hu Y, Zhang H et al. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet 2009;41:783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dykstra B, Kent D, Bowie M et al. Long‐term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 2007;1:218–229. [DOI] [PubMed] [Google Scholar]
- 40. Benz C, Copley MR, Kent DG et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 2012;10:273–283. [DOI] [PubMed] [Google Scholar]
- 41. Zhang B, Li L, Ho Y et al. Heterogeneity of leukemia‐initiating capacity of chronic myelogenous leukemia stem cells. J Clin Invest 2016;126:975–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mahon FX, Rea D, Guilhot J et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010;11:1029–1035. [DOI] [PubMed] [Google Scholar]
- 43. Chomel JC, Bonnet ML, Sorel N et al. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011;118:3657–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamilton AHG, Schemionek M, Zhang B et al. Chronic myeloid leukemia stem cells are not dependent on Bcr‐Abl kinase activity for their survival. Blood 2012;119:1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ross DM, Branford S, Seymour JF et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia 2010;24:1719–1724. [DOI] [PubMed] [Google Scholar]
- 46. Corbin AS, Agarwal A, Loriaux M et al. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR‐ABL activity. J Clin Invest 2011;121:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marley SB, Deininger MW, Davidson RJ et al. The tyrosine kinase inhibitor STI571, like interferon‐alpha, preferentially reduces the capacity for amplification of granulocyte‐macrophage progenitors from patients with chronic myeloid leukemia. Exp Hematol 2000;28:551–557. [DOI] [PubMed] [Google Scholar]
- 48. Hughes TP, Kaeda J, Branford S et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 2003;349:1423–1432. [DOI] [PubMed] [Google Scholar]
- 49. O'Brien SG, Guilhot F, Larson RA et al. Imatinib compared with interferon and low‐dose cytarabine for newly diagnosed chronic‐phase chronic myeloid leukemia. N Engl J Med 2003;348:994–1004. [DOI] [PubMed] [Google Scholar]
- 50. Lin F, Drummond M, O'Brien S et al. Molecular monitoring in chronic myeloid leukemia patients who achieve complete cytogenetic remission on imatinib. Blood 2003;102:1143. [DOI] [PubMed] [Google Scholar]
- 51. Drummond MW, Lush CJ, Vickers MA et al. Imatinib mesylate‐induced molecular remission of Philadelphia chromosome‐positive myelodysplastic syndrome. Leukemia 2003;17:463–465. [DOI] [PubMed] [Google Scholar]
- 52. Cortes J, Rousselot P, Kim DW et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib‐resistant or ‐intolerant chronic myeloid leukemia in blast crisis. Blood 2007;109:3207–3213. [DOI] [PubMed] [Google Scholar]
- 53. Guilhot F, Apperley J, Kim DW et al. Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib‐resistant or ‐intolerant chronic myeloid leukemia in accelerated phase. Blood 2007;109:4143–4150. [DOI] [PubMed] [Google Scholar]
- 54. Hochhaus A, Kantarjian HM, Baccarani M et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic‐phase chronic myeloid leukemia after failure of imatinib therapy. Blood 2007;109:2303–2309. [DOI] [PubMed] [Google Scholar]
- 55. Ottmann O, Dombret H, Martinelli G et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: Interim results of a phase 2 study. Blood 2007;110:2309–2315. [DOI] [PubMed] [Google Scholar]
- 56. Cortes J, O'Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood 2004;104:2204–2205. [DOI] [PubMed] [Google Scholar]
- 57. Bhatia R, Holtz M, Niu N et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003;101:4701–4707. [DOI] [PubMed] [Google Scholar]
- 58. Hu Y, Chen Y, Douglas L et al. beta‐Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR‐ABL‐induced chronic myeloid leukemia. Leukemia 2009;23:109–116. [DOI] [PubMed] [Google Scholar]
- 59. Zhang H, Peng C, Hu Y et al. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat Genet 2012;44:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arrigoni E, Del Re M, Galimberti S et al. Concise Review: Chronic myeloid leukemia: Stem cell niche and response to pharmacologic treatment. Stem Cells Translational Medicine 2018;7:305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lessard J, Sauvageau G. Bmi‐1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003;423:255–260. [DOI] [PubMed] [Google Scholar]
- 62. Park IK, Qian D, Kiel M et al. Bmi‐1 is required for maintenance of adult self‐renewing haematopoietic stem cells. Nature 2003;423:302–305. [DOI] [PubMed] [Google Scholar]
- 63. Molofsky AV, Pardal R, Morrison SJ. Diverse mechanisms regulate stem cell self‐renewal. Curr Opin Cell Biol 2004;16:700–707. [DOI] [PubMed] [Google Scholar]
- 64. Lowe SW, Sherr CJ. Tumor suppression by Ink4a‐Arf: Progress and puzzles. Curr Opin Genet Dev 2003;13:77–83. [DOI] [PubMed] [Google Scholar]
- 65. Molofsky AV, He S, Bydon M et al. Bmi‐1 promotes neural stem cell self‐renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev 2005;19:1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yilmaz OH, Valdez R, Theisen BK et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia‐initiating cells. Nature 2006;441:475–482. [DOI] [PubMed] [Google Scholar]
- 67. Ito K, Bernardi R, Morotti A et al. PML targeting eradicates quiescent leukaemia‐initiating cells. Nature 2008;453:1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Neviani P, Santhanam R, Trotta R et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL‐regulated SET protein. Cancer Cell 2005;8:355–368. [DOI] [PubMed] [Google Scholar]
- 69. Ito T, Kwon HY, Zimdahl B et al. Regulation of myeloid leukaemia by the cell‐fate determinant Musashi. Nature 2010;466:765–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li L, Wang L, Li L et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012;21:266–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen Y, Peng C, Abraham SA et al. Arachidonate 15‐lipoxygenase is required for chronic myeloid leukemia stem cell survival. J Clin Invest 2014;124:3847–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dierks C, Beigi R, Guo GR et al. Expansion of Bcr‐Abl‐positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008;14:238–249. [DOI] [PubMed] [Google Scholar]
- 73. Irvine DA, Zhang B, Kinstrie R et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci Rep 2016;6:25476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Irvine DA, Copland M. Targeting hedgehog in hematologic malignancy. Blood 2012;119:2196–2204. [DOI] [PubMed] [Google Scholar]
- 75. Hurtz C, Hatzi K, Cerchietti L et al. BCL6‐mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med 2011;208:2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bellodi C, Lidonnici MR, Hamilton A et al. Targeting autophagy potentiates tyrosine kinase inhibitor‐induced cell death in Philadelphia chromosome‐positive cells, including primary CML stem cells. J Clin Invest 2009;119:1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang Y, Liu Y, Malek SN et al. Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011;8:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Prost S, Relouzat F, Spentchian M et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature 2015;525:380–383. [DOI] [PubMed] [Google Scholar]
- 79. Abraham SA, Hopcroft LE, Carrick E et al. Dual targeting of p53 and c‐MYC selectively eliminates leukaemic stem cells. Nature 2016;534:341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xie H, Peng C, Huang J et al. Chronic myelogenous leukemia‐initiating cells require polycomb group protein EZH2. Cancer Discov 2016;6:1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Scott MT, Korfi K, Saffrey P et al. Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov 2016;6:1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jaras M, Johnels P, Hansen N et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL‐1 receptor accessory protein. Proc Natl Acad Sci USA 2010;107:16280–16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Herrmann H, Cerny‐Reiterer S, Gleixner KV et al. CD34(+)/CD38(−) stem cells in chronic myeloid leukemia express Siglec‐3 (CD33) and are responsive to the CD33‐targeting drug gemtuzumab/ozogamicin. Haematologica 2012;97:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kobayashi CI, Takubo K, Kobayashi H et al. The IL‐2/CD25 axis maintains distinct subsets of chronic myeloid leukemia‐initiating cells. Blood 2014;123:2540–2549. [DOI] [PubMed] [Google Scholar]
- 85. Sadovnik I, Hoelbl‐Kovacic A, Herrmann H et al. Identification of CD25 as STAT5‐dependent growth regulator of leukemic stem cells in Ph+ CML. Clin Cancer Res 2016;22:2051–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]