

Abstract
Chemically modified RNA (cmRNA) has potential as a safe and efficient tool for nucleic acid‐based therapies and regenerative medicine. Modifications in the chemistry of mRNA can enhance stability, reduce immunogenicity, and thus facilitate mRNA‐based nucleic acid therapy, which eliminates risk of insertional mutagenesis. In addition to these valuable advantages, the mRNA‐based method showed significantly higher efficacy for reprogramming somatic cells to pluripotency compared with DNA‐ or protein‐based methods. These findings suggest cmRNA can provide a powerful and safe tool for cell programming and reprogramming. Delivery methods, particularly using lipid nanoparticles, provide strategies for cell and organ‐specific targeting. The present study comprehensively compares studies that have used cmRNAs for cell fate conversion and tissue engineering. The information should be useful for investigators looking to choose the most efficient and straightforward cmRNA‐based strategy and protocol for tissue engineering and regenerative medicine research. stem cells translational medicine 2019;8:833&843
Keywords: Nucleic acid therapy, Cell programming and reprogramming, Differentiation, iPSCs, Nanoparticles, Regenerative medicine
Significance Statement.
Chemically modified mRNA (cmRNA), as a less immunogenic and more stable form of mRNA with no risk of insertional mutagenesis, offers new strategies for the field of nucleic acid therapy. With respect to cell engineering, the safety is accompanied by high efficiency for reprogramming to pluripotency. The present study comprehensively compares a variety of studies that used cmRNAs for cell fate conversion and tissue engineering. The information should be useful for investigators looking to choose a cmRNA‐based protocol for tissue engineering, and could enhance future investigations using cmRNAs for regenerative medicine in a timely and efficient manner.
Introduction
Recent studies have pursued mRNA therapeutics as an alternative strategy for DNA‐based gene therapy, due to outstanding advantages of mRNA. Foremost, introducing mRNA does not harbor the risk of genomic integration, which remains a major concern for DNA‐based gene therapy. Second, in contrast to DNA, delivery to the nucleus is not required for mRNA function, as it is translated in the cytoplasm 1, 2. However, high levels of mRNA instability and immunogenicity hindered its broad use for therapeutic investigation and clinical applications 3. These problems were largely mitigated by introduction of a number of structural modifications in mRNA molecules, including a m7G 5′‐Cap structure, untranslated regions (UTRs), poly(A) tail, and perhaps most importantly, modified nucleotides. The structural modifications significantly improve mRNA stability, reduce its immunogenicity, and enhance translational efficiency 4, 5, 6. The improvement in translational efficiency of modified mRNA depends on the encoded protein and the type of modifications. For example, Warren et al. demonstrated a fourfold increase in fluorescence intensity when using either 5‐methylcytidine or pseudouridine in the structure of GFP mRNAs, whereas the intensity was 10‐fold higher when using both modified nucleotides 7. The modified mRNA, hereafter referred to as chemically modified mRNA (cmRNA), is synthesized via in vitro transcription. Figure 1 illustrates a generic structure of cmRNA. However, the rules for modification remain a work in progress and likely depend on the application. The full range of possible modifications in the structures of mRNAs, especially the type of modified nucleotides, are not yet completely explored and different research groups have used various sets of cmRNA modifications 6. Overall, there remains much chemistry to explore to find the optimal set of modifications useful for a specific cmRNA indication. Direct cmRNA modifications include the following.
Figure 1.

General structure of chemically modified mRNA (cmRNA). Modifications increase the stability, decrease the immunogenicity, and in some cases increase the translational efficiency. Typical components of a cmRNA include, with color‐coding: 7‐methylguanosine (m7G) cap structure (5′‐Cap). Untranslated regions (5′‐UTR and 3′‐UTR), usually derived from β‐globin mRNAs. Open reading frame, coding sequence for the gene of interest, containing optimized codons and/or chemically modified nucleotides. Polyadenylated tail (Poly[A] tail), stretch of 100–200 adenine nucleotides.
5′‐Cap Structure
During natural transcription, the 5′ end of eukaryotic mRNA links to a 7‐methylguanosine (m7G 5′) cap. The cap structure can dramatically affect translational efficiency by two mechanisms. First, binding of cap to translation initiation factor 4E (eIF4E) is essential for mRNA translation. Second, binding of cap to mRNA decapping enzymes initiates mRNA decay. Therefore, choosing the appropriate cap structures that have high affinity for binding to eIF4E and are resistant against decapping enzymes can significantly increase the translational efficiency as well as cmRNA stability 6, 8.
UTRs
Due to the presence of regulatory sequence elements, impacting tertiary structures such as hairpin loops, and interaction with RNA‐binding proteins, 5′‐ and 3′‐UTRs can affect both stability and translational efficiency of cmRNAs. To this end, many studies have used the highly stabilizing UTRs derived from α/β‐globin genes to increase stability and efficiency of translation of desired cmRNAs 9 .
Poly(A) Tail
Eukaryotic mRNAs naturally bear a poly(A) tail structure at the 3′ end, which plays an important role in mRNA stability and translation. The poly(A) tail, with an optimum length of 120–150 nucleotides, can be added to synthetic cmRNA in either of two ways, encoding the poly(A) structure in the template vector of the target cmRNA, or enzymatically adding adenine nucleotides post‐transcription of cmRNA 6.
Modified Nucleotides
Unmodified mRNAs can be detected by a subclass of Toll‐like receptors (TLRs), such as TLR7, and thus are highly immunogenic. Incorporation of modified nucleotides such as 5‐methylcytidine (5mC), pseudouridine (Ψ), 5‐methyluridine (5mU), or N6‐methyladenosine in the structure of cmRNAs has been shown to significantly decrease the immunogenicity by avoiding activation of TLRs 6, 9.
The advent of modified mRNA has substantially accelerated cmRNA‐based therapeutic investigation both in academia and industry. cmRNA is already being investigated for cancer immunotherapy, mRNA vaccines, protein replacement, gene editing, cell fate conversion, and regenerative medicine 6. Application of cmRNA for cancer immunotherapy and development of mRNA‐based vaccines are already in clinical trials, a topic that has been previously reviewed 10, 11.
Here, we focus on application of cmRNAs for cell fate conversion (cell differentiation or reprogramming) and regenerative medicine (Fig. 2). First, we discuss application of cmRNAs for cell reprogramming (a form of dedifferentiation) toward stem cell fate, which has been validated as a safe and efficient method for production of induced pluripotent stem cells (iPSCs). Various studies are compared according to the type of mRNA modifications, delivery vehicles, number of transfections, and the efficiency and duration of iPSC colony formation. Second, we summarize studies that used cmRNAs for directed differentiation or trans‐differentiation of stem/progenitor cells to defined cell lineages, including for tissue regeneration. Again, various criteria are considered, such as types of mRNA modification, delivery vehicles, and type of biomaterials used for tissue engineering, to compare different cmRNA‐applied methods for cell differentiation. Finally, we discuss strategies for in situ applications of cmRNAs encoding anabolic factors to suppress degenerative diseases, as well as using cmRNA to improve homing of stem cells in the injury area. By providing key information about how cmRNA can be applied for cell and tissue engineering at a glance, we hope to inspire future investigations using cmRNAs for regenerative medicine in a timely and efficient manner.
Figure 2.
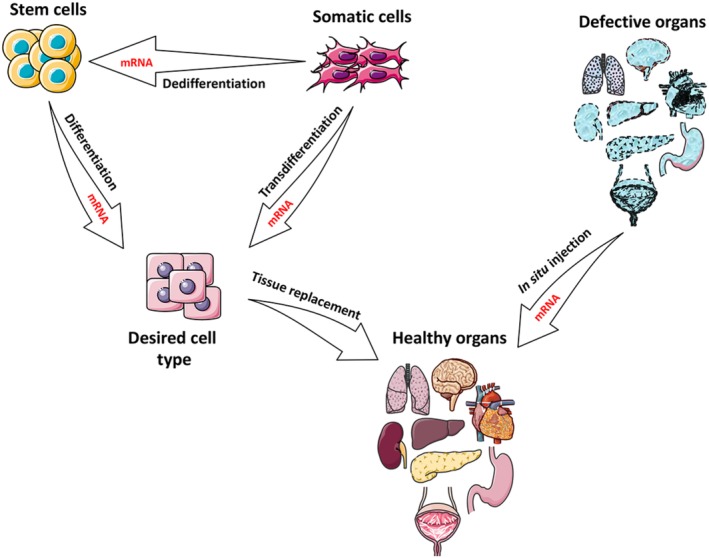
Application of mRNAs in cell and tissue engineering. mRNA can be used for reprogramming (dedifferentiation) of somatic cells to stem cells or directed differentiation of stem cells to the desired cell type. In addition, somatic cells can be directly reprogrammed to a distinct somatic cell type (trans‐differentiation) using mRNAs. Direct injection of therapeutic mRNAs to defective organs (in situ mRNA delivery) may also trigger tissue regeneration. This figure was made in part by using the Servier medical art free image collection.
Cell Reprogramming Using mRNA
Cell reprogramming, also known as cell dedifferentiation, has long been a topic of interest to developmental biologists. In the 1960s, John Gurdon showed for the first time that differentiation of a cell can be reversed by transferring a cell nucleus of an adult frog into an enucleated egg to derive cloned frog embryos 12. In 1996, the research group of Ian Wilmut cloned the first mammal using adult somatic cell nuclei to generate the sheep named Dolly 13. In a similar strategy, using fused (heterochromatic) somatic cells, Blau et al. showed that gene expression in differentiated cells is plastic and can be altered by modulations in the cytoplasm 14.
More recently, this phenomenon has been rediscovered as an important and feasible strategy for biotechnology, whereby a specialized differentiated cell is reverted to a more primitive state of stem or progenitor cell. The self‐renewal properties along with the capability to differentiate to multiple different cell types are features that make stem cells unique tools for regenerative medicine 15. In 2006, Takahashi and Yamanaka demonstrated that forced expression of the four transcription factors OCT3/4, SOX2, KLF4, and c‐MYC (now often referred to as Yamanaka factors) can reprogram mature fibroblasts to pluripotency 16, generating what are referred to as iPSCs.
The discovery of iPSCs was a remarkable achievement in biotechnology with major repercussions for regenerative medicine. iPSCs can be generated from a patient's own cells, and thus should mitigate much of the risk of genetic incompatibility or immune rejection for cellular derivatives that are subsequently transplanted. The patient's own genome is retained in the cells, which can be exploited for patient‐specific disease modeling. Unlike embryonic stem cells (ECSs), production of iPSCs does not require use of embryos, relieving potential ethical concerns 15. However, iPSCs reprogrammed by viral vector delivery of cDNA (for example retroviral, lentiviral, or adenoviral vectors) are prone to insertional mutagenesis and residual viral vector contamination, which in turn limit the clinical application of such iPSCs. Therefore, a number of studies have been performed to establish an integration‐free method for reprogramming to pluripotency. In 2009, for instance, human iPSCs were first generated by direct protein delivery of Yamanaka factors. Although the method presented no risk of genome insertion, low efficiency and lengthy procedure limit the protein‐based reprogramming protocols from most applications (Table 1) 17. Subsequently, the efficiency of this protocol was to some extent improved using partially purified recombinant protein produced in mammalian cells 25. Also in 2009, Sendai virus, a type of RNA virus, was first used for delivery of Yamanaka factors and reprogramming to iPSCs. Although Sendai virus reprogramming is integration‐free and the efficiency was quite improved, the method still suffers from an overall relative low efficiency (maximum 1%) and lengthy procedure (Table 1) 21.
Table 1.
Comparison of different protocols for reprogramming human fibroblasts to iPSCs, based on delivery of proteins, DNAs, and cmRNAs of Yamanaka factors
| Reprogramming protocols | Protein 17 | DNA virus (retrovirus/lentivirus) 18, 19, 20 | RNA virus (Sendai virus) 21, 22, 23 | cmRNA 7, 24 |
|---|---|---|---|---|
| Time course for colony isolation | 8 weeks | 2–4 weeks | 4 weeks | 2 weeks |
| Reprogramming efficiency | 0.001% | 0.01%–0.1% | 0.01%–1% | Up to 4.4% |
| Risk of genome integration | No | Yes | No | No |
Abbreviations: iPSCs, induced pluripotent stem cells; cmRNA, chemically modified RNA.
The first successful reprogramming of somatic cells to iPSCs with cmRNAs encoding Yamanaka factors was performed in 2010 7. With this protocol, fibroblasts underwent a “daily transfection regime” for 14 consecutive days using cationic lipid nonviral vectors for cmRNA delivery, and iPSC colonies were isolated at day 18 24. The resulting transgene‐free iPSCs with no obvious risk of mutagenicity and no viral residual, along with high reprogramming efficiency (up to 4.4%), make this method a candidate preferential procedure for iPSC reprogramming in future clinical applications (Table 1). Accordingly, a number of commercially available kits have been designed, and a human iPSC line has been established, based on the above‐mentioned mRNA reprogramming protocol 26.
Although the cmRNA‐based reprogramming protocol has advantages over protein‐ or DNA‐based strategies with higher safety and efficiency, the protocol is relatively complicated and laborious as it requires a daily transfection regimen over 2 weeks. In addition, the use of feeder cells and in some cases antibiotic‐free culture increases the risk of bacterial and biological contamination 7, 24. As a result, continued efforts have sought to simplify the process and optimize the cmRNA reprogramming protocols. Parameters that have been modified include fewer numbers of transfections, fewer reprogramming factors, shorter time frame for reprogramming, using feeder free culture, various vectors, and various modifications of mRNAs (Table 2). Note that not all of these studies were pursued until colony characterization or testing for capacity of teratoma formation. Indeed, some studies only detected elevated expression levels of endogenous reprogramming genes as markers for initiation of reprogramming.
Table 2.
Summary of studies on reprogramming to iPCS using mRNA
| Transcription factors | Transfection reagent | Duration between first TF and colony formation/pick up | Number of transfections | Using feeder cells | Cell source | Differentiation to three germ layers/teratoma formation | mRNA modifications | References |
|---|---|---|---|---|---|---|---|---|
|
KLF4,
c‐MYC, OCT4, SOX2, LIN28 |
Cationic lipid | Colony formation day 17/colony pick up day 20 | 17 | Yes | BJ1, dH1f, Detroit 551 (D551), and MRC‐5 fibroblast, and skin cells of a cystic fibrosis patient (CF) | Yes | 100% 5mC and Ψ, 5′‐UTR containing Kozak sequence, α‐Globin 3′‐UTR, Poly(A) tail, Cap |
7 |
|
KLF4,
c‐MYC, OCT4, SOX2, LIN28 |
Cationic lipid | Colony formation day 6–9/colony pick up day 15–18 | 14 | Yes | Human primary fibroblasts | No | 100% 5mC and Ψ, 3′‐UTR, 5′‐UTR, Poly(A) tail, Cap |
24 a |
|
OCT‐4, NANOG,
KLF‐4, c‐MYC, SOX‐2, hTERT |
Electroporation and lipofection | Colony formation day 30 | 4 | Yes | Human foreskin cells, adult Huntington fibroblasts, adult skin fibroblasts of healthy donors | Yes | Poly(A) tail, Cap |
27 |
|
OCT4,
SOX2, KLF4, c‐MYC |
Cationic lipids | Not mentioned/not continued | 3 | Yes | Mouse embryonic fibroblasts (MEFs) | No | Poly(A) tail, Cap |
28 |
|
OCT4,
LIN28, SOX2, NANOG |
Cationic lipids | Colony formation day 12–19/colony pick up day 21 | 5 | Yes | Human foreskin Fibroblasts (hFF) |
No | Poly(A) tail, Cap, IRES sequence |
29 |
|
KLF4,
c‐MYC, OCT4, SOX2, LIN28 |
Cationic lipids | Colony formation around day 30 | 18 | No | MSCs derived from adipose tissue of a 50‐year‐old patient | Yes | 100% 5mC and Ψ, Cap |
30 |
|
OCT4,
SOX2, c‐MYC, KLF4, SV40 large T (LT) |
Electroporation | Colony formation day 30 | 1 | No | Human fetal skin fibroblasts (HuF1), Human embryonic lung fibroblasts (MRC5), Human foreskin fibroblasts (HFF) |
No | Poly(A) tail, Cap, 5′‐ and 3′‐UTRs of Xenopus β‐Globin |
31 |
|
OCT4,
SOX2, KLF4, c‐MYC |
Lipofectamine 2000 | Colony pick up day 22 | 5 | No | Goat embryonic fibroblasts (GEFs) | Yes | Poly(A) tail, Cap |
32 |
|
M3O
b
,
SOX2, KLF4, c‐MYC‐T58A, LIN28, NANOG |
Cationic lipids | Colony formation day 9–13 | 9 | No | BJ neonatal Fibroblasts, HDF‐f fetal fibroblasts, HDF‐n neonatal fibroblasts, HDF‐a Adult fibroblasts, XFF xeno‐free neonatal fibroblasts |
Yes | 100% 5mC and Ψ, 5′‐UTR containing Kozak sequence, α‐Globin 3′‐UTR, Poly(A) tail, Cap |
33 c |
|
OCT4, SOX2, c‐MYC,
KLF4 Or Total mRNA or RNA extracted from cells expressing Yamanaka factors |
Graphene oxide‐polyethylenimine (GO‐PEI) using a dynamic suspension culture | Colony formation day 18, Colony pick up day 24 (for hADFs cells) | 3 | Yes | Human adipose tissue‐derived fibroblasts (hADFs), rat ADFs (rADFs), and mouse embryonic fibroblasts (MEFs) | Yes | 3′‐UTR, 5′‐UTR, Poly(A) tail, Cap |
34 |
|
KLF4, c‐MYC,
OCT4, SOX2, LIN28 |
RNAiMAX | Colony formation day 8, Colony pick up day 12–16 |
8–12 | No | BJ human fibroblasts, GM13325 fibroblasts, human adult fibroblasts (HUF1 and HUF58) | Yes | 100% 5mC and Ψ, Cap |
35 |
|
OCT4,
KLF4, SOX2, c‐MYC, LIN28 |
RNAiMAX | Colony formation day 15, colony pick up day 18–21 | 18 | Yes | Bone marrow derived‐MSCs from a patient with β‐thalassemia | Yes | 100% 5mC and Ψ, Poly(A) tail, Cap |
36 |
|
KLF4,
c‐MYC, OCT4, SOX2, LIN28, MBD3 SiRNA |
RNAiMAX | Colony pick up day +7 | 2 | Yes and no (same efficiency) | Adult Human patient‐specific fibroblasts |
Yes | Not mentioned | 37 |
|
KLF4,
c‐MYC, OCT4, SOX2, LIN28 |
RNAiMAX | Colony formation day 18 | 18 | Yes | Human amniotic fluid‐derived stem cells (AFSC) | Yes | 100% 5mC and Ψ, 5′‐UTR containing Kozak sequence, α‐Globin 3′‐UTR, Poly(A) tail, Cap |
38 |
|
M3O, SOX2,
KLF4, cMYC, LIN28A, NANOG, mWasabi, miRNA‐367/302 seconds |
RNAiMAX | Colony formation day 18 | 7 | No | Primary fibroblasts form healthy donors and patientsd | Yes | 100% 5mC and Ψ, 5′‐UTR containing Kozak sequence, α‐Globin 3′‐UTR, Poly(A) tail, Cap |
39 |
5mC and Ψ stand for 5‐methylcytidine and pseudouridine, respectively.
This publication is a protocol based on reference 7.
Oct4 incorporating an N‐terminal MyoD transactivation domain.
Different reprogramming regimens with and without using feeder layer were tested to find the method with highest efficiency and easiest handling.
Primary neonatal fibroblast lines: FN1, FN2, and FN5. Healthy primary adult fibroblast lines: F40, F62, and F50. Fibroblasts derived from patients with inherited skin blistering disorders and Down's syndrome.
In addition to reprogramming to iPSCs, cmRNAs can be used for reprogramming to other multipotent stem cells. To this end, Kim et al. reprogrammed human mesenchymal stem cells (MSCs) to induced neural stem cells (iNSCs) using SOX2 cmRNA 40. This study used the same modifications as the Mandal and Rossi protocol 24. The resulting integration‐free iNSCs can be used for studying neural pathogenesis or potentially for treatment of neurodegenerative disease 40.
Recently, mRNAs have been used not only for making iPSCs, but also for gene editing of the resulting iPSCs using CRISPR/Cas9 technology. In contrast to plasmid DNA, transient delivery of mRNA encoding Cas9 provides an integration‐free platform for further editing cmRNA‐reprogrammed iPSCs, to either introduce a specific gene mutation for disease modeling, or to correct a gene mutation via homology directed repair 41. Both approaches are highly relevant in regenerative medicine.
As mentioned before, other integration‐free reprogramming methods such as using nonintegrative viral vectors and excisable vectors have also been evaluated to find the safest and the most efficient strategy for iPSC production. However, the risk of random genome insertion of segments of viral vectors, the additional efforts to remove the excisable vectors, as well as the low efficiency of these methods limit their applications 42. Using self‐replicating mRNA is another approach to simplify the procedure by elimination of daily transfections. In this method, a single long mRNA encoding all four reprogramming factors was used based on the noninfectious and self‐replicating Venezuelan Equine Encephalitis (VEE) virus RNA replicon. Nonetheless, this method requires conditioned media containing B18R and Puromycin for blocking the innate immune response and retention of VEE RNA, respectively 43. In addition, the time interval for reprogramming to pluripotency using self‐replicating mRNA was almost two times longer compared with the mRNA protocol (iPSC colony isolation on day 25–30 or day 15–18 using self‐replicating mRNA method or cmRNA protocol, respectively 24, 43). Thus, reprogramming to iPSC fate using cmRNAs remains the most efficient and safest method for cell dedifferentiation.
Cell Programming and Tissue Engineering Using mRNA
In parallel with cell reprogramming, cmRNAs have been investigated for their use in cell programming, also known as cell differentiation. In these studies, cmRNAs were used to differentiate stem cells to the desired cell lineages for application in tissue engineering and regenerative medicine (summarized in Table 3).
Table 3.
mRNA applications in cell differentiation and tissue regeneration
| mRNA | Type of cell differentiation or tissue regeneration | mRNA modification | Transfection reagent/method | Biomaterial | Number of transfections | References |
|---|---|---|---|---|---|---|
| BMP2 | Osteogenic diff. Bone reg. |
100% Ψ and 5mC, Poly(A) tail, Cap |
Branched PEI | Collagen | 1 | 44 |
| BMP2 | Osteogenic diff. Bone reg. |
25% 5mC and 2TU, Poly(A) tail, Cap |
In vitro: DreamFect gold (DF‐gold) ± magnetic nanoparticles In vivo: C12‐EPE |
Fibrin gel | 1 | 45 |
| BMP2 or BMP9 | Osteogenic diff. Bone reg. |
100% Ψ and 5mC, Poly(A) tail, Cap |
PEI | Collagen | 1 | 46 |
| BMP2 | Osteogenic diff. Bone reg. |
25% 5mC and 2TU, Poly(A) tail, Cap |
A proprietary lipid | Collagen | 1 | 47 |
| BMP2 | Osteogenic diff. | 25% 5mC and 2TU, Poly(A) tail, Cap |
DF‐gold | Fibrin gel and micro–macro biphasic calcium Phosphate (MBCP) granules |
1 | 48 |
| BMP2 | Osteogenic diff. Bone reg. |
7.5% 5IC and 35% 5 IU Poly(A) tail, Capa |
A proprietary lipid | Collagen | 1 | 49 |
| hVEGF‐A | Vascular reg. Cardiac reg. Cardiovascularendothelial and endovascular diff. |
100% Ψ and 5mC, Poly(A) tail, Cap |
RNAiMAX | None | 1 | 50 |
| VEGF‐A | Endothelial diff. Vascular reg. |
100% 5mC and 2TU Cap Poly(A) tail |
RNAiMAX | Matrigel | 1 or 2 | 51 |
|
NEUROG1
NEUROG2 NEUROG3 NEUROD1 NEUROD2 |
Neuronal cell diff. | 100% Ψ and 5mC, Poly(A) tail, Cap |
Lipofectamine messenger max | None | 2 | 52 |
| ETV2 | Trans‐diff. of fibroblasts to endothelial progenitor cells | Poly(A) tail, Cap |
Nucleofection, FuGENE HD | None | 1 | 53 |
| MYOD | Myogenic diff. | 100% Ψ and 5mC, Poly(A) tail, Cap |
RNAiMAX | None (plate coated with gelatin) | 3 | 7 |
|
Gata4,
Mef2c, Tbx5 |
Trans‐diff. of cardiac fibroblasts to cardiomyocyte‐like cells | 100% Ψ and 5mC, Poly(A) tail, Cap |
c‐lipo (=lipofectamine 2000 + CRPPR‐R9b) | None (plate coated with gelatin) | 14 | 54 |
| Pdx1, Neurogenin3, MafA | Trans‐diff. of pancreatic nonendocrine cells into insulin‐producing β‐cells | 100% Ψ and 5mC, Poly(A) tail, Cap |
Lipofectamine messenger‐MAX mRNA transfection reagent | None | 10 | 55 |
| MAFA c | Trans‐diff. of human pancreatic duct‐derived cells into insulin‐secreting β‐cells | 100% Ψ and 5mC, Poly(A) tail, Cap |
jetPEI | None | 7 | 56 |
5IC and 5 IU stand for 5‐iodo‐cytidine and 5‐iodo‐uridine, respectively. Further modifications in this study are included: omitting an upstream ORF in the 5′‐UTR and a polyadenylation element along with an AU‐rich tract in the 3′‐UTR. Furthermore, a translation initiator of short UTRs (TISU) was incorporated in mRNA structure.
CRPPR‐R9, a polyarginine‐fused heart‐targeting peptide.
V‐Maf musculoaponeurotic fibrosarcoma oncogene homolog A.
Abbreviations: Diff., differentiation; Reg., regeneration.
In most of the studies listed in Table 3, mRNAs contained modified nucleotides, either 100% pseudouridine (Ψ) and 5‐methylcytidine (5mC), or 25% 2‐thiouridine (2TU) and 5mC. Very recently, 7.5% 5‐iodo‐cytidine and 35% 5‐iodo‐uridine modification of BMP2 mRNA has been used for bone regeneration 49. Nonetheless, differentiation to endothelial progenitor cells using ETV2 cmRNA harboring no modified nucleotides has also been reported 53.
Various methods have been used for delivery of cmRNAs to cells in vitro, ex vivo, and in vivo. Mechanical methods such as electroporation and gene gun have been widely used for cmRNA delivery. However, these are expensive methods, to some extent invasive, and clearly not translatable for in vivo clinical applications. Chemical methods for cmRNA delivery, on the other hand, are cheaper, easier, and impose lower toxicity and immunogenicity. Most importantly, chemical methods are capable of noninvasive cmRNA delivery in vivo. In these methods, cationic lipids or polymers bind electrostatically with negatively charged cmRNA molecules and form nanoparticles 9. The use of lipid or polymer nanoparticles can arguably be considered the current gold standard for delivery of nucleic acids including cmRNAs 57. Such complexes are biocompatible, biodegradable, with a natural propensity to interact with cellular membranes to facilitate cargo uptake through an endocytic pathway.
For translation, nanoparticles provide advantages for targeting cmRNA to specific cells or organs, which is a very important consideration for clinical applications, and represents a very active area of current research (Fig. 3). Hydrophilic or electrostatic binding of the cmRNA nanoparticles to the cell membrane can initiate endocytosis and lead to internalization of nanoparticle to the cell cytoplasm 9. The nanoparticle can be delivered systematically via blood injection/administration, in which case the predominant cells of uptake are liver hepatocytes 58. If other cell types are to be targeted in vivo, other strategies can be used, for example direct local tissue injection for muscle 59 or inhalation delivery to target the lung epithelium 60. Modifying the surface charges of the cmRNA nanoparticles is an emerging strategy for targeting cmRNA to defined cell types. Chemical traits that can impact targeting include the specific lipid‐amine compound, the amount of Polyethylene glycol (PEG) included in the formulation, the defined PEG structure, and the molar amount of cholesterol. In an elegant and promising recent study, Sago et al. used a high throughput method to screen over 250 lipid nanoparticle formulations that incorporated DNA barcodes. Following delivery, they could screen organs and cells for biased targeting based on the coincorporation of the bar‐code, and were successful at identifying two nanoparticles that efficiently delivered mRNA to endothelial cells 61. Finally, lipid nanoparticles loaded with mRNA can be decorated with targeting moieties, such as monoclonal antibodies, as was accomplished recently to target expression of Interleukin‐10 in inflammatory leukocytes as a potential therapy for inflammatory bowel disorder 61.
Figure 3.
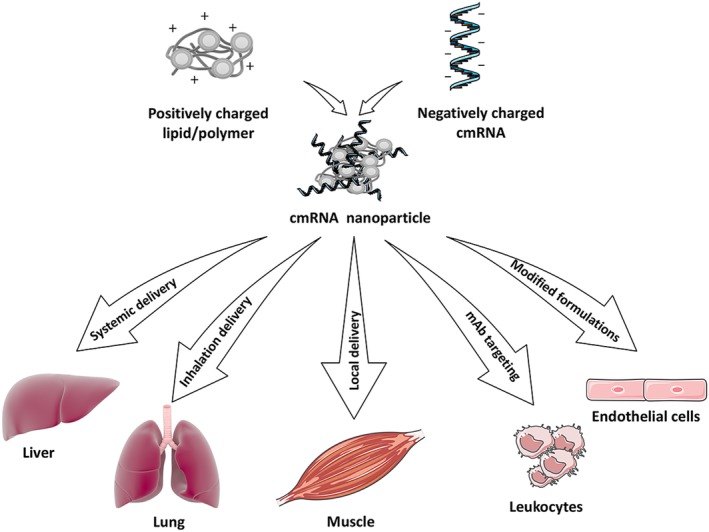
Noninvasive and targeted delivery of chemically modified mRNAs (cmRNAs) in vivo. Negatively charged cmRNA molecules bind electrostatically with positively charged polymers or lipids. Various strategies have been used to target the resulting nanoparticles to certain tissues. Systemic (blood), inhaled, or local injections have been used to preferentially target liver hepatocytes, lung epithelium, or skeletal muscle, respectively. Decorating the nanoparticles with monoclonal antibodies has been used to target leukocytes expressing defined antigens, whereas defined lipid/polymer formulations can bias delivery to distinct cells and tissues, including endothelium. See text for details and relevant references. This figure was made in part by using the Servier medical art free image collection and the library of Science & Medical Illustrations.
Beyond lipid nanoparticles, many in vivo studies have benefited from exploring biomaterials to facilitate the delivery of cmRNAs to a desired tissue. Using biomaterials can reduce the off‐target toxicity and enhance delivery of cmRNA to the specific site. Biomaterials can also work as a “reservoir” for sustained delivery of cmRNA, where the delivery efficiency can be controlled by porosity and/or biodegradability of the biomaterials. To this end, Badieyan et al. applied transcript activated matrices preloaded with cmRNA complexes. They established a sustained cmRNA delivery collagen matrix with a plateau of almost 1 week of protein production for application in tissue engineering 47. Different criteria need to be considered to choose the right biomaterial for each application. These criteria include: biocompatibility, biodegradability, in situ gelation upon injection, and physical and mechanical properties such as permeability and mechanical response. Considering these factors, natural biopolymers such as collagen and fibrin have often been used to deliver cmRNAs to tissues 9.
cmRNAs have been used for differentiation and trans‐differentiation into a variety of cell types including neurons 52, β‐cells 55, endothelial progenitors 53, cardiovascular cells 50, 54, and myogenic cells 7, 54. However, the majority of studies using cmRNA for cell differentiation and tissue engineering have been performed on bone, and to a lesser extent, on heart regeneration. The effect of BMP2 on bone regeneration was previously shown using gene 62 or protein 63 therapy. Recent publications demonstrated successful bone regeneration following application of cmRNAs encoding BMP2 in femur 45, 47 and calvarial 44, 46 bone defect models. Regarding cardiovascular regeneration, Hadas et al. reported successful differentiation of cardiomyocytes and heart regeneration following application of VEGF cmRNA 8. They suggested that cmRNA is an efficient tool for boosting endogenous regeneration after myocardial infarction (MI). This is due to high protein production efficiency of cmRNA as well as fast kinetics of expression that closely meet the time frame of MI pathology. In addition, cmRNA has a transient effect and thus eliminates the risk of malignancy which could occur following induced cell proliferation due to over‐expression of the delivered gene over long periods 8. In addition to cardiovascular regeneration, Lui et al. showed enhancement of engraftment with reduction of apoptosis in human heart progenitor cells after delivery of VEGF‐A mRNA in vivo 51. A recent study characterized the optimal type of modifications as well as doses of cmRNA for cardiovascular delivery. According to this, applying cmRNAs containing N1‐methylpseudouridine‐5′‐triphosphate nucleotide modification with 100 μg per mouse heart (1.6 μg cmRNA per microliter in 60 μl total) in sucrose‐citrate buffer led to the best result for in vivo delivery of cmRNA into mouse heart 64.
In situ delivery of mRNAs encoding anabolic markers has also been used for controlling degenerative diseases. To this end, direct injection of mRNA encoding Runt‐related transcription factor 1 (RUNX1) has been applied to modify osteoarthritis (OA). RUNX1 mRNA formulated with polyplex nanomicelles was injected into knee joints of an OA mouse model. The expression of RUNX1 as a cartilage‐anabolic factor significantly reduced the progression of OA in mice, and suggested in situ mRNA delivery as a potential approach for regenerative medicine 65. Another example of in situ application of mRNA was direct injection of vasopressin mRNA into the hypothalamus to temporarily invert diabetes insipidus in Brattleboro rats. This animal model presents with diabetes insipidus due to a mutation in the propressophysin gene that makes them unable to secret vasopressin in hypothalamic neurons 66.
Another study used in situ delivery of cmRNA encoding IGF1 to damaged myocardial cells after MI. Among other roles, IGF1 can elicit pathways of cell survival and eventually cardiomyocyte repair. The protein production following delivery of nanoparticles of jetPEI and IGF1 cmRNA significantly reduced cell apoptosis and thus promoted the survival of cardiac cells after MI in an in vivo murine mode 67. Kariko et al. also showed mRNA encoding erythropoietin significantly increases production of red blood cells and decreases the risk of anemia 68.
In a number of studies mRNAs were used for migration and homing of MSCs to the site of inflammation and brain tumors. The improvement of homing of MSCs can in turn enhance the capability for regeneration of damaged tissue. To this end, Ryser et al. showed that MSCs transfected with mRNA encoding chemokine (C‐X‐C motif) receptor 4 (CXCR4) can improve migration of MSCs to inflammation sites, and thus can potentially improve cell therapy in the inflammatory area 69. CXCR4 is a chemokine receptor with high affinity to bind to stromal derived factor‐1, which is highly expressed in inflammatory sites, such as an ischemic area. In a similar study, MSCs transfected with mRNA encoding P‐selectin glycoprotein ligand‐1 (PSGL‐1) and α‐(1,3)‐fucosyltransferase (FUT7) improved the tethering and rolling and consequently the homing of MSCs in the inflammatory region 70. FUT7 enzyme increases the secretion of Sialyl‐Lewisx (SLeX). PSGL‐1, further modified with SLeX, facilitates the interaction with P‐selectin that is often upregulated on endothelial cells during inflammation. Likewise, Nowakowski et al. improved MSC migration to the brain ischemic area by using integrin a4 (ITGA4) mRNA‐transfected MSCs 71. Expression of ITGA4 facilitates adhesion of MSCs to the VCAM‐1 ligand of endothelial cells and thus improves migration of MSCs to injured tissue such as ischemic brain.
Conclusion
Although application of mRNA in cell fate conversion and regenerative medicine is a fast progressing field, existing hurdles remain that will need to be managed for eventual translation. The high cost of the modified mRNAs and high sensitivity to heat and ubiquitous nucleases may limit the enthusiasm of industrial and academic groups to invest time and effort on cmRNA‐based applications, as a relatively new field of research. Moreover, the optimal carrier for delivery of mRNA may need to be determined for each single application, particularly for in vivo studies. Even with enhanced engineered delivery vehicles, challenges remain, for example to control release of cmRNA into the cytoplasm following endocytosis, and to control levels of expressed proteins. When reprogramming of cells or regeneration of tissue needs a relatively long time‐course, the ideal biomaterials for prolonged delivery of mRNA should be determined to compensate for the transient effect of mRNA and mitigate the need for multiple transfections. Overall, the accumulating knowledge of enhanced mRNA synthesis, modification, delivery and targeting, should overcome these obstacles and fulfill promise of a bright future for translation of cmRNA toward clinical applications.
Author Contributions
Z.S.B.: conception and design, collection and/or assembly of data, manuscript writing; T.E.: conception and design, financial support, manuscript editing.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Acknowledgments
Z.S.B is supported by a training grant from the New York State Department of Health (NYSTEM C32558GG). T.E. is supported by grants from the National Institutes of Health (R35‐HL135778), the Department of Defense (W81XWH‐17‐1‐0661), The American Heart Association (18CSA34080171), the New York State Department of Health (NYSTEM C32591GG and C32593GG), and the Tri‐Institutional Stem Cell Initiative (2016‐032 and 2016‐004).
References
- 1. Bettinger T, Carlisle RC, Read ML et al. Peptide‐mediated RNA delivery: A novel approach for enhanced transfection of primary and post‐mitotic cells. Nucleic Acids Res 2001;29:3882–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitchell DA, Nair SK. RNA‐transfected dendritic cells in cancer immunotherapy. J Clin Invest 2000;106:1065–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van Tendeloo VF, Ponsaerts P, Berneman ZN. mRNA‐based gene transfer as a tool for gene and cell therapy. Curr Opin Mol Ther 2007;9:423–431. [PubMed] [Google Scholar]
- 4. Kormann MS, Hasenpusch G, Aneja MK et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol 2011;29:154–157. [DOI] [PubMed] [Google Scholar]
- 5. Kariko K, Buckstein M, Ni H et al. Suppression of RNA recognition by Toll‐like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005;23:165–175. [DOI] [PubMed] [Google Scholar]
- 6. Sahin U, Kariko K, Tureci O. mRNA‐based therapeutics—Developing a new class of drugs. Nat Rev Drug Discov 2014;13:759–780. [DOI] [PubMed] [Google Scholar]
- 7. Warren L, Manos PD, Ahfeldt T et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010;7:618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hadas Y, Katz MG, Bridges CR et al. Modified mRNA as a therapeutic tool to induce cardiac regeneration in ischemic heart disease. Wiley Interdiscip Rev Syst Biol Med 2017;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel S, Athirasala A, Menezes PP et al. Messenger RNA delivery for tissue engineering and regenerative medicine applications. Tissue Eng Part A 2018;25:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fiedler K, Lazzaro S, Lutz J et al. mRNA Cancer Vaccines. Recent Results Cancer Res Fortschritte Krebsforschung Progres dans les Recherches Sur le Cancer 2016;209:61–85. [DOI] [PubMed] [Google Scholar]
- 11. Diken M, Kranz LM, Kreiter S et al. mRNA: A versatile molecule for cancer vaccines. Curr Issues Mol Biol 2017;22:113–128. [DOI] [PubMed] [Google Scholar]
- 12. Gurdon JB. The cloning of a frog. Development 2013;140:2446–2448. [DOI] [PubMed] [Google Scholar]
- 13. Campbell KH, McWhir J, Ritchie WA et al. Sheep cloned by nuclear transfer from a cultured cell line. Nature 1996;380:64–66. [DOI] [PubMed] [Google Scholar]
- 14. Blau HM, Pavlath GK, Hardeman EC et al. Plasticity of the differentiated state. Science 1985;230:758–766. [DOI] [PubMed] [Google Scholar]
- 15. Cai S, Fu X, Sheng Z. Dedifferentiation: A new approach in stem cell research. Bioscience 2007;57:655–662. [Google Scholar]
- 16. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- 17. Kim D, Kim CH, Moon JI et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009;4:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park IH, Zhao R, West JA et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008;451:141–146. [DOI] [PubMed] [Google Scholar]
- 19. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 20. Yu J, Vodyanik MA, Smuga‐Otto K et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007;318:1917–1920. [DOI] [PubMed] [Google Scholar]
- 21. Fusaki N, Ban H, Nishiyama A et al. Efficient induction of transgene‐free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 2009;85:348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ban H, Nishishita N, Fusaki N et al. Efficient generation of transgene‐free human induced pluripotent stem cells (iPSCs) by temperature‐sensitive Sendai virus vectors. Proc Natl Acad Sci USA 2011;108:14234–14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ye L, Muench MO, Fusaki N et al. Blood cell‐derived induced pluripotent stem cells free of reprogramming factors generated by Sendai viral vectors. Stem Cells Translational Medicine 2013;2:558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mandal PK, Rossi DJ. Reprogramming human fibroblasts to pluripotency using modified mRNA. Nat Protoc 2013;8:568–582. [DOI] [PubMed] [Google Scholar]
- 25. Park H, Kim D, Kim C‐H et al. Increased genomic integrity of an improved protein‐based mouse induced pluripotent stem cell method compared with current viral‐induced strategies. Stem Cells Translational Medicine 2014;3:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Artero Castro A, Leon M, Del Buey Furio V et al. Generation of a human iPSC line by mRNA reprogramming. Stem Cell Res 2018;28:157–160. [DOI] [PubMed] [Google Scholar]
- 27. Arnold A, Naaldijk YM, Fabian C et al. Reprogramming of human huntington fibroblasts using mRNA. ISRN Cell Biol 2011;2012:1–12. [Google Scholar]
- 28. Tavernier G, Wolfrum K, Demeester J et al. Activation of pluripotency‐associated genes in mouse embryonic fibroblasts by non‐viral transfection with in vitro‐derived mRNAs encoding Oct4, Sox2, Klf4 and cMyc. Biomaterials 2012;33:412–417. [DOI] [PubMed] [Google Scholar]
- 29. Yakubov E, Rechavi G, Rozenblatt S et al. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem Biophys Res Commun 2010;394:189–193. [DOI] [PubMed] [Google Scholar]
- 30. Heng BC, Heinimann K, Miny P et al. mRNA transfection‐based, feeder‐free, induced pluripotent stem cells derived from adipose tissue of a 50‐year‐old patient. Metab Eng 2013;18:9–24. [DOI] [PubMed] [Google Scholar]
- 31. Plews JR, Li J, Jones M et al. Activation of pluripotency genes in human fibroblast cells by a novel mRNA based approach. PLoS One 2010;5:e14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen H, Zuo Q, Wang Y et al. Inducing goat pluripotent stem cells with four transcription factor mRNAs that activate endogenous promoters. BMC Biotechnol 2017;17:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warren L, Ni Y, Wang J et al. Feeder‐free derivation of human induced pluripotent stem cells with messenger RNA. Sci Rep 2012;2:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Choi HY, Lee TJ, Yang GM et al. Efficient mRNA delivery with graphene oxide‐polyethylenimine for generation of footprint‐free human induced pluripotent stem cells. J Control Release 2016;235:222–235. [DOI] [PubMed] [Google Scholar]
- 35. Durruthy‐Durruthy J, Briggs SF, Awe J et al. Rapid and efficient conversion of integration‐free human induced pluripotent stem cells to GMP‐grade culture conditions. PLoS One 2014;9:e94231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Varela I, Karagiannidou A, Oikonomakis V et al. Generation of human beta‐thalassemia induced pluripotent cell lines by reprogramming of bone marrow‐derived mesenchymal stromal cells using modified mRNA. Cell Reprogram 2014;16:447–455. [DOI] [PubMed] [Google Scholar]
- 37. Rais Y, Zviran A, Geula S et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature 2013;502:65–70. [DOI] [PubMed] [Google Scholar]
- 38. Velasquez‐Mao AJ, CJM T, Monroe MN et al. Differentiation of spontaneously contracting cardiomyocytes from non‐virally reprogrammed human amniotic fluid stem cells. PLoS One 2017;12:e0177824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kogut I, McCarthy SM, Pavlova M et al. High‐efficiency RNA‐based reprogramming of human primary fibroblasts. Nat Commun 2018;9:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim BE, Choi SW, Shin JH et al. Single‐factor SOX2 mediates direct neural reprogramming of human mesenchymal stem cells via transfection of in vitro transcribed mRNA. Cell Transplant 2018;27:1154–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kehler J, Greco M, Martino V et al. RNA‐generated and gene‐edited induced pluripotent stem cells for disease modeling and therapy. J Cell Physiol 2017;232:1262–1269. [DOI] [PubMed] [Google Scholar]
- 42. Stadtfeld M, Hochedlinger K. Induced pluripotency: History, mechanisms, and applications. Genes Dev 2010;24:2239–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoshioka N, Gros E, Li HR et al. Efficient generation of human iPSCs by a synthetic self‐replicative RNA. Cell Stem Cell 2013;13:246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Elangovan S, Khorsand B, Do AV et al. Chemically modified RNA activated matrices enhance bone regeneration. J Control Release 2015;218:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balmayor ER, Geiger JP, Aneja MK et al. Chemically modified RNA induces osteogenesis of stem cells and human tissue explants as well as accelerates bone healing in rats. Biomaterials 2016;87:131–146. [DOI] [PubMed] [Google Scholar]
- 46. Khorsand B, Elangovan S, Hong L et al. A comparative study of the bone regenerative effect of chemically modified RNA encoding BMP‐2 or BMP‐9. AAPS J 2017;19:438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Badieyan ZS, Berezhanskyy T, Utzinger M et al. Transcript‐activated collagen matrix as sustained mRNA delivery system for bone regeneration. J Control Release 2016;239:137–148. [DOI] [PubMed] [Google Scholar]
- 48. Balmayor ER, Geiger JP, Koch C et al. Modified mRNA for BMP‐2 in combination with biomaterials serves as a transcript‐activated matrix for effectively inducing osteogenic pathways in stem cells. Stem Cells Dev 2017;26:25–34. [DOI] [PubMed] [Google Scholar]
- 49. Zhang W, De La Vega RE, Coenen MJ et al. An improved, chemically modified RNA encoding BMP‐2 enhances osteogenesis in vitro and in vivo. Tissue Eng Part A 2019;25:131–144. [DOI] [PubMed] [Google Scholar]
- 50. Zangi L, Lui KO, von Gise A et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol 2013;31:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lui KO, Zangi L, Silva EA et al. Driving vascular endothelial cell fate of human multipotent Isl1+ heart progenitors with VEGF modified mRNA. Cell Res 2013;23:1172–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goparaju SK, Kohda K, Ibata K et al. Rapid differentiation of human pluripotent stem cells into functional neurons by mRNAs encoding transcription factors. Sci Rep 2017;7:42367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Van Pham P, Vu NB, Dao TT et al. Production of endothelial progenitor cells from skin fibroblasts by direct reprogramming for clinical usages. In Vitro Cell Dev Biol Anim 2017;53:207–216. [DOI] [PubMed] [Google Scholar]
- 54. Lee K, Yu P, Lingampalli N et al. Peptide‐enhanced mRNA transfection in cultured mouse cardiac fibroblasts and direct reprogramming towards cardiomyocyte‐like cells. Int J Nanomedicine 2015;10:1841–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koblas T, Leontovyc I, Loukotova S et al. Reprogramming of pancreatic exocrine cells AR42J into insulin‐producing cells using mRNAs for Pdx1, Ngn3, and MafA transcription factors. Mol Ther Nucleic Acids 2016;5:e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Corritore E, Lee YS, Pasquale V et al. V‐Maf musculoaponeurotic fibrosarcoma oncogene homolog a synthetic modified mrna drives reprogramming of human pancreatic duct‐derived cells into insulin‐secreting cells. Stem Cells Translational Medicine 2016;5:1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xue HY, Guo P, Wen WC et al. Lipid‐based nanocarriers for RNA delivery. Curr Pharm Des 2015;21:3140–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Coelho T, Adams D, Silva A et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013;369:819–829. [DOI] [PubMed] [Google Scholar]
- 59. Bahl K, Senn JJ, Yuzhakov O et al. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol Ther 2017;25:1316–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kuzmov A, Minko T. Nanotechnology approaches for inhalation treatment of lung diseases. J Control Release 2015;219:500–518. [DOI] [PubMed] [Google Scholar]
- 61. Sago CD, Lokugamage MP, Paunovska K et al. High‐throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc Natl Acad Sci USA 2018;115:E9944–E9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Partridge K, Yang X, Clarke NM et al. Adenoviral BMP‐2 gene transfer in mesenchymal stem cells: in vitro and in vivo bone formation on biodegradable polymer scaffolds. Biochem Biophys Res Commun 2002;292:144–152. [DOI] [PubMed] [Google Scholar]
- 63. Geiger M, Li RH, Friess W. Collagen sponges for bone regeneration with rhBMP‐2. Adv Drug Deliv Rev 2003;55:1613–1629. [DOI] [PubMed] [Google Scholar]
- 64. Sultana N, Magadum A, Hadas Y et al. Optimizing cardiac delivery of modified mRNA. Mol Ther 2017;25:1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aini H, Itaka K, Fujisawa A et al. Messenger RNA delivery of a cartilage‐anabolic transcription factor as a disease‐modifying strategy for osteoarthritis treatment. Sci Rep 2016;6:18743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jirikowski GF, Sanna PP, Maciejewski‐Lenoir D et al. Reversal of diabetes insipidus in Brattleboro rats: Intrahypothalamic injection of vasopressin mRNA. Science 1992;255:996–998. [DOI] [PubMed] [Google Scholar]
- 67. Huang CL, Leblond AL, Turner EC et al. Synthetic chemically modified mrna‐based delivery of cytoprotective factor promotes early cardiomyocyte survival post‐acute myocardial infarction. Mol Pharm 2015;12:991–996. [DOI] [PubMed] [Google Scholar]
- 68. Kariko K, Muramatsu H, Keller JM et al. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine‐containing mRNA encoding erythropoietin. Mol Ther 2012;20:948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ryser MF, Ugarte F, Thieme S et al. mRNA transfection of CXCR4‐GFP fusion—Simply generated by PCR‐results in efficient migration of primary human mesenchymal stem cells. Tissue Eng Part C Methods 2008;14:179–184. [DOI] [PubMed] [Google Scholar]
- 70. Levy O, Zhao W, Mortensen LJ et al. mRNA‐engineered mesenchymal stem cells for targeted delivery of interleukin‐10 to sites of inflammation. Blood 2013;122:e23–e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nowakowski A, Andrzejewska A, Boltze J et al. Translation, but not transfection limits clinically relevant, exogenous mRNA based induction of alpha‐4 integrin expression on human mesenchymal stem cells. Sci Rep 2017;7:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.