

Abstract
Scar formation is the most common cause for failure of glaucoma filtration surgery because of increased fibroblast proliferation and activation. We have now examined the effect of Y‐27632, a Rho‐associated protein kinase (ROCK) inhibitor, on postsurgical scarring formation in human Tenon fibroblasts (HTFs). Collagen gel contraction assay was used to compare contractility activity of Y‐27632 with several antiglaucoma drugs. Immunofluorescence and western blotting were used to examine expression of scar formation–related factors. We found that Y‐27632 inhibited collagen gel contraction, as well as α‐smooth muscle actin and vimentin expression; these were promoted by treatment with latanoprost, timolol, or transforming growth factor (TGF)–β. To investigate the effect of Y‐27632 in postsurgical scarring, we mimicked TGF‐β secretion by stimulating HTFs with TGF‐β prior to Y‐27632 treatment. HTFs cultured in the presence of TGF‐β significantly increased gel contraction. In contrast, when HTFs were treated with 10μM Y‐27632, contraction was significantly inhibited. Furthermore, Y‐27632 reduced TGF‐β–induced phosphorylation of mitogen‐activated protein kinase signalling. These results suggest that ROCK inhibitors may inhibit fibrosis by inhibiting transdifferentiation of Tenon fibroblasts into myofibroblasts and by inhibiting TGF‐β signalling after surgery through mitogen‐activated protein kinase pathway suppression. These results implicate that ROCK inhibitors may improve outcomes after filtering surgery with a potential antiscarring effect, while latanoprost and timolol may induce fibrosis.
Significance of the study
Scar formation is the primary cause of failure after glaucoma filtration surgery. A ROCK inhibitor, Y‐27632, has been introduced as a novel potential antiglaucoma treatment to reduce intraocular pressure. The aim of our study was to elucidate the effect of Y‐27632 on scarring formation after glaucoma filtration surgery, in direct comparison with other antiglaucoma drugs. Our findings thus suggested that Y‐27632 may inhibit fibrosis and improve outcome after glaucoma filtration surgery through inhibition of transdifferentiation of Tenon fibroblasts into myofibroblasts, and the TGF‐β and MAPK signalling after surgery, while latanoprost and timolol may induce fibrosis.
Keywords: glaucoma, scarring formation, Tenon fibroblast, wound healing, Y‐27632
1. INTRODUCTION
Glaucoma, the second leading cause of blindness, is an optic neuropathy characterized by damage to the optic nerve head and defects in the visual field.1, 2, 3, 4 Increased intraocular pressure (IOP) is frequently related to glaucoma; reduction of IOP is the primary target treatment for prevention of glaucoma progression.2, 5
Medical therapy is the main treatment approach for glaucoma; initial monotherapy commonly comprises topical prostaglandins or β‐blockers, which are used to reduce IOP by respectively increased humour flow through the uveoscleral pathway or reduced aqueous humour production.2, 6, 7 When monotherapy is ineffective for the reduction of IOP, multidrug therapy is used. If medical therapy cannot sufficiently reduce IOP, laser or surgical treatment is indicated.2, 7 Glaucoma filtration surgery is regularly performed and remains the gold standard for lowering IOP; trabeculectomy is the most effective incisional surgery for uncontrolled glaucoma.2, 8, 9
However, scar formation is the primary cause of failure after glaucoma filtration surgery, due to increased fibroblast proliferation, activation, and collagen deposition.9, 10, 11 This scarring results from an unphysiological healing response to surgical injury. Fibroblasts in the Tenon capsule are target cells in the initiation of wound healing and scar formation after trabeculectomy.11, 12, 13 Astonishingly, long‐term use of classical antiglaucoma medication increases the risk of filtering failure caused by scar formation in patients who have undergone filtering surgery.11, 14
In addition to fibroblasts, another factor that plays a crucial role in scar formation is the excessive secretion of transforming growth factor (TGF)–β after glaucoma surgery.15, 16, 17 TGF‐β causes activation and proliferation of resident fibroblasts, as well as their migration into damaged tissue11, 18, 19; this causes a change to the myofibroblast phenotype.18, 20 Myofibroblasts, classically identified by expression of α‐smooth muscle actin (SMA) and vimentin,21, 22, 23, 24, 25 are characterized by actin stress formation, increased cell contractility, and uncontrolled production and degradation of the extracellular matrix (ECM), which leads to scarring or fibrosis.19, 26, 27, 28 In addition, mitogen‐activated protein kinase (MAPK), associated with the development of renal fibrosis,29 pulmonary fibrosis,30 and latanoprost‐induced human Tenon fibroblast (HTF) contractility,14 is activated by TGF‐β.
Rho/ROCK signal transduction is a key mediator of the actin cytoskeleton, as well as cell contractility, proliferation, shape, and motility.31, 32, 33, 34 Recent studies have indicated that blockage of the ROCK pathway with a ROCK inhibitor effectively reduces IOP by directly targeting trabecular meshwork (TM) cells (retraction and rounding of cell bodies,26, 35 disruption of actin bundles and focal adhesion,35 and reduction of TM cell contraction31) and Schlemm canal endothelial (SCE) cells (disruption of tight junctions26 and an increased number of giant vacuoles in SCEs36, 37), and by creating a large empty space in juxtacanalicular tissue.37, 38 Y‐27632 is the first known specific inhibitor of ROCK/Rho family protein kinases5; its use in rabbits resulted in a significant reduction of IOP in a dose‐dependent manner.35 In addition to their IOP‐lowering effect, ROCK inhibitors can prevent optic nerve head damage through increased optic nerve head blood flow39, 40 and the protection of neurons against stress.41, 42 Previous research showed that ROCK inhibitors may protect against postoperative scarring by changing collagen contraction.27, 43 However, the mechanisms of these changes and efficacy of ROCK inhibitors, compared with other antiglaucoma drugs, are not yet clear. Therefore, in this study, we investigated the effect of the ROCK inhibitor, Y‐27632, on scar formation after glaucoma filtration surgery through direct comparison with other antiglaucoma drugs.
2. MATERIALS AND METHODS
2.1. Reagent and antibodies
The ROCK inhibitor Y‐27632 was purchased from EMD Millipore Corp (Billerica, Massachusetts). Latanoprost was purchased from Cayman Chemical (Ann Arbor, Michigan), and timolol maleate was obtained from LKT Laboratories, Inc (St. Paul, Minnesota). TGF‐β1 was purchased from R&D Systems, Inc (Minneapolis, Minnesota). All drugs and cytokine were used in concentration 10μM.
Minimum Essential Medium (MEM), foetal bovine serum, 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), trypsin‐EDTA, and penicillin (100 U/mL)/streptomycin (100 μg/mL) were supplied by Gibco‐Invitrogen (Carlsbad, California). Phosphate‐buffered saline (PBS), radioimmunoprecipitation assay buffer, and phosphatase inhibitor solution were from Wako Pure Chemical Industries, Ltd (Osaka, Japan). Type I‐A collagen, 5X Dulbecco's Modified Eagle Medium (DMEM), and reconstitution buffer were from Nitta Gelatin, Inc (Osaka, Japan). An apoptotic/necrotic cell detection kit was purchased from PromoCell GmbH (Heidelberg, Germany).
The following rabbit monoclonal antibodies were used: anti–α‐SMA (ab124964), antivimentin (D21H3; for western blotting only), anti–phospho‐p44/42 MAPK (extracellular signal–regulated kinase [ERK] 1/2) (4370), anti‐ERK 1/2 (4695), and anti–phospho‐p38 MAPK (4511) antibodies were obtained from Cell Signaling Technology (Danvers, Massachusetts); as a loading control, anti–α‐tubulin (T9026) antibody was supplied by Sigma‐Aldrich (St. Louis, Missouri).
A rabbit polyclonal antibody to p38 MAPK (9212) was obtained from Cell Signaling Technology; a guinea pig polyclonal antibody to vimentin (GP53; for immunofluorescence staining only) was from Progen Biotechnik GmbH (Heidelberg, Germany).
The following mouse monoclonal antibodies were used: anti–p‐c‐Jun N‐terminal kinase (JNK) (sc‐6254) and anti‐JNK (sc‐7345) were from Santa Cruz Biotechnology (Dallas, Texas). The secondary antibody/horseradish peroxidase conjugate for western blotting was supplied by Promega (Madison, Wisconsin). Alexa Fluor 488–conjugated secondary antibodies and 4′,6‐diamidino‐2‐phenylindole (DAPI) were purchased from Invitrogen (Molecular Probes, Eugene, Oregon).
2.2. Isolation and culture of HTFs
This study was conducted in accordance with the principles of the Declaration of Helsinki. HTFs were obtained from patients who met the following criteria: They were undergoing strabismus surgery, had provided written informed consent, had no previous topical ocular medications, had no ocular disease except strabismus, and had no prior history of ocular surgery/trauma. The Tenon tissue from each patient was excised and digested as previously described.14, 44 Cells from patients were then cultured in MEM supplemented with 10% foetal bovine serum and penicillin (100 U/mL)/streptomycin (100 μg/mL) and maintained in the exponential growth phase of culture. When cells reached approximately 80% confluence, they were passaged by using the trypsin‐EDTA method. Cells from passages 3 to 7 were used for this study. Cell numbers were determined by using the indicated methods.
2.3. Collagen gel contraction assay (three‐dimensional cell culture)
Collagen gel contraction was used for evaluating HTF contraction activity induced by antiglaucoma drugs. The assay was conducted as previously described14, 31, 43, 45 with some modifications. HTF cultures were washed twice with PBS, detached with trypsin‐EDTA, and counted by using a hematocytometer. HTFs (2 × 106/mL) were centrifuged at 12.000 revolutions per minute for 10 minutes and resuspended in serum‐free MEM. Type I‐A collagen, 5X DMEM, reconstitution buffer, and HTFs were mixed in an ice bath at a volume ratio of 7:2:1:1. Immediately, 0.5 mL of the mixture was transferred to each well of a 24‐well plate (final cell density: 1 × 106 cells/well) and incubated at 37°C under 5% CO2 for 30 minutes to polymerize the gel. After gel polymerization, various conditioned media (0.5 mL) with 10μM final concentrations of drugs and TGF‐β were added on the top of the gel; incubation was continued at 37°C. After incubation for 1 hour, gels were dissociated from the wells by using microspatulas. Gels were incubated for 24 hours before measurement with a ruler and then photographed before stimulation with other drugs for 24 hours. Gels were then prepared as samples for western blotting after additional measurements and photographs were obtained. For each figure, experiments were performed independently at least three times. Estimated contraction is expressed as diameter changes before and after treatment.
2.4. Immunofluorescence assay
HTFs were plated on coverslips in 24‐well plates (5 × 104 cells/well) and incubated for 24 hours. The medium was then exchanged with starvation medium for 1 day, followed by overnight stimulation with Y‐27632, latanoprost, timolol, TGF‐β, or a combination of these drugs with Y‐27632. Subsequently, cells were washed three times with PBS, fixed with 100% methanol for 30 minutes at 20°C, washed again with PBS three times, and blocked with 1% bovine serum albumin (Sigma‐Aldrich) in PBS. Cells were then incubated with primary antibodies to α‐SMA (1:1000) and vimentin (1:200) in blocking buffer overnight at 4°C, washed with PBS, and incubated with secondary antibodies conjugated to Alexa Fluor 488 for 30 minutes in blocking solution. DAPI was used to stain nuclei. Signals of antigen detection were observed by using a laser‐scanning confocal microscope (Zeiss Axio Observer D1 Inverted microscope, Carl Zeiss GmbH, Jena, Germany) with AxioVision software (version 4.8; Carl Zeiss GmbH).
2.5. Detection of apoptotic cells
Apoptotic cells were detected by an apoptotic/necrotic cell detection kit, in accordance with the manufacturer's protocol. Briefly, cells treated as described in the immunofluorescence procedure (above) were washed twice with 1X binding buffer, incubated with staining solution (5‐μL FITC‐Annexin V and 5‐μL ethidium homodimer III in 100‐μL 1X binding buffer) for 15 minutes at room temperature in the dark. After staining, cells were washed twice and covered with 50‐μL 1X binding buffer and then observed with a laser‐scanning confocal microscope (Zeiss Axio Observer).
2.6. Western blotting
Western blotting was performed as previously described,14, 45 with minor modifications. We used gels from collagen gel assays that had been treated with various drugs. Each gel was lysed with 200‐μL radioimmunoprecipitation assay buffer (50mM Tris‐HCl pH 8.0, 150mM NaCl, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% Nonidet P‐40 substitute). Samples were then sonicated and centrifuged at 12.000 revolutions per minute for 10 minutes. Supernatants were collected for use as western blotting samples. Equal amounts of proteins were separated by electrophoresis. Separated proteins were transferred to nitrocellulose membranes, blocked with 5% skim milk in Tris‐buffered saline with Tween for 1 hour, and probed with primary antibodies. Antibodies targeting α‐SMA, vimentin, ERK, p38, JNK, p‐ERK, p‐p38, and p‐JNK were diluted in accordance with manufacturer guidelines. Tubulin was used for normalization. Horseradish peroxidase–conjugated secondary antibodies were used in the dilution ranges suggested by the manufacturer. Proteins were visualized by using ECL Western Blotting Detection Reagent (GE Healthcare) and developed with X‐ray film (Hyperfilm, GE Healthcare).
2.7. Statistical analysis
All sets of data are expressed as mean ± SD. Statistical analyses were performed by using one‐way analysis of variance, followed by the Dunnett test (two‐sided) for comparing multiple groups and the Student unpaired t test for comparisons between two groups. The Levene test for equality of variances was performed prior to multiple‐comparisons tests to ensure that variances among groups were homogenous. When variances differed among the groups, logarithmic, root, or reciprocal transformations were applied. SPSS statistical analysis software (SPSS Inc, Version 22.0, Chicago, Illinois) was performed to determine the statistical significance of differences between mean values. P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Effect of Y‐27632, timolol, latanoprost, and TGF‐β on collagen gel contraction
Collagen gel contraction assays were used to compare the HTF‐mediated contractility effects of several antiglaucoma drugs and TGF‐β on collagen gel. Cells were cultured in a three‐dimensional collagen gel, in the absence or presence of antiglaucoma drugs and TGF‐β.
Our results showed increased contraction of cells stimulated with timolol, latanoprost, or TGF‐β, relative to control; cells stimulated with Y‐27632 showed reduced contraction. Differences were statistically significant between control and each of Y‐27632–, latanoprost‐, and TGF‐β–stimulated groups; no significant difference was observed between control and the timolol‐stimulated group. Y‐27632 significantly inhibited gel contraction, in contrast to timolol, latanoprost, or TGF‐ β (Figure 1A and 1B).
Figure 1.
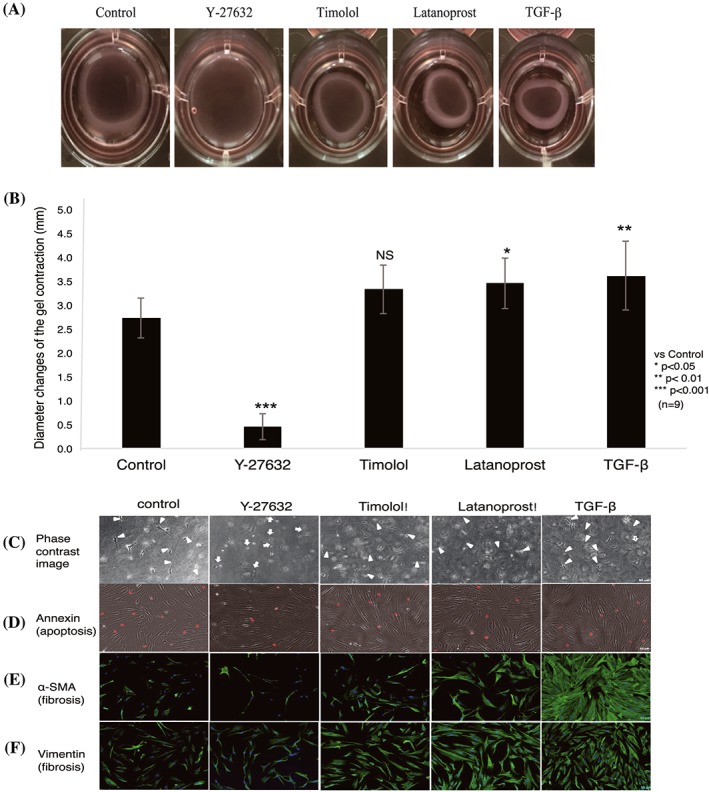
Effects of Y‐27632 and other antiglaucoma drugs on collagen gel contraction. A, Representative collagen gel photographs are shown after 24 h of treatment with 10μM Y‐27632, timolol, latanoprost, or TGF‐β. B, Gel contraction (diameter) induced by Y‐27632–treated cells and other drugs was compared with control. Y‐27632 significantly reduced collagen gel contraction, compared with control, while other groups exhibited contraction. Data shown are mean ± SD of three independent experiments in triplicate; data were analysed by one‐way ANOVA, followed by the Dunnett post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001 versus control (NS: nonsignificant). C, Effects of Y‐27632 on human Tenon fibroblast cell morphology. Representative phase contrast images are shown. Y‐27632 induced rounding and retraction of cells (white arrow), while other treatments resulted in spindle‐shaped/elongated fibroblasts (white arrowhead). D, Effect of Y‐27632 on apoptotic cells. Representative images show that Y‐27632 had no effect on apoptotic cells, compared with control. E, F, Effect of Y‐27632 on α‐SMA and vimentin expression. Timolol‐, latanoprost‐, and TGF‐β–treated cells showed increased expression of α‐SMA and vimentin, compared with control. In contrast, Y‐27632 reduced the expression of both proteins. Composite images (E, F) show α‐SMA and vimentin (green) and DAPI‐stained nuclei (blue). Scale bar: 50 μm, n = 3. ANOVA, analysis of variance; DAPI, 4′,6‐diamidino‐2‐phenylindole; TGF‐β, transforming growth factor‐β; α‐SMA, α‐smooth muscle actin
3.2. Effects of Y‐27632 and other antiglaucoma drugs on α‐SMA and vimentin expression in HTFs
To determine whether Y‐27632–induced inhibition of collagen gel contraction correlated with morphological changes and proliferation of HTFs, collagen gels (three‐dimensional cell culture) that were treated with various drugs were observed by phase contrast microscopy (Figure 1C). In contrast to HTFs treated with latanoprost, timolol, or TGF‐β, which preserved their native morphologies, cells treated with Y‐27632 exhibited morphological changes, including a rounded shape (Figure 1C, white arrows). Similarly, fibroblast proliferation was reduced in Y‐27632–treated cells, compared with control; in contrast, latanoprost‐, timolol‐, and TGF‐β–treated cells showed fibroblast proliferation, as indicated by an increase in the number of elongated fibroblasts.
To confirm that the inhibition of collagen gel contraction after Y‐27632 treatment was not correlated with apoptosis, we performed cell apoptosis assays (Figure 1D). HTFs treated with Y‐27632 showed no increase in the number of apoptotic cells, compared with control. Thus, inhibition of collagen gel contraction by cells treated with Y‐27632 was indeed due to fibroblast contraction, rather than a change in the number of apoptotic cells.
We next examined the effects of antiglaucoma drugs on the expression of α‐SMA and vimentin, associated with fibrosis (Figure 1E and 1F), in HTFs cultured in collagen gels. Immunofluorescence analysis showed increased expression of α‐SMA and vimentin in cells treated with latanoprost, timolol, or TGF‐β, which served to induce fibrosis. In contrast, α‐SMA and vimentin were both inhibited in Y‐27632–treated cells.
3.3. Dose dependence of Y‐27632 inhibition of gel contraction
To determine whether Y‐27632 inhibited gel contraction in a dose‐dependent manner, and to ascertain the optimal concentration of Y‐27632 in vitro, HTFs were treated with various concentrations of Y‐27632 (0μM through 100μM). Y‐27632 significantly suppressed the contraction of collagen gel in a dose‐dependent manner (Figure 2). The inhibitory effect was statistically significant at 5μM; the maximum effect was observed at 100μM. Latanoprost, timolol, and TGF‐β also promoted contraction in a dose‐dependent manner (Figure 2), suggesting that collagen gel contraction was strongly affected by latanoprost, timolol, and TGF‐β.
Figure 2.
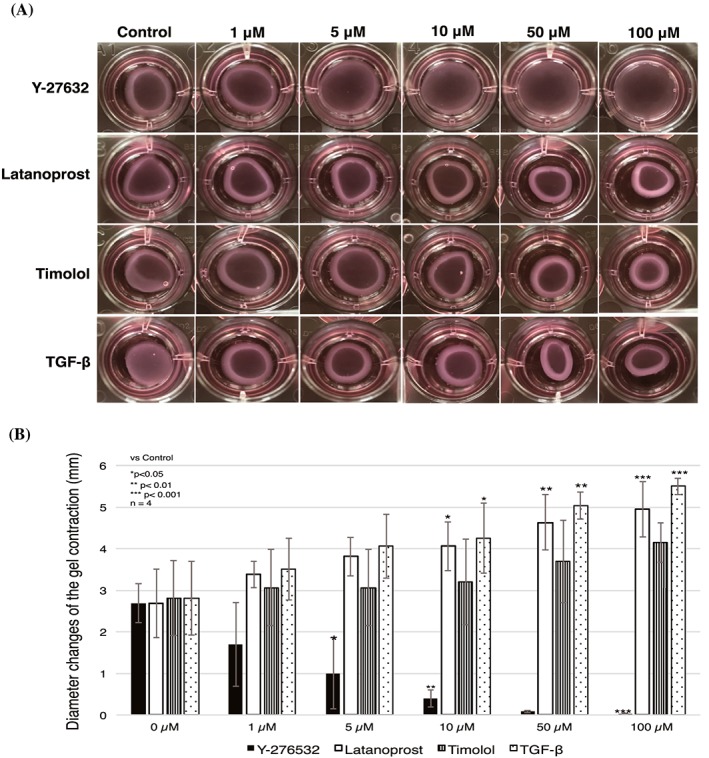
Y‐27632 induced significant dose‐dependent inhibition of human Tenon fibroblast cell‐mediated collagen gel contraction. The dose dependence of collagen gel contraction was tested with various concentrations of antiglaucoma drugs (latanoprost and timolol maleate), Y‐27632, and TGF‐β, from 0μM through 100μM, for 24 h. A, Images show representative pictures of the collagen gel. B, Contraction was significantly reduced by a minimum treatment of 5μM Y‐27632; it reached a maximum at 100μM Y‐27632. Data represent the mean ± SD (n = 4) and were analysed by one‐way ANOVA, followed by the Dunnett post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001. ANOVA, analysis of variance; TGF‐β, transforming growth factor‐β
3.4. Effect of Y‐27632 in combination with latanoprost, timolol, and TGF‐β
Next, we studied the ability of Y‐27632 to inhibit collagen gel contraction combined with timolol or latanoprost. Collagen gel diameters decreased after combined treatment with latanoprost or timolol, compared with negative control; slightly similar diameters were observed after combined treatment with TGF‐β. These results indicated that latanoprost and timolol induced contraction, while combined treatment with Y‐27632 inhibited contractility (Figure 3A).
Figure 3.
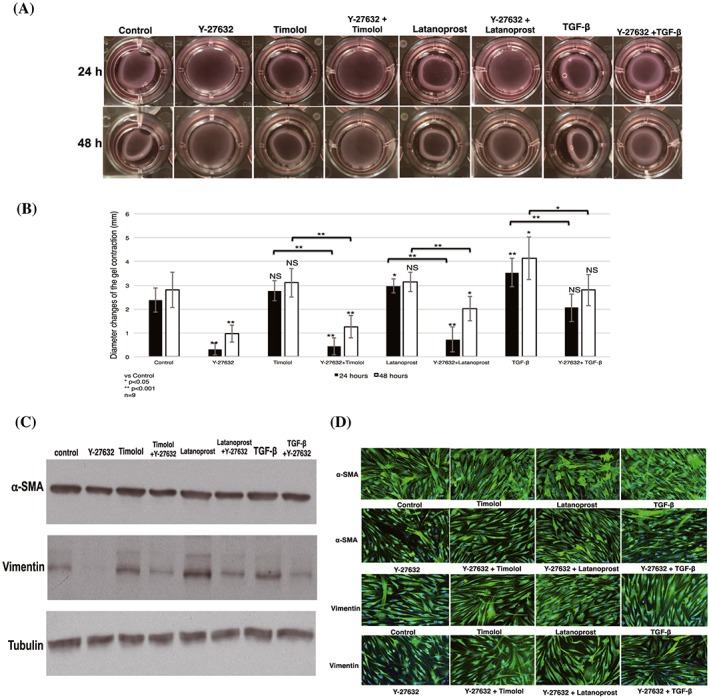
Contractility inhibitory effect of Y‐27632 on collagen gel in combination with other antiglaucoma drugs. A, Representative photographs show changes in collagen gel contraction after administration of Y‐27632 with a combination of timolol, latanoprost, and TGF‐β, compared with single treatment with timolol, latanoprost, or TGF‐β for 24 and 48 h. B, The findings of contraction analysis were analysed; results shown represent the mean ± SD (n = 9). Significant differences were determined by one‐way ANOVA, followed by the Dunnett post hoc test versus control; differences between two groups were determined with the Student unpaired t test. *P < 0.05, **P < 0.001 (NS: nonsignificant). C, D, Expression of α‐SMA and vimentin by cells stimulated with combination treatment plus Y‐27632. Total protein was extracted from collagen gels (shown in Figure 3A). Blots were analysed by western blotting (C) with anti–α‐SMA and antivimentin antibodies and verified by immunofluorescence analysis (D). Blots were probed with antitubulin antibody to confirm equal loading and transfer. Both western blotting and immunofluorescence analyses showed increased α‐SMA and vimentin expression after a single treatment with latanoprost, timolol, and TGF‐β; addition of Y‐27632 prevented expression of each protein. ANOVA, analysis of variance; TGF‐β, transforming growth factor‐β; α‐SMA, α‐smooth muscle actin
Statistical analysis showed significant reduction of contraction in cells treated with Y‐27632 in a combined treatment, compared with a single drug treatment. Combined treatment with Y‐27632 resulted in significant reductions in contraction, compared with single treatments, at both 24 and 48 hours after stimulation. A single administration of Y‐27632 yielded the maximum and most consistent inhibitory effect, compared with control (Figure 3B).
To confirm these results, we tested the effect of Y‐27632, in combination with latanoprost, timolol, and TGF‐β, on HTFs by fibrosis marker protein analysis. Western blotting showed that Y‐27632 significantly decreased α‐SMA and vimentin expression, markers of fibrosis, by HTFs (Figure 3C). Consistent with western blotting results, immunofluorescence analysis confirmed that addition of Y‐27632, in combination with latanoprost, timolol, and TGF‐β, significantly downregulated the expression of α‐SMA and vimentin (Figure 3D).
3.5. Effect of Y‐27632 and other antiglaucoma drugs on TGF‐β–induced collagen gel contraction
We then examined whether Y‐27632 could block contractility activity of TGF‐β secreted in the wound healing process. We mimicked the mechanism by initially inducing TGF‐β to HTFs for 24 hours, followed by addition of Y‐27632 and other antiglaucoma drugs. Additional treatment with Y‐27632 significantly blocked contraction induced by TGF‐β (Figure 4A). A similar pattern of results was obtained by immunoblotting. Increased α‐SMA and vimentin expression was observed in the presence of TGF‐β. In contrast, cells treated with Y‐27632 after TGF‐β stimulation for 24 hours showed reduced α‐SMA and vimentin expression, while addition of latanoprost and timolol enhanced protein expression (Figure 4B).
Figure 4.
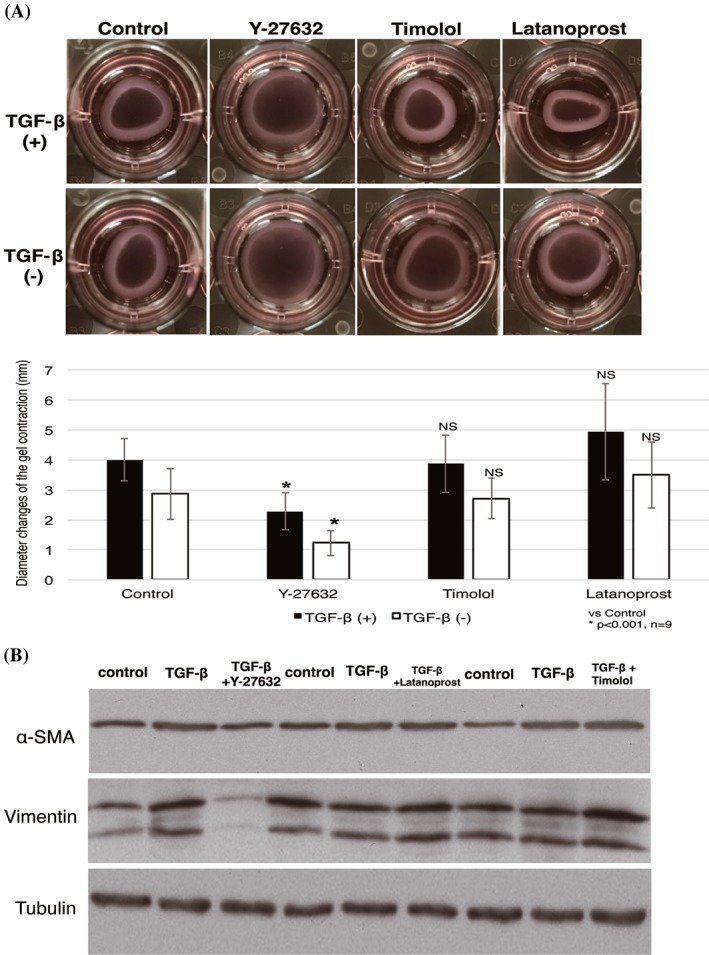
Effect of Y‐27632 on TGF‐β–induced fibrosis. A, HTFs were cultured on collagen gels before treatment with Y‐27632, timolol, and latanoprost for 24 h; HTFs were then incubated with TGF‐β (top) and without TGF‐β (bottom) for 1 day to mimic postoperative excretion of TGF‐β and to assess the ability of Y‐27632 to suppress the TGF‐β effect. The extent of contraction was then measured. Data shown are the mean ± SD; *P < 0.001 by one‐way ANOVA, followed by the Dunnett post hoc test. B, Lysates of collagen gels were prepared for western blotting with antibodies to α‐SMA, vimentin, and tubulin (loading control). Data are representative of three independent experiments performed in triplicate (NS: nonsignificant). ANOVA, analysis of variance; HTF, human Tenon fibroblast; TGF‐β, transforming growth factor‐β; α‐SMA, α‐smooth muscle actin
3.6. Effect of Y‐27632 on latanoprost‐ and timolol‐induced collagen gel contraction
The previous experiment indicated the ability of latanoprost and timolol to increase contraction. To investigate the ability of Y‐27632 for blocking contraction induced by the classical antiglaucoma drugs, latanoprost and timolol, HTFs were cultured in the presence of latanoprost, timolol, and a combination of latanoprost with timolol for 24 hours; they were subsequently treated with and without Y‐27632 (Figure 5A).
Figure 5.
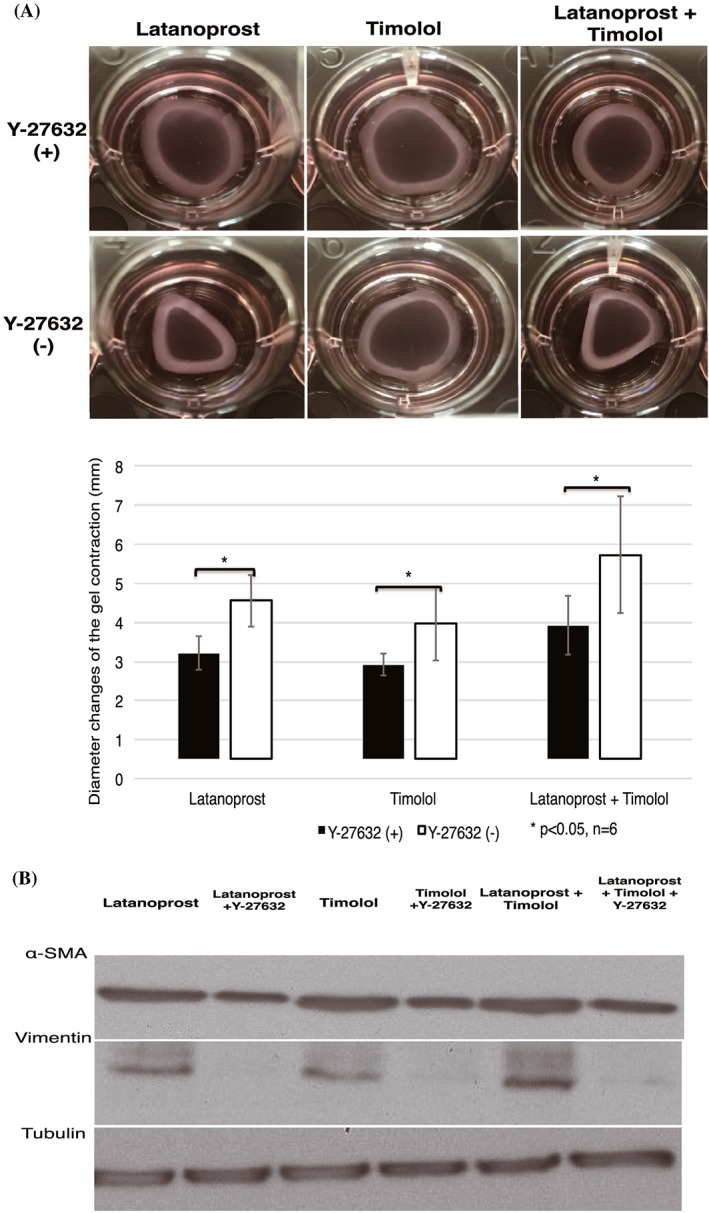
ROCK inhibitor decreased latanoprost‐, timolol‐, and combination latanoprost/timolol–induced collagen gel contraction and expression of α‐SMA and vimentin. A, HTFs were serum‐starved overnight, stimulated with antiglaucoma drugs for 24 h, and then treated with and without Y‐27632 afterwards. Diameter changes of gel contraction were observed 24 h after Y‐27632 stimulation. B, Gels were then lysed and analysed by western blotting. Blots were reprobed for tubulin as a loading control. The statistical significance of differences between groups treated and untreated with Y‐27632 was determined by the Student unpaired t test (n = 6). Differences were considered statistically significant when *P < 0.05 (NS: nonsignificant). HTF, human Tenon fibroblast; ROCK, Rho‐associated protein kinase; α‐SMA, α‐smooth muscle actin
Contractions were observed in HTFs that underwent single stimulations with latanoprost, timolol, and a combination of latanoprost with timolol. When HTFs were treated with Y‐27632, contraction was inhibited, compared with that without Y‐27632. Y‐27632 significantly suppressed contraction in latanoprost, timolol, and combination latanoprost/timolol groups. Consistent with collagen gel results, upregulation of α‐SMA and vimentin expression was observed in cells treated with latanoprost, timolol, and combination latanoprost/timolol, whereas addition of Y‐27632 downregulated expression of these proteins (Figure 5B).
3.7. Effect of Y‐27632 on TGF‐β–induced MAPK phosphorylation in HTF cells
Given that MAPKs have been implicated in the regulation of TGF‐β–induced epithelial‐mesenchymal transition (EMT) (fibrosis), we examined the effect of Y‐27632 on phosphorylation of MAPK to determine whether Y‐27632 might reduce activation of MAPK on TGF‐β–induced collagen contraction, which might mediate its ability to block collagen gel contraction. HTFs were cultured in a serum‐free medium for 24 hours before stimulation with TGF‐β, without and with Y‐27632 (Figure 6). Cells were lysed and analysed by immunoblotting. TGF‐β induced activation of ERK 1/2, p38, and JNK, as indicated by phosphorylation of MAPK compared with control, which was blocked by Y‐27632. The ability of Y‐27632 to block activation of MAPK began at 1 hour, peaked at 6 hours, and persisted until 24 hours. These data suggest that Y‐27632 blocked the contractility effect of TGF‐β by blocking the MAPK.
Figure 6.
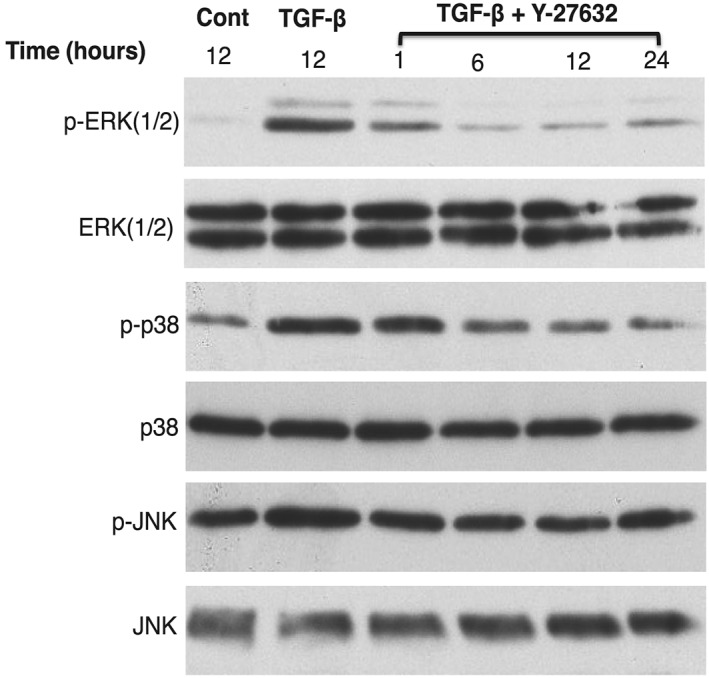
Inhibition of MAPK on TGF‐β–induced MAPK signalling by Y‐27632. Serum‐starved HTFs were untreated (negative control), treated with TGF‐β for 12 h (positive control), and treated with TGF‐β with Y‐27632 for 1 through 24 h (sample test). Cells were lysed and then analysed by western blotting. Treatment with Y‐27632 resulted in inhibition of phosphorylated (p‐) ERK 1/2, p38, and JNK, compared with the positive control. Data are representative of three independent experiments. ERK, extracellular signal–regulated kinase; HTF, human Tenon fibroblast; JNK, c‐Jun N‐terminal Kinase; MAPK, mitogen‐activated protein kinase; TGF‐β, transforming growth factor‐β
4. DISCUSSION
Medical therapy is frequently the initial method used for reduction of IOP associated with glaucoma. Prostaglandins or β‐blockers are typically selected as the initial agent among the various classes of antiglaucoma drugs.2, 5, 7 Since 1978, β‐blockers, such as timolol, have become the drug of choice. In 1996, prostaglandins replaced β‐blockers as the most commonly used agents for initial therapy.46 When IOP is insufficiently reduced by medical therapy, surgical intervention becomes an option.2, 7 However, recently, several studies have indicated that some antiglaucoma drugs may be risk factors for postoperative failure. Latanoprost (a prostaglandin) was shown to mediate contraction of HTFs,14 increase subconjunctival fibroblast proliferation with the presence of myofibroblasts,47 and provoke an inflammatory reaction.48 Timolol also caused proliferation of HTFs11 and conjunctival fibroblasts,49 as well as recruitment of inflammatory cells,50 both of which correlate with wound healing.
In recent years, ROCK inhibitors have been introduced as a potential antiglaucoma therapy. Several studies have investigated whether ROCK inhibitors can control IOP,31, 35, 38 in addition to their neuroprotective effects.40, 41, 42 The present study showed the effect of a ROCK inhibitor on scar formation. Our results revealed that Y‐27632 significantly suppresses the HTF‐mediated contraction of collagen gel, expression of α‐SMA and vimentin (markers of fibrosis), and proliferation of fibroblasts without the induction of apoptosis; in contrast, latanoprost, timolol, and TGF‐β show the reverse effects (Figure 1). Consistent with our results, a previous study with another ROCK inhibitor, ripasudil, also showed attenuated activation of human conjunctival fibroblasts and inhibition of TGF‐β2–induced ECM production with a barely detectable cytotoxic effect.43 Scar formation is the result of fibroblasts from EMT, or resident fibroblasts, migrating to and proliferating within damaged tissue, which leads to excessive synthesis of ECM in connective tissue and contributes to scar formation.24, 25, 28 Fibroblasts are associated with many diseases, either through a direct contribution to disease aetiology or through accompanying damage to other cell types.51
Rho GTPases regulate actin dynamics and control actin rearrangement, which is known to regulate cell shape through interactions with integrin.24, 27 Our results showed blockage of the Rho GTPase pathway with the ROCK inhibitor, Y‐27632, which led to a rounded cell shape in HTFs. This morphologic event was similar to a previous report regarding Y‐27632, where TM cells showed retraction and rounding of cell bodies, as well as impairment of focal adhesion and actin bundles35; this implies that blockage of this Rho pathway with Y‐27632 induces relaxation of the cell, rather than contraction.
Postoperative, TGF‐β is induced to promote wound healing, recruit fibroblasts, and increase fibroblast proliferation.10, 11 Fibroblasts subsequently differentiate into the contractile phenotype, myofibroblasts, during TGF‐β–induced scar formation.20, 22 To mimic TGF‐β secretion after glaucoma surgery and determine the ability of Y‐27632 to block the TGF‐β effect, we stimulated HTFs prior to Y‐27632 treatment. The results showed that Y‐27632 significantly blocked TGF‐β–induced collagen gel contraction, as well as the expression of myofibroblast markers (Figure 5). A previous study showed that Y‐27632 treatment of HTFs blocked the assembly and contraction of TGF‐β–induced stress fibres that are used in myofibroblast transdifferentiation.15 This finding implies that ROCK inhibitors may suppress scar formation after glaucoma filtration surgery.
TGF‐β also activates conventional MAPK (ERK, p38, and JNK) intracellular signal transduction pathways, which are important in the production of proinflammatory and profibrotic mediators, as well as ECM involved in scar formation.29, 30 Inhibition of ERK or p38 MAPK activity also repressed TGF‐β–induced EMT,30, 52, 53 indicating that TGF‐β may be a precursor of the MAPK signalling pathway. In this study, we examined whether Y‐27632 could inhibit the activation of MAPK signalling in TGF‐β–induced HTF cells. Our results showed that Y‐27632 significantly inhibits the activation of MAPK in the early response, suggesting that the MAPK signalling pathway is involved after glaucoma filtering surgery and that Y‐27632 can suppress this effect. However, further investigation of the role of the MAPK pathway in scar formation after glaucoma surgery is needed, along with a greater understanding of the ability of Y‐27632 to modulate this process.
In conclusion, our results showed that Y‐27632 may improve outcomes after glaucoma filtration surgery by the reduction of collagen gel contraction and proliferation of fibroblasts, suppression of fibroblast transdifferentiation into myofibroblasts, induction of morphological changes and relaxation of cells, and suppression of TGF‐β and MAPK pathways. These findings suggest that Y‐27632 may aid in the attenuation of fibrosis after glaucoma surgery, while other antiglaucoma drugs promote such fibrosis. Further exploration is necessary to determine the full effect of ROCK inhibitor usage in vivo.
ACKNOWLEDGEMENTS
The authors thank Chinami Hiraoka for excellent technical assistance and Indonesia Endowment Fund for Education (LPDP), Ministry of Finance, Republic of Indonesia, for providing PhD scholarship and financial support to the first author. We also thank Ryan Chastain‐Gross, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Ibrahim DG, Ko J‐A, Iwata W, Okumichi H, Kiuchi Y. An in vitro study of scarring formation mediated by human Tenon fibroblasts: Effect of Y‐27632, a Rho kinase inhibitor. Cell Biochem Funct. 2019;37:113–124. 10.1002/cbf.3382
This work was supported by a grant (no. 15K10868) from the Japan Society for the Promotion of Science (KAKENHI).
REFERENCES
- 1. Agarwal R, Gupta SK, Agarwal P, Saxena R, Agrawal SS. Current concepts in the pathophysiology of glaucoma. Indian J Ophthalmol. 2009;57(4):257‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311(18):1901‐1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foster PJ, Buhrmann R, Quigley HA, Johnson GJ. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol. 2002;86(2):238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cholkar K, Trinh HM, Pal D, Mitra AK. Discovery of novel inhibitors for the treatment of glaucoma. Expert Opin Drug Discovery. 2015;10(3):293‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gaton DD, Sagara T, Lindsey JD, Gabelt BT, Kaufman PL, Weinreb RN. Increased matrix metalloproteinases 1, 2, and 3 in the monkey uveoscleral outflow pathway after topical prostaglandin F(2 alpha)‐isopropyl ester treatment. Arch Ophthalmol (Chicago, Ill: 1960). 2001;119(8):1165‐1170. [DOI] [PubMed] [Google Scholar]
- 7. Prum BE Jr, Rosenberg LF, Gedde SJ, et al. Primary open‐angle glaucoma preferred practice pattern(®) guidelines. Ophthalmology. 2016;123(1):P41‐p111. [DOI] [PubMed] [Google Scholar]
- 8. Sharpe RA, Kammerdiener LL, Williams DB, Das SK, Nutaitis MJ. Efficacy of selective laser trabeculoplasty following incisional glaucoma surgery. Int J Ophthalmol. 2018;11(1):71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salim S. Current variations of glaucoma filtration surgery. Curr Opin Ophthalmol. 2012;23(2):89‐95. [DOI] [PubMed] [Google Scholar]
- 10. Lukowski ZL, Min J, Beattie AR, et al. Prevention of ocular scarring after glaucoma filtering surgery using the monoclonal antibody LT1009 (Sonepcizumab) in a rabbit model. J Glaucoma. 2013;22(2):145‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skuta GL, Parrish RK 2nd. Wound healing in glaucoma filtering surgery. Surv Ophthalmol. 1987;32(3):149‐170. [DOI] [PubMed] [Google Scholar]
- 12. Xi X, McMillan DH, Lehmann GM, et al. Ocular fibroblast diversity: implications for inflammation and ocular wound healing. Invest Ophthalmol Vis Sci. 2011;52(7):4859‐4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan Gaskin JC, Nguyen DQ, Soon Ang G, O'Connor J, Crowston JG. Wound healing modulation in Glaucoma filtration surgery‐conventional practices and new perspectives: the role of antifibrotic agents (part I). J Curr Glaucoma Pract. 2014;8(2):37‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Y, Ko JA, Yanai R, et al. Induction by latanoprost of collagen gel contraction mediated by human tenon fibroblasts: role of intracellular signaling molecules. Invest Ophthalmol Vis Sci. 2008;49(4):1429‐1436. [DOI] [PubMed] [Google Scholar]
- 15. Meyer‐ter‐Vehn T, Sieprath S, Katzenberger B, Gebhardt S, Grehn F, Schlunck G. Contractility as a prerequisite for TGF‐beta‐induced myofibroblast transdifferentiation in human Tenon fibroblasts. Invest Ophthalmol Vis Sci. 2006;47(11):4895‐4904. [DOI] [PubMed] [Google Scholar]
- 16. Sime PJ, O'Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol (Orlando, Fla). 2001;99(3):308‐319. [DOI] [PubMed] [Google Scholar]
- 17. Cordeiro MF, Chang L, Lim KS, et al. Modulating conjunctival wound healing. Eye (Lond). 2000;14(Pt 3B):536‐547. [DOI] [PubMed] [Google Scholar]
- 18. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton‐Piallat M‐L, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807‐1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526‐537. [DOI] [PubMed] [Google Scholar]
- 20. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor‐beta 1 induces alpha‐smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122(1):103‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masur SK, Dewal HS, Dinh TT, Erenburg I, Petridou S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc Natl Acad Sci U S A. 1996;93(9):4219‐4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamanaka O, Kitano‐Izutani A, Tomoyose K, Reinach PS. Pathobiology of wound healing after glaucoma filtration surgery. BMC Ophthalmol. 2015;15(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeisberg M, Neilson EG. Biomarkers for epithelial‐mesenchymal transitions. J Clin Invest. 2009;119(6):1429‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest. 2009;119(6):1420‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaneko Y, Ohta M, Inoue T, et al. Effects of K‐115 (Ripasudil), a novel ROCK inhibitor, on trabecular meshwork and Schlemm's canal endothelial cells. Sci Rep. 2016;6(1):19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Honjo M, Tanihara H, Kameda T, Kawaji T, Yoshimura N, Araie M. Potential role of Rho‐associated protein kinase inhibitor Y‐27632 in glaucoma filtration surgery. Invest Ophthalmol Vis Sci. 2007;48(12):5549‐5557. [DOI] [PubMed] [Google Scholar]
- 28. Bagalad BS, Mohan Kumar KP, Puneeth HK. Myofibroblasts: master of disguise. J Oral Maxillofac Pathol: JOMFP. 2017;21(3):462‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stambe C, Atkins RC, Tesch GH, Masaki T, Schreiner GF, Nikolic‐Paterson DJ. The role of p38alpha mitogen‐activated protein kinase activation in renal fibrosis. J Am Soc Nephrol: JASN. 2004;15(2):370‐379. [DOI] [PubMed] [Google Scholar]
- 30. Matsuoka H, Arai T, Mori M, et al. A p38 MAPK inhibitor, FR‐167653, ameliorates murine bleomycin‐induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2002;283(1):L103‐L112. [DOI] [PubMed] [Google Scholar]
- 31. Koga T, Koga T, Awai M, Tsutsui J, Yue BY, Tanihara H. Rho‐associated protein kinase inhibitor, Y‐27632, induces alterations in adhesion, contraction and motility in cultured human trabecular meshwork cells. Exp Eye Res. 2006;82(3):362‐370. [DOI] [PubMed] [Google Scholar]
- 32. Etienne‐Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629‐635. [DOI] [PubMed] [Google Scholar]
- 33. Liao JK, Seto M, Noma K. Rho kinase (ROCK) inhibitors. J Cardiovasc Pharmacol. 2007;50(1):17‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J, Liu X, Zhong Y. Rho/Rho‐associated kinase pathway in glaucoma (Review). Int J Oncol. 2013;43(5):1357‐1367. [DOI] [PubMed] [Google Scholar]
- 35. Honjo M, Tanihara H, Inatani M, et al. Effects of Rho‐associated protein kinase inhibitor Y‐27632 on intraocular pressure and outflow facility. Invest Ophthalmol Vis Sci. 2001;42(1):137‐144. [PubMed] [Google Scholar]
- 36. Kameda T, Inoue T, Inatani M, et al. The effect of Rho‐associated protein kinase inhibitor on monkey Schlemm's canal endothelial cells. Invest Ophthalmol Vis Sci. 2012;53(6):3092‐3103. [DOI] [PubMed] [Google Scholar]
- 37. Rao PV, Deng PF, Kumar J, Epstein DL. Modulation of aqueous humor outflow facility by the Rho kinase‐specific inhibitor Y‐27632. Invest Ophthalmol Vis Sci. 2001;42(5):1029‐1037. [PubMed] [Google Scholar]
- 38. Inoue T, Tanihara H. Rho‐associated kinase inhibitors: a novel glaucoma therapy. Prog Retin Eye Res. 2013;37:1‐12. [DOI] [PubMed] [Google Scholar]
- 39. Sugiyama T, Shibata M, Kajiura S, et al. Effects of fasudil, a Rho‐associated protein kinase inhibitor, on optic nerve head blood flow in rabbits. Invest Ophthalmol Vis Sci. 2011;52(1):64‐69. [DOI] [PubMed] [Google Scholar]
- 40. Tokushige H, Waki M, Takayama Y, Tanihara H. Effects of Y‐39983, a selective Rho‐associated protein kinase inhibitor, on blood flow in optic nerve head in rabbits and axonal regeneration of retinal ganglion cells in rats. Curr Eye Res. 2011;36(10):964‐970. [DOI] [PubMed] [Google Scholar]
- 41. Kitaoka Y, Kitaoka Y, Kumai T, et al. Involvement of RhoA and possible neuroprotective effect of fasudil, a Rho kinase inhibitor, in NMDA‐induced neurotoxicity in the rat retina. Brain Res. 2004;1018(1):111‐118. [DOI] [PubMed] [Google Scholar]
- 42. Hirata A, Inatani M, Inomata Y, et al. Y‐27632, a Rho‐associated protein kinase inhibitor, attenuates neuronal cell death after transient retinal ischemia. Graefes Arch Clin Exp Ophthalmol = Albrecht von Graefes Archiv Fur Klinische Und Experimentelle Ophthalmologie. 2008;246(1):51‐59. [DOI] [PubMed] [Google Scholar]
- 43. Futakuchi A, Inoue T, Fujimoto T, Inoue‐Mochita M, Kawai M, Tanihara H. The effects of ripasudil (K‐115), a Rho kinase inhibitor, on activation of human conjunctival fibroblasts. Exp Eye Res. 2016;149:107‐115. [DOI] [PubMed] [Google Scholar]
- 44. Fujitsu Y, Fukuda K, Kumagai N, Nishida T. IL‐4‐induced cell proliferation and production of extracellular matrix proteins in human conjunctival fibroblasts. Exp Eye Res. 2003;76(1):107‐114. [DOI] [PubMed] [Google Scholar]
- 45. Ko JA, Sotani Y, Ibrahim DG, Kiuchi Y. Role of macrophage migration inhibitory factor (MIF) in the effects of oxidative stress on human retinal pigment epithelial cells. Cell Biochem Funct. 2017;35(7):426‐432. [DOI] [PubMed] [Google Scholar]
- 46. Daneshvar R, Amini N. Rho‐associated kinase inhibitors: potential future treatments for glaucoma. J Ophthalmic Vis Res. 2014;9(3):395‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lark KK, Pasha AS, Yan X, Edward DP. The effect of latanoprost and brimonidine on rabbit subconjunctival fibroblasts. J Glaucoma. 1999;8(1):72‐76. [PubMed] [Google Scholar]
- 48. Rodrigues Mde L, Felipe Crosta DP, Soares CP, et al. Immunohistochemical expression of HLA‐DR in the conjunctiva of patients under topical prostaglandin analogs treatment. J Glaucoma. 2009;18(3):197‐200. [DOI] [PubMed] [Google Scholar]
- 49. Young TL, Higginbotham EJ, Zou XL, Farber MD. Effects of topical glaucoma drugs on fistulized rabbit conjunctiva. Ophthalmology. 1990;97(11):1423‐1427. [DOI] [PubMed] [Google Scholar]
- 50. Sherwood MB, Grierson I, Millar L, Hitchings RA. Long‐term morphologic effects of antiglaucoma drugs on the conjunctiva and Tenon's capsule in glaucomatous patients. Ophthalmology. 1989;96(3):327‐335. [DOI] [PubMed] [Google Scholar]
- 51. Chang HY, Chi JT, Dudoit S, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99(20):12877‐12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meyer‐Ter‐Vehn T, Gebhardt S, Sebald W, et al. p38 inhibitors prevent TGF‐beta‐induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci. 2006;47(4):1500‐1509. [DOI] [PubMed] [Google Scholar]
- 53. Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD. MEK‐ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol. 2012;46(3):380‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]