

Abstract
H2S is a gasotransmitter with several physiological roles, but its reactivity and short half-life in biological media make it difficult to deliver in a controlled manner. For biological applications of the gas, hydrogels have the potential to deliver H2S with several advantages over other donor systems, including localized delivery, controlled release rates, biodegradation, and variable mechanical properties. In this study, we designed and evaluated peptide-based H2S-releasing hydrogels with controllable H2S delivery. The hydrogels were prepared from short, self-assembling aromatic peptide amphiphiles (APAs), functionalized on their N-terminus with S-aroylthiooximes (SATOs), which release H2S in response to a thiol trigger. The APAs were studied both in solution and in gel forms, with gelation initiated by addition of CaCl2. Various substituents were included on the SATO component of the APAs in order to evaluate their effect on self-assembled morphology and H2S release rate in both the solution and gel phases. Transmission electron microscopy (TEM) images confirmed that all H2S-releasing APAs self-assembled into nanofibers above a critical aggregation concentration (CAC) of ~0.5 mg/mL. Below the CAC, substituents on the SATO group affected H2S release rates predictably in line with electronic effects (Hammett σ values) according to a linear free energy relationship. Above the CAC, circular dichroism, infrared, and fluorescence spectroscopies demonstrated that substituents influenced the self-assembled structures by affecting hydrogen bonding (β-sheet formation) and π-π stacking. At these concentrations, electronic control over release rates diminished, both in solution and in the gel form. Rather, the release rate depended primarily on the degree of organization in the β-sheets and the amount of π-π stacking. This supramolecular control over release rate may enable the evaluation of H2S-releasing hydrogels with different release rates in biological applications.
Keywords: peptide amphiphiles, aromatic stacking, controlled drug delivery, beta-sheets
Graphical Abstract
INTRODUCTION
Gasotransmitters are endogenously produced gases that can freely permeate membranes and induce certain physiological or biochemical changes in organisms, tissues, or cells.1–3 Nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S) are the three compounds currently regarded as gasotransmitters, while others including nitroxyl (HNO), sulfur dioxide (SO2), and ammonia (NH3) may join this classification with more study.4 Of the three known gasotransmitters, H2S was the last to be discovered but has recently received much attention.5 H2S is produced endogenously by cystathionine β-synthase (CBS), cystathionine γ-lyse (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST) in mammalian tissues.6, 7 It plays important biological roles and regulates various physiological processes, most notably sGC/cGMP pathways and KATP channels.8, 9 H2S biology has typically been studied either by inhibition of CBS/CSE or by exogenous application of small molecules designed to release the gas, termed H2S donors. Physiological effects have been demonstrated using these methods, with studies establishing that H2S dilates blood vessels, promotes proliferation of vascular endothelial cells, inhibits inflammation, and decreases oxidative stress in tissues.5, 10, 11 Due to these physiological effects, H2S is recognized as a potential treatment for conditions including heart disease,12 ischemia,13 cancer,14 and wound healing,15 among others.
Despite substantial progress in recent years, the delivery of H2S remains a hurdle in tapping its therapeutic potential. Inhalation is the simplest way to deliver H2S, but storage and dosage control of this flammable, noxious gas are difficult, and target specificity is absent. To address the limitations and experimental difficulties associated with administering gaseous H2S, small molecule H2S donors have been developed that can deliver H2S in vitro and in vivo under physiological conditions.16, 17 These include GYY4137,18 N-benzoylthiobenzamides,19 S-aroylthiooximes (SATOs),20 perthiol-based H2S donors21 and others (Figure 1).22–26 Among the various H2S donors, SATOs are easy to synthesize from an S-aroylthiohydroxylamine (SATHA) and an aldehyde or ketone, and they release the gas in response to a thiol trigger. Moreover, the half-life of H2S release from SATOs can be controlled by tuning the electronics of the SATO structure.20 The simple synthesis and tunable reactivity of SATOs makes them well suited for derivatizing other compounds, enabling the construction of H2S-donating polymers and peptides.27
Figure 1.
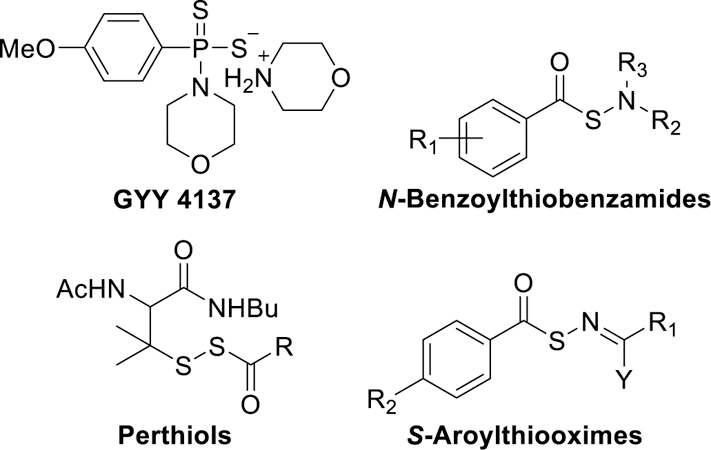
Examples of H2S donors.
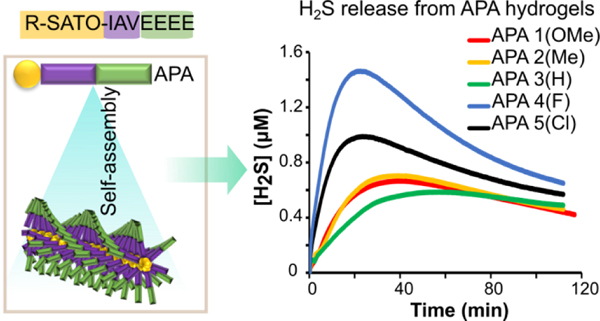
While the release of H2S by small molecules is in some cases tunable, most H2S donors lack target specificity because they are delivered systemically. A solution to this limitation is to deliver H2S locally using a polymer or hydrogel.28 While this technique has been used for both NO and CO, only a handful materials are available for localized H2S delivery.27, 29–32 An interesting class of materials that has been used for delivery of gases, small molecules, and proteins is self-assembling peptides. Intrinsically biodegradable, a wide variety of hydrogel platforms based on self-assembling peptides have been explored.33 Of the many platforms available, of particular interest to us are aromatic peptide amphiphiles (APAs). APAs contain an N-terminal aromatic group linked to a short peptide sequence. Aromatic stacking and β-sheet H-bonding interactions govern the self-assembly of APAs in water, with nanostructure morphologies including spheres, cylindrical micelles, sheets, ribbons, tapes, and nanofibers.34 When designed appropriately, APA nanostructures can entangle to form gels upon changes in ionic strength, pH, solvent, or addition of specific salts.
We recently reported on an APA that contained a SATO functionality as the aromatic unit.30 This APA self-assembled in water and underwent gelation upon addition of CaCl2, forming the first H2S releasing hydrogel. The APA hydrogel exhibited slow release of H2S for more than 5 h. This preliminary work demonstrated that the APA platform could be a potential strategy for sustained H2S delivery. However, no control over the release rate of the hydrogel was observed. Considering that the electronic structure of SATOs can be manipulated through the use of substituents on the aroyl ring to tune the kinetics of H2S release in small molecules,20 we reasoned that we might be able to exploit this element of the SATO group to control release from APAs in solution. We also hypothesized that substituents on the aroyl ring might affect self-assembly, enabling control over H2S release from APA hydrogels through controlling supramolecular packing and therefore access of the cysteine (Cys) trigger to the aromatic, H2S-releasing SATO unit. In this work, we report on substituted SATO-containing APAs with the peptide sequence Ile-Ala-Val-Glu-Glu-Glu-Glu (IAVE4), and we evaluate the effects of different substituents in the SATO component on self-assembly in aqueous solution and on H2S release rates.
EXPERIMENTAL SECTION
Materials.
Rink amide MBHA resin and Fmoc-protected amino acids were purchased from P3 Biosystems and used as received. All other reagents were purchased from commercial vendors and used as received without further purification unless noted.
Preparation of APAs.
S-Aroylthiohydroxylamines (SATHAs) were prepared according to literature procedures.20 4-Formylbenzoyl-IAVE4 peptide (FBA-IAVE4) was prepared by Fmoc-based solid-phase peptide synthesis. After cleavage, the FBA-IAVE4 peptide was purified by preparative HPLC.30 SATHAs (0.1 mmol) and FBA-IAVE4 (0.05 mmol) were dissolved in DMSO (400 µL), and a catalytic amount of trifluoroacetic acid (TFA, 15 µL) was added to the solution. Reaction progress was monitored by mass spectrometry using an Advion ExpressIon Compact Mass Spectrometer. Once complete, acetonitrile (ACN, 4 mL) and 0.1 M phosphate buffer (pH 7.4, 2 mL) were added to the reaction mixture, then the resulting solution was filtered through a 0.45 µm PTFE syringe filter. The solution was injected onto an Agilent Technologies 1260 Infinity preparative HPLC system with an Agilent PLRP-S column (particle size 100 Å, 25 mm x 150 mm), using a gradient of 2% to 90% ACN in milliQ water. Absorbance was monitored at 220 nm, and fractions were collected in test tubes as controlled by PrepLC software. The fractions were analyzed by mass spectrometry, and product-containing fractions were combined before lyophilization (LabConco).
Critical Aggregation Concentration (CAC) Measurements.
A 1 mg/mL Nile Red stock solution was prepared in acetone and then diluted in milliQ water to a final concentration of 0.01 mg/mL. The 0.01 mg/mL Nile Red solution was used in all dissolving and diluting operations to keep Nile Red concentration consistent in all samples. A series of peptide solutions (4, 3, 2, 1, 0.5, 0.25, 0.1, 0.01, 0.001, 0.0001 mg/mL peptide) were prepared from and diluted with 0.01 mg/mL Nile Red solution. Samples from the dilution series were transferred via micropipette into a 96-well plate and analyzed by fluorescence spectroscopy (λex = 550 nm and λem = 648 nm with slit widths varied from 10–20 nm depending on the sample series). Analysis of the data was carried out as previously reported.30
Hydrolysis of APAs.
APAs were dissolved in phosphate buffer (pH 7.4 10 mM) at a concentration of 45 µM. UV-vis spectra were taken in a 1 cm quartz cuvette at predetermined time points on a Cary 100 UV-vis spectrophotometer. The scanning rate was 200 nm/min from 400 to 200 nm. Phosphate buffer solution was used as the blank before each scan. The absorbance indicative of the SATO group was observed near 330 nm for each peptide. Kinetics analyses were carried out using a previously published method.35
TEM.
Peptide solutions (1 wt% in milliQ water) were cast onto carbon-coated copper TEM grids (300 mesh, Electron Microscopy Sciences) and allowed to stand for 30 min before removing excess solution through wicking with filter paper. Samples were stained with a 0.5% uranyl acetate aqueous solution for 2 min, then this solution was wicked away. Finally, each grid was washed with milliQ water once using the same wicking procedure and allowed to dry under air for at least 12 h. Images were taken on a Philips EM420 TEM with a slow scan CCD camera.
Methylene Blue Assay.
Methylene blue assays were used to determine the kinetics of H2S release in the presence of cysteine (Cys) using procedures similar to literature reports.36 In these experiments on APA solutions, 0.01 M phosphate buffer saline (1x PBS) was prepared by dissolving Na2HPO4 (1.420 g), KH2PO4 (0.245 g), NaCl (8.007 g), and KCl (0.201 g) in 800 mL of milliQ water, followed by adjusting to pH 7.4 with HCl or NaOH and adding additional water to reach 1 L. FeCl3 (30 mM in 1.2 M HCl) and N,N-dimethyl-p-phenylenediamine (20 mM in 7.2 M HCl) solutions were also prepared, as were solutions of Zn(OAc)2 (40 mM in H2O) and Cys (500 mM in H2O). To carry out methylene blue kinetics assays, SATO-peptide (400 µg) was dissolved in 3.792 mL of PBS solution, then 200 µL of Zn(OAc)2 was added. Cys solution (8 µL) was then injected to the peptide solution to trigger H2S release. A blank PBS solution was made by adding 100 µL of Zn(OAc)2 and 4 µL of Cys to 1.896 mL PBS solution. At room temperature (rt), reactions for methylene blue were run in quadruplicate with one blank. Aliquots were taken out at each timepoint. For each replicate, 100 µL of releasing solution/blank was removed from the reaction vial and added to a plastic 1.5 mL microcentrifuge tube. Each aliquot was then quickly diluted with 100 µL of N,N-dimethyl-p-phenylenediamine solution and 100 µL of FeCl3 solution. These aliquot solutions were stored overnight before transferring 280 µL of each into 96-well plates. The spectra of peptide solutions and background solutions were collected from 500 to 800 nm on a plate reader (Synergy 2 Multi-Mode Reader, BioTek Instruments). Kinetic analysis was done by subtracting the absorbance of the blank solution from each time point at 750 nm. The first-order half-life of H2S release was determined by plotting time vs. ln[1/(1-conversion)], with t1/2=ln(2)/slope.
H2S Release Profile of APA Hydrogels.
A special glass vial with a gel-holding insert well at the bottom was designed, and it was used for detecting H2S released from APA solutions (Figure S5) or hydrogels (Figure S8). APA peptide solution or gel (100 µL) was transferred into the inner well, then 10 µL Cys was added to the well to trigger H2S release. The holder was covered immediately using a Breathe-EASIER membrane (Diversified Biotech) and sealed with an O-ring. Next, 5 mL of 1x PBS was added to the vial to absorb H2S. An H2S-sensitive electrode probe was submerged into the PBS, and the evolution of H2S was monitored over time at rt.
Fluorescence Spectra.
Fluorescence spectroscopy was performed on an Agilent Cary Eclipse fluorescence spectrophotometer (Agilent Technologies) with a scanning speed of 120 nm/min, a 1 nm data pitch, an excitation slit width of 20 nm, an emission slit width of 10 nm, and λex = 290 nm. Measurements were taken in a 1 cm quartz cuvette.
IR Spectra.
IR spectra were recorded on a Nicolet 8700 FT-IR Spectrometer equipped with an attenuated total reflectance (ATR) sampling accessory (Thermo Fisher Scientific). Peptide hydrogels were made with D2O, and all spectra were recorded as an average of 64 scans from 1800 to 1200 cm−1. A background spectrum of a blank was collected and subtracted from the sample spectra.
Circular Dichroism (CD) Spectra.
CD spectra were obtained using a Jasco J-815 CD spectrometer (Jasco Inc.) at rt with a constant N2 flow set to 120 mL/min. The range of wavelengths employed was 250 to 190 nm (50 nm/min) with a response time of 8 s. Samples were prepared freshly before analysis at 0.25 mg/mL in water in a 1 mm quartz cuvette. Samples at 10 mg/mL were analyzed in a 0.2 mm quartz cuvette. Raw data was converted to mean residual ellipticity for comparison.
Statistics.
The H2S peaking times and peaking concentrations of APA solutions and hydrogels were collected from three trials under the same experimental conditions. Mean values are shown with error bars indicating standard deviations from three trials. All data were analyzed by a one-way analysis of variance (ANOVA) with the Tukey-Kramer HSD test, where n=3 and p < 0.05 denotes statistical significance. ANOVA analysis was performed using JMP® software (version 10.0.2, Copyright © 2012 SAS Institute Inc.).
RESULTS AND DISCUSSION
Results
Synthesis of APAs.
Substituted APAs (APAs 1–5) were successfully synthesized following the route in Scheme 1. Briefly, an aldehyde-containing peptide with the sequence FBA-IAVE4 (FBA = 4-formylbenzoic acid) was prepared by Fmoc-based solid-phase peptide synthesis, adding the aromatic aldehyde unit by coupling FBA to the peptide N-terminus on resin. After cleavage, the FBA-IAVE4 peptide was purified by preparative HPLC. Next, substituted SATHAs were added in a condensation reaction catalyzed by trifluoroacetic acid (TFA). Product peptides were isolated and purified by preparative HPLC. Purity was confirmed by analytical HPLC and mass spectrometry (Figures S1 and S2).
Scheme 1.
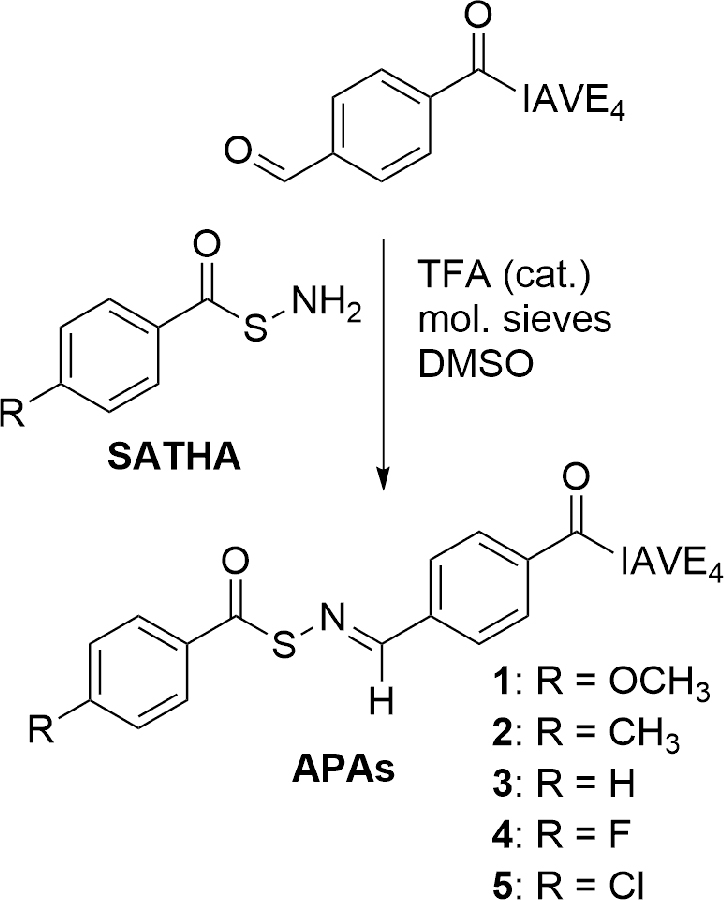
Synthesis of SATO-containing APAs 1–5a. aIAVE4 indicates peptide Ile-Ala-Vla-Glu-Glu-Glu-Glu.
Critical Aggregation Concentration (CAC) of APAs.
The CACs of APAs 1–5 were determined by the Nile Red assay, as we have previously used for self-assembled peptides.30 The assay revealed that the CAC values (Table S1) for all APAs hovered near 0.5 mg/mL. Based on these data, all subsequent experiments were conducted either well below (0.1 or 0.25 mg/mL) or above (10 mg/mL) the CAC to evaluate the characteristics of the APAs in their molecularly dissolved state or in self-assembled aggregates, respectively.
Hydrolysis of APAs.
With pure APAs in hand, we next considered their hydrolytic stability. Therapeutic or bioengineering applications of APAs will require aqueous solution under physiological conditions, and a hydrolytically unstable peptide may limit its practical applications. Hydrolysis experiments were conducted in phosphate buffer at pH 7.4 at concentrations below the CAC, as we have done previously on other SATO peptides.35 We found that solutions of APAs 1–5 had hydrolysis half-lives ranging from 9 to 14 h (Table S1). Thus, the APAs were relatively stable at physiological pH. A linear free energy relationship (rate of hydrolysis vs. Hammett σ values) revealed a correlation between substituents and their electronic effects, with ρ=0.3 (Figure S3). Electron-donating groups improved the hydrolytic stability of the APAs, consistent with a hydrolysis mechanism involving an electrophilic C=N carbon. The low value for ρ is consistent with the relatively long distance from the aroyl ring substituent to the reactive C=N group.
Transmission Electron Microscopy (TEM) Imaging.
APAs 1–5 were imaged by conventional TEM after depositing from 10 mg/mL solutions and staining with uranyl acetate (Figures 2A–E). In all cases, one-dimensional nanostructures were observed, albeit with some differences between the individual APAs. APA 1 formed short, fibrillar structures along with some poorly defined aggregates, while APA 4 formed bundles of nanofibers. APAs 2, 3, and 5 formed long, thin nanofibers. These differences may be related to variability in packing of the self-assembled structures. The fibrillar structures and nanofibers of APAs 1–5 had lengths in the μm scale and widths of about 10 nm, while the lengths of individual APA molecules were around 5 nm, as calculated by Chemdraw.37 Thus, the diameters of the nanofibers were approximately twice the widths of the APA molecules, indicating that the self-assembled structures are likely cylindrical micelles (Figure 2F).
Figure 2.

TEM images of self-assembled APAs 1–5 (A-E) and a schematic illustration of the proposed nanofiber structure (F). 1 wt% peptide solutions in water were cast onto TEM grids before staining with 2 wt % uranyl acetate and then allowed to dry under air for 12 h before imaging.
H2S Release from Dilute APA Solutions.
We evaluated the release kinetics of the APAs in dilute solution (below the CAC) using both the methylene blue method and an electrode probe method at rt. The methylene blue method is a widely employed colorimetric assay to detect H2S release.22, 36, 38 Although it can lead to erroneous data in biological media,39 we have found that it works well for measuring cumulative kinetics of H2S release in aqueous solution.38 By converting N,N-dimethyl-p-phenylenediamine into methylene blue in the presence of trapped H2S, the absorbance at 750 nm indicates H2S concentration in solution.39 In contrast to the methylene blue method, the H2S-sensitive electrode probe detects a wider concentration range of H2S and shows H2S release data in real-time. However, because H2S is constantly oxidizing and volatilizing from solution, it cannot measure cumulative release. Peaking time and concentration are typically used to compare data from release curves generated using this method.
The H2S release half-lives of dilute (0.1 mg/mL) APA solutions were determined by the methylene blue assay using previously reported conditions.38 By fitting the cumulative release data to pseudo-first-order kinetics (Table S2), we found that half-lives of release ranged from 13 min (APA 5) to 31 min (APA 1). The data revealed a correlation between H2S release kinetics and Hammett σ values of the para-substituents (Figure S4), with the Hammett plot showing a ρ value of 0.77, which is close to our previously reported ρ value of 1.05 for small molecule analogues of these APAs.20
The electrode probe method was also applied to monitor the H2S release profiles from dilute APA solutions. APA solutions were prepared and then transferred to the inner well in a specially designed vial. A solution of Cys (2 equiv with respect to APA) was added to the inner well, and the well was sealed with a gas permeable membrane. A large volume of PBS was then added to the vial to cover the inner well and absorb H2S, which was detected by the electrode probe. When the substituents changed from electron-donating OMe (APA 1) to electron-withdrawing Cl (APA 5), the peaking time decreased from 270 min to 120 min. The trend of H2S peaking time from APAs 1 to 5 matched the decreasing trend of H2S release half-lives determined by the methylene blue assay.
H2S Release from Self-assembled APA Solutions.
To measure H2S release kinetics from APA nanostructures, APAs were dissolved in 1x PBS at 10 mg/mL (above the CAC), and then these solutions were transferred to the specially designed vial. Cys (2 equiv with respect to APA) was added to trigger H2S release to the gel-holding inner well containing the peptide, the inner well was sealed, and the experiments were carried out under the same conditions as for the dilute solutions. We found that this method provided more consistent results than methylene blue for measuring H2S release from concentrated solutions and gels, so methylene blue was not used in these experiments.
The peaking times were in the same range (~2–4 h) for the self-assembled APA solutions (10 mg/mL) and dilute APA solutions (0.25 mg/mL), but the correlation found in dilute peptide solutions between Hammett σ values and peaking time was lost at 10 mg/mL (Table 1). APAs 1 and 3 had significantly longer peaking times than the other three APAs at 10 mg/mL. Peaking concentrations were nearly an order of magnitude higher than in dilute solutions. We attribute the loss in electronic control over release rate to variations in packing in the self-assembled nanofibers, as described in the Discussion section.
Table 1.
H2S release profiles of APAs 1–5a.
| APA | R group | Dilute solution | Assembled solution | Hydrogel | |||
|---|---|---|---|---|---|---|---|
| Peaking time (min) | Cmax (µM) | Peaking time (min) | Cmax (µM) | Peaking time (min) | Cmax (µM) | ||
| APA 1 | OMe | 270±20 | 0.5±0.2 | 240±30 | 3.8±0.6 | 42±3 | 0.9±0.3 |
| APA 2 | Me | 240±10 | 0.3±0.1 | 160±20 | 3.4±0.4 | 40±6 | 0.9±0.1 |
| APA 3 | H | 230±5 | 0.5±0.2 | 250±10 | 3.3±0.7 | 56±5 | 0.8±0.1 |
| APA 4 | F | 180±30 | 0.4±0.1 | 100±20 | 4.5±0.5 | 26±7 | 1.3±0.1 |
| APA 5 | Cl | 120±10 | 0.3±0.0 | 160±30 | 6.7±0.9 | 23±3 | 1.5±0.1 |
H2S release peaking time and peaking concentration (Cmax) from APAs as dilute solutions (0.25 mg/mL), self-assembled solutions (10 mg/mL), and hydrogels (10 mg/mL), as determined by the electrode probe method. Cys (2 equiv with respect to SATO groups) was used as the trigger for H2S release in all cases. Mean values are shown with error bars indicating standard deviations from three trials. See Figures S10–12 for full statistical analyses.
Gel Preparation and Rheology.
Hydrogels were prepared from each APA from self-assembled solutions (10 mg/mL). Addition of CaCl2 led to instantaneous gelation, and time-sweep rheology experiments revealed an increase in storage moduli over time, with each beginning to plateau after 1 h (Figure S6). The storage moduli of the APA hydrogels after 1 h ranged from 50 to 300 Pa (Figure S6 and Table S3). APAs 1, 3, and 4 had storage moduli around 50 Pa, lower than our previously reported 10 mg/mL SATO-IAVE3 peptide hydrogel (320 Pa).30 We attribute this difference to the additional Glu unit in the APAs reported here, as this increases the hydrophilicity of the APA, decreasing the driving force for self-assembly. Hydrogels made from APAs 2 and 5 were stiffer than the other APAs, with storage moduli near 300 Pa.
H2S Release from Gels.
We also measured H2S release from hydrogels prepared from APAs 1–5. To quantify H2S release from APA hydrogels, we used a similar experimental setup as described for the electrode probe studies on APA solutions. In this case, APA solution (10 mg/mL) was added to the inner well of the specially designed vial, followed by CaCl2. After allowing 2 h for hydrogel maturation, Cys was added to trigger release of H2S. The inner well was then quickly sealed, PBS solution was added to the vial, and H2S escaping through the gas-permeable membrane was detected using the electrochemical probe at rt.
In general, the release profiles from the hydrogels were quite different from the self-assembled solutions. The peaking time of APA 3 was significantly longer than other APAs, and the peaking concentration of APA 3 was significantly smaller than other APAs. Though the trend is similar to self-assembled APA solutions, the peaking times were considerably shorter, and peaking concentrations were lower. We noticed that the gels remained at least partially intact throughout the release experiments, even though the products after release are soluble.30 We attribute this to slow diffusion of Cys throughout the gel, such that release occurred only from the top layer. However, despite these differences in release profiles, the overall trends in peaking times were similar to the self-assembled solutions.
Circular Dichroism Spectroscopy.
Several spectroscopic methods were used to provide molecular-level characterization of the APAs. We began with circular dichroism (CD). Each APA was analyzed in solution at 0.25 mg/mL (below the CAC) and 10 mg/mL (above the CAC), as well as at 10 mg/mL in gel form after addition of CaCl2. All CD spectra were recorded in unbuffered water as buffer salts prevent data acquisition at low wavelengths.
In a typical CD spectrum for a protein, an α-helix structure presents two negative bands at 208 and 222 nm, a β-sheet conformation has a positive peak at 195 nm and a negative peak at 218 nm, and a random coil shows a single negative band just below 200 nm.40, 41 At 0.25 mg/mL, all of the APAs showed negative peaks between 195–200 nm (Figure 3), signifying a random coil conformation, which is consistent with the Nile Red data indicating that this concentration is below the CAC. In contrast, at 10 mg/mL the CD spectra displayed no peaks at 195 nm, while the absorption at 222–226 nm dominated (Table 2), which is consistent with β-sheet formation. A thioflavin T (ThT) assay (Figure S7) on each APA also confirmed the presence of β-sheets. When 20 mM CaCl2 was added to the solutions to form hydrogels, the absorptions changed marginally. The β-sheet absorption peaks shifted to lower wavelengths, to 219–223 nm, with small changes in the relative intensity of the band among the different APAs. Compared with classical β-sheet peaks (218 nm), the β-sheet peaks of 10 mg/mL APA solutions and hydrogels were shifted to higher wavelength. This shift likely indicates twisted β-sheet packing in the self-assembled structures, as addressed in the Discussion section.42
Figure 3.

Circular dichroism spectra of APAs 1–5 at 0.25 mg/mL in H2O, 10 mg/mL in H2O, and 10 mg/mL in gel form (H2O with 20 mM CaCl2).
Table 2.
Spectroscopic data of APAs 1–5 in solution and in hydrogel form.
| Name | R group | CD peak (nm)a | FTIR peak (cm−1) b | Fluorescence redshift (nm)c | |
|---|---|---|---|---|---|
| Solution | Hydrogel | ||||
| APA 1 | OMe | 224 | 223 | 1618 | 4 |
| APA 2 | Me | 226 | 221 | 1624 | 2 |
| APA 3 | H | 223 | 220 | 1626 | 2 |
| APA 4 | F | 223 | 220 | 1624 | 2 |
| APA 5 | Cl | 222 | 219 | 1632 | 10 |
APA 1–5 solutions (10 mg/mL in H2O) and hydrogels (10 mg/mL in H2O with addition of 20 mM CaCl2) were prepared for CD experiments. All measurements were taken in a 0.2 mm quartz cuvette.
APAs 1–5 in hydrogel form (10 mg/mL with 20 mM CaCl2) were prepared in D2O for IR experiments.
Indicates shift in emission maximum upon concentration change from 0.25 to 10 mg/mL with an excitation wavelength of 290 nm.
FTIR spectroscopy.
Fourier-transform infrared (FTIR) spectroscopy was used to complement the CD spectra for hydrogels prepared from APAs 1–5. Instead of H2O, D2O was used for these experiments because H2O absorbs at 1645 cm−1, which prevents observation of the amide I peak in β-sheet-forming peptides. In these experiments, APA hydrogels (10 mg/mL) were prepared as before, and FTIR spectra were recorded using attenuated total reflectance mode.
In Figure 4, low frequency amide I peaks ranging from 1615 to 1630 cm−1 were assigned to β-sheet conformations,43,44 and the lack of high frequency amide I peaks around 1680–1690 cm−1 suggested no antiparallel arrangement in the self-assembled structures.45, 46 This signature indicates that the APA self-assembled structures adopted parallel β-sheet packing. As observed via TEM, APAs self-assembled into cylindrical micelles (Figure 2). In this model, hydrophobic aromatic groups stack in the core, while hydrophilic peptides face outward, consistent with parallel β-sheet packing. Thus, the FTIR results matched the cylindrical micelle model, furthur supporting the TEM observations.
Figure 4.
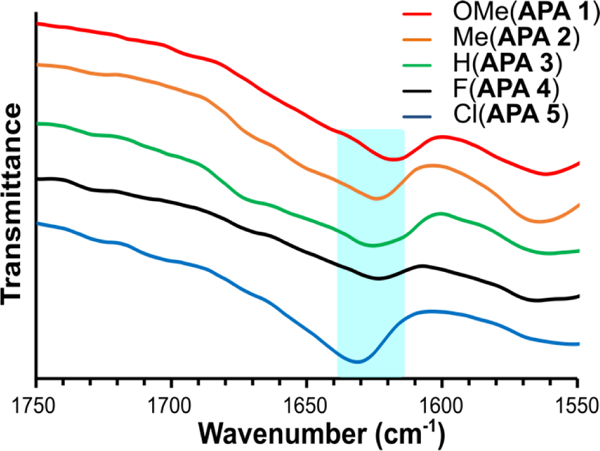
FTIR spectra of hydrogels prepared from APAs 1–5 (10 mg/mL APA with 20 mM CaCl2 in D2O). The blue band highlights the amide I absorption range where a peak is expected in β-sheet-forming peptides. Traces are offset for sake of clarity.
Although the IR spectra for the APAs were similar, the peak absorptions in the low frequency amide I peaks varied somewhat. For example, APA 1 had a β-sheet peak at 1618 cm−1, while APA 5 had β-sheet peak at 1632 cm−1. APAs 2–4 all had similar peak positions, ranging from 1624 to 1626 cm−1. These differences likely indicate changes in β-sheet packing among the five APAs. This is addressed further in the Discussion section.
Fluorescence spectroscopy.
Fluorescence spectroscopy was employed to further evaluate the aromatic stacking behavior of the APAs. The APAs in this study contain both electron-donating (OMe, Me) and electron-withdrawing (Cl, F) substituents, leading to variable electron densities in the APA aromatic ring systems. Dilute (0.25 mg/mL) and self-assembled (10 mg/mL) APA solutions, as well as 10 mg/mL APA hydrogels formed after adding CaCl2, were prepared. Fluorescence spectra were measured with an excitation wavelength of 290 nm.
At concentrations below the CAC (0.25 mg/mL), APA emission peaks were observed at approximately 380 nm (Figure 5). The spectra had similar shapes, but emission maxima varied somewhat among the APAs. At concentrations above the CAC (10 mg/mL), redshifts were observed compared to the dilute solution spectra (0.25 mg/mL), which indicates the onset of aromatic stacking.47 The extent of the redshift varied among the different APAs. Finally, no obvious shifts in emission maxima were found when CaCl2 was added to induce hydrogel formation; however, the overall emission intensity increased dramatically.
Figure 5.
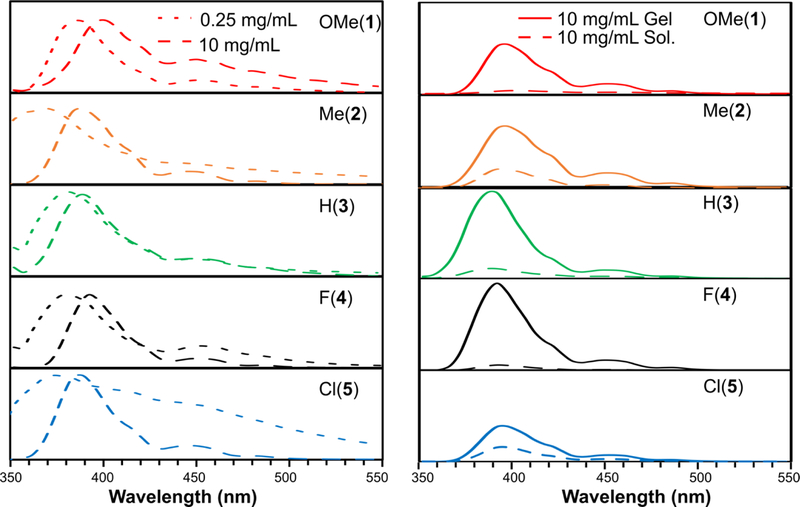
Fluorescence spectra of APAs 1–5 (λex=290 nm). Solutions were prepared in 10 mM PBS, and hydrogels were formed by addition of 20 mM CaCl2. The graphs on the left show dilute solution (0.25 mg/mL) and self-assembled solution (10 mg/mL) spectra, with peak intensities normalized to highlight changes in peak emission. The spectra on the right show self-assembled solutions and hydrogels, both at 10 mg/mL. The intensities are not normalized in these spectra.
Discussion
Substituent Effects on Self-Assembly.
The TEM images of APAs 1–5 indicated that the subtle chemical differences among the peptides impacted molecular packing. APA 1 formed fibrillar structures along with poorly defined aggregates, and APAs 2–5 formed nanofibers. Although all APAs showed primarily cylindrical micelles, the electronic and steric differences among them led to differences in the length of nanostructures. For example, TEM images of APA 1 (OMe) revealed mostly fibrillar structures, but small, poorly defined aggregates were also observed. These aggregates likely arose as a result of a greater steric bulk of OMe group relative to the other substituents. These differences also became apparent while examining the effect of substituents on hydrogen bonding in each of the APAs. We used FTIR spectroscopy to assess the amide I peak of the β-sheets, where lower absorption frequency indicates a greater average hydrogen bond length.48 We observed a trend of shifting to higher frequency from APA 1 (1618 cm−1) to APA 5 (1632 cm−1) with APAs 2–4 all absorbing in the range of 1624–1626 cm−1 (Figure 4). APA 1 (OMe) showed the most elongated β-sheet hydrogen bonding, consistent with the cylindrical micelle model in which the presence of the bulky OMe group twisted the alignment of the β-sheets and affected the hydrogen bond length, resulting in the shifted amide I peak compared to that of the typical β-sheet peak range (1630 cm−1).49 Similarly, the highest IR absorption, indicative of the shortest average hydrogen bond lengths and least-twisted β-sheets, was found for APA 5 (Cl). CD results also supported the presence of twisted β-sheets for the APA hydrogels (Figure 3). The negative β-sheet peaks were shifted from their typical position at 218 nm to 219–223 nm. Again, APA 1 showed the largest shift (peak absorption at 223 nm), while APA 5 showed the smallest shift (peak absorption at 219 nm). Collectively, the TEM, FTIR, and CD results support a twisted β-sheet model where the bulky OMe substituent on APA 1 causes a greater twist and shorter fibrillar structures than the other substituents, while the Cl substituent on APA 5 appears to favor tighter packing than the other APAs.
Consistent with their effects on supramolecular packing in the peptide region, the substituents also affected π-π stacking of the aromatic SATO units of the peptides. Redshifts were observed in the fluorescence spectra upon crossing the CAC, i.e., going from 0.25 mg/mL to 10 mg/mL for APAs 1–5 (Figure 5). Two packing patterns exist in parallel π-π stacking: the displaced stacking conformation (J-aggregation) and the sandwich stacking conformation (H-aggregation).50 We found that APAs 1 and 5 had redshifts of 4 nm and 10 nm, respectively, while APAs 2–4 all had redshifts of 2 nm. As redshift is an indicator for J-aggregation in π-π stacking,51 the higher redshifts of APAs 1 and 5 compared with APAs 2–4 suggest that self-assembled APAs 1 and 5 exhibit more J-aggregation in the aromatic region than APAs 2–4. The enhanced J-aggregation in APAs 1 and 5 is likely related to both steric hindrances (APA 1) and electronic effects (APA 5). For APA 1, the steric bulk of the OMe group can induce more J-aggregation than H-aggregation. In contrast, for APA 5, the electron-withdrawing Cl substituent likely decreases the energy barrier for the displaced conformation and leads to more J-aggregation.52 The results from fluorescence spectroscopy are consistent with the results from FTIR spectroscopy, with APAs 2–4 all exhibiting similar spectral features and APAs 1 and 5 showing distinct IR absorptions and fluorescence redshifts. Based on these observations, we conclude that the substituents not only affected the H-bonding and packing of β-sheets among the APAs, but also affected their π-π stacking.
Tunable H2S Release from APAs.
In our previous work with small molecule SATOs, we demonstrated that by manipulating the electronics of the SATO unit, we could tune the H2S release rate over an order of magnitude.20 We expected that the electronic control of H2S release kinetics observed in small molecule SATOs would be preserved in dilute APA solutions (below the CAC), but that the trend would be lost in self-assembled APAs (above the CAC), where the nanostructure could limit Cys diffusion. To assess this hypothesis, we evaluated H2S release kinetics in dilute solutions, self-assembled solutions, and hydrogels. The results revealed noticeable differences in the H2S release profiles of the APAs under these different conditions. For dilute APA solutions (0.1 and 0.25 mg/mL), substituent electronics largely controlled H2S release rates, whereas molecular packing influenced H2S release behavior in concentrated APA solutions (10 mg/mL) and hydrogels (10 mg/mL). Below we discuss these different general trends as well as the effect of specific substituents on release rates.
The H2S release rates for dilute APA solutions at 0.1 mg/mL determined by the methylene blue method fit well to a Hammett plot (Figure S4). APAs 1 and 2, which have electron-donating substituents on the SATO unit, had longer release half-lives (31 min and 22 min, respectively) than unsubstituted APA 3 (19 min), while APAs 4 and 5 with electron-withdrawing substituents had shorter release half-lives (14 min and 13 min, respectively). The Hammett ρ value was 0.77, demonstrating a strong dependence of H2S release rate on substituent electronics. Related experiments using an electrochemical probe method to measure H2S release from 0.25 mg/mL solutions (Table 1) showed similar results. The trend of H2S release peaking times detected by the electrode probe was the same as the methylene blue assay, with APAs 4–5 significantly faster than APAs 1–3. Based on these two methods of evaluating H2S release rates, we conclude that H2S release from APAs at concentrations below the CAC value was regulated by substituent electronic effects.
The correlation between substituent Hammett σ values and reaction rates in dilute APA solutions weakened upon moving to higher concentration (Table 1 and Figure 6), which we attribute to self-assembled nanostructure formation. In peptide amphiphiles and other short, self-assembling peptides, small changes in chemical structure can dramatically impact supramolecular assembly and drug release.53–58 As is evident from the TEM images, peptides self-assembled to form rigid nanofibers at 10 mg/mL. The packing of peptide units into nanofibers could slow down the diffusion of Cys to the hydrophobic SATO core, thereby decelerating the reaction rate. We recently saw similar effects in SATO-containing polymer micelles, which released H2S ~10-fold more slowly than analogous small SATO molecules.38
Figure 6.
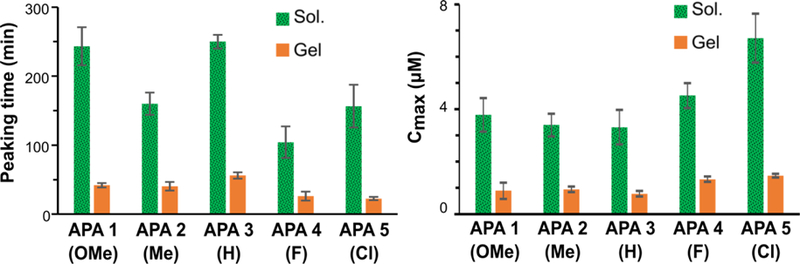
Peaking time and peak concentration (Cmax) of 10 mg/mL APA solutions and gels. Mean values were averaged from three trials with error bars indicating standard deviations of three separate trials. Please refer for Figures S11 and S12 for a full statistical analysis of the data.
In concentrated solution experiments (10 mg/mL), APAs 1–2 showed longer H2S peaking times (160–240 min) compared to APAs 4–5 (100–160 min), a trend that was consistent with the dilute solution release results. APAs 4 and 5 also had higher peaking concentrations, which tends to occur with faster-releasing H2S donors because more H2S builds up before it oxidizes and volatilizes. However, APA 3 (H) showed the longest peaking time of all (250 min), indicating that the correlation between Hammett σ values and release rate was not as strong as for the dilute solution experiments. Therefore, we propose that the differences among molecularly packed structures of APAs also translated into their H2S release rates. For example, a high degree of twist in the nanostructures could facilitate the penetration of Cys into the aromatic core, accelerating the release rate, while aromatic stacking might stabilize the SATO core, decreasing the release rate.
These differences in packing are highlighted by several examples. First, self-assembled APA 1 (OMe) had a shorter H2S release peaking time than APA 3 (H), even though the electronics of the OMe group favor slow release. We attribute this observation to the short fibrillar morphology of APA 1 compared with the nanofiber morphology of APA 3 as observed by TEM and its more twisted structure as indicated by CD and FTIR results. The high degree of twist in APA 1 likely facilitated Cys diffusion into the nanostructure core, accelerating the reaction rate. In another example, APA 5 (Cl), which showed the fastest reaction rate in dilute solution (Table S2), exhibited a peaking time comparable to APA 2 (Me) in concentrated solution. APA 5 showed the smallest degree of β-sheet twist among all APAs based on CD and FTIR data, along with the greatest degree of J-aggregation based on fluorescence data. These two effects appear to contribute to the slower than expected H2S release rate for APA 5 in concentrated solution. Finally, APAs 2–4 had similar β-sheet twist and J-aggregation levels, but the H2S release rate for APA 3 was slower than expected based on electronic differences, suggesting that additional molecular packing effects contributed to these differences that were not immediately apparent in our FTIR, CD, and fluorescence experiments. Thus, in general the SATO substituents were less effective in controlling H2S release rates in concentrated solution than in dilute solution because differences in molecular packing influenced release rates.
Collectively, the H2S release profiles from 10 mg/mL APA hydrogels were quite different than the profiles for 10 mg/mL solutions. Concentrated APA solutions allowed for free diffusion of Cys throughout the releasing solution, which was not true for hydrogels, where only the top layer of the gel appeared to degrade and release H2S (the gel becomes soluble after complete H2S release). Penetration of Cys into the hydrogels could be affected by many factors, including the rigidity of the supramolecular nanostructures and gel mesh size.38, 59 Overall, limited Cys penetration led to lower peaking concentrations (Cmax) and much shorter peaking times for the gels than for the solutions. While the data appear to show much less overall release from the gels, the only difference between the two is the CaCl2 gelating agent. We attribute this difference between solution and gel release profiles to the experimental setup, where we can only add Cys once before sealing the inner well of the vial. Continuous availability of Cys or other reduced thiols, as would be expected in vivo, would likely lead to full release from the gels.
Despite the collective differences in release profiles between self-assembled APA solutions and their corresponding gels, the trends among APAs were generally consistent between the two data sets. For example, APA 3 had longest peaking time (56 min) among the APA hydrogels and among the APA solutions. Also similar to the solution data, the peaking times of APAs 1 and 2 (42 min and 40 min, respectively) were significantly greater than those for APAs 4 and 5 (26 min and 24 min, respectively). As in the concentrated solutions, peaking concentration was higher for the faster-releasing gels, with APAs 1–3 showing peaking concentrations of 0.8–0.9 μM and APAs 4–5 showing peaking concentrations of 1.3–1.5 μM. Gel stiffness did not appear to have a large impact on H2S release rates.
Conclusions:
This work explores substituent effects on the supramolecular packing of SATO-conjugated APAs, as well as the influence of self-assembled structures on H2S release profiles. Electron-withdrawing and donating substituents on the SATO groups on these APAs affected both reactivity and molecular packing. In dilute solution (below the CAC), H2S release half-lives correlated linearly with substituent Hammett σ values in a Hammett plot, with electron-withdrawing substituents accelerating the H2S release rate and electron-donating substituents decreasing it. The steric and electronic components of the substituents played a different role in self-assembled solutions (above the CAC) and the resulting hydrogels, dictating β-sheet packing, π-π stacking, and nanostructure formation. Overall, APAs with electron-donating substituents and tightly packed self-assembled structures elongated the H2S release profile. These results highlight the complex interplay between molecular-level effects (i.e., substituent electronics) and supramolecular effects (i.e., supramolecular packing) in drug releasing systems, expanding the design parameters for supramolecular materials with applications in biology and medicine.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Tijana Grove, Dr. Charles Frazier, Dr. Ann Norris, Dr. Jatinder Josan, and Justin Grams for their help with CD, rheology and analytical HPLC. We thank Mingjun Zhou for assistance with peptide synthesis and characterization, Yin Wang for helpful discussions, and Anastasia Volokohova and Sarah Blosch for critical readings of the manuscript.
Funding Sources
This work was supported by the National Science Foundation (DMR-1454754) and the National Institutes of Health (R01GM123508). We also thank 3M for support of this work through a Non-Tenured Faculty Award. Yun Qian was supported by a doctoral fellowship from the Virginia Tech Institute for Critical Technology and Applied Science.
Footnotes
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI:
Detailed synthesis and characterization, UV-vis hydrolysis data, ThT assays, rheology experiments for APAs 1–5, additional H2S release rate data for APA 1–5 solutions and hydrogels (Hammett plot and H2S release profiles), and statistical analysis of peaking times and peaking concentrations.
The authors declare no competing financial interest.
REFERENCES
- (1).Motterlini R; Otterbein LE The Therapeutic Potential of Carbon Monoxide. Nat. Rev. Drug Discov 2010, 9, 728–743. [DOI] [PubMed] [Google Scholar]
- (2).Carpenter AW; Schoenfisch MH Nitric Oxide Release: Part Ii. Therapeutic Applications. Chem. Soc. Rev 2012, 41, 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang R Shared Signaling Pathways among Gasotransmitters. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 8801–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wang R Gasotransmitters: Growing Pains and Joys. Trends Biochem. Sci 2014, 39, 227–232. [DOI] [PubMed] [Google Scholar]
- (5).Wang R Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed. Physiol. Rev 2012, 92, 791–896. [DOI] [PubMed] [Google Scholar]
- (6).Abe K; Kimura H The Possible Role of Hydrogen Sulfide as an Endogenous Neuromodulator. J. Neurosci 1996, 16, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kimura H Hydrogen Sulfide: From Brain to Gut. Antioxid. Redox Signal 2010, 12, 1111–1123. [DOI] [PubMed] [Google Scholar]
- (8).Taniguchi S; Kang L; Kimura T; Niki I Hydrogen Sulphide Protects Mouse Pancreatic Β-Cells from Cell Death Induced by Oxidative Stress, but Not by Endoplasmic Reticulum Stress. Br. J. Pharmacol 2011, 162, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhao W; Zhang J; Lu Y; Wang R The Vasorelaxant Effect of H2s as a Novel Endogenous Gaseous Katp Channel Opener. EMBO J 2001, 20, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wagner F; Asfar P; Calzia E; Radermacher P; Szabo C Bench-to-Bedside Review: Hydrogen Sulfide - the Third Gaseous Transmitter: Applications for Critical Care. Crit. Care 2009, 13, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Polhemus DJ; Lefer DJ Emergence of Hydrogen Sulfide as an Endogenous Gaseous Signaling Molecule in Cardiovascular Disease. Circ. Res 2014, 114, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shen Y; Shen Z; Luo S; Guo W; Zhu YZ The Cardioprotective Effects of Hydrogen Sulfide in Heart Diseases: From Molecular Mechanisms to Therapeutic Potential. Oxid. Med. Cell Longev 2015, 2015. [DOI] [PMC free article] [PubMed]
- (13).Elrod JW; Calvert JW; Morrison J; Doeller JE; Kraus DW; Tao L; Jiao X; Scalia R; Kiss L; Szabo C; Kimura H; Chow CW; Lefer DJ Hydrogen Sulfide Attenuates Myocardial Ischemia-Reperfusion Injury by Preservation of Mitochondrial Function. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 15560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chattopadhyay M; Kodela R; Olson KR; Kashfi K Nosh-Aspirin (Nbs-1120), a Novel Nitric Oxide- and Hydrogen Sulfide-Releasing Hybrid Is a Potent Inhibitor of Colon Cancer Cell Growth in Vitro and in a Xenograft Mouse Model. Biochem. Biophys. Res. Commun 2012, 419, 523–8. [DOI] [PubMed] [Google Scholar]
- (15).Papapetropoulos A; Pyriochou A; Altaany Z; Yang G; Marazioti A; Zhou Z; Jeschke MG; Branski LK; Herndon DN; Wang R Hydrogen Sulfide Is an Endogenous Stimulator of Angiogenesis. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhao Y; Pacheco A; Xian M, Medicinal Chemistry: Insights into the Development of Novel H2S Donors. In Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide, Springer: 2015; pp 365–388. [DOI] [PubMed] [Google Scholar]
- (17).Powell CR; Dillon KM; Matson JB A Review of Hydrogen Sulfide (H2s) Donors: Chemistry and Potential Therapeutic Applications. Biochem Pharmacol 2018, 149, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li L; Whiteman M; Guan YY; Neo KL; Cheng Y; Lee SW; Zhao Y; Baskar R; Tan C; Moore PK Characterization of a Novel, Water-Soluble Hydrogen Sulfide–Releasing Molecule (GYY4137) New Insights into the Biology of Hydrogen Sulfide. Circulation 2008, 117, 2351–2360. [DOI] [PubMed] [Google Scholar]
- (19).Zhao Y; Wang H; Xian M Cysteine-Activated Hydrogen Sulfide (H2s) Donors. J. Am. Chem. Soc 2010, 133, 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Foster JC; Powell CR; Radzinski SC; Matson JB S-Aroylthiooximes: A Facile Route to Hydrogen Sulfide Releasing Compounds with Structure-Dependent Release Kinetics. Org. Lett 2014, 16, 1558–1561. [DOI] [PubMed] [Google Scholar]
- (21).Zhao Y; Bhushan S; Yang C; Otsuka H; Stein JD; Pacheco A; Peng B; Devarie-Baez NO; Aguilar HC; Lefer DJ Controllable Hydrogen Sulfide Donors and Their Activity against Myocardial Ischemia-Reperfusion Injury. ACS Chem. Biol 2013, 8, 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Powell CR; Foster JC; Okyere B; Theus MH; Matson JB Therapeutic Delivery of H2s Via Cos: Small Molecule and Polymeric Donors with Benign Byproducts. J. Am. Chem. Soc 2016, 138, 13477–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zheng Y; Yu B; Ji K; Pan Z; Chittavong V; Wang B Esterase-Sensitive Prodrugs with Tunable Release Rates and Direct Generation of Hydrogen Sulfide. Angew. Chem. Int. Ed 2016, 55, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhao Y; Pluth MD Hydrogen Sulfide Donors Activated by Reactive Oxygen Species. Angew. Chem. Int. Ed 2016, 55, 14638–14642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhao Y; Bolton SG; Pluth MD Light-Activated Cos/H2S Donation from Photocaged Thiocarbamates. Org. Lett 2017, 19, 2278–2281. [DOI] [PubMed] [Google Scholar]
- (26).Chauhan P; Bora P; Ravikumar G; Jos S; Chakrapani H Esterase Activated Carbonyl Sulfide/Hydrogen Sulfide (H2s) Donors. Org. Lett 2017, 19, 62–65. [DOI] [PubMed] [Google Scholar]
- (27).Qian Y; Matson JB Gasotransmitter Delivery Via Self-Assembling Peptides: Treating Diseases with Natural Signaling Gases. Adv. Drug Deliv. Rev 2016, 110–111, 137–156. [DOI] [PubMed]
- (28).Urquhart MC; Ercole F; Whittaker MR; Boyd BJ; Davis TP; Quinn JF Recent Advances in the Delivery of Hydrogen Sulfide Via a Macromolecular Approach. Polym. Chem 2018, 9, 4431–4439. [Google Scholar]
- (29).Wu J; Li Y; He C; Kang J; Ye J; Xiao Z; Zhu J; Chen A; Feng S; Li X Novel H2s Releasing Nanofibrous Coating for in Vivo Dermal Wound Regeneration. ACS Appl. Mater. Interfaces 2016, 8, 27474–27481. [DOI] [PubMed] [Google Scholar]
- (30).Carter JM; Qian Y; Foster JC; Matson JB Peptide-Based Hydrogen Sulphide-Releasing Gels. Chem. Commun 2015, 51, 13131–13134. [DOI] [PubMed] [Google Scholar]
- (31).Xiao Z; Bonnard T; Shakouri-Motlagh A; Wylie RAL; Collins J; White J; Heath DE; Hagemeyer CE; Connal LA Triggered and Tunable Hydrogen Sulfide Release from Photogenerated Thiobenzaldehydes. Chem. Eur. J 2017, 23, 11294–11300. [DOI] [PubMed] [Google Scholar]
- (32).Yu SH; Ercole F; Veldhuis NA; Whittaker MR; Davis TP; Quinn JF Polymers with Acyl-Protected Perthiol Chain Termini as Convenient Building Blocks for Doubly Responsive H2s-Donating Nanoparticles. Polym. Chem 2017, 8, 6362–6367. [Google Scholar]
- (33).Matson JB; Stupp SI Self-Assembling Peptide Scaffolds for Regenerative Medicine. Chem. Commun 2012, 48, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fleming S; Ulijn RV Design of Nanostructures Based on Aromatic Peptide Amphiphiles. Chem. Soc. Rev 2014, 43, 8150–8177. [DOI] [PubMed] [Google Scholar]
- (35).Kaur K; Qian Y; Gandour RD; Matson JB Hydrolytic Decomposition of S-Aroylthiooximes: Effect of pH and N-Arylidene Substitution on Reaction Rate. J. Org. Chem 2018, 83, 13363–13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Siegel LM A Direct Microdetermination for Sulfide. Anal. Biochem 1965, 11, 126–132. [DOI] [PubMed] [Google Scholar]
- (37). Chemdraw Professional; Perkinelmer: Waltham, Ma (Accessed November, 2018); Molecule Length Calculated Using Distance (3d Property), Software Version 16.0.1.4 (61) (1985–2017 Perkinelmer Information, Inc.)
- (38).Foster JC; Radzinski SC; Zou X; Finkielstein CV; Matson JB H2S-Releasing Polymer Micelles for Studying Selective Cell Toxicity. Mol. Pharmaceutics 2017, 14, 1300–1306. [DOI] [PubMed] [Google Scholar]
- (39).Cline JD Spectrophotometric Determination of Hydrogen Sulfide in Natural Waters. Limnol. Oceanogr 1969, 14, 454–458. [Google Scholar]
- (40).Manning MC; Illangasekare M; Woody RW Circular Dichroism Studies of Distorted Α-Helices, Twisted β-Sheets, and β-Turns. Biophys. Chem 1988, 31, 77–86. [DOI] [PubMed] [Google Scholar]
- (41).Greenfield NJ Using Circular Dichroism Spectra to Estimate Protein Secondary Structure. Nat. Protoc 2006, 1, 2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Micsonai A; Wien F; Kernya L; Lee Y-H; Goto Y; Réfrégiers M; Kardos J Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc. Natl. Acad. Sci. U.S.A 2015, 112, E3095–E3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Fleming S; Debnath S; Frederix PWJM; Hunt NT; Ulijn RV Insights into the Coassembly of Hydrogelators and Surfactants Based on Aromatic Peptide Amphiphiles. Biomacromolecules 2014, 15, 1171–1184. [DOI] [PubMed] [Google Scholar]
- (44).Abul-Haija YM; Ulijn RV Sequence Adaptive Peptide–Polysaccharide Nanostructures by Biocatalytic Self-Assembly. Biomacromolecules 2015, 16, 3473–3479. [DOI] [PubMed] [Google Scholar]
- (45).Matsuzawa Y; Ueki K; Yoshida M; Tamaoki N; Nakamura T; Sakai H; Abe M Assembly and Photoinduced Organization of Mono- and Oligopeptide Molecules Containing an Azobenzene Moiety. Adv. Funct. Mater 2007, 17, 1507–1514. [Google Scholar]
- (46).Debnath S; Shome A; Das D; Das PK Hydrogelation through Self-Assembly of Fmoc-Peptide Functionalized Cationic Amphiphiles: Potent Antibacterial Agent. J. Phys. Chem. B 2010, 114, 4407–4415. [DOI] [PubMed] [Google Scholar]
- (47).Ma M; Kuang Y; Gao Y; Zhang Y; Gao P; Xu B Aromatic−Aromatic Interactions Induce the Self-Assembly of Pentapeptidic Derivatives in Water to Form Nanofibers and Supramolecular Hydrogels. J. Am. Chem. Soc 2010, 132, 2719–2728. [DOI] [PubMed] [Google Scholar]
- (48).Kong J; Yu S Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures. Acta Biochim. Biophys. Sin 2007, 39, 549–559. [DOI] [PubMed] [Google Scholar]
- (49).Pashuck ET; Cui HG; Stupp SI Tuning Supramolecular Rigidity of Peptide Fibers through Molecular Structure. J. Am. Chem. Soc 2010, 132, 6041–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Eisfeld A; Briggs JS The J- and H-Bands of Organic Dye Aggregates. Chem. Phys 2006, 324, 376–384. [Google Scholar]
- (51).Deng Y; Yuan W; Jia Z; Liu G H-and J-Aggregation of Fluorene-Based Chromophores. J. Phys. Chem. B 2014, 118, 14536–14545. [DOI] [PubMed] [Google Scholar]
- (52).Arnstein SA; Sherrill CD Substituent Effects in Parallel-Displaced Pi-Pi Interactions. Phys. Chem. Chem. Phys 2008, 10, 2646–55. [DOI] [PubMed] [Google Scholar]
- (53).Matson JB; Newcomb CJ; Bitton R; Stupp SI Nanostructure-Templated Control of Drug Release from Peptide Amphiphile Nanofiber Gels. Soft Matter 2012, 8, 3586–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wang Y; Kaur K; Scannelli SJ; Bitton R; Matson JB Self-Assembled Nanostructures Regulate H2S Release from Constitutionally Isomeric Peptides. J. Am. Chem. Soc 2018, 140, 14945–14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kalafatovic D; Nobis M; Son J; Anderson KI; Ulijn RV MMP-9 Triggered Self-Assembly of Doxorubicin Nanofiber Depots Halts Tumor Growth. Biomaterials 2016, 98, 192–202. [DOI] [PubMed] [Google Scholar]
- (56).Pappas CG; Abul-Haija YM; Flack A; Frederix PWJM; Ulijn RV Tuneable Fmoc–Phe–(4-X)–Phe–NH2 Nanostructures by Variable Electronic Substitution. Chem. Commun 2014, 50, 10630–10633. [DOI] [PubMed] [Google Scholar]
- (57).Lin Y-A; Kang M; Chen W-C; Ou Y-C; Cheetham AG; Wu P-H; Wirtz D; Loverde SM; Cui H Isomeric Control of the Mechanical Properties of Supramolecular Filament Hydrogels. Biomater. Sci 2018, 6, 216–224. [DOI] [PubMed] [Google Scholar]
- (58).Su H; Zhang P; Cheetham AG; Koo JM; Lin R; Masood A; Schiapparelli P; Quinones-Hinojosa A; Cui H Supramolecular Crafting of Self-Assembling Camptothecin Prodrugs with Enhanced Efficacy against Primary Cancer Cells. Theranostics 2016, 6, 1065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Branco MC; Pochan DJ; Wagner NJ; Schneider JP Macromolecular Diffusion and Release from Self-Assembled Beta-Hairpin Peptide Hydrogels. Biomaterials 2009, 30, 1339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.