

Abstract
Monoclonal antibodies, fusion proteins including the immunoglobulin fragment c (Ig Fc) CH2‐CH3 domains, and engineered antibodies are prominent representatives of an important class of drugs and drug candidates, which are referred to as biotherapeutics or biopharmaceuticals. These recombinant proteins are highly heterogeneous due to their glycosylation pattern. In addition, enzyme‐independent reactions, like deamidation, dehydration, and oxidation of sensitive side chains, may contribute to their heterogeneity in a minor amount. To investigate the biological impact of a spontaneous chemical modification, especially if found to be recurrent in a biotherapeutic, it would be necessary to reproduce it in a homogeneous manner. Herein, we undertook an explorative study towards the chemical synthesis of the IgG1 Fc CH3 domain, which has been shown to undergo spontaneous changes like succinimide formation and methionine oxidation. We used Fmoc‐solid‐phase peptide synthesis (SPPS) and native chemical ligation (NCL) to test the accessibility of large fragments of the IgG1 Fc CH3 domain. In general, the incorporation of pseudoproline dipeptides improved the quality of the crude peptide precursors; however, sequences larger than 44 residues could not be achieved by standard stepwise elongation with Fmoc‐SPPS. In contrast, the application of NCL with cysteine residues, which were either native or introduced ad hoc, allowed the assembly of the C‐terminal IgG1 Fc CH3 sequence 371 to 450. The syntheses reported here show advantages and limitations of the chemical approaches chosen for the preparation of the synthetic IgG1 Fc CH3 domain and will guide future plans towards the synthesis of both the native and selectively modified full‐length domain.
Keywords: Fc CH3 domain, native chemical ligation, pseudoproline, solid phase peptide synthesis
1. INTRODUCTION
Monoclonal antibodies (mAbs) and immunoglobulin fragment c‐fusion proteins are the most prominent representatives of the rapidly emerging class of biopharmaceuticals, which are applied to treat life‐threatening diseases like cancer and autoimmune disorders (eg, MabThera/Rituximab for B‐cell lymphoma, Herceptin/Trastuzumab for breast cancer, Enbrel/Etanercept, and Remicade/Infliximab for rheumatoid arthritis).1, 2, 3 Still in the context of cancer therapy, Ab engineering, including minibodies that are formed by the dimer of a single polypeptide chain reassembling the Ab VH and VL regions with the immunoglobulin fragment c CH3 domain,4, 5 has become a valuable approach, as it allows designing Ab‐related molecules with tuned pharmacokinetic and immunogenic properties.6, 7
The analytical and biological characterization of mAbs and, in general, of large biomolecules is a challenging but indispensable task. Indeed, the quality of a protein‐based drug may be affected by the existence of multiple variants displaying altered biophysical and biochemical properties; such chemical diversity mainly arises from post‐translational modifications (PTMs), like glycosylation, and spontaneous degradation during the production and formulation processes.8, 9 To this regard, the most commonly encountered enzyme‐independent modifications are methionine oxidation,10, 11, 12 deamidation of asparagine coupled to succinimide (Snn), aspartate and isoaspartate (isoAsp) formation,13, 14, 15, 16 N‐terminal glutamate or glutamine cyclization to pyroglutamate,17, 18, 19 and arginine glycation or carbonylation.20, 21 Spontaneous degradation products due to pyroglutamate formation, methionine oxidation, and asparagine deamidation have been observed in therapeutic mAbs.22 In particular, the IgG1 Fc domain contains two conserved and solvent‐exposed methionines at positions 432 in the CH3 domain (Figure 1) and 256 in the CH2 domain, which have been shown to be susceptible to oxidation upon manufacturing and storage.11 Moreover, it has been demonstrated by surface plasmon resonance that oxidation of Met‐432 and Met‐256 impacts the binding affinity to the human neonatal Fc receptor (FcRn),23 protein A, and protein G,24 which has been attributed to the alteration of the secondary structure surrounding the residues involved in the binding.25
Figure 1.

Sequence of the IgG1 Fc CH3 domain and detected, spontaneously occurring PTMs at Met and Asn sites
Asparagine deamidation and related products (Asp, isoAsp, and Snn) have been detected in both the Fc and Fab regions of mAbs, causing alterations of their secondary structure, potency, and binding affinity, especially when occurring across the flexible complementarity‐determining region,26, 27, 28 thus having direct consequences on the Ab‐mediated immune response. In the IgG1 Fc region, such PTM has been reported at Asn‐319 within the sequence Leu‐Asn‐Gly‐Lys in the CH2 domain, as well as at Asn‐388, Asn‐393, and Asn‐438 within the CH3 domain29, 30, 31 (Figure 1). The biological impact of deamidation has been studied in the Fab region of mAbs,26, 27, 28 whereas its effect on the Fc region remains to date unclear. However, the results of an alanine‐scan on human IgG1 have revealed that replacement of Asn‐438 influences the binding affinity for FcRn.32 Therefore, also the structural change caused by deamidation at Asn‐438 might potentially affect the FcRn binding.
Thus, oxidation and deamidation events may be considered two of the most important sources of enzyme‐independent PTMs in drugs based on mAbs and Fc‐fusion proteins. Moreover, both changes may influence the Ig binding to FcRn, thus affecting the half‐life of Igs and Fc‐fusion proteins in the blood.33, 34, 35 Although spontaneous PTMs are fortunately present only as minimal impurities, which may be hardly detectable even by highly sophisticated MS analysis,36, 37 nevertheless, the assessment of their structural and biological effects is of high significance. For this reason, it is often necessary to treat the protein chemically, in order to induce and investigate the desired PTM. For example, H2O2 treatments are usually performed to increase the amount of oxidized methionine. This, however, results in multiple combinations of reduced and oxidized methionine residues.11, 23, 38 In contrast, chemical protein synthesis (CPS) based on the native chemical ligation (NCL) of synthetic fragment precursors would, in principle, allow obtaining only one species at the time by replacing only the desired methionine with methionine sulfoxide. CPS has been successfully applied for small and medium‐size proteins,39, 40, 41, 42, 43 whereas other approaches have been developed to overcome the protein‐size limitation, which rely on the NCL between synthetic and recombinant fragment precursors (so‐called expressed protein ligation44, 45, 46), or on the incorporation of unnatural amino acids (AAs) into a protein by genetic‐code expansion.47, 48, 49
At the light of the important role played by the CH3 domain in the binding of IgG molecules to FcRn50, 51 as well as in the dimerization process of minibodies,4 we undertook the present work to assess the accessibility of this domain by chemical synthesis, which, in turn, would make variants containing, for example, oxidized methionine or d‐amino acids at the desired position also accessible. In particular, we focused on the preparation of IgG1 CH3 fragment precursors for two possible NCL strategies that rely on threonine‐based52, 53 and cysteine‐based43, 54 chemoselective reactions (Scheme 1). The C‐terminal fragment precursor (either 1, 4, or 6) contains oxidized methionine at position 432. Problems encountered during the solid‐phase peptide synthesis (SPPS) of the precursors and some solutions to them, as well as NCL attempts, are discussed below. Although the total chemical synthesis of the IgG1 Fc CH3 domain could not be achieved in this work, the conducted explorative study provides useful insights into the Fmoc‐based SPPS of IgG1 Fc CH3 sequences, including C‐terminally activated intermediates bearing the salicyladehyde ester (Sal) or a thioester.
Scheme 1.

Proposed strategies for the synthesis of the IgG1 Fc CH3 domain based on two (A and B) or three (C) ligation points. The * in fragments 1, 4, and 6 indicates the presence of Met(O)‐432. Sal, salicylaldehyde ester. Peptide 4 is also referred to as 1b in the following schemes. The ligations indicated by green arrows were successfully performed in this work
2. MATERIALS AND METHODS
2.1. Chemicals
All protected AAs, Fmoc‐Rink amide MBHA resin, N‐(9‐fluorenylmethyloxycarbonyloxy)‐succinimide (Fmoc‐OSu), N,N‐dimethylformamide (DMF), 1‐methyl‐2‐pyrrolidinone (NMP), dichloromethane (DCM), diethylether (Et2O), 2,2,2‐trifluoroethanol (TFE), and N,N‐diisopropylethylamine (DIPEA) were purchased from Iris Biotech GmbH (Marktredwitz, Germany). Pseudoproline dipeptides, O‐(7‐azabenzotriazol‐1‐yl)‐N,N,N′,N′‐tetramethyluronium‐hexafluorophosphate (HATU), H‐Gly‐2‐Cl‐trityl resin, H‐Thr(tBu)‐2‐Cl‐trityl resin, Fmoc‐Gly‐NovaSyn‐TGT resin, Fmoc‐Dbz‐NovaSyn‐TGR resin, 4‐nitrophenylchloroformate, and disodiumhydrogenphosphate were from Merck (Darmstadt, Germany). 1‐Hydroxybenzotriazole (HOBt), 2‐(1H‐benzotriazol‐1‐yl)‐1,1,3,3‐tetramethyluronium hexafluorophosphate (HBTU), N,N′‐diisopropylcarbodiimide (DIC), trifluoroacetic acid (TFA), and piperidine were obtained from Biosolve (Valkenswaard, The Netherlands). HPLC‐grade acetonitrile (ACN), thiophenol, 4‐mercaptophenylacetic acid (MPAA), triisopropylsilane (TIS), thioanisole (TIA), tris(2‐carboxyethyl) phosphine hydrochloride (TCEP), guanidine hydrochloride (GuHCl), and salicylaldehyde were obtained from Sigma Aldrich (Vienna, Austria). HPLC‐grade trifluoroacetic acid (TFA) was from Alfa‐Aesar (Karlsruhe, Germany).
2.2. Methods
Analytical RP‐HPLC was performed using a Thermo Fisher Scientific Dionex UltiMate 3000 UHPLC system (Germering, Germany) and either a Syncronics C‐18 column (100 Å, 5 μm, 250 × 4.6 mm, Thermo Fisher Scientific) at a flow rate of 1.5 mL/min or a Nucleosil C‐18 column (100 Å, 5 μm, 250 × 4 mm, Macherey‐Nagel, Düren, Germany) at a flow rate of 1 mL/min. Unless specifically stated, the first one was used. The UV detection was set at 220 nm. The elution system was (A) 0.06% (v/v) TFA in water, and (B) 0.05% (v/v) TFA in ACN. The crude products were dissolved in ACN/H2O (10:90, v/v) containing 0.1% TFA. Analytical chromatograms were obtained with the following gradients: method A: 10% B for 5 minutes, 10% to 70% B in 40 minutes; method B: 20% B for 5 minutes, 20% to 70% B in 30 minutes; method C: 25% B for 5 minutes, 25% to 75% B in 40 minutes; method D: 20% B for 5 minutes, 20% to 60% B in 40 minutes. Mass spectra were recorded on an Autoflex Speed MALDI‐TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) by using α‐cyano‐4‐hydroxycinnamic acid as matrix.
2.3. Stepwise synthesis of [Met(O)‐432]‐IgG1‐Fc 419‐450 (1aTRT) and Fmoc‐[Met(O)‐432]‐IgG1‐Fc 407‐450 (1aTGT+ψ)
Solid‐phase peptide synthesis was automatically performed on a Syro‐I (Biotage, Uppsala, Sweden) peptide synthesizer by using the Fmoc/tBu strategy. Sequence assembly was carried out on H‐Gly‐2‐Cl‐trityl (loading: 0.63 mmol/g; 18‐μmol scale) or Fmoc‐Gly‐NovaSyn‐TGT resin (loading: 0.2 mmol/g; 8‐μmol scale). The couplings of the protected AAs were carried out using AA/HOBt 1:1 (5 equivalents each), HBTU (4.9 equivalents), and DIPEA (10 equivalents), in DMF/NMP (7:3, v/v). Each coupling was performed twice (2 × 45 minutes). The couplings of the pseudoproline dipeptides were performed manually by employing (AAψAA)/HOBt 1:1 (2 equivalents each), HBTU (1.9 equivalents), and DIPEA (4 equivalents), in DMF/NMP (7:3, v/v) for 1 hour. The completeness of the coupling was then checked by the ninhydrin test, and, when needed, the coupling was repeated under the same conditions to assure complete acylation. Nα‐deprotection was obtained with a 3‐minute treatment with 30% piperidine in DMF, followed by a 12‐minute treatment with 15% piperidine in DMF. After the coupling of the last residue, the peptidyl‐resin was washed with DMF, DCM, and Et2O (three times each) and vacuum‐dried overnight. The resin‐bound peptide was treated with TFA/H2O/TIA/TIS/EDT (90:3:2:2:3, v/v) for 3 hours at room temperature (rt), then the peptide was precipitated with cold Et2O, recovered by centrifugation, and washed three times with the same solvent to remove the residual scavengers. The crude products were characterized by analytical RP‐HPLC and MALDI‐TOF‐MS.
2.4. Synthesis of [Met(O)‐432]‐IgG1‐Fc 398‐450 (1) by chemoselective ligation
Peptide 1b was assembled on the H‐Gly‐2‐Cl‐trityl resin (loading: 0.63 mmol/g; 18‐μmol scale) by using the SPPS protocol described in the subsection 2.3. The crude product was characterized by analytical RP‐HPLC using method A and by MALDI‐TOF‐MS. Peptide 1c was synthetized with the commercially available Fmoc‐Dbz‐NovaSyn‐TGR resin (loading: 0.2 mmol/g; 9‐μmol scale). The Fmoc group of the Dbz linker was cleaved with 20% piperidine in DMF for 20 minutes, then the resin was treated with 5 equivalents Fmoc‐Ser(tBu)‐OH, 5 equivalents HATU, and 10 equivalents DIPEA in DMF for 1 hour at rt. Then, the resin was washed with DMF, DCM, and Et2O (three times each) and dried under vacuum. The new loading was determined by measurement of the UV absorbance of the dibenzofulvene‐piperidine adduct at 301 nm. The peptide chain was assembled with the SPPS protocol described in the subsection 2.3 and the use of the pseudoproline dipeptides Fmoc‐Lys(Boc)‐Ser(ψMe,Mepro)‐OH, Fmoc‐Tyr(tBu)‐Ser(ψMe,Mepro)‐OH, and Fmoc‐Asp(tBu)‐Ser(ψMe,Mepro)‐OH. The coupling of Gly‐424 was performed manually with Fmoc‐Gly‐OPfp/HOBt 1:1 (6 equivalents) for 1 hour in DMF at rt, and the progress of the reaction was monitored by the ninhydrin test. After coupling the last residue as Boc‐AA, the resin was swollen in DCM for 30 minutes and treated with 5 equivalents p‐nitrophenylchloroformate dissolved in DCM (50 mM) for 1 hour under nitrogen at rt. The solvent was sucked off, the resin was washed with DCM, DMF, and again DCM (three times each), and finally subjected to a 30‐minute treatment with 0.5 M DIPEA in DMF at rt. After washing cycles with DMF, DCM, and Et2O, the peptide was cleaved from the resin with TFA/H2O/TIS (95:2.5:2.5, v/v) for 2.5 hours at rt, precipitated and successively washed with cold Et2O, and finally dried under vacuum. The crude product was characterized by analytical RP‐HPLC using method B and by MALDI‐TOF‐MS. Peptides 1b and 1c were dissolved in nitrogen‐purged ligation buffer (0.2 M phosphate buffer at pH 7, 6 M GuHCl, 20 mM TCEP, and 1% thiophenol). The mixture was shaken for 2.5 hours, then acidified by using 0.5% TFA in water and lyophilized. The ligation product was isolated by analytical RP‐HPLC using method D. The fraction containing the peak of interest was collected and lyophilized.
2.5. Synthesis of [Thz‐371, Thr‐Sal‐397]‐IgG1‐Fc 371‐397 (2)
Peptide chain elongation was performed by Fmoc/tBu chemistry on H‐Thr(tBu)‐2‐Cl‐trityl resin (loading: 0.73 mmol/g; 18‐μmol scale) as described in the subsection 2.3. The protected peptide was cleaved from the resin by using six 30‐minute treatments with TFE/DCM (2:8, v/v). The combined filtrates resulting from each treatment were collected and concentrated in vacuum, and the fully protected peptide was precipitated with a cold mixture of Et2O/hexane (2:8, v/v), recovered by centrifugation and vacuum‐dried overnight. The solid was dissolved in DCM/DMF (9:1, v/v), and the resulting solution was cooled to 0°C before 8 equivalents DIC and 10 equivalents salicylaldehyde dimethyl acetal were added. The mixture was stirred first at 0°C for 3 hours, and then at rt for 16 hours. The solvent was evaporated, and the product was precipitated from cold Et2O/hexane (2:8, v/v) and washed twice with the same mixture. The resulting white solid was then treated with a mixture of TFA/H2O/TIA (95:2.5:2.5, v/v) for 2 hours at rt, and the deprotected peptide was recovered by centrifugation from cold Et2O. The crude product was characterized by analytical RP‐HPLC using method B and by MALDI‐TOF‐MS.
2.6. Synthesis of [Thr‐Dbz‐370]‐IgG1‐Fc 345‐370 (3a)
Mono‐Fmoc‐Dbz‐OH was prepared starting from commercially available 3,4‐diaminobenzoic acid, then coupled to Rink amide MBHA resin (loading: 0.45 mmol) as previously described.55 Briefly 3,4‐diaminobenzoic acid (0.5 g, 3.3 mmol) was suspended in 15‐mL 0.1 M NaHCO3 (aq.)/ACN (1:1), and Fmoc‐OSu (1.1 g, 3.3 mmol) was added over 20 minutes. The reaction was stirred for 6 hours, then the pH was brought to 2 by adding HCl (aq.). The resulting white precipitate was filtered, washed with water, cold Et2O, hexane, and dried under vacuum to afford the expected mono‐Fmoc‐Dbz‐OH (0.6 g, 48%). The latter (0.04 g, 0.11 mmol) was dissolved in DMF, and the resulting solution was added to pre‐swelled Rink amide MBHA resin (0.05 g, 0.022 mmol) together with HBTU (0.042 g, 0.11 mmol) and DIPEA (40 μl, 0.22 mmol) and stirred for 2 hours. The resin was washed three times with DCM and DMF and subjected to Fmoc deprotection of the linker with 20% piperidine in DMF for 20 minutes. Subsequently, the resin was treated with 5 equivalents Fmoc‐Thr(tBu)‐OH, 5 equivalents HATU and 10 equivalents DIPEA, for 1 hour at rt; the coupling was performed a second time to assure complete acylation. The loading was determined by measurement of the UV absorbance of the dibenzofulvene‐piperidine adduct at 301 nm. The peptide chain was elongated by Fmoc/tBu chemistry as described in the subsection 2.3. The resin‐bound peptide was then treated with a mixture of TFA/TIS/TIA (95:2.5:2.5, v/v) for 2.5 hours at rt, and the peptide was recovered by centrifugation from cold Et2O. The crude product was characterized by analytical RP‐HPLC using method B and by MALDI‐TOF‐MS.
2.7. Synthesis of [Thz‐371, Ser‐Nbz‐428]‐IgG1‐Fc 371‐428 (5)
Fragment 5 was synthesized with the protocol that was used for 1c. An additional pseudoproline dipeptide, Fmoc‐Glu(tBu)‐Ser (ψMe,Mepro)‐OH, was used in the SPPS. The crude product was characterized by analytical RP‐HPLC using method B and by MALDI‐TOF‐MS.
2.8. Synthesis of [Cys(Acm)‐371,429, Cys‐390,428]‐IgG1‐Fc 371‐450 by chemoselective ligation (10)
Peptides 6 and 8 were assembled on a H‐Gly‐2‐Cl‐trityl resin (loading: 0.63 mmol/g; 18‐μmol scale) with the protocol described in the subsection 2.3. Peptide 6 was cleaved from the resin and deprotected by using a mixture of TFA/H2O/TIA/TIS/EDT (90:3:2:2:3, v/v) for 3 hours at rt and then recovered by precipitation from cold Et2O and centrifugation. The crude product was characterized by analytical RP‐HPLC using method A and by MALDI‐TOF‐MS. For peptide 8, the peptidyl‐resin was treated for 30 minutes with a mixture of TFE/DCM (2:8, v/v), and the procedure was repeated six times. The combined filtrates resulting from each treatment were collected, concentrated in vacuum, and the fully protected peptide was precipitated with cold Et2O. After centrifugation and overnight drying under vacuum, the solid was dissolved in DCM/DMF (7:3, v/v), and the resulting solution was cooled at 0°C before 3 equivalents DIC and 5 equivalents benzylmercaptan were added. The mixture was stirred at 0°C for 1 hour, and at rt for 16 hours. Afterwards, the solvent was evaporated, then the product was precipitated with cold Et2O and washed twice with the same solvent. The resulting white solid was dried under vacuum and then treated with a mixture of TFA/H2O/TIA (95:2.5:2.5, v/v) for 2 hours, at rt, and the peptide was recovered by precipitation from cold Et2O and centrifugation. The crude product was characterized by analytical RP‐HPLC using method B and by MALDI‐TOF‐MS. Peptide 7a was assembled on the commercially available Fmoc‐Dbz‐NovaSyn TGR resin (loading: 0.2 mmol/g; 9‐μmol scale). The Fmoc group of the linker was cleaved with 20% piperidine in DMF for 20 minutes, and the resin was treated with 5 equivalents Fmoc‐Phe‐OH, 5 equivalents HATU, and 10 equivalents DIPEA for 1 hour at rt. The resin was then washed with DMF, DCM, and Et2O (three times each). After drying under vacuum, the degree of substitution was determined by measurement of the UV absorbance of the dibenzofulvene‐piperidine adduct at 301 nm. The peptide chain was assembled by Fmoc/tBu chemistry as described in the subsection 2.3, with the use of the pseudoproline dipeptides Fmoc‐Lys(Boc)‐Ser(ψMe,Mepro)‐OH, Fmoc‐Tyr(tBu)‐Ser(ψMe,Mepro)‐OH and Fmoc‐Asp(tBu)‐Ser(ψMe,Mepro)‐OH within the SPPS. The coupling of Gly‐424 was performed manually with Fmoc‐Gly‐OPfp/HOBt 1:1 (6 equivalents) for 1 hour in DMF at rt, and the progress of the reaction was monitored by the ninhydrin test. Once the synthesis was completed, the resin was swollen in DCM for 30 minutes and treated with 5 equivalents p‐nitrophenylchloroformate dissolved in DCM (50 mM) for 1 hour under nitrogen at rt. The solvent was sucked off, the resin was washed with DCM, DMF, and again DCM (three times each), and finally subjected to a 30‐minute treatment with 0.5 M DIPEA in DMF at rt. After washing cycles with DMF, DCM, and Et2O, the peptide was cleaved from the resin with TFA/H2O/TIS (95.2.5:2.5, v/v) for 2.5 hours at rt, precipitated and successively washed with cold Et2O, and finally dried under vacuum. The crude product was characterized by analytical RP‐HPLC using method B and by MALDI‐TOF‐MS. Peptides 6 and 7a were dissolved in nitrogen‐purged ligation buffer (0.2 M phosphate buffer at pH 7, 6 M GuHCl, 90 mM TCEP, and 180 mM MPAA). The mixture was shaken for 2.5 hours, then 20 μL of a nitrogen‐flushed 1.4 M methoxylamine solution containing 200 μM TCEP dissolved in 0.2 M Na2HPO4 and 6 M GuHCl were added, and the resulting mixture was acidified to pH 4 and stirred for 6 hours at rt. The mixture was then eluted through a column prepacked with Sephadex G‐25 (NAP GE Healthcare) using 1 M GuHCl as elution system. After lyophilization, the solid and peptide 8 were dissolved in nitrogen‐flushed ligation buffer (0.2 M phosphate buffer at pH 6.8, 6 M GuHCl containing 50 mM TCEP and 100 mM MPAA). After 2.5 hours, the reaction mixture was acidified by using 0.5% TFA in water and lyophilized. The ligation product was isolated by analytical RP‐HPLC using method B. The fraction containing the peak of interest was collected, lyophilized, and characterized by analytical RP‐HPLC using method B and by MALDI‐TOF‐MS.
3. RESULTS AND DISCUSSION
3.1. Ligation routes
Our work toward the chemical synthesis of the IgG1 Fc CH3 domain consisted of the preparation of fragment precursors that were designed on the base of the three ligation routes shown in Scheme 1. In the first route, the 53‐residue long fragment precursor 1 should be ligated to the 27‐residue long fragment 2 by salicylaldehyde ester (Sal)‐Thr ligation: the chemoselective reaction between the C‐terminal Sal of 2 and the N‐terminal Thr of 1 in pyridine/AcOH forms an N‐acyl oxazolidine intermediate that can be cleaved by acidic hydrolysis to afford the native peptide bond at the ligation site52, 53 (Scheme 1A). The resulting ligation product should be subjected to thiazolidine (Thz) ring opening, followed by Cys‐thioester ligation43 with the 26‐residue long fragment 3. The second route exploits the presence of native Cys‐429 and Cys‐371 and the sequential NCL between the precursors 3, 4 and 5 (Scheme 1B). Both routes involve the synthesis of one large precursor (either 1 with 53 residues or 5 with 58 residues), which may represent a hurdle for the synthesis of the precursor as well as for the ligation reaction. Moreover, the presence of a C‐terminal threonine at the ligation site may slow down the reaction rate, as shown by a systematic study conducted by Dawson and coworkers42 on all 20 proteinogenic AAs, which classified threonine as one of the slowest ligation sites after valine, isoleucine, and proline. Then, we envisioned a third route that contemplates the Cys‐thioester ligation of four fragments, the largest one containing 38 residues (7), at two fast‐reacting (Gly‐389 and Phe‐427) and one slow‐reacting (Thr‐370) C‐terminal thioester (Scheme 1C). This, however, requires the temporary replacement of two native residues, Gln‐390 and Ser‐428, with Cys, and the reconversion of the two non‐native residues to the native ones with suitable methods (see section 3.7).
3.2. Attempts of stepwise synthesis of [Met(O)‐432]‐IgG1‐Fc 398‐450 (1)
Fragment 1 contains the C‐terminal part of the IgG1 Fc CH3 domain, covering 53 residues and including the PTM site Met(O)‐432. At first, we proved the feasibility of this fragment to be assembled by stepwise SPPS with Fmoc/tBu chemistry. We started the synthesis on a polystyrene‐divinylbenzene‐2‐chlorotrityl resin preloaded with glycine (0.63 mmol/g) (Scheme 2A). Met‐432 was inserted as Met(O) to take into account the PTM. We checked the growing chain after 22 and 32 cycles: unfortunately, the homogeneity of the crude product dropped from ~80% to ~20% (1a TRT in Scheme 2A). Thus, we decided to repeat the synthesis of 1 by choosing a polar resin56, 57 with low loading (Fmoc‐Gly‐NovaSyn TGT resin, based on low cross‐linked hydroxyethylpolystyrene‐polyethylene glycol, with a loading of 0.2 mmol/g). However, these two parameters (polarity and low loading of the solid support) were not sufficient to accomplish the synthesis of the desired 53‐residue long fragment, as the growing chain was highly inhomogeneous after 44 cycles. Neither the addition of 2% DBU to the conventional 20% piperidine in DMF,58 nor the use of HATU in place of HBTU for the activation of selected AAs improved the synthesis (data not shown).
Scheme 2.

Attempts of stepwise synthesis of [Met(O)‐432]‐IgG1‐Fc 398–450 (1). A, Assembly attempt on polystyrene‐divinylbenzene 2‐chlorotrityl resin preloaded with glycine (0.63 mmol/g). The RP‐HPLC profiles of the crude peptide acids with free N‐terminus at cycle 22 (till Cys‐429) and 32 (till Ser‐419, 1a TRT) were obtained by using method A. B, Assembly attempt on the polar and low‐loaded resin NovaSyn TGT (0.2 mmol/g Fmoc‐glycine) by using three pseudoproline dipeptides (in red). The RP‐HPLC profile of the Fmoc‐protected crude peptide acid at cycle 44 (till Ser‐407, 1a TGT+ψ) was obtained by using method C with the Nucleosil C‐18 column (MALDI‐TOF‐MS peaks for M + H+ and [M + 2H+]/2. Mcalc. for C242H352N62O69S2: 5298.01 Da)
Considering the presence of quite regularly distributed Thr and Ser residues in the sequence, we envisioned the possibility to increase the homogeneity of fragment 1 by incorporation of pseudoproline dipeptides, which are well known to reduce the on‐resin self‐association propensity of the growing peptide and, consequently, to improve the quality of the crude product.59, 60, 61 Accordingly, by using the combination of a polar and low‐loaded resin with the employment of the pseudoproline dipeptides Leu‐445‐ψSer‐446, Phe‐427‐ψSer‐428, and Tyr‐411‐ψSer‐412, the growing chain was ~60% homogeneous after 44 couplings (1a TGT+ψ in Scheme 2B). Nevertheless, despite this encouraging result, the attempt to elongate this fragment with additional nine residues to obtain the desired precursor 1 failed (data not shown).
3.3. Synthesis of [Met(O)‐432]‐IgG1‐Fc 398‐450 (1) by chemoselective ligation
Given the difficulties encountered in the standard stepwise Fmoc‐SPPS of the fragment precursor 1, we considered the chemoselective ligation approach to accomplish the 53‐residue long fragment by using the natural Cys‐429 as ligation point between the two fragments 1b (= 4 in Scheme 1) and 1c (Scheme 3). As shown in Scheme 2A, the C‐terminal fragment 1b could be successfully prepared by standard Fmoc‐SPPS on the polystyrene‐divinylbenzene 2‐chlorotrityl resin preloaded with glycine (0.63 mmol/g). For the synthesis of the peptide thioester 1c, we chose the N‐acyl‐urea approach,55 in which the peptide is synthesized on a diaminobenzoic (Dbz) acid linker by Fmoc‐SPPS, followed by treatment with p‐nitrophenylchloroformate and, then, a tertiary base to afford the corresponding N‐acyl‐benzimidazolinone (Nbz). The latter can be either used directly for the NCL62, 63 or subjected to thiol exchange reaction providing access to the corresponding peptide thioester.64, 65 Thus, we accomplished the synthesis of 1c starting from the low‐loaded Fmoc‐Dbz NovaSyn TGR resin (0.2 mmol/g). The first residue, Fmoc‐Ser(tBu)‐OH, was attached manually by using HATU/DIPEA. Then, the peptide chain was elongated by automated SPPS using the HBTU/HOBt/DIPEA coupling protocol, except for Gly‐424 that was coupled manually using Fmoc‐Gly‐OPfp (Pfp = pentafluorophenyl) to reduce the formation of branched by‐products arising from the double acylation of the Dbz moiety. Also, in this case, we used pseudoproline dipeptides (Lys‐418‐ψSer‐419, Tyr‐411‐ψSer‐412, and Asp‐403‐ψSer‐404), as the IgG1 Fc CH3 sequence 398‐428 resulted to be otherwise very difficult to assemble (data not shown). After the coupling of the last residue as Nα‐Boc‐protected AA, the C‐terminal Dbz group was converted into the Nbz group, and TFA cleavage gave the desired C‐terminal Nbz peptide 1c with satisfactory homogeneity (~60%) (Scheme 3A).
Scheme 3.
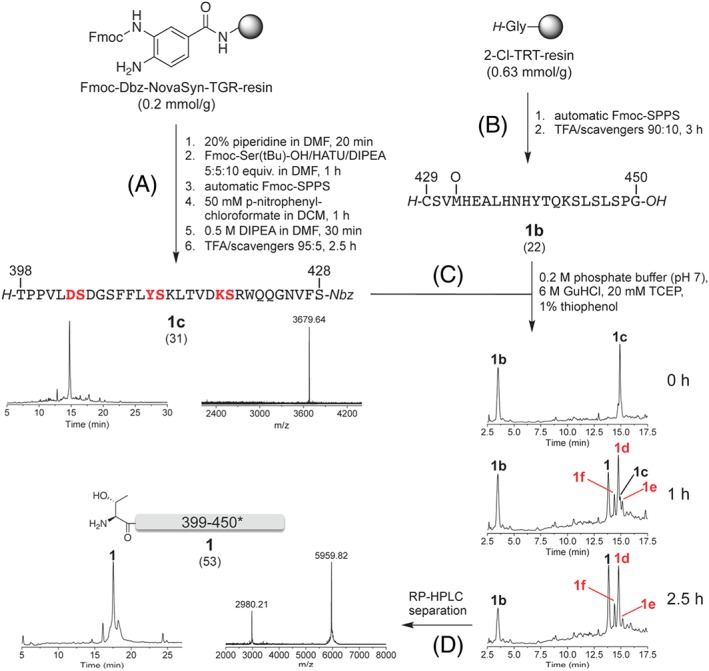
Synthesis of [Met(O)‐432]‐IgG1‐Fc 398–450 (1) by chemoselective ligation. A, Fragment 1c was prepared on a Fmoc‐Dbz NovaSyn TGR resin by using three pseudoproline dipeptides (in red). The RP‐HPLC profile of the crude product was obtained with method B (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C169H245N43O50: 3679.10 Da). B, Fragment 1b was assembled on a 2‐chlorotrityl resin preloaded with glycine (the RP‐HPLC profile is shown in Scheme 2A). C, Ligation of 1c and 1b was followed by RP‐HPLC over 2.5 h. The peaks labelled with 1d‐f are degradation products of 1c due to hydrolysis (1d: MALDI‐TOF‐MS peak for M + H+: 3521.35 Da. Mcalc. for C161H240N40O49: 3519.95 Da), intramolecular aminolysis (1e: MALDI‐TOF‐MS peak for M + H+: 3503.52 Da. Mcalc. for C161H238N40O48: 3501.93 Da), and N‐acylation of guanidine with the Nbz‐peptide (1f: MALDI‐TOF‐MS peak for M + H+: 3562.72 Da. Mcalc. for C162H243N43O48: 3561.0 Da). The HPLC runs were performed by using method B. D, The ligation product 1 was separated from the impurities by HPLC fraction collection. The corresponding RP‐HPLC profile was obtained by using method D (MALDI‐TOF‐MS peaks for M + H+ and [M + 2H+]/2. Mcalc. for C265H401N71O82S2: 5957.71 Da)
Fragments 1b and 1c were used for the NCL without any further purification (Scheme 3C). After 1 hour, a significant amount of the desired ligation product 1 could be detected; however, also a large amount of the acid arising from the hydrolysis of 1c was formed (1d). Moreover, two other by‐products were identified, having a mass difference of −18 Da (1e) and +41 Da (1f) with respect to the acid 1d (Scheme 3C): the first one (1e) might arise from an intramolecular cyclization of the thioester precursor 1c or of the corresponding phenyl thioester formed in situ. The second one (1f) might be the result of the reaction of 1c or of the corresponding phenyl thioester formed in situ with guanidine.66
After 2.5 hours, the C‐terminal Nbz peptide 1c was completely consumed, and no further changes were observed with respect to the previous control (Scheme 3C). Therefore, the reaction mixture was acidified, and the HPLC fraction containing the ligation product 1 was collected and lyophilized. The mass spectrum confirmed the successful separation of the desired ligation product 1 from all impurities present in the NCL mixture. However, an additional peak at slightly higher retention time appeared in the HPLC profile, whose identity could not be assessed by mass spectrometry.
3.4. Synthesis of [Thz‐371, Thr‐Sal‐397]‐IgG1‐Fc 371‐397 (2)
Initially, we attempted to prepare fragment 2 by on‐resin phenolysis of the corresponding C‐terminal Nbz peptide with salicylaldehyde dimethyl acetal.52, 67 However, despite successful elongation of the peptide chain on the Fmoc‐Dbz Rink amide MBHA resin (0.45 mmol/g), both the activation of the Dbz linker with p‐nitrophenylchloroformate and the subsequent on‐resin phenolysis were poorly efficient, probably due to the sterically hindered C‐terminal Thr(tBu)‐397. Therefore, we looked for alternative solutions. Owing to the fact that the presence of a tryptophan residue in the sequence excluded the use of any protocol involving aldehyde generation by means of post‐SPPS oxidative processes,68 the esterification in solution was chosen. The peptide was assembled on the H‐Thr(tBu)‐chlorotrityl resin (0.73 mmol/g) and released from the solid support under mildly acidic conditions (Scheme 4). The fully protected peptide acid was esterified with salicylaldehyde dimethyl acetal by using DIC as condensation agent, followed by TFA treatment for the removal of the side‐chain protecting groups. HPLC and MS analysis showed that the desired peptide could be obtained with low homogeneity (~30% for the major HPLC peak in Scheme 4). Indeed, some impurities due to deleted sequences and double coupling of salicylaldehyde were present (MS in Scheme 4). We decided not to purify the crude product for further use in the ligation, as solubility tests of 2 under the conditions reported for Sal‐Ser/Thr ligation (at least 1 mM in pyridine/AcOH52, 53, 69) were unsatisfactory (good solubility of 2 was obtained in pure AcOH, which, however, would not allow for efficient ligation). Another concern for the use of 2 in the ligation strategy was related to the possibility of 5(4H)‐oxazolone‐mediated epimerization of the C‐terminal threonine during the esterification step of the fully protected fragment (Scheme 4). To this regard, it would have been of benefit to incorporate the C‐terminal threonine as pseudoproline,70 or to couple it as aldehyde‐protected amino salicylaldehyde ester to the fully protected peptide by using the Sakakibara conditions.71, 72
Scheme 4.
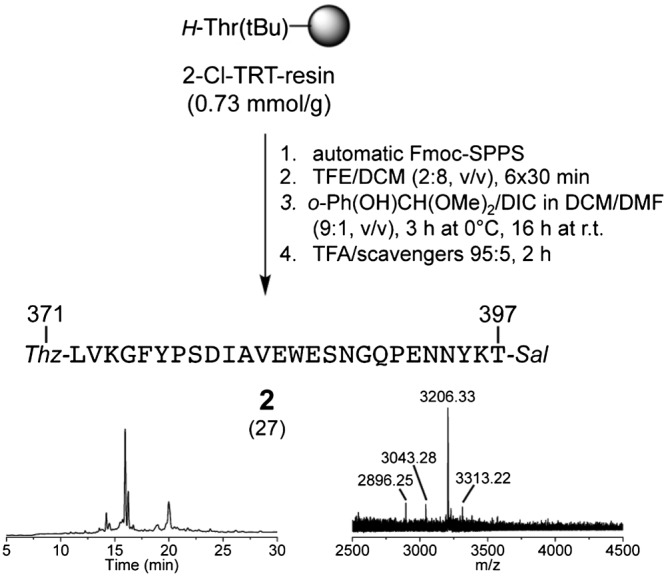
Synthesis of [Thz‐371, Thr‐Sal‐397]‐IgG1‐Fc 371–397 (2) by esterification in solution. The RP‐HPLC profile of the crude product was obtained with method B (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C146H206N34O46S1: 3205.53 Da. The masses at 2896.25, 3043.28, and 3313.22 Da were attributed to impurities with ΔFY, ΔY, or condensation with an additional salicylaldehyde)
3.5. Synthesis of [Thr‐Dbz‐370]‐IgG1‐Fc 345‐370 (3a)
For the synthesis of the C‐terminal Nbz peptide 3, we first functionalized the Rink amide MBHA resin (0.45 mmol/g) with mono‐Fmoc‐diaminobenzoic acid synthetized from commercially available 3,4‐diaminobenzoic acid. The first residue, Fmoc‐Thr(tBu)‐OH, was coupled manually with HATU/DIPEA, followed by spectrophotometric determination of the Fmoc group, which confirmed the success of the coupling. The peptide chain was then elongated by automated SPPS with the HBTU/HOBt/DIPEA coupling protocol. After the assembly was completed, we attempted to convert the Dbz group into the Nbz group. However, the conversion was incomplete, probably due to the sterically hindered C‐terminal Thr(tBu)‐370. Thus, we decided to cleave the peptide from the resin in the Dbz form 3a, which is more stable than the Nbz form 3 but may be orthogonally converted into a benzotriazole moiety with NaNO2 and then displaced by the thiol additive in the ligation buffer to afford the peptide thioester in situ.73 The TFA‐cleaved product 3a showed satisfactory homogeneity (~60%) (Scheme 5).
Scheme 5.

Synthesis of [Thr‐Dbz‐370]‐IgG1‐Fc 345‐370 (3a). The RP‐HPLC profile of the crude product was obtained with method B (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C135H216N40O43: 3087.47 Da)
3.6. Synthesis of [Thz‐371, Ser‐Nbz‐428]‐IgG1‐Fc 371‐428 (5)
Fragment 5 was prepared by following the same protocol used for the synthesis of fragment 1c (Scheme 3). To maintain a periodic distribution of the pseudoprolines, an additional pseudoproline dipeptide (Glu‐386‐ψSer‐387) was incorporated. After activation of the Dbz linker and TFA cleavage, the desired 58‐residue long C‐terminal Nbz peptide was obtained with poor homogeneity (~25%), as shown in Scheme 6. In an attempt to purify 5 by HPLC, we encountered some difficulties that were associated with the tendency of the C‐terminal Nbz moiety to hydrolyze as well as with the poor solubility of the crude peptide in the HPLC elution system (TFA/ACN/H2O).
Scheme 6.

Synthesis of [Thz‐371, Ser‐Nbz‐428]‐IgG1‐Fc 371‐428 (5). The fragment was prepared on a Fmoc‐Dbz NovaSyn TGR resin by using four pseudoproline dipeptides (in red). The RP‐HPLC profile of the crude product was obtained with method B (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C308H445N77O94S1: 6762.50 Da)
3.7. Assembly of [Cys(Acm)‐371,429, Cys‐390,428]‐IgG1‐Fc 371‐450 (10)
Although known strategies addressing the synthesis of C‐terminal salicylaldehyde peptide esters offer a solution to the epimerization risk of fragment 2,71, 72 its very poor solubility in pyridine/AcOH prompted us to explore a way to perform the assembly of the IgG1 fragment 371 to 450 in aqueous solution by Cys‐based NCL. However, this requires the presence of cysteine residues at suitable ligation sites, which is not the case for the native Cys‐371 and Cys‐429 (see Scheme 3 for ligation at Cys‐429: although the ligation product was quickly built, concurrent side‐reactions leading to thioester degradation were also fast). Therefore, Ser‐428 and Gln‐390 were substituted with cysteine, in order to exploit highly ligation‐favourable sites at Phe‐427 and Gly‐38942 and also to have the possibility to achieve the assembly of the native IgG1 CH3 domain in the future (Scheme 1C). Indeed, desulfurization/deselenization protocols allow performing NCL also in the absence of native cysteine residues. In this specific case, selenocysteine54 and γ‐mercapto‐glutamine74 would be suitable precursors for Ser‐428 and Gln‐390. With this in mind, we incorporated the native Cys‐429 and Cys‐371 in the Acm‐protected form.
For the synthesis of [Cys(Acm)‐371,429, Cys‐390,428]‐IgG1‐Fc 371–450 (10), we prepared three new fragments (6–8) as shown in Scheme 7. Fragment 6 was obtained with ~60% homogeneity. Fragment 7a (the N‐terminally Thz‐protected version of 7) was obtained by Fmoc‐SPPS on low‐loaded Fmoc‐Dbz NovaSyn TGR resin that was preloaded manually with Fmoc‐Phe‐OH. Three pseudoproline dipeptides were incorporated (Lys‐418‐ψSer‐419, Tyr‐411‐ψSer‐412, and Asp‐403‐ψSer‐404). Fragment 8 was prepared by C‐terminal thioesterification of the fully protected peptide acid in solution, and its homogeneity was ~60%.
Scheme 7.

Synthesis of the fragment precursors 6, 7a, and 8 for the NCL‐mediated assembly of [Cys(Acm)‐371,429, Cys‐390,428]‐IgG1‐Fc 371‐450 (10). A, Fragment 6 was prepared on a 2‐chlorotrityl resin preloaded with glycine. The RP‐HPLC profile of the crude product was obtained with method A (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C110H173N33O36S3: 2630.00 Da. The other annotated masses correspond to sodium and potassium adducts and plus tBu group. Lower masses were also detected, indicating the presence of deleted sequences). B, Fragment 8 was prepared on a 2‐chlorotrityl resin preloaded with glycine. The RP‐HPLC profile of the crude product was obtained with method B (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C106H151N23O30S2: 2291.65 Da. The other annotated masses indicate the presence of deleted sequences and a tBu group). C, Fragment 7a was prepared on a Fmoc‐Dbz NovaSyn TGR resin by using three pseudoproline dipeptides (in red). The RP‐HPLC profile of the crude product was obtained with method B (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C207H299N53O62S1: 4554.07 Da)
Then, we proceeded with the ligation of the three peptides, starting with 6 and 7a that were mixed together in phosphate buffer (pH 7) containing 6 M GuHCl (Scheme 8A). MPAA was added for in situ conversion of the N‐acyl‐Nbz moiety into an aryl thioester. After 2‐hour reaction, methoxylamine was added at pH 4 and left react for 6 hours to convert the N‐terminal thiazolidine into cysteine and deliver peptide 9. Some impurities were present, which arose from the hydrolysis and methoxylaminolysis of the residual amount of unreacted peptide 7a. Before performing the second ligation with the peptide thioester 8, the mixture was eluted through a pre‐packed gel‐filtration column to remove the excess of methoxylamine that, otherwise, would have reacted with 8. The eluate was lyophilized, re‐dissolved in the ligation cocktail at pH 6.8 in the presence of MPAA, and left react with the peptide thioester 8 till full consumption of the latter (Scheme 8B). The ligation product 10 was finally isolated by RP‐HPLC; however, some MPAA as well as some impurities due to deleted sequences were still present.
Scheme 8.

Synthesis of [Cys(Acm)‐371,429, Cys‐390,428]‐IgG1‐Fc 371‐450 (10) by tandem chemoselective ligation. A, Fragments 6 and 7a were ligated in the presence of MPAA. The ligation product 9 was obtained after treatment with NH2OMe. The RP‐HPLC profile of the crude product was obtained with method B. The mass spectrum of the major HPLC peak is shown (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C308H465N83O96S4: 6994.89 Da). The HPLC peaks labelled with * were attributed to the N‐terminally deprotected peptide acid deriving from hydrolysis of peptide 7a (MALDI‐TOF‐MS peak for M + H+ = 4384.76 Da. Mcalc. for C198H294N50O61S1: 4382.91 Da) and its C‐terminal methoxylamide derivative (MALDI‐TOF‐MS peak for M + H+ = 4414.20 Da. Mcalc. for C199H297N51O61S1: 4411.96 Da). B, The second ligation between 9 and 8 was performed after removal of excess NH2OMe by gel filtration. Also, in this case, MPAA was added to convert the benzyl thioester into an aryl thioester in situ. After 2.5 h, the ligation product 10 was isolated by RP‐HPLC. The RP‐HPLC profile of the collected fraction was obtained with method B (MALDI‐TOF‐MS peak for M + H+. Mcalc. for C407H608N106O126S5: 9162.34 Da. The annotated lower masses indicate the presence of deleted sequences)
4. CONCLUSIONS
In this work, we evaluated the synthesis of different fragments of the IgG1 Fc CH3 domain with regard to peptide‐chain length and preparation of C‐terminal thioesters or thioester precursors. Stepwise elongation from the C‐terminal Gly‐450 by Fmoc‐SPPS became challenging after Asn‐425. However, the simultaneous use of pseudoproline dipeptides and a polar, low‐loaded resin allowed reaching Ser‐407 (fragment 1a TGT+ψ with 44 residues). Unfortunately, this fragment could not be elongated further, but it was possible to assemble the C‐terminal fragment 398 to 450 (1) by using NCL between an Nbz‐activated peptide (1c, 398‐428) and an N‐terminal Cys‐peptide (1b, 429‐450). These results suggest that the C‐terminal region, once reached a length of more than 22 residues, starts to aggregate, thus preventing an efficient stepwise elongation. The presence of β‐sheet breakers like pseudoprolines and the use of low‐loaded, highly swelling resins partially reduced the formation of aggregates, but this was not sufficient to obtain sequences larger than 44 residues, as also shown for the 58‐residue long fragment 371 to 428 (5) (Figure 2). However, optimization of the protocol for the stepwise Fmoc‐SPPS might still be possible by the application of polyethylene glycol resins like ChemMatrix,75 backbone‐amide protecting groups like Dmb (dimethoxybenzyl)76, 77 and Hmb (hydroxymethoxybenzyl),78 or the O‐acyl isopeptide method.79, 80, 81, 82, 83 Moreover, heat‐assisted SPPS has been recently very successful in the synthesis of very difficult sequences.84, 85, 86, 87, 88, 89, 90, 91
Figure 2.
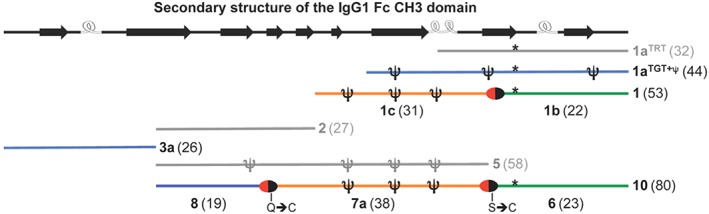
Summary of the synthetic IgG1 Fc CH3 fragments of this study. All fragment precursors were obtained with homogeneity of ~60% as crude products, except those colored in grey that were less homogeneous. The red/black ovals represent the ligation sites. The * represents Met(O)‐432. On the top, the secondary structure (β‐strands and helical turns) composition of the IgG1 Fc CH3 domain is shown (based on the crystal structure with PDB ID 1OQO)
We did not investigate the accessibility of the N‐terminal fragment 345 to 397 by stepwise elongation, but rather prepared the two shorter fragments 2 and 3. Unfortunately, the Sal‐activated peptide 2 was poorly soluble in the ligation solvent (pyridine/AcOH). Thus, the assembly route to obtain the IgG1 Fc CH3 sequence was redesigned on the base of only Cys‐based NCL. To test the feasibility of this route, besides using the native Cys‐371, we decided to incorporate two non‐native Cys residues in place of Ser‐428 and Gln‐390. The choice of the ligation points at these sites was dictated by the following reasons: (1) the length of the four fragment precursors was between 19 and 38; (2) the presence of the non‐native Cys‐428 allows the ligation with a fast‐reacting C‐terminal glycine thioester; (3) the ligation at the native Cys‐429 was not ideal due to fast degradation of the thioester precursor, which led us move the ligation site at the non‐native Cys‐428; and (4) in future, the use of Sec‐428 and γ‐mercapto‐Gln‐390 would allow obtaining the native Ser‐428 and Gln‐390 residues upon deselenization/desulfurization. Three of these fragments were ligated in a tandem reaction to give the sequence 371–450 (10). Although this tandem ligation looks promising, the third ligation might be challenging, due to the presence of threonine at the ligation site (Scheme 1). We conducted preliminary ligation experiments between 3a (Scheme 5) and the C‐terminal free acid of 8 (Scheme 7B). Peptide 3a contains the Dbz moiety at the C‐terminal Thr‐370. As the Dbz peptide precursor 3a could not be converted into the Nbz form by using 4‐nitrophenylchloroformate in DCM, we repeated the reaction in DMF: indeed, Brik and coworkers reported that the use of DMF instead of DCM could convert Dbz into Nbz on a peptide bearing a C‐terminal leucine.63 Unfortunately, in our case, the use of DMF did not solve the problem, and no Nbz peptide was formed (probably, because of the presence of the β‐branched threonine). Thus, we activated the Dbz peptide 3a by converting it into an N‐acyl‐benzotriazole peptide by using NaNO2 at pH~3, as previously shown by Liu and coworkers.73 For the Acm cleavage from the C‐terminal free acid of 8, we used the Pd‐catalyzed reaction reported by Brik and coworkers.92 Both reactions were successful, but, unfortunately, no ligation product was detected after 18‐hour reaction. We did not try to change further the protocol, as the slow ligation rate of the C‐terminal threonine is likely to prevent an efficient synthesis. An alternative would be to move the third ligation site between Gly‐375 and Phe‐376, which would be advantageous for the preparation of the C‐terminal Nbz peptide precursor as well as for the ligation rate. However, β‐mercapto‐Phe must be used, whose synthesis and use in NCL have been previously reported.93 Another way would be the stepwise synthesis of the segment 345 to 389, which would avoid a third ligation.
In conclusion, this explorative study has shed light on the behaviour of different fragments of the IgG1 Fc CH3 domain during the standard Fmoc‐SPPS as well as NCL, which will be useful to plan future syntheses. Once completed the CPS of the CH3 domain, it will be necessary to test, if the synthetic bis(cysteinyl)‐polypeptide chain can undergo oxidative folding and build the expected non‐covalent homodimer.
ACKNOWLEDGEMENTS
C. C. kindly acknowledges the Land Salzburg and the University of Salzburg for financial support. The financial support by the Austrian Federal Ministry of Science, Research, and Economy and by a Start‐up Grant of the State of Salzburg is gratefully acknowledged.
Grassi L, Roschger C, Stanojlović V, Cabrele C. An explorative study towards the chemical synthesis of the immunoglobulin G1 Fc CH3 domain. J Pep Sci. 2018;24:e3126 10.1002/psc.3126
REFERENCES
- 1. Bhupinder SS. Biopharmaceuticals: an overview. Thai J Pharm Sci. 2010;34:1‐19. [Google Scholar]
- 2. Czajkowsky DM, Hu J, Zhifeng S, Pleass RJ. Fc‐fusion proteins: new developments and future perspectives. EMBO Mol Med. 2012;4(10):1015‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu‐Justin KH. The history of monoclonal antibody development—progress, remaining challenges and future innovations. Ann Med Surg. 2014;3(4):113‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu SZ, Shively L, Raubitschek A, et al. Minibody: a novel engineered anti‐carcinoembryonic antigen antibody fragment (single‐chain Fv‐CH3) which exhibits rapid, high‐level targeting of xenografts. Cancer Res. 1996;56(13):3055‐3061. [PubMed] [Google Scholar]
- 5. Beckman RA, Weiner LM, Davis HM. Antibody constructs in cancer therapy. Cancer. 2007;109(2):170‐179. [DOI] [PubMed] [Google Scholar]
- 6. Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6(5):343‐357. [DOI] [PubMed] [Google Scholar]
- 7. Haussner C, Lach J, Eichler J. Synthetic antibody mimics for the inhibition of protein‐ligand interactions. Curr Opin Chem Biol. 2017;40:72‐77. [DOI] [PubMed] [Google Scholar]
- 8. Lee H, Pardridge W. Monoclonal antibody radiopharmaceuticals: cationization, pegylation, radiometal chelation, pharmacokinetics, and tumor imaging. Bioconjug Chem. 2003;14(3):546‐553. [DOI] [PubMed] [Google Scholar]
- 9. Rodwell JD, Alvarez VL, Lee C, et al. Site‐specific covalent modification of monoclonal antibodies: in vitro and in vivo evaluations. Proc Natl Acad Sci U S A. 1986;83(8):2632‐2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang T, Bourret J, Cano T. Isolation and characterization of therapeutic antibody charge variants using cation exchange displacement chromatography. J Chromatogr B. 2011;1218(31):5079‐5086. [DOI] [PubMed] [Google Scholar]
- 11. Chumsae C, Gaza‐Bulseco G, Sun J, Liu H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibody. J Chromatogr B. 2007;850(1‐2):285‐294. [DOI] [PubMed] [Google Scholar]
- 12. Teshima G, Li M, Danishmand R, et al. Separation of oxidized variants of a monoclonal antibody by anion‐exchange. J Chromatogr A. 2011;1218(15):2091‐2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim J, Jones L, Taylor L, et al. Characterization of a unique IgG1 mAb CEX profile by limited Lys‐C proteolysis/CEX separation coupled with mass spectrometry and structural analysis. J Chromatogr B. 2010;878(22):1973‐1981. [DOI] [PubMed] [Google Scholar]
- 14. Zheng JY, Janis LJ. Influence of pH, buffer species, and storage temperature on physicochemical stability of a humanized monoclonal antibody LA298. Int J Pharm. 2006;308(1‐2):46‐51. [DOI] [PubMed] [Google Scholar]
- 15. Zhang YT, Hu J, Pace AL, Wong R, Wang YJ, Kao Y‐H. Characterization of asparagine 330 deamidation in an Fc‐fragment of IgG1 using cation exchange chromatography and peptide mapping. J Chromatogr B. 2014;965:65‐71. [DOI] [PubMed] [Google Scholar]
- 16. Grassi L, Regl C, Wildner S, et al. Complete NMR assignment of succinimide and its detection and quantification in peptides and intact proteins. Anal Chem. 2017;89(22):11962‐11970. [DOI] [PubMed] [Google Scholar]
- 17. Moorhouse KG, Nashabeh W, Deveney J, Bjork NS, Mulkerrin MG, Ryskamp T. Validation of an HPLC method for the analysis of the charge heterogeneity of the recombinant monoclonal antibody IDEC‐C2B8 after papain digestion. J Pharm Biomed Anal. 1997;16(4):593‐603. [DOI] [PubMed] [Google Scholar]
- 18. Yu L, Vizel A, Huff MB, Young M, Remmele RL Jr, He B. Investigation of N‐terminal glutamate cyclization of recombinant monoclonal antibody in formulation development. J Pharm Biomed Anal. 2006;42(4):455‐463. [DOI] [PubMed] [Google Scholar]
- 19. Liu YD, Goetze AM, Bass RB, Flynn GC. N‐terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. J Biol Chem. 2011;286(13):11211‐11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Slade DJ, Subramanian V, Fuhrmann J, Thompson PR. Chemical and biological methods to detect post‐translational modifications of arginine. Biopolymers. 2014;101(2):133‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matveenko M, Cichero E, Fossa P, Becker CF. Impaired chaperone activity of human heat shock protein Hsp27 site‐specifically modified with argpyrimidine. Angew Chem Int Ed. 2016;55(38):11397‐11402. [DOI] [PubMed] [Google Scholar]
- 22. Beck A, Wagner‐Rousset E, Ayoub D, Van Dorsselaer A, Sanglier‐Cianferani S. Characterization of therapeutic antibodies and related products. Anal Chem. 2013;85(2):715‐736. [DOI] [PubMed] [Google Scholar]
- 23. Bertolotti‐Ciarlet A, Wang W, Lownes R, et al. Impact of methionine oxidation on the binding of human IgG1 to FcRn and Fcγ receptors. Mol Immunol. 2009;46(8‐9):1878‐1882. [DOI] [PubMed] [Google Scholar]
- 24. Gaza‐Bulseco G, Faldu S, Hurkmans K, Chumsae C, Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J Chromatogr B. 2008;870(1):55‐62. [DOI] [PubMed] [Google Scholar]
- 25. Liu D, Ren D, Huang H, et al. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry. 2008;47(18):5088‐5100. [DOI] [PubMed] [Google Scholar]
- 26. Vlasak J, Bussat MC, Wang S, et al. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem. 2009;392(2):145‐154. [DOI] [PubMed] [Google Scholar]
- 27. Harris R, Kabakoff B, Macchi FD, et al. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl. 2001;752(2):233‐245. [DOI] [PubMed] [Google Scholar]
- 28. Huang L, Lu J, Wroblewski VJ, Beals JM, Riggin RM. In vivo deamidation characterization of monoclonal antibody by LC/MS/MS. Anal Chem. 2005;77(5):1432‐1439. [DOI] [PubMed] [Google Scholar]
- 29. Chelius D, Rehder DS, Bondarenko PV. Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal Chem. 2005;77(18):6004‐6011. [DOI] [PubMed] [Google Scholar]
- 30. Gaza‐Bulseco G, Li B, Bulseco A, Liu H. Method to differentiate Asn deamidation that occurred prior to and during sample preparation of a monoclonal antibody. Anal Chem. 2008;80(24):9491‐9498. [DOI] [PubMed] [Google Scholar]
- 31. Mukherjee R, Adhikary L, Khedkar A, Iyer H. Probing deamidation in therapeutic immunoglobulin gamma (IgG1) by ‘bottom‐up’ mass spectrometry with electron transfer dissociation. Rapid Commun Mass Spectrom. 2010;24(7):879‐884. [DOI] [PubMed] [Google Scholar]
- 32. Shields RL, Namenuk AK, Hong K, et al. High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J Biol Chem. 2001;276(9):6591‐6604. [DOI] [PubMed] [Google Scholar]
- 33. Firan M, Bawdon R, Radu C, et al. The MHC class I‐related receptor, FcRn, plays an essential role in the maternofetal transfer of γ‐globulin in humans. Int Immunol. 2001;13(8):993‐1002. [DOI] [PubMed] [Google Scholar]
- 34. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715‐725. [DOI] [PubMed] [Google Scholar]
- 35. Petkova SB, Akilesh S, Sproule TJ, et al. Enhanced half‐life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18(12):1759‐1769. [DOI] [PubMed] [Google Scholar]
- 36. Woods RJ, Xie MH, Spreter von Kreudenstein T, Ng GY, Dixit SB. LC‐MS characterization and purity assessment of a prototype bispecific antibody. MAbs. 2013;5(5):711‐722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang D, Wynne C, Gu F, et al. Characterization of drug‐product‐related impurities and variants of a therapeutic monoclonal antibody by higher energy C‐trap dissociation mass spectrometry. Anal Chem. 2015;87(2):914‐921. [DOI] [PubMed] [Google Scholar]
- 38. Haberger M, Heidenreich AK, Schlothauer T, et al. Functional assessment of antibody oxidation by native mass spectrometry. MAbs. 2015;7(5):891‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weinstock MT, Jacobsen MT, Kay MS. Synthesis and folding of a mirror‐image enzyme reveals ambidextrous chaperone activity. Proc Natl Acad Sci U S A. 2014;111(32):11679‐11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar KSA, Bavikar SN, Spasser L, Moyal T, Ohayon S, Brik A. Total chemical synthesis of a 304 amino acid K48‐linked tetraubiquitin protein. Angew Chem Int Ed. 2011;50(27):6137‐6141. [DOI] [PubMed] [Google Scholar]
- 41. Torbeev VY, Kent SB. Convergent chemical synthesis and crystal structure of a 203 amino acid “covalent dimer” HIV‐1 protease enzyme molecule. Angew Chem Int Ed. 2007;46(10):1667‐1670. [DOI] [PubMed] [Google Scholar]
- 42. Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proc Natl Acad Sci U S A. 1999;96(18):10068‐10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dawson P, Muir T, Clark‐Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266(5186):776‐779. [DOI] [PubMed] [Google Scholar]
- 44. Berrade L, Camarero JA. Expressed protein ligation: a resourceful tool to study protein structure and function. Cell Mol Life Sci. 2009;66(24):3909‐3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muir TW. Semisynthesis of proteins by expressed protein ligation. Annu Rev Biochem. 2003;72(1):249‐289. [DOI] [PubMed] [Google Scholar]
- 46. Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci USA. 1998;95(12):6705‐6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu WS, Brock A, Chen S, Chen SB, Schultz PG. Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat Methods. 2007;4(3):239‐244. [DOI] [PubMed] [Google Scholar]
- 48. Xie J, Schultz PG. A chemical toolkit for proteins‐an expanded genetic code. Nat Rev Mol Cell Biol. 2006;7(10):775‐782. [DOI] [PubMed] [Google Scholar]
- 49. Young DD, Schultz PG. Playing with the molecules of life. ACS Chem Biol. 2018;13(4):854‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ying T, Ju TW, Wang Y, Prabakaran P, Dimitrov DS. Interactions of IgG1 CH2 and CH3 domains with FcRn. Front Immunol. 2014;5:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ying T, Chen W, Feng Y, Wang Y, Gong R, Dimitrov DS. Engineered soluble monomeric IgG1 CH3 domain: generation, mechanisms of function, and implications for design of biological therapeutics. J Biol Chem. 2013;288(35):25154‐25164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y, Xu C, Lam HY, Lee CL, Li X. Protein chemical synthesis by serine and threonine ligation. Proc Natl Acad Sci. 2013;110(17):6657‐6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee CL, Li X. Serine/threonine ligation for the chemical synthesis of proteins. Curr Opin Chem Biol. 2014;22:108‐114. [DOI] [PubMed] [Google Scholar]
- 54. Malins LR, Payne RJ. Recent extensions to native chemical ligation for the chemical synthesis of peptides and proteins. Curr Opin Chem Biol. 2014;22:70‐78. [DOI] [PubMed] [Google Scholar]
- 55. Blanco‐Canosa JB, Dawson PE. An efficient Fmoc‐SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew Chem Int Ed. 2008;47(36):6851‐6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bayer E. Towards the chemical synthesis of proteins. Angew Chem Int Ed. 1991;30(2):113‐216. [Google Scholar]
- 57. Li W, Xiao X, Czarnik AW. Kinetic comparison of amide formation on various cross‐linked polystyrene resins. J Comb Chem. 1999;1(2):127‐129. [Google Scholar]
- 58. Kates SA, Sole NA, Beyermann M, Barany G, Albericio F. Optimized preparation of deca (L‐alanyl)‐L‐valinamide by 9‐fluorenylmethyloxycarbonyl (Fmoc) solid‐phase synthesis on polyethylene glycol‐polystyrene (PEG‐PS) graft supports, with 1,8‐diazobicyclo [5.4.0]‐undec‐7‐ene (DBU) deprotection. Pept Res. 1996;9(3):106‐113. [PubMed] [Google Scholar]
- 59. Goncalves V, Gautier B, Huguenot F, et al. Total chemical synthesis of the D2 domain of human VEGF receptor 1. J Pept Sci. 2009;15(6):417‐422. [DOI] [PubMed] [Google Scholar]
- 60. Wöhr T, Wahl F, Nefzi A, et al. Pseudo‐prolines as a solubilizing, structure‐disrupting protection technique in peptide synthesis. J Am Chem Soc. 1996;118(39):9218‐9227. [Google Scholar]
- 61. White P, Keyte JW, Bailey K, Bloomberg G. Expediting the Fmoc solid phase synthesis of long peptides through the application of dimethyloxazolidine dipeptides. J Pept Sci. 2004;10(1):18‐26. [DOI] [PubMed] [Google Scholar]
- 62. Siman P, Karthikeyan SV, Nikolov M, Fischle W, Brik A. Convergent chemical synthesis of histone H2B protein for the site‐specific ubiquitination at Lys34. Angew Chem Int Ed. 2013;52(31):8059‐8063. [DOI] [PubMed] [Google Scholar]
- 63. Jbara M, Maity SK, Morgan M, Wolberger C, Brik A. Chemical synthesis of phosphorylated histone H2A at Tyr57 reveals insight into the inhibition mode of the SAGA deubiquitinating module. Angew Chem Int Ed. 2016;55(16):4972‐4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Siman P, Blatt O, Moyal T, et al. Chemical synthesis and expression of the HIV‐1 Rev protein. Chembiochem. 2011;12(7):1097‐1104. [DOI] [PubMed] [Google Scholar]
- 65. Kumar KSA, Spasser L, Erlich LA, Bavikar SN, Brik A. Total chemical synthesis of di‐ubiquitin chains. Angew Chem Int Ed. 2010;49(48):9126‐9131. [DOI] [PubMed] [Google Scholar]
- 66. Gates ZP, Stephan JR, Lee DJ, Kent SBH. Rapid formal hydrolysis of peptide‐α‐thioesters. Chem Commun. 2013;49(8):786‐788. [DOI] [PubMed] [Google Scholar]
- 67. Zhang Y, Li T, Li X. Synthesis of human growth hormone‐releasing hormone via three‐fragment serine/threonine ligation (STL). Org Biomol Chem. 2013;11(34):5584‐5587. [DOI] [PubMed] [Google Scholar]
- 68. Zhao J‐F, Zhang X‐H, Ding Y‐J, Yang Y‐S, Bi X‐B, Liu C‐F. Facile synthesis of peptidyl salicylaldehyde esters and its use in cyclic peptide synthesis. Org Lett. 2013;15(20):5182‐5185. [DOI] [PubMed] [Google Scholar]
- 69. Wong C, Li T, Lam HY, Zhang Y, Li H. Realizing serine/threonine ligation: scope and limitations and mechanistic implication thereof. Front Chem. 2014;2:28 10.3389/fchem.2014.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ullmann V, Radisch M, Boos I, et al. Convergent solid‐phase synthesis of N‐glycopeptides facilitated by pseudoprolines at consensus‐sequence Ser/Thr residues. Angew Chem Int Ed. 2012;51(46):11566‐11570. [DOI] [PubMed] [Google Scholar]
- 71. Sakakibara S. Synthesis of large peptides in solution. Biopolymers. 1995;37(1):17‐28. [DOI] [PubMed] [Google Scholar]
- 72. Lee CL, Liu H, Wong CTT, Chow HY, Li X. Enabling N‐to‐C Ser/Thr ligation for convergent protein synthesis via combining chemical ligation approaches. J Am Chem Soc. 2016;138(33):10477‐10484. [DOI] [PubMed] [Google Scholar]
- 73. Wang J‐X, Fang G‐M, He Y, et al. Peptide O‐aminoanilides as crypto‐thioesters for protein chemical synthesis. Angew Chem Int Ed. 2015;54(7):2194‐2198. [DOI] [PubMed] [Google Scholar]
- 74. Siman P, Karthikeyan SV, Brik A. Native chemical ligation at glutamine. Org Lett. 2012;14(6):1520‐1523. [DOI] [PubMed] [Google Scholar]
- 75. Garcia‐Martin F, Quintanar‐Audelo M, Garcia‐Ramos Y, et al. Chemmatrix, a poly(ethylene glycol)‐based support for the solid‐phase synthesis of complex peptides. J Comb Chem. 2006;8(2):213‐220. [DOI] [PubMed] [Google Scholar]
- 76. Narita M, Ishikawa K, Nakano H, Isokawa S. Tertiary peptide bond containing‐oligo (Leu)s. Conformational studies in solution of oligo (l‐leucine)s with l‐proline residue and glycyl‐N‐(2,4‐dimethoxybenzyl)‐l‐leucine sequence. Int J Pept Protein Res. 1984;24:14‐24. [PubMed] [Google Scholar]
- 77. Weygand F, Steglich W, Bjarnason J, Akhtar R, Khan NM. Leicht abspaltbare Schutzgruppen für Säureamidfunktionen 1. Mitteilung. Tetrahedron Lett. 1966;7(29):3483‐3487. [Google Scholar]
- 78. Johnson T, Quibell M, Owen D, Sheppard RC. A reversible protecting group for the amide bond in peptides ‐ use in the synthesis of difficult sequences. J Chem Soc Chem Commun. 1993;(4):369‐372. [Google Scholar]
- 79. Beisswenger M, Yoshiya T, Kiso Y, Cabrele C. Synthesis and conformation of an analog of the helix‐loop‐helix domain of the Id1 protein containing the O‐acyl iso‐prolyl‐seryl switch motif. J Pept Sci. 2010;16(6):303‐308. [DOI] [PubMed] [Google Scholar]
- 80. Carpino LA, Krause E, Sferdean CD, et al. Synthesis of 'difficult' peptide sequences: application of a depsipeptide technique to the Jung‐Redemann 10‐ and 26‐mers and the amyloid peptide A beta(1‐42). Tetrahedron Lett. 2004;45(40):7519‐7523. [Google Scholar]
- 81. Kiewitz SD, Kakizawa T, Kiso Y, Cabrele C. Switching from the unfolded to the folded state of the helix‐loop‐helix domain of the Id proteins based on the O‐acyl isopeptide method. J Pept Sci. 2008;14(11):1209‐1215. [DOI] [PubMed] [Google Scholar]
- 82. Mutter M, Chandravarkar A, Boyat C, et al. Switch peptides in statu nascendi: induction of conformational transitions relevant to degenerative diseases. Angew Chem Int Ed. 2004;43(32):4172‐4178. [DOI] [PubMed] [Google Scholar]
- 83. Sohma Y, Sasaki M, Hayashi Y, Kimura T, Kiso Y. Novel and efficient synthesis of difficult sequence‐containing peptides through O‐N intramolecular acyl migration reaction of O‐acyl isopeptides. Chem Commun. 2004;(1):124‐125. [DOI] [PubMed] [Google Scholar]
- 84. Bacsa B, Bosze S, Kappe CO. Direct solid‐phase synthesis of the beta‐amyloid (1‐42) peptide using controlled microwave heating. J Org Chem. 2010;75(6):2103‐2106. [DOI] [PubMed] [Google Scholar]
- 85. Masuda K, Ooyama H, Shikano K, et al. Microwave‐assisted solid‐phase peptide synthesis of neurosecretory protein GL composed of 80 amino acid residues. J Pept Sci. 2015;21(6):454‐460. [DOI] [PubMed] [Google Scholar]
- 86. Murray JK, Gellman SH. Application of microwave irradiation to the synthesis of 14‐helical beta‐peptides. Org Lett. 2005;7(8):1517‐1520. [DOI] [PubMed] [Google Scholar]
- 87. Muthusamy K, Albericio F, Arvidsson PI, et al. Microwave assisted SPPS of amylin and its toxicity of the pure product to RIN‐5F cells. Biopolymers. 2010;94(3):323‐330. [DOI] [PubMed] [Google Scholar]
- 88. Pedersen SL, Tofteng AP, Malik L, Jensen KJ. Microwave heating in solid‐phase peptide synthesis. Chem Soc Rev. 2012;41(5):1826‐1844. [DOI] [PubMed] [Google Scholar]
- 89. Sabatino G, Papini AM. Advances in automatic, manual and microwave‐assisted solid‐phase peptide synthesis. Curr Opin Drug Discov Devel. 2008;11(6):762‐770. [PubMed] [Google Scholar]
- 90. Singer D, Zauner T, Genz M, Hoffmann R, Zuchner T. Synthesis of pathological and nonpathological human exon 1 huntingtin. J Pept Sci. 2010;16(7):358‐363. [DOI] [PubMed] [Google Scholar]
- 91. Yu HM, Chen ST, Wang KT. Enhanced coupling efficiency in solid‐phase peptide‐synthesis by microwave irradiation. J Org Chem. 1992;57(18):4781‐4784. [Google Scholar]
- 92. Maity SK, Jbara M, Laps S, Brik A. Efficient palladium‐assisted one‐pot deprotection of (acetamidomethyl) cysteine following native chemical ligation and/or desulfurization to expedite chemical protein synthesis. Angew Chem Int Ed. 2016;55(28):8108‐8112. [DOI] [PubMed] [Google Scholar]
- 93. Malins LR, Giltrap AM, Dowman LJ, Payne RJ. Synthesis of beta‐thiol phenylalanine for applications in one‐pot ligation‐desulfurization chemistry. Org Lett. 2015;17(9):2070‐2073. [DOI] [PubMed] [Google Scholar]