

Summary
Background
Tofacitinib is an oral, small molecule Janus kinase inhibitor that is being investigated for inflammatory bowel disease.
Aims
This 48‐week open‐label extension study primarily investigated long‐term safety of tofacitinib 5 and 10 mg b.d. and secondarily investigated efficacy as maintenance therapy in patients with Crohn's disease.
Methods
Patients who had completed the phase 2b maintenance study, or withdrawn due to treatment failure, were enrolled. Patients in remission (Crohn's disease activity index <150) at baseline received tofacitinib 5 mg b.d.; all others received 10 mg b.d. A single dose adjustment was allowed after 8 weeks’ fixed, open‐label treatment.
Results
Sixty‐two patients received tofacitinib 5 mg b.d.; 88 received 10 mg b.d. Both groups had similar rates of adverse events and serious infections. Crohn's disease worsening was the most frequent adverse event for tofacitinib 5 (33.9%) and 10 mg b.d. (19.3%). Patients not in remission at baseline, receiving 10 mg b.d., had higher rates of serious adverse events (19.3%) and discontinuation attributed to insufficient clinical response (30.7%) vs 5 mg b.d. (8.1% and 9.7%, respectively). At week 48, of patients with baseline remission receiving 5 mg b.d., 87.9% maintained remission and 75.0% sustained remission as observed (46.8% and 38.7%, respectively, by non‐responder imputation). Study design prevented between‐dose efficacy comparisons.
Conclusions
No new safety signals emerged. Although both doses showed generally similar safety outcomes for overall adverse events, serious adverse events were more frequent for tofacitinib 10 than 5 mg b.d. Discontinuation due to insufficient clinical response was lower among patients in remission at baseline. ClinicalTrials.gov: NCT01470599.
1. INTRODUCTION
Crohn's disease is a chronic, progressive inflammatory disease of the gastrointestinal tract1 that has a significant impact on patients’ quality of life.2 Current therapies for Crohn's disease include corticosteroids, thiopurines, methotrexate, anti‐tumour necrosis factor α antibodies, anti‐integrin antibodies and anti‐p40 antibodies.3 However, not all patients respond to these medications, leaving an unmet need for novel therapies.4
Tofacitinib is an oral, small molecule Janus kinase inhibitor approved in several countries for the treatment of ulcerative colitis. It has also been investigated for Crohn's disease. The efficacy and safety of tofacitinib for inducing and maintaining clinical remission (defined as Crohn's disease activity index [CDAI] score <150) in patients with moderate‐to‐severe Crohn's disease have previously been investigated in two phase 2b studies.5 These induction and maintenance studies showed a modest treatment effect of tofacitinib compared with placebo, although the primary induction efficacy endpoint of clinical remission at week 8 was not significantly different from placebo. After 26 weeks of maintenance therapy, a higher proportion of patients receiving tofacitinib 10 mg twice daily (b.d.) showed clinical response‐100 (defined as a CDAI score reduction of at least 100 points from baseline) or remission vs placebo, although these differences were also not statistically significant.5
Here, we present results of a phase 2b open‐label extension study that evaluated the safety and tolerability of tofacitinib, and exploratory efficacy over 48 weeks of treatment and 4 weeks of follow‐up.
2. MATERIALS AND METHODS
2.1. Study design
This was a phase 2b, open‐label, multicentre, 48‐week extension study, followed by a 4‐week safety follow up (ClinicalTrials.gov: NCT01470599). Patients in clinical remission at week 26 of the phase 2b maintenance study (NCT01393899; baseline of open‐label extension study), who were receiving either placebo, tofacitinib 5 or 10 mg b.d., were assigned tofacitinib 5 mg b.d. in this open‐label extension study. All other patients, who were not in clinical remission at week 26 of the phase 2b maintenance study, including those with an early termination visit due to treatment failure, received tofacitinib 10 mg b.d., regardless of blinded prior maintenance study treatment. A single dose adjustment from 5 to 10 mg b.d. or vice versa was allowed at the physician's discretion after the initial 8 weeks of fixed, open‐label treatment (Figure 1). This study was conducted at 57 centres in 17 countries (File S1), between 23 April 2012 and 25 July 2016. Details of protocol amendments are presented in File S2.
Figure 1.
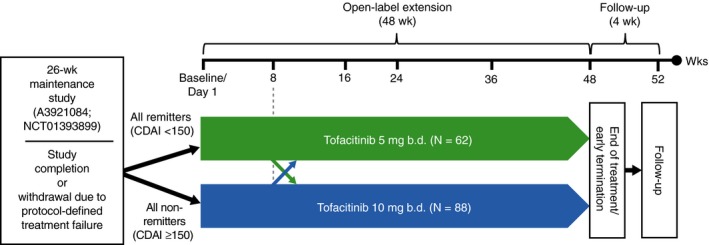
Open‐label extension study design. Patients were permitted to switch dose after week 8. If a patient discontinued early from the open‐label therapy, study procedures were to be completed as per end of treatment/early termination. b.d., twice daily; CDAI, Crohn's disease activity index
This study was approved by the institutional review board or independent ethics committee for each centre and conducted in accordance with the Declaration of Helsinki and in compliance with all International Conference on Harmonisation Good Clinical Practice Guidelines. All patients provided written informed consent.
2.2. Study patients
To be eligible for this study, patients with Crohn's disease had either completed the 26‐week, double‐blind, maintenance study or had withdrawn from the maintenance study after meeting pre‐specified treatment failure criteria. Maintenance study treatment failure was defined as meeting both of the following criteria on two consecutive visits, at least 2 weeks apart, in patients who had completed ≥4 weeks of treatment in the maintenance study: an increase in CDAI ≥ 100 points from maintenance study baseline value and an absolute CDAI score ≥175 points. Patients with active (draining) fistulae, intra‐abdominal or perineal collection, or abscess were excluded. Full details of eligibility criteria and prohibited concomitant medications are included in Files S3 and S4.
All patients who withdrew early or completed the open‐label extension study received a safety follow‐up visit approximately 4 weeks after the last study medication dose. For patients who discontinued, study procedures were completed as per end of treatment/early termination.
2.3. Primary objective: safety outcomes
The primary objective was to investigate the long‐term safety and tolerability of tofacitinib 5 and 10 mg b.d. in patients with Crohn's disease during the 48‐week study and the 4‐week follow‐up. Safety outcomes included incidence and severity of adverse events, classified by Medical Dictionary for Regulatory Activities version 19.0 coding. Abnormalities and changes from baseline were monitored for clinical laboratory values, vital sign measurements, electrocardiogram measurements and clinically significant changes in physical examination. Safety endpoints of special interest were adjudicated, including major adverse cardiovascular events, hepatic injury, opportunistic infection, malignancy, gastrointestinal perforation and interstitial lung disease. There was a histopathological central over‐read of specimens from potential malignancies, and the malignancy adjudication committee reviewed clinical and histopathological results. Safety outcomes were assessed at baseline, weeks 8, 16, 24, 36, 48 and 52. The 4‐week safety follow‐up visit at week 52 represented the end of the study.
2.4. Secondary objective and secondary efficacy outcomes
The secondary objective was to investigate the effects of tofacitinib maintenance therapy on clinical remission, patient quality of life, and biomarkers measured by C‐reactive protein and faecal calprotectin, through 48 weeks.
Secondary binary efficacy outcomes included clinical remission, sustained clinical remission (achieved clinical remission at both weeks 24 and 48) and steroid‐free clinical remission at week 48 among patients receiving steroids at baseline. Other endpoints included CDAI scores over time, time to relapse, serum high‐sensitivity C‐reactive protein and faecal calprotectin over time, changes from baseline, and corticosteroid use. Relapse was defined as an increase in CDAI of >100 points from baseline, and an absolute CDAI score of >220 points. The proportion of patients who relapsed, and the time to relapse, was analysed for patients in clinical remission at baseline who initially received tofacitinib 5 mg b.d. Also evaluated was the proportion of patients switching (at the investigator's discretion) from tofacitinib 5 to 10 mg b.d., or vice versa. Efficacy outcomes were monitored through 48 weeks, to end of treatment, or to early termination for those who discontinued the study.
2.5. Patient‐reported outcomes
The following patient‐reported outcomes were collected during this open‐label extension study: the Inflammatory Bowel Disease Questionnaire (IBDQ),6 the Short Form‐36 v2 (SF‐36v2; physical and mental component summary scores [PCS/MCS] and eight domain scores, with a 1‐week recall)7 and the European Quality of Life Five Dimensions Questionnaire (EQ‐5D) and Visual Analogue Scale (VAS) EQ‐5D/VAS.8 The inflammatory bowel disease Patient‐Reported Treatment Impact (PRTI; v2) comprised three endpoints (patient satisfaction, preference for the study medication over their previous treatment, and willingness to use the study treatment again).9, 10
Endpoints for IBDQ included: absolute scores and change from baseline in IBDQ total score and domain scores (ie, bowel symptoms, systemic symptoms, emotional function and social function) over time, IBDQ clinical remission (total score ≥170) at week 48, absolute scores and change from baseline in SF‐36v2, EQ‐5D/VAS over time, and absolute scores for PRTI at week 48. IBDQ scores were recorded at baseline, weeks 8, 24 and 48. SF‐36v2 and EQ‐5D/VAS scores were recorded at baseline, weeks 24 and 48, and PRTI was recorded at week 48. The number of patients hospitalised, and the length of hospitalisations related to Crohn's disease, was collected at every study visit.
2.6. Statistical analyses
Descriptive summary statistics, including two‐sided 95% confidence intervals for efficacy, biomarkers and patient‐reported outcome endpoints, were calculated based on observed data (no imputation of missing data) by initially assigned dose group from the full analysis set, defined as all patients who enrolled in the open‐label extension study. Additional summaries for clinical remission and sustained clinical remission were also completed, based on nonresponder imputation and last observation carried forward. Kaplan‐Meier estimates and the median time to relapse for patients in clinical remission at baseline were also reported. In accordance with the study design, which assigned patients who were not in clinical remission at baseline to tofacitinib 10 mg b.d., no statistical comparisons between 5 and 10 mg b.d. treatment groups were performed, due to the difference in study population. Safety endpoints were summarised for the safety analysis set, defined as all patients enrolled in the open‐label extension study who received ≥1 dose of study medication. All analyses were performed using SAS® 9.0 in Linux.
3. RESULTS
3.1. Patients
A total of 150 patients were enrolled in this study. Of these, 62 patients in remission at entry were assigned tofacitinib 5 mg b.d., and 88 patients with active disease were assigned 10 mg b.d. Of those enrolled, 19 (30.6%) and 43 (48.9%) patients discontinued in the 5 and 10 mg b.d. groups, respectively (Figure 2).
Figure 2.
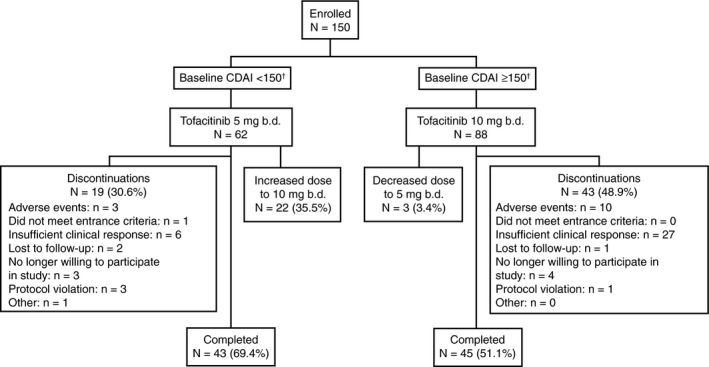
Patient disposition diagram. Safety analyses patient population. Includes enrolled patients who received ≥1 dose of study medication. N represents the total number of patients in each group; n represents the number of patients with events. †Dose assignment was determined based on last available haematocrit result prior to open‐label extension study entry. Baseline CDAI scores were recalculated using baseline haematocrit results received after treatment assignment was determined. Therefore, one patient in the tofacitinib 5 mg b.d. group and four patients in the 10 mg b.d. group had remission status that did not correspond to their initial dose assignment. b.d., twice daily; CDAI, Crohn's disease activity index
Patient demographics were similar between the tofacitinib groups. There was a geographical spread of patients recruited to the study from across the study site locations (geographical regions shown in Table S1). A greater proportion of patients assigned 10 mg b.d. were taking corticosteroids and had higher mean CDAI scores and higher median C‐reactive protein and faecal calprotectin concentrations, compared with those receiving 5 mg b.d. (Table 1). Ileocolonic disease location was recorded in a numerically higher proportion of patients receiving tofacitinib 10 mg b.d. than 5 mg b.d.
Table 1.
Patient demographics and baseline characteristics
|
Tofacitinib 5 mg b.d. (N = 62)a |
Tofacitinib 10 mg b.d. (N = 88)a |
|
|---|---|---|
| Female, n (%) | 30 (48.4) | 41 (46.6) |
| Race, n (%) | ||
| White | 49 (79.0) | 71 (80.7) |
| Black | 2 (3.2) | 1 (1.1) |
| Asian | 11 (17.7) | 14 (15.9) |
| Other | 0 (0.0) | 2 (2.3) |
| Mean age, y (SD) | 41.0 (12.6) | 38.2 (11.6) |
| Mean age of onset of Crohn's disease, y (SD)b | 28.0 (9.8) | 26.6 (10.9) |
| Duration of Crohn's disease, n (%)b | ||
| <5 y | 17 (27.4) | 26 (29.5) |
| 5‐<10 y | 13 (21.0) | 19 (21.6) |
| ≥10 y | 32 (51.6) | 43 (48.9) |
| Disease location, n (%)b | ||
| Ileal alone | 18 (29.0) | 21 (23.9) |
| Colonic alone | 22 (35.5) | 23 (26.1) |
| Ileocolonic | 22 (35.5) | 43 (48.9) |
| Missing | 0 (0.0) | 1 (1.1) |
| Prior tumour necrosis factor inhibitor failure, n (%)b | 42 (67.7) | 69 (78.4) |
| Prior immunosuppressant failure, n (%)b | 43 (69.4) | 68 (77.3) |
| Use of corticosteroids at study entry, n (%)a | 2 (3.2) | 22 (25.0) |
| Baseline CDAI score, mean (SD)c | 77.1 (43.2) | 291 (95.8) |
|
Baseline C‐reactive protein concentration, mg/L; median (SD)a |
3.6 (18.6) | 8.4 (30.1) |
|
Baseline faecal calprotectin concentration, mg/kg; median (SD)a |
255.0 (483.3) | 512.0 (305.4) |
Only patients not in remission were assigned tofacitinib 10 mg b.d.
b.d., twice daily; CDAI, Crohn's disease activity index; SD, standard deviation.
Baseline characteristics at open‐label extension study entry.
Baseline information from the induction study (NCT01393626).
Baseline information from the maintenance study week 26 central laboratory measurement.
3.2. Safety outcomes
The proportion of patients with adverse events was 79.0% and 76.1% for tofacitinib 5 and 10 mg b.d., respectively (Table 2), and the most frequently observed adverse events were Crohn's disease worsening (33.9% and 19.3%, respectively). Nasopharyngitis and urinary tract infection were the most frequently occurring infections for tofacitinib 5 mg b.d. (both adverse events, 12.9%) and 10 mg b.d. (both adverse events, 8.0%). Serious adverse events were recorded in 8.1% and 19.3% of patients’ assigned tofacitinib 5 and 10 mg b.d., of which Crohn's disease worsening accounted for 4.8% and 10.2%, respectively. Both tofacitinib 5 and 10 mg b.d. showed similar rates of serious infections, with 3.2% and 2.3%, respectively.
Table 2.
Adverse events and events of special interest in the open‐label extension study
|
Tofacitinib 5 mg b.d. (N = 62)a |
Tofacitinib 10 mg b.d. (N = 88)a |
|
|---|---|---|
| Duration of treatment, d; median (SD) | 333.5 (95.1) | 284.5 (114.7) |
| Exposure, patient‐years | 47.0 | 57.1 |
| Switched dose groups after week 8, n (%) | 22 (35.5) | 3 (3.4) |
| Treatment‐emergent adverse events, all causalities | ||
| Patients with adverse events, n (%) | 49 (79.0) | 67 (76.1) |
| Patients with serious adverse events, n (%) | 5 (8.1) | 17 (19.3)b |
| Discontinuations | ||
| Due to adverse events, n (%)c | 3 (4.8) | 10 (11.4) |
| Due to infections | 2 (3.2) | 3 (3.4) |
| Due to insufficient clinical response | 6 (9.7) | 27 (30.7) |
| Infections, n (%) | 31 (50.0) | 42 (47.7) |
| Serious infections, n (%) | 2 (3.2) | 2 (2.3) |
| Diarrhoea infectious | 1 (1.6) | 0 (0.0) |
| Pyelonephritis | 1 (1.6) | 0 (0.0) |
| Anal abscess | 0 (0.0) | 1 (1.1) |
| Vulvar abscess | 0 (0.0) | 1 (1.1) |
| Adverse events of special of interest, n (%) | ||
| Death | 0 (0.0) | 0 (0.0) |
| Herpes zosterd | 1 (1.6) | 2 (2.3) |
| Opportunistic infectione , f | 1 (1.6) | 1 (1.1) |
| Malignancy (excluding NMSC)e | 0 (0.0) | 0 (0.0) |
| NMSCe | 0 (0.0) | 1 (1.1)g |
| Hepatic injurye | 0 (0.0) | 0 (0.0)h |
| Intestinal perforatione , i | 0 (0.0) | 0 (0.0) |
| Interstitial lung diseasee | 0 (0.0) | 0 (0.0) |
| Most frequently observed adverse events by preferred term (frequency ≥8.0%), n (%) | ||
| Crohn's disease worsening | 21 (33.9) | 17 (19.3) |
| Nasopharyngitis | 8 (12.9) | 7 (8.0) |
| Urinary tract infection | 8 (12.9) | 7 (8.0) |
| Abdominal pain | 7 (11.3) | 7 (8.0) |
| Gastroenteritis | 7 (11.3) | 2 (2.3) |
| Influenza | 5 (8.1) | 8 (9.1) |
| Nausea | 2 (3.2) | 9 (10.2) |
| Pyrexia | 1 (1.6) | 7 (8.0) |
| Arthralgia | 5 (8.1) | 8 (9.1) |
| Most frequently observed serious adverse events by preferred term (frequency ≥8.0%), n (%) | ||
| Crohn's disease worsening | 3 (4.8) | 9 (10.2) |
| Laboratory changes of interest | ||
| Alanine aminotransferase increased | 1 (1.6) | 0 (0.0) |
| Blood alkaline phosphatase increased | 2 (3.2) | 0 (0.0) |
| Blood cholesterol increased | 1 (1.6) | 0 (0.0) |
| Blood creatine phosphokinase increased | 3 (4.8) | 5 (5.7) |
| Liver function test increased | 0 (0.0) | 1 (1.1) |
| Two sequential laboratory measurements, meeting criteria for discontinuationj | ||
| Neutrophil counts <750 neutrophils/mm3 | 0 (0.0) | 0 (0.0) |
| Lymphocyte counts <500 lymphocytes/mm3 | 0 (0.0) | 1 (1.1) |
| Creatinine kinase elevation >10 × ULN | 0 (0.0) | 0 (0.0) |
| Haemoglobin <0.8 g/L or a decrease of >30% from baseline value | 1 (1.6) | 1 (1.1) |
The adverse event of worsening Crohn's disease leading to discontinuation was termed insufficient clinical response. Only patients who were not in remission were assigned tofacitinib 10 mg b.d.
Infections that were reported as a serious adverse event are described in the table as “serious infections.”
b.d., twice daily; NMSC, non‐melanoma skin cancer; SD, standard deviation; ULN, upper limit of normal.
Patients evaluable for adverse events by initially assigned dose group.
Three out of 17 patients experienced serious adverse events after the 4‐week safety follow‐up.
Excluding worsened Crohn's disease attributed to insufficient clinical response.
No serious cases of herpes zoster were reported.
Events based upon adjudication.
Adjudicated opportunistic infections included the single case of herpes zoster multidermatomal (non‐adjacent or >2‐6 adjacent dermatomes) in the tofacitinib 5 mg b.d. group, and one case of primary varicella infection in the tofacitinib 10 mg b.d. group.
Adjudicated malignancy events, including one case of basal cell carcinoma.
One case of biliary colic was adjudicated as unrelated to drug‐induced liver injury.
Adverse event preferred term, including “perforation.” However, one event of perineal fistula and one event of anal abscess in the tofacitinib 10 mg b.d. group were adjudicated as intestinal perforation.
Laboratory values meeting predetermined criteria for discontinuation.
Discontinuation due to insufficient clinical response was more frequent for patients who entered the study not in remission, receiving 10 mg b.d. (30.7%), compared with 5 mg b.d. (9.7%). By investigator decision, some cases of discontinuation due to insufficient clinical response did not have an associated adverse event of worsening Crohn's disease recorded, and one patient discontinued due to an adverse event of worsening Crohn's disease but did not have this specified as a reason for insufficient clinical response. Laboratory values meeting predetermined laboratory criteria for re‐test or discontinuation were similarly low for both tofacitinib groups.
During this study, there were no deaths, adjudicated cases of cardiovascular events, interstitial lung disease or hepatic injury, or malignancy (excluding non‐melanoma skin cancer [NMSC]) (Table 2). One case of basal cell carcinoma of the skin was reported in the tofacitinib 10 mg b.d. group, and no cases of squamous cell carcinoma. Three cases of nonserious herpes zoster (investigator‐determined) occurred; one in the 5 mg b.d. group, and two in the 10 mg b.d. group (one of which was a case of acute, primary varicella infection, confirmed by the study site, that was misadjudicated as a multidermatomal herpes zoster reactivated infection). There were two opportunistic infections: a single, mild (investigator‐determined) case of multidermatomal (non‐adjacent or >2‐6 adjacent dermatomes) herpes zoster reported in the 5 mg b.d. group (study drug continued, infection resolved with antiviral treatment); and one case of moderate, systemic, disseminated, multidermatomal primary varicella infection in the 10 mg b.d. group (study drug discontinued, infection resolved with antiviral therapy).
No cases with a preferred term of “intestinal perforation” were reported, but one perineal fistula and one anal abscess in the tofacitinib 10 mg b.d. group were adjudicated as gastrointestinal perforation.
3.3. Exploratory efficacy outcomes
This study had no primary efficacy endpoints; all efficacy endpoints, patient‐reported outcomes and biomarkers were exploratory endpoints. Dose adjustments were permitted post‐week 8 at the investigator's discretion. Twenty‐two patients (35.5%) adjusted from tofacitinib 5 to 10 mg b.d.: 16 patients at week 8, 4 patients at week 16 and 2 patients at week 24. Three patients (3.4%) adjusted from tofacitinib 10 to 5 mg b.d.: two at week 24 and one at week 36.
Among patients receiving tofacitinib 5 mg b.d. at week 48, 87.9% were in clinical remission and 75.0% in sustained clinical remission, as observed (Figure 3A, B). At week 48, when analysed by last observation carried forward and nonresponder imputation, 85.5% and 46.8% of patients, respectively, achieved clinical remission; and 77.4% and 38.7%, respectively, achieved sustained clinical remission. In the 5 mg b.d. group, median baseline C‐reactive protein was 3.6 mg/L. At week 48, of the 27 patients with baseline high‐sensitivity C‐reactive protein <3 mg/L, 14 patients (51.9%) maintained clinical remission, and of the 35 patients with baseline high‐sensitivity C‐reactive protein ≥3 mg/L, 15 patients (42.9%), maintained clinical remission. Median baseline faecal calprotectin was 255.0 mg/kg. Similarly, at week 48, of the 31 patients with baseline faecal calprotectin <250 mg/kg, and the 31 patients with baseline faecal calprotectin ≥250 mg/kg, 15 and 14 patients (48.4% and 45.2%), respectively, maintained clinical remission for tofacitinib 5 mg b.d.
Figure 3.
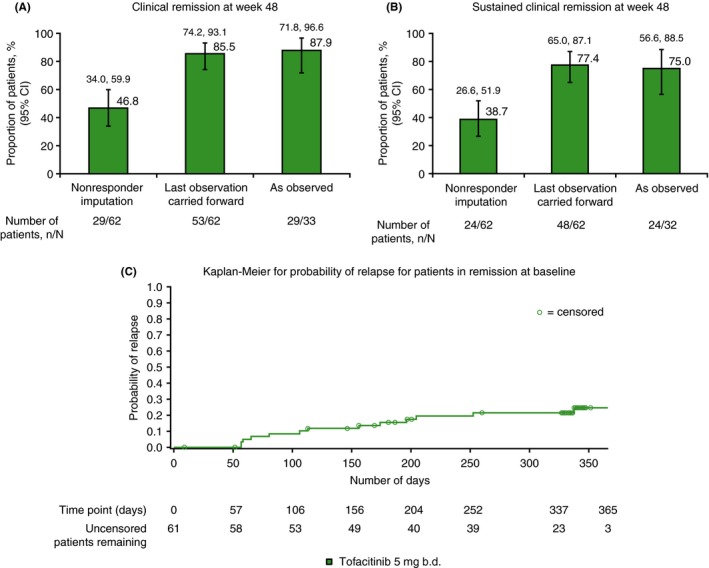
Proportion of patients in clinical remission at baseline and assigned tofacitinib 5 mg b.d., who had A, clinical remission at week 48, and B, sustained clinical remission at weeks 24 and 48, and also C, Kaplan‐Meier plot showing probability of relapse among those in clinical remission at baseline. Clinical remission was defined as CDAI <150. Sustained clinical remission was defined as remission at both weeks 24 and 48. Relapse was defined as an increase in CDAI of >100 points from baseline, and an absolute CDAI score of >220 points. Patients who discontinued due to lack of efficacy were treated as a relapse event, with an event time at the time of drop out. All data represent full analysis set. N represents the total number of patients analysed, n represents the number of patients achieving clinical remission or sustained clinical remission. Clopper‐Pearson 95% confidence intervals are shown in panels A and B. The Kaplan‐Meier plot for relapse is shown to 365 days, as there were three patients or fewer at risk from days 365 through 458. b.d., twice daily; CDAI, Crohn's disease activity index; SE, standard error
The median time to relapse for patients in clinical remission at baseline was 366 days for patients receiving tofacitinib 5 mg b.d. The estimated relapse rate increased gradually over time for patients in remission at baseline, and was greatest at week 48 (24.7%). A Kaplan‐Meier plot of probability of relapse is shown up to day 365 for patients receiving 5 mg b.d. who were in remission at baseline (Figure 3C). Among all patients receiving 5 mg b.d., seven (11.3%) were hospitalised due to Crohn's disease during the study.
At week 48, patients receiving tofacitinib 5 mg b.d. showed generally consistent mean CDAI scores through week 48 (excepting week 8), showing a median CDAI score at week 48 of 67.0, and a week 48 mean change from baseline in CDAI score of −4.8. Decreases from baseline in C‐reactive protein (–4.6 mg/L) and faecal calprotectin levels (−189.6 mg/kg) (Figure 4) were also observed at week 48.
Figure 4.
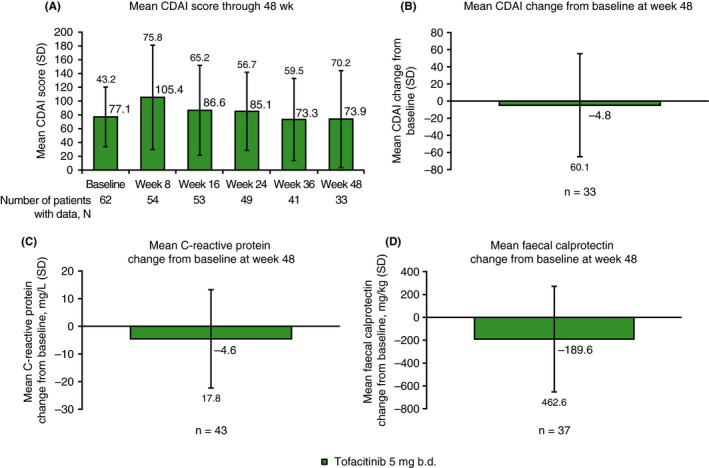
A, Mean CDAI score through week 48, and mean changes from baseline at week 48 for B, CDAI, C, C‐reactive protein and D, faecal calprotectin, for tofacitinib 5 mg b.d. All data represent full analysis set (observed). N represents the total number of patients analysed at each time point, n represents the number of patients with data at week 48 or at baseline and week 48. b.d., twice daily; CDAI, Crohn's disease activity index; SD, standard deviation
For patients receiving tofacitinib 10 mg b.d., 55.6% achieved clinical remission and 34.3% achieved sustained clinical remission at week 48, as observed (Figure 5). At week 48, when analysed by last observation carried forward and nonresponder imputation, respectively; 39.8% and 22.7% of patients achieved clinical remission, and 29.6% and 13.6% achieved sustained clinical remission. In the tofacitinib 10 mg b.d. group, in which patients were not in remission at baseline, 45 patients (51.1%) achieved remission at some point during the study. The median time to remission, for those who achieved it, was 60 days. Patients showed improvements in mean CDAI score from baseline (−121.9), with a median CDAI score of 147.5 at week 48, and decreases from baseline in C‐reactive protein (−11.5 mg/L) and faecal calprotectin levels (−123.9 mg/kg) (Figure 6). Fourteen patients (15.9%) receiving tofacitinib 10 mg b.d. were hospitalised due to Crohn's disease during the study.
Figure 5.
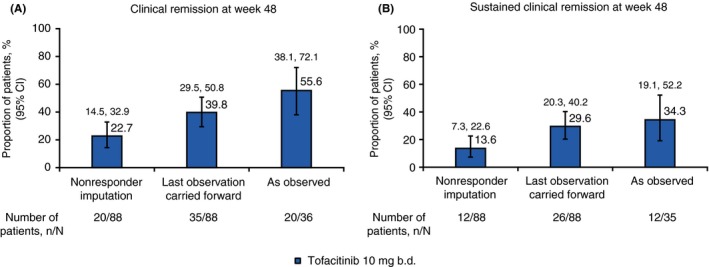
Proportion of patients assigned tofacitinib 10 mg b.d. at baseline who had A, clinical remission at week 48 and B, sustained clinical remission at weeks 24 and 48. Clinical remission was defined as CDAI <150. Sustained clinical remission was defined as remission at both week 24 and week 48. All data represent full analysis set. N represents the total number of patients analysed, n represents the number of patients achieving clinical remission or sustained clinical remission. Clopper‐Pearson 95% confidence intervals are shown. b.d., twice daily; CDAI, Crohn's disease activity index
Figure 6.
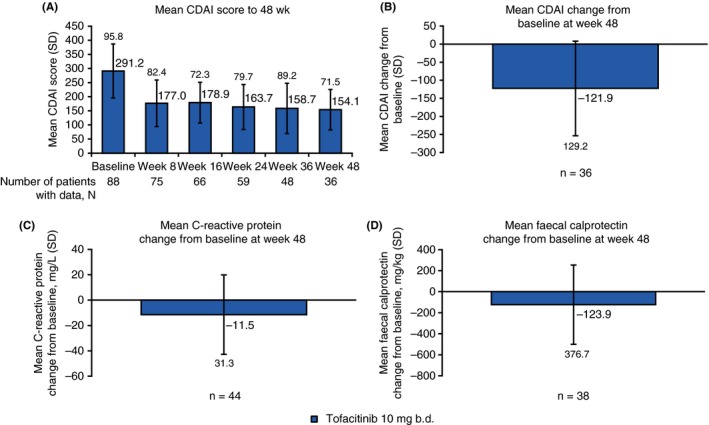
A, Mean CDAI score through week 48, and mean changes from baseline at week 48 for B, CDAI, C, C‐reactive protein and D, faecal calprotectin for tofacitinib 10 mg b.d. All data represent full analysis set (observed). N represents the total number of patients analysed at each time point, n represents the number of patients with data at week 48, or at baseline and week 48. b.d., twice daily; CDAI, Crohn's disease activity index; SD, standard deviation
For tofacitinib 5 and 10 mg b.d. groups in this open‐label extension study, remission data over time were summarised by doses previously received in the maintenance study (data for baseline through week 48 are shown in Table S2). For patients initially assigned tofacitinib 5 mg b.d. in this open‐label extension study, who by study design were in remission at baseline, there were 15, 20 and 27 patients who received placebo, tofacitinib 5 mg b.d. and 10 mg b.d. in the maintenance study, respectively. Of these patients, 11, 11 and 11 patients remained in this open‐label extension study to week 48; and 10 (90.9%), 9 (81.8%) and 10 (90.9%) of these patients were in remission at week 48, respectively.
For patients initially assigned tofacitinib 10 mg b.d. in this open‐label extension study, who by study design were not in remission at baseline, there were 33, 32 and 23 patients who received placebo, tofacitinib 5 and 10 mg b.d. in the maintenance study, respectively. Among them, 14, 10 and 12 patients remained in this open‐label extension study to week 48; and 7 (50.0%), 8 (80.0%) and 5 (41.7%) of these patients were in remission at week 48, respectively. These data support the potential efficacy of regaining therapeutic benefit with retreatment or dose escalation of therapy with 10 mg b.d., in achieving remission in those who had previously failed in the maintenance study with placebo or 5 mg b.d., respectively.
At baseline of the open‐label extension study, 2 (3.2%) and 22 (25.0%) patients in the tofacitinib 5 and 10 mg b.d. groups, respectively, received corticosteroids. As expected, this proportion was low for the 5 mg b.d. group, as these patients were in clinical remission, and corticosteroids had been tapered during the parent study. For tofacitinib 5 and 10 mg b.d., 3 (4.8%) and 19 (21.6%) patients, respectively, received corticosteroids at week 8, and 1 (1.6%) and 7 (8.0%) patients, respectively, at week 48.
3.4. Patient‐reported outcomes
For patients who entered the study in remission receiving tofacitinib 5 mg b.d., the majority (range: 69.5%‐81.0%) showed high (≥170) IBDQ total scores at baseline, weeks 8, 24 and 48 (Figure 7). At week 48, mean change from baseline IBDQ total score (−2.8) and individual IBDQ domains showed a general maintenance of values over time, with change from baseline range −3.1‐0.1 (see Table S3). At week 48, small mean changes from baseline were observed for SF‐36v2 domains (−0.2 and −1.2 for PCS and MCS, respectively), and for EQ‐5D/VAS score (1.2). For PRTI assessment, 66.7% and 33.3% of patients reported being “extremely satisfied” or “satisfied,” respectively, with their study drug, 88.1% of patients reported they preferred their study drug, and 83.3% reported they would use the same drug again.
Figure 7.
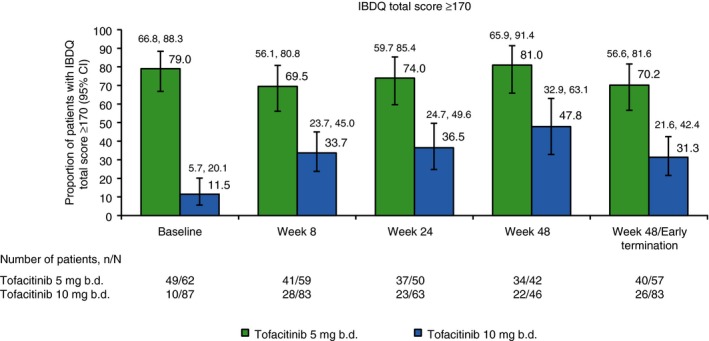
Proportion of patients with IBDQ clinical remission (total score ≥170) patient‐reported outcome endpoint, by baseline and weeks 8, 24 and 48. N represents the total number of patients analysed at each time point, n represents the number of patients achieving IBDQ clinical remission. 95% Clopper‐Pearson exact confidence intervals are shown for proportions. BD, twice daily; IBDQ, Inflammatory Bowel Disease Questionnaire
In the tofacitinib 10 mg b.d. group, comprising patients who entered the study not in remission, there was a numerically increasing proportion of patients with high (≥170) total IBDQ score through week 48, among those who remained in the study (see Table S3). Substantial improvements (37.2) in mean change from baseline for IBDQ total score were observed at week 48. Each individual IBDQ domain showed increases in values (range: 6.6‐12.6) at week 48.
At week 48 with tofacitinib 10 mg b.d., there were clinically relevant improvements in PCS and MCS of 8.4 and 7.6, respectively, greater than the previously published minimal clinically important differences of 1.6‐4.0 and 2.3‐5.7, respectively, using either a distribution‐ or effect size anchor‐based approach.11 At week 48, there were clinically relevant increments from baseline for the EQ‐5D/VAS score of 21.2, greater than the minimal clinically important differences of 4.2‐14.8.11 For the PRTI assessment, 32.6% and 41.3% of patients reported being “extremely satisfied” or “satisfied” with their study drug, respectively. 56.5% of patients reported they preferred their study drug, and 60.9% reported they would use the same drug again.
4. DISCUSSION
No new or unexpected safety signals were observed during this study. Rates of adverse events were generally similar for patients receiving tofacitinib 5 and 10 mg b.d. and were consistent with the previous 26‐week maintenance study.5 In the maintenance study, Crohn's disease worsening was also the most frequently occurring adverse event,5 although rates were lower than those reported here. Patient‐years of tofacitinib exposure in the maintenance study were lower for tofacitinib 5 and 10 mg b.d., at 22.9 patient‐years and 23.6 patient‐years, respectively, compared with the longer extension study shown here. As such, although only open‐label extension events are reported here, the longer study duration and length of exposure might impact upon rates of safety events.
Consistent with the maintenance study, adverse events of special interest were similarly infrequent among both treatment groups, and there were no deaths during the study. There was one case of colon perforation (microperforation of the sigmoid) in the maintenance study.5 Additionally, the rates of adverse events of special interest were generally similar between this open‐label extension study and the maintenance, 52‐week OCTAVE Sustain study in patients with ulcerative colitis and clinical response to induction therapy, with neither study observing cases of intestinal perforation, malignancies (excluding NMSC) and generally similar proportions of NMSC and herpes zoster, for tofacitinib 5 and 10 mg b.d.12
Infections were reported in approximately half of patients receiving tofacitinib in this study. The safety profile reported here is generally consistent with previous studies in rheumatoid arthritis with up to 114 months’ observation.13 No new safety signals emerged and, as in the rheumatoid arthritis programme, the most frequently occurring adverse events were infections, including nasopharyngitis.13
Rates of discontinuation due to adverse events and insufficient clinical response were higher for tofacitinib 10 mg b.d. than 5 mg b.d., indicating that patients who entered the open‐label extension study in remission had a lower probability of experiencing insufficient clinical response resulting in discontinuation. Compared with the maintenance study, rates of discontinuation due to adverse events and discontinuation due to insufficient clinical response reported here were generally similar for tofacitinib 10 mg b.d. and lower for 5 mg b.d.5
In this open‐label extension study, rates of serious adverse events were higher with tofacitinib 10 mg b.d. than 5 mg b.d., and compared with rates observed in the maintenance study.5 However, it is important to note that patients assigned 10 mg b.d. were not in remission at baseline, and were therefore likely to have more severe Crohn's disease symptoms, compared with patients in remission at baseline receiving 5 mg b.d.
In comparison, a long‐term extension study of adalimumab (ADHERE) included patients with moderately‐to‐severely active Crohn's disease who had completed the maintenance period of the 56‐week CHARM study, which provides some context for this open‐label extension study.14, 15 Analyses were based on the intent‐to‐treat population, comprising patients who entered CHARM (maintenance study) after open‐label induction with adalimumab and achieved CDAI ≥70 decrease while receiving adalimumab. Rates of remission (CDAI <150) at week 104 from the CHARM maintenance study baseline, for patients in remission at ADHERE baseline, were 77.2%, 86.2% and 86.8%, as nonresponder imputation, last observation carried forward and observed data, respectively.14 Median CDAI scores at week 92 from the CHARM baseline, as observed, were 107 and 94 for adalimumab 40 mg dosed every other week and weekly, respectively.15 The rates of remission presented in this open‐label extension study at week 48 (90 weeks from maintenance study baseline) for tofacitinib 5 mg b.d. (patients in remission at open‐label extension baseline) were 46.8%, 85.5% and 87.9%, for nonresponder imputation, last observation carried forward and observed data, respectively, and median CDAI scores at week 48, as observed, were 67.0 and 147.5 for tofacitinib 5 and 10 mg b.d., respectively.
However, there were several key differences between study entry criteria for the CHARM study and the study presented here, which precludes direct comparisons of efficacy data. Safety is the primary consideration for this open‐label extension study. Any comparisons of efficacy data are provided for context purposes only. The results presented for this open‐label extension study included patients who had previously withdrawn in the maintenance study due to treatment failure; whereas in the ADHERE study, only patients who had completed CHARM were included in the ADHERE extension, which might exclude some patients with severe disease activity who were nonresponders to adalimumab. As such, some patients in this open‐label extension study might have been more refractory than those in the CHARM study. Additionally, differences between patient group sizes mean that it is not appropriate to directly compare the results between these studies, as for tofacitinib 5 and 10 mg b.d. only 33 and 36 patients, respectively, had nonmissing data at week 48, vs a range of 129‐154 patients for efficacy outcomes at 104 weeks from CHARM baseline. In addition, there were dose adjustments for patients in both treatment groups.
Among patients initially assigned tofacitinib 5 mg b.d. who were in clinical remission at baseline, and remained in the study at week 48, approximately three‐quarters achieved sustained clinical remission at week 48, as observed and with last observation carried forward, and approximately one‐third as nonresponder imputation. By week 48, three‐quarters of patients had not relapsed, and there were decreases in C‐reactive protein and faecal calprotectin from baseline at each time point through week 48. The aggregate data for time to relapse, clinical remission rate and sustained clinical remission rate, CDAI value, and C‐reactive protein and faecal calprotectin values appear to show maintenance of efficacy for some patients receiving tofacitinib 5 mg b.d.
For patients initially assigned tofacitinib 10 mg b.d., because they were not in remission at baseline, approximately one‐third achieved sustained remission, as observed or with last observation carried forward. Decreases in C‐reactive protein and faecal calprotectin from baseline were observed at each time point through week 48, and the magnitude of the decrease in C‐reactive protein from baseline was greater at each time point for 10 mg b.d. than for 5 mg b.d. However, patients assigned 10 mg b.d. were not in remission at baseline by study design, and had higher values of C‐reactive protein and faecal calprotectin. This patient population included patients who had previously received either placebo, or tofacitinib 5 or 10 mg b.d., in the maintenance study; these data might indicate treatment effect with 10 mg b.d., although no control group data are available.
Changes from baseline for patient‐reported outcome endpoints over time were generally maintained for patients receiving tofacitinib 5 mg b.d. For patients receiving tofacitinib 10 mg b.d., clinically relevant improvements from baseline in patient‐reported outcome endpoints were observed in SF‐36v2 domains and EQ‐5D, and improvements in PCS and MCS and the proportions of patients with IBDQ clinical remission increased over time.
The inflammatory bowel disease PRTI tool has previously been used to evaluate patient preference in tofacitinib studies for patients with ulcerative colitis, where it was shown to have approximately linear associations with Mayo scores.9, 10 The majority of patients receiving tofacitinib reported positive outcomes of “extremely satisfied” and “satisfied” for the PRTI assessments of patient satisfaction. Proportions of patients responding positively were slightly higher for 5 mg b.d. than 10 mg b.d.
This study had several limitations. Only patients not in remission were assigned tofacitinib 10 mg b.d.; therefore, as the two patient populations had different disease activity at the outset, this precluded definitive comparison of efficacy outcomes between doses. Patients were permitted to switch doses after 8 weeks of receiving their initially assigned study dose and, as such, efficacy and safety outcomes were analysed by initial dose assignment, not taking dose changes into account. Additionally, this was an open‐label study, without a control group.
In summary, no new safety signals were observed through week 52 of this open‐label extension study of tofacitinib in patients with Crohn's disease. Rates of adverse events were generally similar among both groups, and were consistent with rates observed in the previous tofacitinib Crohn's disease maintenance study. There were higher rates of serious adverse events and discontinuation due to adverse events in the tofacitinib 10 mg b.d. group. However, as patients were only assigned tofacitinib 10 mg b.d. if they were not in remission at baseline and therefore had more active disease than those receiving 5 mg b.d., and because dose switching was permitted, direct comparisons between dose groups were not conducted. Patients in remission at open‐label extension study entry had a lower rate of discontinuation due to insufficient clinical response than those not in remission at study entry. Exploratory efficacy analyses at week 48 appeared to show that many patients receiving tofacitinib 5 mg b.d. maintained remission and achieved sustained remission by week 48, and although fewer patients receiving 10 mg b.d. achieved remission (approximately one‐quarter by nonresponder imputation), these patients did show improvement in mean CDAI scores from baseline. For those patients who had received placebo or 5 mg b.d. in the maintenance study (but entered this open‐label extension study not in remission), 50.0% and 80.0%, respectively, who subsequently received retreatment or dose escalation with 10 mg b.d., were able to achieve remission by week 48 in this open‐label extension study.
5. DATA SHARING STATEMENT
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (a) for indications that have been approved in the US and/or EU or (b) in programmes that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Supporting information
ACKNOWLEDGEMENTS
Declaration of personal interests: The authors would like to thank the patients, investigators and study teams who were involved in this study. J. Panés has received consulting fees from AbbVie, Amgen, Boehringer Ingelheim, Celgene, Ferring, Genentech/Roche, GSK, Janssen, MSD, Oppilan, Pfizer Inc, Robarts, Shire, Takeda, Theravance and TiGenix. G. R. D'Haens has served as an advisor for AbbVie, Ablynx, Amakem, AM Pharma, Avaxia, Biogen, Bristol‐Myers Squibb, Boehringer Ingelheim, Celgene, Celltrion, Cosmo, Covidien, Dr. Falk Pharma, Engene, Ferring, Galapagos, Gilead, GSK, Hospira, Johnson and Johnson, Medimetrics, Millennium/Takeda, Mitsubishi Pharma, MSD, Mundipharma, Novo Nordisk, Pfizer Inc, Prometheus Laboratories/Nestle, Receptos, Robarts Clinical Trials, Salix, Sandoz, Setpoint, Shire, Teva, TiGenix, Tillotts, Topivert, Versant and Vifor; and has received speaker fees from AbbVie, Ferring, Johnson and Johnson, Millennium/Takeda, MSD, Mundipharma, Norgine, Pfizer Inc, Shire, Tillotts and Vifor. P. D. R. Higgins has received consulting fees from AbbVie, Genentech/Roche, GSK, Takeda, PRIME Medical Education and Lycera. L. Mele, M. Moscariello, G. Chan, W. Wang, W. Niezychowski, C. Su and E. Maller are all employees and stockholders of Pfizer Inc.
AUTHORSHIP
Guarantor of the article: Julián Panés.
Author contributions: All authors substantially contributed in the acquisition, analysis or interpretation of data. All authors drafted the work and/or revised it critically for important intellectual content; approved the final published version of the manuscript; and are accountable for all aspects of this work.
Panés J, D’Haens GR, Higgins PDR, et al. Long‐term safety and tolerability of oral tofacitinib in patients with Crohn’s disease: results from a phase 2, open‐label, 48‐week extension study. Aliment Pharmacol Ther. 2019;49:265–276. 10.1111/apt.15072
Funding information
This study was funded in full by Pfizer Inc. The writing of this paper was funded by Pfizer Inc. Medical writing support, under the guidance of the authors, was provided by Rebecca Douglas, PhD, at CMC Connect, a division of Complete Medical Communications Ltd, Macclesfield, UK and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461‐464).
The Handling Editor for this article was Dr Nicholas Kennedy, and it was accepted for publication after full peer‐review.
REFERENCES
- 1. Torres J, Mehandru S, Colombel JF, Peyrin‐Biroulet L. Crohn's disease. Lancet. 2017;389:1741‐1755. [DOI] [PubMed] [Google Scholar]
- 2. Love JR, Irvine EJ, Fedorak RN. Quality of life in inflammatory bowel disease. J Clin Gastroenterol. 1992;14:15‐19. [DOI] [PubMed] [Google Scholar]
- 3. Lichtenstein GR, Abreu MT, Cohen R, Tremaine W. American Gastroenterological Association Institute technical review on corticosteroids, immunomodulators, and infliximab in inflammatory bowel disease. Gastroenterology. 2006;130:940‐987. [DOI] [PubMed] [Google Scholar]
- 4. Gordon JP, McEwan PC, Maguire A, Sugrue DM, Puelles J. Characterizing unmet medical need and the potential role of new biologic treatment options in patients with ulcerative colitis and Crohn's disease: a systematic review and clinician surveys. Eur J Gastroenterol Hepatol. 2015;27:804‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panés J, Sandborn WJ, Schreiber S, et al. Tofacitinib for induction and maintenance therapy of Crohn's disease: results of two phase IIb randomised placebo‐controlled trials. Gut. 2017;66:1049‐1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96:804‐810. [PubMed] [Google Scholar]
- 7. Ware JE Jr, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). I. Conceptual framework and item selection. Med Care. 1992;30:473‐483. [PubMed] [Google Scholar]
- 8. Rabin R, de Charro F. EQ‐5D: a measure of health status from the EuroQol Group. Ann Med. 2001;33:337‐343. [DOI] [PubMed] [Google Scholar]
- 9. Panés J, Su C, Bushmakin AG, Cappelleri JC, Mamolo C, Healey P. Randomized trial of tofacitinib in active ulcerative colitis: analysis of efficacy based on patient‐reported outcomes. BMC Gastroenterol. 2015;15:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Panés J, Su C, Bushmakin AG, Cappelleri JC, Healey P. Direct and indirect effects of tofacitinib on treatment satisfaction in patients with ulcerative colitis. J Crohns Colitis. 2016;10:1310‐1315. [DOI] [PubMed] [Google Scholar]
- 11. Coteur G, Feagan B, Keininger DL, Kosinski M. Evaluation of the meaningfulness of health‐related quality of life improvements as assessed by the SF‐36 and the EQ‐5D VAS in patients with active Crohn's disease. Aliment Pharmacol Ther. 2009;29:1032‐1041. [DOI] [PubMed] [Google Scholar]
- 12. Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376:1723‐1736. [DOI] [PubMed] [Google Scholar]
- 13. Wollenhaupt J, Silverfield J, Lee EB, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: safety and efficacy in open‐label, long‐term extension studies over 9 years. Arthritis Rheumatol. 2017;69:683‐684. [DOI] [PubMed] [Google Scholar]
- 14. Panaccione R, Colombel JF, Sandborn WJ, et al. Adalimumab maintains remission of Crohn's disease after up to 4 years of treatment: data from CHARM and ADHERE. Aliment Pharmacol Ther. 2013;38:1236‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panaccione R, Colombel JF, Sandborn WJ, et al. Adalimumab sustains clinical remission and overall clinical benefit after 2 years of therapy for Crohn's disease. Aliment Pharmacol Ther. 2010;31:1296‐1309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials