

Abstract
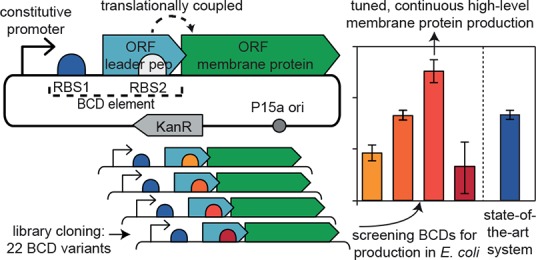
Escherichia coli has been widely used as a platform microorganism for both membrane protein production and cell factory engineering. The current methods to produce membrane proteins in this organism require the induction of target gene expression and often result in unstable, low yields. Here, we present a method combining a constitutive promoter with a library of bicistronic design (BCD) elements, which enables inducer-free, tuned translation initiation for optimal protein production. Our system mediates stable, constitutive production of bacterial membrane proteins at yields that outperform those obtained with E. coli Lemo21(DE3), the current gold standard for bacterial membrane protein production. We envisage that the continuous, fine-tunable, and high-level production of membrane proteins by our method will greatly facilitate their study and their utilization in engineering cell factories.
Keywords: protein production, membrane proteins, translational coupling, bicistronic design, Escherichia coli
The high-level heterologous production of membrane proteins in E. coli and other hosts has proven challenging, especially due to oversaturation of the membrane protein biogenesis machinery.1 Common systems for recombinant protein production, such as those based on the strong T7 promoter, often lead to the jamming of chaperones and membrane translocation systems, consequently making it impossible to produce correctly folded membrane proteins at high levels.1,2
Several E. coli strains and expression systems have been developed to improve the production of especially bacterial membrane proteins. Commonly used systems include the E. coli Walker strains (C41(DE3),C43(DE3)),3E. coli BL21-AI,4 and the more recently developed E. coli Lemo21(DE3).5−7 These systems rely on downregulating the levels of T7 RNA polymerase (T7RNAP), consequently reducing expression rates to better accommodate translocation and folding of membrane proteins.8 Particularly, E. coli Lemo21(DE3) has been constructed to fine-tune transcription through an indirect control of T7RNAP activity through l-rhamnose-inducible production of its inhibitor, T7-lysozyme (LysY).5−7 The system has proven successful and has been recently streamlined into a one-plasmid system named pReX,9 but it requires the properly timed addition of two different inducer compounds: l-rhamnose and the expensive IPTG (isopropyl β-d-1-thiogalactopyranoside). Additionally, to date, neither Lemo21(DE3) nor any other currently available system has successfully demonstrated long-term (>24 h) continuous production of membrane proteins in E. coli. Realizing inducer-free, stable production remains a major challenge, which is relevant for many synthetic biology applications. This includes, for example, the heterologous production of transporter membrane proteins in microbial cell factories to be used in (continuous) production processes, or signal-transduction membrane proteins in strains that need to function as a biosensor over a long time.
Another limitation in heterologous protein production relates to ribosome binding site (RBS) accessibility.10−12 Several proteins, including membrane proteins, have been categorized as “difficult-to-produce” due to strong mRNA secondary structures in the 5′ untranslated region (5′-UTR) and in the start of the coding sequence (CDS), which impede the proper translation initiation at the RBS.13,14 Some efforts aimed at resolving such structures rely on fusing well-expressed short peptide tags to the N-terminus of membrane proteins.13,15,16 However, fusion peptides can affect protein stability, structure, and function.13 Furthermore, translation initiation has been successfully improved by randomly mutating nucleotides around the start codon,13,14,17 which enhanced the production of several “difficult-to-produce” membrane proteins, but this method requires a high-throughput screening or selection approach.
To overcome the limitations of state-of-the-art membrane protein production, we explored an alternative method for protein expression: the so-called bicistronic design elements (BCDs).18 The system is based on a constitutive promoter and tuning by using two RBSs that are translationally coupled. The first RBS mediates strong translation initiation of a short leader peptide, while the second RBS, which is located within the leader peptide’s CDS and drives the translation of the protein of interest, has a tunable strength (Figure 1). It is hypothesized that the intrinsic helicase activity of the ribosomes translating the leader peptide can unwind potential secondary structures of the mRNA, thereby eliminating translation initiation problems near the second RBS and the start of the target CDS.19
Figure 1.
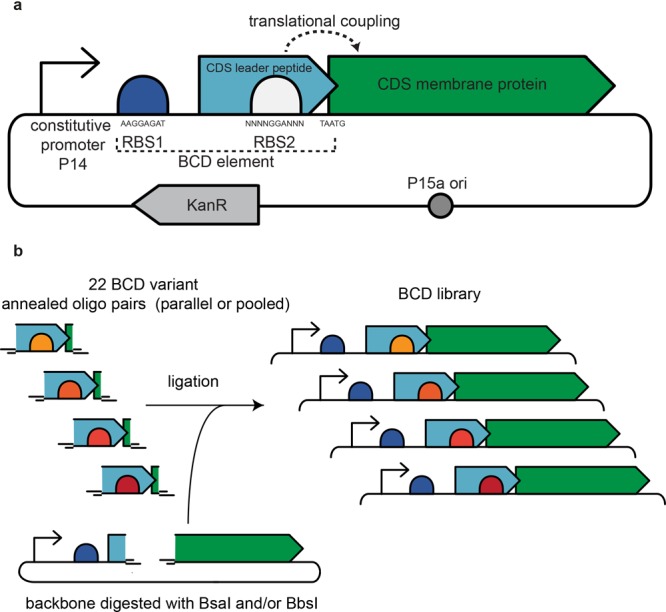
Expression vector design and assembly. (a) The standard expression vector contains a medium-strength constitutive promoter, RBS1, which allows for strong translation initiation of a leader peptide, and a translationally coupled, variable RBS2, mediating translation initiation of the coding sequence (CDS) of the membrane protein of interest.18 (b) Vectors are assembled with different BCD elements. First, the vector is amplified by PCR, subsequently it is digested by type IIS restriction enzymes. The latter allows for seamless assembly with a library of annealed oligo pairs (encoding the different BCD variants), which have overhangs complementary to the digested vector.
In this work, we employ a library of translational coupling elements to tune and optimize the constitutive production of several bacterial membrane proteins. A medium-strength, constitutive promoter (P14) and 22 variable-strength translational coupling elements (BCDs) were selected from the work of Mutalik et al.;18 the BCD elements can be inserted seamlessly in the expression vector using a simple golden gate-based cloning method20 (Figure 1).
We tested our system for the expression of four different membrane proteins: YidC, AraH, and two rhodopsins. YidC is a membrane-translocation chaperone in E. coli, which has been frequently used as a model for studying membrane protein production.6,21,22 AraH is the integral membrane component of the E. coli arabinose ABC transporter, which is considered a “difficult-to-produce” protein because of its translation initiation limitations in a typical pET vector;13,14 GR is a proton-pumping rhodopsin photosystem from the cyanobacterium Gloeobacter violaceus, and TR is a thermophilic rhodopsin from Thermus thermophilus.
For the two first proteins, YidC and AraH, GFP was fused to their C-terminus, rendering YidC-GFP and AraH-GFP. All the 22 BCD variants were cloned separately into expression vectors carrying YidC-GFP and AraH-GFP. We then estimated levels of membrane inserted YidC and AraH by measuring fluorescence levels. C-terminal GFP only folds properly and results in fluorescence when membrane proteins fused to the GFP are integrated into the membrane and do not end up in inclusion bodies, hence providing a quantitative approximation for membrane-embedded expression.23,24 The expression of YidC-GFP and AraH-GFP by different BCD elements was ranked in order of the translation initiation strengths previously observed for each BCD element in the work by Mutalik et al.,18 in order to allow for a systematic analysis of their fluorescence (Figure 2a,e).
Figure 2.

Production of YidC-GFP, AraH-GFP, and rhodopsins by BCD elements and comparison to state-of-the art systems. (a) Volumetric YidC-GFP production based on whole-cell fluorescence measurements and final growth yields for the BCD constructs, and a comparison to Lemo21(DE3)-based production at optimized (2 mM l-rhamnose) and nonoptimized (0 mM l-rhamnose) conditions. BCD variants are ordered in the X-axis based on previously reported translation initiation strength.18 (b) Western blots performed with antihis-tag antibody (upper panels) to visualize both inclusion body and well-folded YidC-GFP-his, and anti-IbpB6,21 (lower panels) to visualize inclusion body binding protein B. (c) Single-cell production of YidC-GFP analyzed by flow cytometry for several increasing strength BCD elements and optimized Lemo21(DE3). (d) Single-cell production of YidC-GFP by BCD19 and BCD2 in a 72 h stability experiment. (e) Volumetric AraH-GFP production based on whole-cell fluorescence measurements and final growth yields for the BCD constructs, and a comparison to pET-opt-AraH-GFP.14 BCD variants are ordered in the X-axis based on previously reported variant strength.18 (f) Western blots performed with antihis-tag antibody (upper panels) to visualize both AraH-GFP-his, and anti-IbpB6,21 (lower panels) to visualize inclusion body binding protein. (g) Single-cell production of AraH-GFP analyzed by flow cytometry for several increasing strength BCD elements and pET-opt-AraH-GFP. (h) Single-cell production of AraH-GFP by BCD19 and BCD2 in a 72 h stability experiment. (i) Volumetric Gloeobacter Rhodopsin (GR) and (j) Thermophilic Rhodopsin (TR) production determined by spectroscopy, and pictures of red-pigmented pellets. All cultivations were performed in 10 mL of medium in 50 mL tubes, for YidC-AraH and AraH-GFP at 30 °C, for rhodopsins GR and TR at 37 °C, and for pET-opt-AraH-GFP in E. coli BL21(DE3) pLysS at 25 °C (as optimized for in original work). BCD-based production was measured after 22 h of cultivation, while Lemo21(DE3) and pET based production was measured after 22 h of induction. Whole-cell fluorescence or rhodopsin quantification data are based on at least three biological replicates. For 72 h stability experiments E. coli BL21(DE3) harboring BCD vectors were reinoculated 1:50 into fresh LB kanamycin medium every 24 h. Notation: RFU, relative fluorescence units; OD600, optimal density of 600 nm.
For both fusion proteins, the tested BCD variants resulted in a range of GFP fluorescence-signals, suggesting different levels of functional membrane protein production. In the case of YidC-GFP, a rough pattern was observed considering the correlation between the fluorescence and the expected translation initiation strength of the different BCD constructs. BCD elements up to BCD19 generally resulted in increased levels of production, whereas elements stronger than BCD19 mostly resulted in lower production levels (Figure 2a). Some of the strongest translation initiation variants resulted in negligible and/or highly irreproducible production levels. For example, in some replicate cultures the strong BCD2 gave high expression but in several other cultures expression was completely absent (ranging from no expression to 70 000 RFU/mL). When the BCD-based expression was compared to the optimized Lemo21(DE3)-based expression of YidC-GFP, the latter gave rise to an emerging nonproducing subpopulation of cells after 22 h, as indicated by flow cytometry analysis, whereas the production by the highest-producing BCD19-YidC-GFP remained homogeneous (Figure 2c). In agreement with this observation, Western blot analysis revealed that the formation of YidC-GFP in inclusion bodies was reduced for the BCD19 versus optimized Lemo21(DE3)-expression (Figure 2b). Moreover, the medium-strength BCD19 yielded approximately twice as much production per cell than E. coli Lemo21(DE3), which was previously proven to be a superior production system for YidC-GFP over other commonly used systems such as E. coli C41(DE3) and C43(DE3).6
In the case of AraH-GFP, a similar trend was observed, that is, increasing levels of fluorescence were measured up to BCD19. However, unlike for YidC-GFP, no large decrease or unstable AraH-GFP production was observed for stronger BCD elements, and the strongest BCD2 produced at similar high levels as BCD19 (Figure 2e). AraH-GFP production by BCD19 and BCD2 was compared to the production by a previously optimized pET vector (pET-opt-AraH-GFP), which was obtained by screening a large library of vectors with mutations around the start codon.14 The volumetric production by both BCDs was found to be higher than that of pET-opt-AraH-GFP under the originally optimized conditions, mainly due to a higher biomass yield (Figure 2e, Figure S2). Production by the BCDs also resulted in less inclusion body binding protein than pET-based production, while a nonfolded protein could not be clearly detected in Western blot for any construct (Figure 2f). Additionally, production by pET-opt-AraH-GFP resulted in a small emerging nonproducing population of cells after 22 h of cultivation, as revealed by flow cytometry analysis, while production by the BCD elements was still fully homogeneous (Figure 2g). Notably, by screening a library of only 22 BCD variants, we were able to find clones for which the production was comparable (per cell) or even better (per volume) than that of the previously optimized pET-opt-AraH-GFP, which required the high-throughput screening of a large library of variants (1.6 × 104) through fluorescence-activated cell sorting (FACS).14
To further estimate the production stability of the medium-strength, high-producing BCD19 and the strongest translation initiation element BCD2, we assessed production per cell by flow cytometry during longer serial cultivation experiments up to 72 h. Remarkably, BCD19-YidC-GFP, and for AraH both BCD19 and BCD2, result in stable homogeneously producing populations, even after 72 h (Figure 2d,h, Figure S3). For BCD2-YidC-GFP, however, only 2 out of 4 precultures prepared for the stability experiment maintained their initial production level, despite the fact that the colonies used for initial inoculation were selected on the basis of high fluorescence. This demonstrated again an unstable production phenotype for the strong BCD2 with YidC, as observed we observed in earlier experiments. The two stably producing precultures were further inoculated for the long-term experiment in fresh medium, and their production decreased over the course of time (Figure 2d, Figure S3). The strong translation initiation of YidC-GFP driven by BCD2 seemed to stress the cells, favoring the emergence of nonexpressing cells in the population (Figure 2d), which may have been caused by suppressing mutations in plasmids or the genome (not further characterized).
The BCD system was further employed to optimize the production of bacterial rhodopsin proteins. These simple membrane-bound light-harvesting energy systems can be employed for light-driven cell-factories25 or optogenetic regulation.26 When properly folded in the cytoplasmic membrane, rhodopsins are known to bind the retinal pigment, leading to red pigmentation of the host cells.27,28 Rather than cloning all the BCDs in parallel, this time the 22 BCD variants were pooled, cloned into the vectors containing GR or TR, and transformed as a library. Clones of the resulting transformation were then randomly picked and grown in 96-well plates with retinal (Figure S4). For both GR and TR, 11 clearly red-pigmented pellets were identified out of 89 and 96 screened clones, respectively. For each of the rhodopsin proteins, three intensely red clones were selected for further characterization. In the case of GR, one of the selected clones contained BCD14 and the other two carried BCD15. For TR, BCD9, BCD12, and BCD21 were identified. All these variants are in the medium-strength range of the BCD system.
Production of GR and TR by the best rhodopsin-producing BCD variants was scaled up from deep-well plates to 10 mL cultures in 50 mL tubes. While all elements rendered very similar levels of GR expression (Figure 2i), the production of TR significantly differed from one BCD to another; BCD9 performed significantly better than BCD12, while BCD21 gave the lowest production (Figure 2j). These three variants performed very similar when grown in deep-96-well plates, while the results of the production assays in 50 mL tubes are significantly different. This indicates that results are not always comparable when scaling-up from deep-well plates (0.5 mL culture) to test tubes (10 mL culture). The previously discussed strains of YidC-GFP and AraH-GFP were initially grown directly in 10 mL cultures in 50 mL tubes, and now compared with their performance in deep-well plates (0.5 mL culture) (Figure S5), confirming differences in performance depending on culture conditions. This reflects the common challenge of optimizing and scaling up recombinant production. However, the optimization for the BCD system in scaled-up conditions is quite feasible given the limited number of tuning variants that need to be tested.
We then compared the production of GR and TR to the levels produced by the Lemo21(DE3) system, which was optimized in tubes. Compared to rhodopsin production by the Lemo21(DE3) system, the best performing BCD variants for GR and TR resulted in at least 2-fold and 3-fold higher volumetric rhodopsin production, respectively (Figure 2i,j).
The here employed BCD system outperforms the membrane protein productivities of previously established approaches. Moreover, the BCD system provides stable membrane protein production for at least 72 h, a stability never reported to date. While most current systems for membrane protein production are limited to specific E. coli strains, the applied P14 constitutive promoter allows our system to be generally applicable in all E. coli strains. By applying the principles of our method and potentially including different promoters and/or BCDs, our system is likely feasible for bacterial membrane protein production in other bacterial hosts as well. The BCD system may also be applicable for producing certain eukaryotic membrane proteins in E. coli, although expression of eukaryotic proteins in bacterial hosts may lead to issues that cannot be solved just by tuning expression strength and tackling RBS-accessibility, this includes issues as glycosylation or the requirement of eukaryotic-like membrane lipids.1 Overall, it is anticipated that the here described approach for bacterial membrane protein production will be useful for many future studies, ranging from biochemical characterization to cell factory and biosensor engineering.
Acknowledgments
We thank Wendy van der Vliet and Snezana Gegic for their kind assistance with the flow cytometry experiments. We thank Daniel Daley for sharing pGFPe-AraH-his. All BIOFAB plasmids were a kind gift of Drew Endy. We gratefully acknowledge financial support from the Wageningen University IP/OP Systems Biology (Grant KB-17-003.02-024) and The Netherlands Organization for Scientific Research (NWO) via the Spinoza and SIAM Gravitation Grant (Project 024.002.002) to W.M.d.V., and a Rubicon Grant to N.C. (019.163LW.035), M.F.B and J.v.d.O are supported by the NWO Gravitation Grant BaSyC (024.003.019). The funders had no role in study design, data collection and analysis, or preparation of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.9b00101.
Methods; Supplementary figures; Codon-adapted gene sequences; Supplementary references (PDF)
Author Contributions
The idea for this study was conceived by N.J.C., and experiments were designed by N.J.C., M.F.B., J.W.d.G., W.M.d.V., and J.v.d.O. N.J.C., M.F.B., B.S., F.M., and J.d.G. performed the experimental work and data analysis. N.J.C. and M.F.B. wrote the manuscript, which was critically revised by B.S., J.W.d.G., W.M.d.V., and J.v.d. and approved by all authors.
Author Contributions
# N.J.C. and M.F.-B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Schlegel S.; Hjelm A.; Baumgarten T.; Vikström D.; de Gier J.-W. (2014) Bacterial-based membrane protein production. Biochim. Biophys. Acta, Mol. Cell Res. 1843, 1739–49. 10.1016/j.bbamcr.2013.10.023. [DOI] [PubMed] [Google Scholar]
- Wagner S.; Baars L.; Ytterberg A. J.; Klussmeier A.; Wagner C. S.; Nord O.; Nygren P.-A.; van Wijk K. J.; de Gier J.-W. (2007) Consequences of Membrane Protein Overexpression in Escherichia coli. Mol. Cell. Proteomics 6, 1527–1550. 10.1074/mcp.M600431-MCP200. [DOI] [PubMed] [Google Scholar]
- Miroux B.; Walker J. E. (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–98. 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Narayanan A.; Ridilla M.; Yernool D. A. (2011) Restrained expression, a method to overproduce toxic membrane proteins by exploiting operator – repressor interactions. Protein Sci. 20, 51–61. 10.1002/pro.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel S.; Löfblom J.; Lee C.; Hjelm A.; Klepsch M.; Strous M.; Drew D.; Slotboom D. J.; De Gier J.-W. (2012) Optimizing Membrane Protein Overexpression in the Escherichia coli strain Lemo21(DE3). J. Mol. Biol. 423, 648–659. 10.1016/j.jmb.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Wagner S.; Klepsch M. M.; Schlegel S.; Appel A.; Draheim R.; Tarry M.; Hogbom M.; van Wijk K. J.; Slotboom D. J.; Persson J. O.; de Gier J.-W. (2008) Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. U. S. A. 105, 14371–14376. 10.1073/pnas.0804090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelm A.; Schlegel S.; Baumgarten T.; Klepsch M.; Wickström D.; Drew D.; De Gier J.-W. (2013) Optimizing E. coli-Based Membrane Protein Production Using Lemo21(DE3) and GFP-Fusions. Methods Mol. Biol. 1033, 381–400. 10.1007/978-1-62703-487-6_24. [DOI] [PubMed] [Google Scholar]
- Schlegel S.; Genevaux P.; de Gier J.-W. (2015) De-convoluting the Genetic Adaptations of E. coli C41(DE3) in Real Time Reveals How Alleviating Protein Production Stress Improves Yields. Cell Rep. 10, 1758–1766. 10.1016/j.celrep.2015.02.029. [DOI] [PubMed] [Google Scholar]
- Kuipers G.; Karyolaimos A.; Zhang Z.; Ismail N.; Trinco G.; Vikström D.; Slotboom D. J.; de Gier J.-W. (2017) The tunable pReX expression vector enables optimizing the T7-based production of membrane and secretory proteins in E. coli. Microb. Cell Fact. 16, 226. 10.1186/s12934-017-0840-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G.; Murray A. W.; Tollervey D.; Plotkin J. B. (2009) Coding-Sequence Determinants of Gene Expression in Escherichia coli. Science (Washington, DC, U. S.) 324, 255–258. 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambray G.; Guimaraes J. C.; Arkin A. P. (2018) Evaluation of 244,000 synthetic sequences reveals design principles to optimize translation in Escherichia coli. Nat. Biotechnol. 36, 1005–1015. 10.1038/nbt.4238. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop T.; Claassens N. J.; van der Oost J. (2019) Improved protein production and codon optimization analyses in Escherichia coli by bicistronic design. Microb. Biotechnol. 12, 173–179. 10.1111/1751-7915.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørholm M. H. H.; Toddo S.; Virkki M. T. I.; Light S.; von Heijne G.; Daley D. O. (2013) Improved production of membrane proteins in Escherichia coli by selective codon substitutions. FEBS Lett. 587, 2352–8. 10.1016/j.febslet.2013.05.063. [DOI] [PubMed] [Google Scholar]
- Mirzadeh K.; Martinez V.; Toddo S.; Guntur S.; Herrgard M.; Elofsson A.; Nørholm M. H. H.; Daley D. O. (2015) Enhanced protein production in Escherichia coli by optimization of cloning scars at the vector:coding sequence junction. ACS Synth. Biol. 4, 959–965. 10.1021/acssynbio.5b00033. [DOI] [PubMed] [Google Scholar]
- Kim H. S.; Ernst J. a; Brown C.; Bostrom J.; Fuh G.; Lee C. V.; Huang A.; Vandlen R. L.; Yansura D. G. (2012) Translation levels control multi-spanning membrane protein expression. PLoS One 7, e35844. 10.1371/journal.pone.0035844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Albacete D.; Cavaleiro A. M.; Christensen U.; Seppälä S.; Møller B. L.; Nørholm M. H. H. (2017) An expression tag toolbox for microbial production of membrane bound plant cytochromes P450. Biotechnol. Bioeng. 114, 751–760. 10.1002/bit.26203. [DOI] [PubMed] [Google Scholar]
- Rennig M.; Martinez V.; Mirzadeh K.; Dunas F.; Röjsäter B.; Daley D. O. O.; Nørholm M. H. H. H. H. (2018) TARSyn: Tuneable antibiotic resistance devices enabling bacterial synthetic evolution and protein production. ACS Synth. Biol. 7, 432–442. 10.1021/acssynbio.7b00200. [DOI] [PubMed] [Google Scholar]
- Mutalik V. K.; Guimaraes J. C.; Cambray G.; Lam C.; Christoffersen M. J.; Mai Q.-A.; Tran A. B.; Paull M.; Keasling J. D.; Arkin A. P.; Endy D.; Hoynes-O’Connor A.; Moon T. S. (2013) Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods 10, 354–360. 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- Takyar S.; Hickerson R. P.; Noller H. F. (2005) mRNA helicase activity of the ribosome. Cell 120, 49–58. 10.1016/j.cell.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Engler C.; Kandzia R.; Marillonnet S. (2008) A one pot, one step, precision cloning method with high throughput capability. PLoS One 3, e3647. 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel S.; Löfblom J.; Lee C.; Hjelm A.; Klepsch M.; Strous M.; Drew D.; Slotboom D. J.; de Gier J.-W. (2012) Optimizing membrane protein overexpression in the Escherichia coli strain Lemo21(DE3). J. Mol. Biol. 423, 648–59. 10.1016/j.jmb.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Gialama D.; Kostelidou K.; Michou M.; Delivoria D. C.; Kolisis F. N.; Skretas G. (2017) Development of Escherichia coli strains that withstand membrane protein-induced toxicity and achieve high-level recombinant membrane protein production. ACS Synth. Biol. 6, 284–300. 10.1021/acssynbio.6b00174. [DOI] [PubMed] [Google Scholar]
- Drew D.; Slotboom D.-J.; Friso G.; Reda T.; Genevaux P.; Rapp M.; Meindl-Beinker N. M.; Lambert W.; Lerch M.; Daley D. O.; Van Wijk K.-J.; Hirst J.; Kunji E.; De Gier J.-W. (2005) A scalable, GFP-based pipeline for membrane protein overexpression screening and purification. Protein Sci. 14, 2011–2017. 10.1110/ps.051466205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew D. E; von Heijne G.; Nordlund P.; de Gier J.-W. L (2001) Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 507, 220–224. 10.1016/S0014-5793(01)02980-5. [DOI] [PubMed] [Google Scholar]
- Claassens N. J.; Volpers M.; Martins dos Santos V. A. P.; van der Oost J.; de Vos W. M. (2013) Potential of proton-pumping rhodopsins: engineering photosystems into microorganisms. Trends Biotechnol. 31, 633–642. 10.1016/j.tibtech.2013.08.006. [DOI] [PubMed] [Google Scholar]
- Deisseroth K. (2015) Nat. Neurosci. 18, 1213–1225. 10.1038/nn.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdon P.; Alfredsson A.; Pedersen A.; Malmerberg E.; Nyblom M.; Widell M.; Berntsson R.; Pinhassi J.; Braiman M.; Hansson Ö.; Bonander N.; Karlsson G.; Neutze R. (2008) Optimized in vitro and in vivo expression of proteorhodopsin: A seven-transmembrane proton pump. Protein Expression Purif. 58, 103–113. 10.1016/j.pep.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Martinez A.; Bradley A. S.; Waldbauer J. R.; Summons R. E.; DeLong E. F. (2007) Proteorhodopsin photosystem gene expression enables photophosphorylation in a heterologous host. Proc. Natl. Acad. Sci. U. S. A. 104, 5590–5595. 10.1073/pnas.0611470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.