

Abstract
Purpose:
Patients with BRAF V600 wild-type melanoma whose tumors progress on checkpoint inhibition currently have limited therapeutic options, and additional rational treatment targets are needed. ERBB2 alterations may be amenable to targeted inhibition, but the rate of ERBB2 alterations across melanoma subtypes is not well described.
Patients and Methods:
All patients with nonuveal melanoma (cutaneous, acral, mucosal, and unknown primary) whose tumors underwent multigene sequencing with MSK-IMPACT at Memorial Sloan Kettering Cancer Center (New York, NY) from 2014 to 2018 were reviewed for known or likely oncogenic somatic alterations in ERBB2 and the other known canonical driver genes BRAF, NRAS, KIT, NF1, GNAQ, and GNA11.
Results:
A patient with acral melanoma resistant to checkpoint inhibition was found to have ERBB2 amplification and achieved a durable complete response to trastuzumab emtansine. Tumor sequencing results from 732 melanoma cases were analyzed for ERBB2 and canonical driver gene alterations. ERBB2 amplifications were detected in acral (3%) and mucosal (3%) melanomas. ERBB2 mutations were found in cutaneous (1%), acral (2%), and mucosal (2%) subtypes and frequently cooccurred with NF1 alterations. Among the 140 patients whose tumors lacked canonical driver alterations, ERBB2 amplifications were detected in acral (7%) and mucosal (6%) melanomas.
Conclusions:
ERBB2 amplification is present in a minority of acral lentiginous and mucosal melanomas. Activating mutations in ERBB2 were identified in nonuveal melanoma subtypes and are frequently comutated with canonical drivers. HER2 could represent a therapeutically relevant target across melanoma subtypes.
Introduction
The introduction of programmed death-1 (PD-1)–based checkpoint inhibition and BRAF V600–directed therapies has been crucial advancements in the treatment of melanoma (1). However, for patients whose tumors progress on first-line therapies, existing options have limited utility, particularly for patients whose tumors are BRAF V600 wild-type (1). Studies enrolling patients with melanoma that have progressed on PD-1–based therapy are ongoing, but early results of novel checkpoint inhibitor combinations suggest objective response rate (ORR) similar to traditional cytotoxic chemotherapy (2). Acral lentiginous melanoma is a rare subtype of melanoma arising from the palms, soles, and nail beds. BRAF V600 mutations are less prevalent in acral melanoma than in cutaneous melanoma (~15% vs. ~45%) suggesting that the pathogenesis of these melanomas is distinct (3). Prior studies suggest that the majority of acral melanomas harbor alterations in cyclin-dependent kinase pathways such as CDKN2A/CDK4/ CCND1 that are less easily targetable by currently available therapies (4). Identifying novel actionable driver alterations would provide important new opportunities for therapeutic intervention.
Alterations in ERBB2, the gene that encodes the HER2 receptor tyrosine kinase, can lead to uncontrolled cellular proliferation and oncogenesis through various mechanisms (5). The therapeutic implications of ERBB2 amplifications are best understood in breast adenocarcinomas, where they are predictive of clinical benefit from HER2-directed mAbs (trastuzumab and pertuzumab), antibody-drug conjugates (trastuzumab emtansine), and HER2 kinase inhibitors (lapatinib; ref. 6). ERBB2 amplifications are also predictive of response to trastuzumab in gastroesophageal junction adeno-carcinoma (7). In addition, it is now recognized that some cancers activate ERBB2 through mutation rather than amplification of the wild-type gene (8). Recent clinical data suggest that a subset of patients with ERBB2 mutant tumors can achieve durable responses to the irreversible pan-HER kinase inhibitor neratinib (9).
Trastuzumab emtansine (T-DM1) acts through the binding of the trastuzumab antibody component to the HER2 receptor, which triggers endocytosis of the HER2-T-DM1 complex and subsequent release of the microtubule assembly-inhibiting emtansine component (10). A phase III clinical trial has shown 44% ORR to TDM-1 in advanced breast cancers (11). We report the case of a patient with acral melanoma refractory to checkpoint inhibition found to harbor ERBB2 amplification who was treated with HER2-directed T-DM1 therapy. We also report the frequency of ERBB2 alterations across multiple melanoma subtypes at a single institution.
Case Report
A 58-year-old Caucasian male was diagnosed in 2012 with T4bN1a acral melanoma of the left great toe that was BRAF/ KIT/NRAS wild-type using targeted exon sequencing. He was treated with wide local excision, full lymph node dissection, and adjuvant high-dose interferon alpha-2b. Six months into adjuvant therapy, he was found to have isolated lung metastases in the right middle lobe and opted to undergo wedge resection. Eleven months later, he had a recurrence in the lung and enrolled on a clinical trial of tumor infiltrating lymphocyte (TIL) therapy with primary progression. He was then treated with ipilimumab 3 mg/kg plus nivolumab 1 mg/kg within the context of a clinical trial. He received three doses with a partial response complicated by grade 4 hemolytic anemia. For 18 months, he received nivolumab 3 mg/kg every 2 weeks. During this time, he had two local interventions for isolated progressive disease in the lung and mediastinum (10 and 14 months into nivolumab therapy, respectively) with eventual progression in the pleura. He then received one cycle of temozolomide 175 mg/m2 × 5 days, which was discontinued due to grade 4 pancytopenia. Within 1 month of receiving temozolomide, the patient’s disease had progressed with an enlarging pleural mass and the development of right pleural and pericardial effusions (Fig. 1A).
Figure 1.
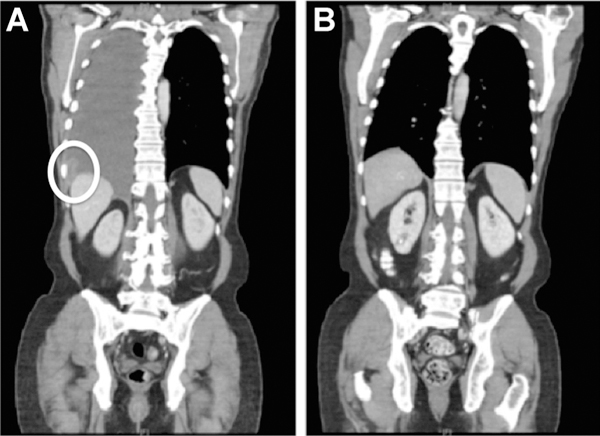
A, Coronal CT image of the chest, abdomen, and pelvis 2 weeks prior to T-DM1 treatment is notable for pleural/mediastinal disease with symptomatic pleural effusions. B, After 8 months of T-DM1 therapy, a comparison image shows a complete response in the pleural/mediastinal disease and resolution of the effusions that is maintained for 27+ months.
As the patient’s prior limited tumor molecular analysis had identified no known driver alteration, a lung metastasis was collected following disease progression on immune checkpoint inhibitor therapy and analyzed using a hybridization capture-based panel of 410 cancer-related genes (MSK-IMPACT; ref. 12). The tumor had a low mutation rate with only a single missense mutation, a missense mutation of unknown significance in the AURKB gene. The tumor did, however, harbor 26-fold amplification of ERBB2. CDK12, also located on Chromosome 17q12, was amplified 7.3-fold, and CDK4 and MDM2 were amplified 3.8- and 4.2-fold.
On the basis of the finding of ERBB2 amplification, the patient was treated off-label with T-DM1 at a dose of 2.7 mg/kg every 3 weeks, which represented an upfront 25% dose reduction due to baseline thrombocytopenia. After 2 months of T-DM1 treatment, a repeat CT scan showed a significant reduction in both the pleural and pericardial effusions and otherwise stable pleural disease. Following 8 months of T-DM1, there was a radiographic complete response with resolution of effusions (Fig. 1B). This response has persisted for 28 months, and treatment is ongoing without evidence of disease progression. The patient’s treatment course has been notable for grade 2 transaminitis with regenerative hepatic nodules, which was managed with occasional dose delays. The patient has consented to the publication of this report.
Retrospective Assessment of ERBB2 Alterations in Melanoma Subtypes
Given the index patient’s profound and durable response to HER2-directed therapy, we sought to characterize the rate of ERBB2 aberrations across melanoma subtypes. After Institutional Review Board approval of methods in accordance with the Belmont Report, we clinically annotated and reviewed genomic data for all 732 patients with nonuveal melanoma who had undergone tumor genomic profiling using MSK-IMPACT at our institution between 2014 and 2018. Mutations in ERBB2 were assessed for functional significance using the annotated database OncoKB (13). The primary site was cutaneous in 438 patients, acral in 58, mucosal in 118, or an unknown primary in 118. Alterations detected included nonsynonymous single nucleotide variants, small indels, copy-number alterations (log2 fold change >2 or <−2), and structural rearrangements. All patients without detected oncogenic alterations in BRAF, NRAS, KIT, NF1, GNAQ, or GNA11 were considered “canonical wild-type” cases.
In patients with melanoma who had undergone MSK-IMPACT testing, the rate of ERBB2 amplification was 3% in acral (range, 3.5 to 26.0-fold), 3% in mucosal (range, 2.1 to 35.8-fold), and 0% in other subtypes. Rates of ERBB2 missense mutation were 1% in cutaneous, 2% in acral, 3% in mucosal, and 0% in melanoma of unknown primary. Previously validated hotspot mutations were found in both the extracellular and kinase domains (Fig. 2). No melanomas had both ERBB2 amplification and coding alterations. In the subgroup of canonical wild-type tumors (N = 140), rates of ERBB2 amplification were 7% in acral (range, 3.5 to 26.0-fold) and 6% in mucosal melanoma (range, 2.4 to 25.8-fold). In contrast, among canonical wild-type tumors, ERBB2 missense mutations were only present in one cutaneous melanoma (Table 1). The remaining 6 ERBB2 mutant melanomas had comutation of NF1 (N = 4), KIT (N = 1), and NFI with BRAF (N = 1; Fig. 3).
Figure 2.
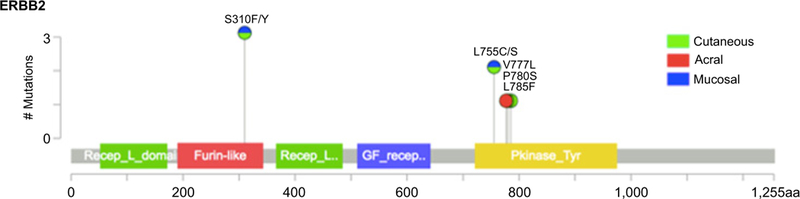
Lollipop plot of ERBB2-coding variants detected by MSK-IMPACT in seven patients with various subtypes of melanoma. One tumor was found to have two coding variants (S310F and L785F).
Table 1.
Oncogenic ERBB2 alterations detected by MSK-IMPACT in 732 patients with various subtypes of melanoma and the subset of tumors wild-type for BRAF, NRAS, KIT, NF1, GNAQ, and GNA11
| All tumors |
Canonical wild-type tumors |
|||||
|---|---|---|---|---|---|---|
| Melanoma subtype | N | ERBB2 amplification | ERBB2 mutation | N, % of total | ERBB2 amplification | ERBB2 mutation |
| Cutaneous | 438 | 0 (0%) | 4 (1%) | 56 (13%) | 0 (0%) | 1 (2%) |
| Acral | 58 | 2 (3%) | 1 (2%) | 28 (48%) | 2 (7%) | 0 (0%) |
| Mucosal | 118 | 4 (3%) | 2 (2%) | 35 (30%) | 2 (6%) | 0 (0%) |
| Unknown primary | 118 | 0 (0%) | 0 (0%) | 18 (15%) | 0 (0%) | 0 (0%) |
Figure 3.
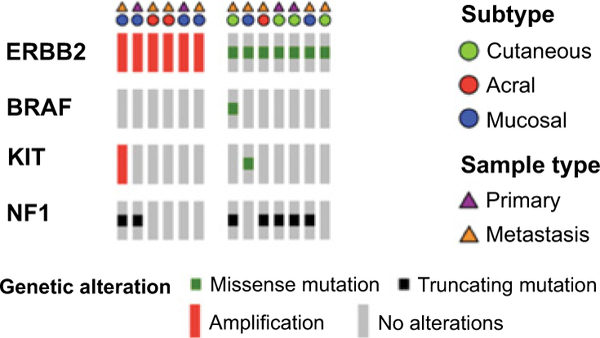
Oncoprint of ERBB2-altered cases (six amplifications and seven mutations) across melanoma subtypes. Canonical driver genes with concurrent alterations are listed. No alterations in NRAS, GNAQ, and GNA11 were detected in these cases.
Discussion
This case represents, to our knowledge, the first successful use of a HER2-targeted therapy in a patient with melanoma harboring ERBB2 amplification. The patient had progressed on ipilimumab plus nivolumab, investigational TIL therapy, had received multiple local interventions, and was intolerant to cytotoxic therapy. Treatment of this patient with trastuzumab emtansine for over 2 years represents meaningful therapeutic benefit derived from broad genomic sequencing that incorporates copy-number amplifications.
In contrast with cutaneous and unknown primary melanomas, acral and mucosal melanomas are less likely to have BRAF and NRAS driver alterations (3). A minority will have KIT alterations, although KIT-directed therapy has proven less effective than BRAF V600–directed therapy in patients with melanoma (14). As a result, most patients with acral and mucosal melanomas that progress on checkpoint inhibitors lack targeted therapy options. A prior study of 600 melanomas that included 18 acral and mucosal primaries found a rate of HER2 overexpression of 5% by IHC, but did not report rates by primary site (15). Our data suggest that ERBB2 amplifications are present at a rate of 3% in unselected acral lentiginous and mucosal melanomas, and are enriched (6%–7%) among the subset wild-type for BRAF, NRAS, KIT, NF1, GNAQ, and GNA11. Thus, clinical testing for ERBB2 amplification should be considered in acral or mucosal melanomas, particularly those that are wild-type for these canonical drivers. In contrast, ERBB2 mutations occur at low rates across cutaneous, mucosal, and acral melanomas and appear to cooccur with canonical alterations, most frequently NF1. The comutation pattern could potentially impact the sensitivity of these tumors to both HER2-targeted therapies as well as therapies directed against other canonical driver such as KIT. The 1% to 3% rate of ERBB2 mutations in this report may be an underestimate of the true rate of oncogenic ERBB2 alterations across melanoma subtypes as OncoKB periodically incorporates future investigations that validate additional pathogenic mutations.
ERBB2 amplifications and mutations appear to be distinct molecular events with little overlap across melanoma subtypes, consistent with findings from a recent pan-cancer analysis (16). The molecular basis for this distinction remains unclear, although responses to therapy appear to vary by type of ERBB2 alteration. ERBB2 amplifications have been successfully targeted in breast cancer by T-DM1, trastuzumab, and pertuzumab with and without conventional chemotherapy, resulting in overall response rates (ORR) of 44% to 80% (6). Targeted therapy in HER2-amplified metastatic colon cancers has also shown efficacy in phase II trials, with a 30% ORR with the combination of trastuzumab and lapatinib (17). Two preliminary reports of prospective trials of T-DM1 in HER2-amplified tumors suggest varying efficacy by histology, with several responses seen in salivary cancers but fewer seen in colon cancers (18, 19). No data on melanoma were reported from either trial. HER2-directed therapy has generally shown less benefit in ERBB2 mutant patients in prior clinical trials, although it also likely varies by histologic subtype. A phase II “basket trial” of neratinib for patients with ERBB2- or ERBB3-mutant tumors of any histology demonstrated an ORR of 32% in 25 patients with breast cancer, but a similar sized cohort of patients with lung cancer had a response rate of only 4% (9).
To our knowledge, the only other patient with melanoma treated thus far with HER2-directed therapy was a patient with cutaneous melanoma harboring an ERBB2 S310F mutation enrolled on the neratinib “basket trial.” The ERBB2 mutation in this tumor was the “sole driver” identified by MSK-IMPACT tumor profiling. Similar to the patient in this report, that patient had previously received ipilimumab-nivolumab and dacarbazine; in contrast to our case, that patient did not benefit to neratinib and progressed after 1.3 months (9). Notably, that patient’s tumor was of insufficient purity to allow for an assessment of the clonality of that alteration. Given the frequency of coaltered canonical drivers with ERBB2 missense mutations in this report, it is reasonable to hypothesize that the patient’s tumor may have harbored another undetected driver alteration. The recently reported MyPathway study enrolling patients with ERBB2 amplifications to receive trastuzumab plus pertuzumab did not specify the number of enrolled patients with melanoma, but no objective responses in this subtype were reported (20).
In our patient, the primary and untreated baseline metastatic tumors were not available for sequencing, so it is not clear whether the ERBB2 amplification was present at baseline or arose under the selective pressure of therapy. The depth and durability of response, however, strongly suggest that ERBB2 amplification represents either a true driver of this acral melanoma or a clinically relevant resistance mechanism to checkpoint inhibitor therapy. The presence of ERBB2 alterations in other baseline, pretreatment acral and melanoma samples in our cohort also suggests they represent recurrent drivers. Two molecular features are worth noting. First, HER2-directed therapy was successful in our patient despite the presence of CDK4 and MDM2 amplification, two alterations that are thought to be clinically relevant driver alterations in acral melanoma and in well-and dedifferentiated liposarcomas (21, 22). Second, the magnitude of ERBB2 amplification (log2 of 26-fold) was near the highest in the entire melanoma cohort. In a large meta-analysis of patients with ERBB2-amplified breast cancer treated with adjuvant trastuzumab; however, the magnitude of ERBB2 amplification measured by fluorescence in situ hybridization was not associated with disease-free survival (23).
Overall, this case demonstrates the potential clinical utility of detecting ERBB2 copy-number alterations in acral and mucosal melanomas. These results from a single exceptional responder are not a substitute for prospective clinical trial data. Testing the validity of this N = 1 observation with a standard phase II trial would be difficult, unfortunately: approximately 550 patients with these rare melanomas would have to be screened in the checkpoint inhibitor refractory setting to successfully treat approximately 18 patients. Ongoing “basket trials” such as SUMMIT (NCT01953926), TAPUR (NCT02693535), NCI-MATCH (NCT02465060), and My Pathway (NCT02091141) represent crucial prospective assessments of the broader utility of this clinical observation without the strict requirement for enrolling a specific number of cases of a rare histology. Thus, every effort should be made to enroll eligible patients with ERBB2-altered melanomas on these ongoing protocols. Clinicians treating patients ineligible for clinical trials with HER2-targeted agents should be encouraged to share efficacy data with the testing entity for periodic publication of “real-world” outcomes data. Together, these data will help us to understand how pathologic features like histology as well as molecular features such as magnitude of amplification, mutant allele frequency, and presence of concurrent driver alterations may influence the chance of clinical benefit to HER2-directed therapy.
Translational Relevance.
There are few treatment options with durable benefit for patients with BRAF V600 wild-type melanomas that progress despite checkpoint inhibition. This report describes a case of durable complete response in a patient with ERBB2-amplified acral melanoma to trastuzumab emtansine (T-DM1), a HER2-directed antibody–drug conjugate approved for breast adeno-carcinomas. Upon review of a single-left cohort of 732 patients with various nonuveal melanoma subtypes, distinct patterns of ERBB2 amplifications and mutations were seen. ERBB2 amplifications were detected at a modest rate (6%–7%) in acral and mucosal melanomas that lack canonical driver alterations (BRAF, NRAS, KIT, NF1, GNAQ, and GNA11). ERBB2 mutations were detected across multiple melanoma subtypes, most often comutated with canonical drivers such as NF1. These data suggest ERBB2 testing should be considered for patients with acral and mucosal melanomas and ERBB2 mutations may contribute to tumor growth in a subset of melanomas with a known driver alteration.
Acknowledgments
We gratefully acknowledge support from the Marie-Josee and Henry R. Kravis Center for Molecular Oncology, Cycle for Survival and the MSKCC Core Grant (P30 CA008748).
Footnotes
Disclosure of Potential Conflicts of Interest
D.M. Hyman is a consultant/advisory board member for Bayer, Boehringer Ingelheim, Chugai, Debiopharm, Genentech, and Pfizer, and reports receiving commercial research grants from Puma Biotechnology and Loxo Incology. A.N. Shoushtari is a consultant/advisory board member for Bristol-Myers Squibb, Castle Biosciences and Immunocore. No potential conflicts of interest were disclosed by the other authors.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Pasquali S, Hadjinicolaou AV, Chiarion Sileni V, Rossi CR, Mocellin S. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database Syst Rev 2018;2:CD011123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascierto PA, Melerò I, Bhatia S, Bono P, Sanborn RE, Lipson EJ, et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J Clin Oncol 2017;35: 9520. [Google Scholar]
- 3.Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 2014;9:239–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong Y, Sheng X, Wu X, Yan J, Ma M, Yu J, et al. Frequent genetic aberrations in the CDK4 pathway in acral melanoma indicate the potential for CDK4/6 inhibitors in targeted therapy. Clin Cancer Res 2017;23:6946–57. [DOI] [PubMed] [Google Scholar]
- 5.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005;5:341. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed S, Sami A, Xiang J. HER2-directed therapy: current treatment options for HER2-positive breast cancer. Breast Cancer 2015;22: 101–16. [DOI] [PubMed] [Google Scholar]
- 7.Bang Y-J, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A,et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97. [DOI] [PubMed] [Google Scholar]
- 8.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 2013;3:224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition inpatients with HER2- and HER3-mutantcancers. Nature 2018;554:189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res 2014;16:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017; 2017:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011;305:2327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kluger HM, DiVito K, Berger AJ, Halaban R, Ariyan S, Camp RL, et al. Her2/neu is not a commonly expressed therapeutic target in melanoma - a large cohort tissue microarray study. Melanoma Res 2004;14:207–10. [DOI] [PubMed] [Google Scholar]
- 16.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of10,000 patients. Nat Med 2017;23:703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:738–46. [DOI] [PubMed] [Google Scholar]
- 18.Jhaveri KL, Makker V, Wang XV, Chen AP, Flaherty K, Conley BA, et al. Ado-trastuzumab emtansine (T-DM1) in patients (pts) with HER2 amplified (amp) tumors excluding breast and gastric/gastro-esophageal junction (GEJ) adenocarcinomas: results from the National Cancer Institute (NCI) Molecular Analysis for Therapy Choice (MATCH) trial. J Clin Oncol 2018;36:100. [Google Scholar]
- 19.Li BT, Makker V, Buonocore DJ, Offin MD, Olah ZT, Panora E, et al. Amulti-histology basket trial of ado-trastuzumab emtansine in patients with HER2-amplified cancers. J Clin Oncol 2018;36:2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, et al. Targeted therapy for advanced solid tumors onthe basis of molecular profiles: results from mypathway, an open-label, Phase IIa multiple basket study. J Clin Oncol 2018;36:536–42. [DOI] [PubMed] [Google Scholar]
- 21.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353: 2135–47. [DOI] [PubMed] [Google Scholar]
- 22.Crago AM, Dickson MA. Liposarcoma: multimodality management and future targeted therapies. Surg Oncol Clin N Am 2016;25:761–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu QQ, Pan B, Wang CJ, Zhou YD, Mao F, Lin Y, et al. HER2 amplification level is not a prognostic factor for HER2-positive breast cancer with trastuzumab-based adjuvant treatment: a systematic review and meta-analysis. Oncotarget 2016;7:63571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]