

Abstract
Congenital disorder of glycosylation type Ic (CDG‐Ic) is caused by mutations in hALG6 gene encoding α‐1,3 glucosyltransferase (NP_037471.2), an enzyme that catalyzes the addition of the first glucose residue to the growing lipid‐linked oligosaccharide precursor in N‐glycosylation process. The most frequent mutation in hALG6 gene causing CDG‐Ic is c.998C>T that results in p.A333V substitution. Up‐to‐date, no CDG‐Ic patient has been detected in Croatia. However, as a part of the comprehensive project undertaken with the aim to estimate the frequencies of the carriers for specific mutations and polymorphisms related to particular CDGs in Croatian population, we screened genomic DNA samples obtained from 600 healthy nonconsanguineous Croatian residents to determine the frequency of the A333V mutation. For that purpose, we established the conditions for polymerase chain reaction‐based single‐strand conformation polymorphism analysis that is suitable for primary screening and in population studies, especially when the initial sample volume is small or DNA quantity is limited. None of the analyzed samples carried this mutation, indicating that the frequency of the patients carrying this homozygous mutation in Croatian population would be <1 in 1.4×106. J. Clin. Lab. Anal. 25:65;–70, 2011. © 2011 Wiley‐Liss, Inc.
Keywords: congenital disorder of glycosylation (CDG) Ic, hALG6 gene, A333V mutation, PCR‐SSCP
INTRODUCTION
Congenital disorders of glycosylation (CDG) are a rapidly growing group of inherited metabolic disorders caused by defects in the synthesis of glycans or their attachment to proteins and lipids, with more than 40 diseases reported so far. More than half of them is caused by mutations in genes encoding proteins involved in either the formation of the lipid‐linked oligosaccharide (LLO) precursor (CDGs type I) or processing of the protein N‐linked carbohydrate chains (CDG type II) 1, 2. Up‐to‐date, 16 disease‐causing defects of protein N‐glycosylation have been characterized and enormous diversity regarding the symptoms and degree of severity were described 3, 4, 5. Although some of the syndromes are characterized with mild hepatointestinal problems and coagulopathy, in some of them mental symptoms with seizures, hypotonia, profound developmental delay, and even death in early childhood were described. Although CDG are very rare diseases (less than 1,000 patients have been diagnosed so far), the real incidence is probably much higher because they can be easily mis‐ or underdiagnosed 1, 6. Therefore, the improvement of already existing 7, 8, 9 and development of new simple, cheap, and user‐friendly methods for CDG screening is of utmost importance.
CDG‐Ic (OMIM ♯603147) is caused by mutations in human ortholog of yeast Alg6 (hALG6) gene that encodes dolichyl‐p‐glucose:mannose9‐n‐acetyl glucosamine2‐pyrophosphate‐dolichyl (Dol‐P‐Glc:Man9GlcNAc2‐P‐P‐Dol) α‐1,3 glucosyltransferase, an enzyme catalyzing the transfer of the first glucose residue to the growing LLO precursor 10 (Fig. 1). Defects in hALG6 lead to the accumulation of mannose9‐N‐acetyl glucosamine2‐pyrophosphate‐dolichol (Man9GlcNAc2‐P‐P‐Dol) in the endoplasmic reticulum and inefficient transfer of this oligosaccharide to proteins 10, 11, 12. The patients affected by this syndrome are clinically characterized by psychomotor retardation, dysmorphic features, muscular hypotonia, seizures, epilepsy, and occasionally proteinlosing enteropathy 13, 14, 15, 16, 17, 18. Up‐to‐date, more than 30 cases of CDG‐Ic were recognized worldwide; thus, this syndrome represents the second most frequent CDG subtype 5. Twenty different hALG6 mutations have been described so far 5, where C998T mutation resulting in A333V substitution accounts for the majority of the alleles 12, 15.
Figure 1.
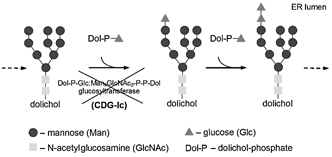
A part of N‐glycosylation pathway involving dolichyl‐p‐glucose: mannose9‐N‐acetyl glucosamine2‐pyrophosphate‐dolichyl (Dol‐P‐Glc:Man9GlcNAc2‐P‐P‐Dol) α‐1,3 glucosyltransferase. Dol‐P‐Glc:Man9GlcNAc2‐P‐P‐Dol α‐1,3 glucosyltransferase is an enzyme that catalyzes the transfer of the first glucose residue to the growing LLO precursor (Man9GlcNAc2‐P‐P‐Dol). The reduced activity of this enzyme caused by mutations in hALG6 gene results in congenital disorder of glycosylation Ic (CDG‐Ic).
Among the various screening methods, polymerase chain reaction‐based single‐strand conformation polymorphism (PCR‐SSCP) analysis is the most widely used, owing to its simplicity and effectiveness. It had been used in mutation detection for many diseases, including some other CDG types 19, because it is relatively rapid and generally less expensive than other screening methods.
Here, we report the conditions for the PCR‐SSCP analysis for the detection of A333V mutation, as well as the results of the screening for the carriers in Croatian population, using this newly established protocol.
MATERIALS AND METHODS
Materials
Most materials were obtained from Sigma (St. Louis, MO), with the exception of the following: boric acid (Roth, Karlsruhe, Germany); proteinase K, Platinum Taq polymerase, primer and dNTP set (Invitrogen, Carlsbad, CA); Mass RulerTM DNA Ladder Low Range (Fermentas, Vilnius, Lithuania); Chelex®100 Molecular Biology Grade Resin (Bio‐Rad Laboratories, Hercules, CA); and Blood Stain Cards (Whatman, Brentford, Middlesex, UK). For the PCR product purification, QIAquick PCR Purification Kit from Qiagen (Valencia, CA) was purchased. All reagents used for sequencing were purchased from Applied Biosystems (Foster City, CA), except for the CENTRI‐SEP Columns (Princeton preparations, Adelphia). All materials used in this study were of analytical grade.
Participants and Samples
This study was reviewed and approved by the institutional Ethical Committees of the Faculty of Pharmacy and Biochemistry, University of Zagreb, and the hospitals where the samples were collected. The total study population comprised 600 healthy nonconsanguineous Croatian residents, 131 females (age median: 31 years; interquartile range: 24–44) and 469 males (age median: 38 years; interquartile range: 29–50). Blood samples were collected on Blood Stain Cards®, in the Transfusion Medicine Departments of the Clinical or General Hospitals, between September 2005 and September 2007. To comprise whole Croatian population, ten regional centers were enrolled in the study: the Croatian Institute of Transfusion Medicine (210 participants), Clinical Hospital Osijek (80 participants), Clinical Hospital Split (80 participants), General Hospital Varaždin (40 participants), General Hospital Pula (40 participants), General Hospital Sisak (40 participants), General Hospital Zabok (40 participants), General Hospital Bjelovar (30 participants), General Hospital Dubrovnik (20 participants), and General Hospital Šibenik (20 participants). Written informed consent, based on the Helsinki declaration, was obtained from all volunteers before enrolment. As a positive control genomic DNA obtained from heterozygote for A333V mutation was used (obtained by the courtesy of H. H. Freeze, Burnham Institute, San Diego, CA).
Genomic DNA Isolation
The following version of the standard Chelex® extraction procedure was used for the isolation of genomic DNA13; the punch of whole blood (2×2 mm) was cut from Blood Stain Cards® and transferred into 1.5 ml tube. 1 ml of sterile deionized water was added and the mixture was incubated at room temperature for 20 min, vortexed for 10 sec every 5 min, and centrifuged for 3 min at 15,700×g. 950 μl of supernatant was discarded and 50 μl proteinase K solution (10 g/l in 10 mmol/l Tris‐HCl, pH 7.5, 20 mmol/l CaCl2, 50% glycerol) and 150 μl of a freshly prepared, continuously stirred 5% suspension of Chelex®100 in 1 mol/l Tris‐HCl, pH 7.5, was added. The solution was incubated overnight in a heat block at 56°C, vortexed for a few seconds, boiled for 8 min, and centrifuged for 3 min at 15,700×g. The supernatant containing genomic DNA was stored at 4°C until use.
PCR Amplification
PCR amplifications of exon 11 and parts of intronic adjacent sequences (−115 bp) of intron 10 and (+101 bp) of intron 11 were performed, using specific primers (forward primer 5′‐GCTTTAATAAACTTTCAACTTTCATTTG‐3′ and reverse primer 5′‐CATTTGTGTAGTTTTGTTTTGCATTC‐3′) as described earlier 20 with some modifications regarding the concentration of the primers and the amount of the DNA template.
The PCR products were amplified in a final volume of 50 μl containing the components of the reaction mixture in the optimal proportion, which were determined experimentally: 2.5 U Platinum Taq polymerase, 5 μl sample containing genomic DNA prepared by Chelex® 100 extraction method, PCR buffer (50 mmol/l KCl, 10 mmol/l Tris‐HCl, pH 8.3), 0.2 mmol/l dNTPs, 1.5 mmol/l MgCl2, 0.5 μM of each primer.
The PCR was conducted (in Gene Amp® PCR System 2700; Applied Biosystems) for 35 cycles under the following conditions: 45 sec of denaturation at 95°C, 30 sec of annealing at 56°C, and 1 min of extension at 72°C, followed by a final extension for 7 min at 72°C and cooling down to 4°C. The purity of PCR products was confirmed on 1.8% agarose electrophoresis, and additionally, the quantity of amplified DNA fragments in PCR mixtures was estimated using DNA mass ruler (Fermentas).
SSCP Analysis of Amplified Fragments of hALG6 Gene
The samples for SSCP were prepared as follows: 10–25 μl of PCR products (containing approximately 50 ng of DNA) were diluted in a ratio of 1:2 with formamide dye (95% (v/v) formamide, 10 mmol/l NaOH, 0.1% (w/v) xylene cyanol, and 0.1% (w/v) bromphenol blue). The mixtures were denatured at 94°C for 6 min, immediately cooled on ice, and subsequently loaded onto prepared nondenaturing polyacrylamide gel. To obtain optimal separation of the ssDNA molecules, concentration of polyacrylamide gel (6, 8, and 10% (w/v), acrylamide:bisacrylamide (37.5:1)) and gel temperature (4, 15, and 25°C) during electrophoresis was experimentally determined.
The electrophoreses were carried out in a 18 cm long HoeferTM SE 600 Ruby standard dual cooled gel electrophoresis unit (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK), in 1.5 mm thick gels under the following conditions: TBE buffer (45 mmol/l Tris‐HCl, 45 mmol/l boric acid, 1 mmol/l Na2EDTA, pH 8.3) at 200 V (30 mA) for 5 hr and 30 min. Finally, all samples were analyzed under optimized electrophoretical conditions (6% (w/v), acrylamide:bisacrylamide (37.5:1) at 4°C).
After electrophoresis, the gels were silver stained as follows: gel fixation in 50% methanol/10% acetic acid for 30 min was followed by incubation in 10% ethanol/7% acetic acid overnight. After washing two times for 10 min in 10% ethanol/7% acetic acid and five times for 5 min in bidistilled water gels were stained for 30 min in 0.1% AgNO3. The gels were briefly washed in bidistilled water and incubated in 2.5% Na2CO3/0.02% formaldehyde until the stained bands appeared. The reaction was stopped by washing in 1% acetic acid for a few minutes. All solutions were prepared immediately before use.
DNA Sequencing Analysis
Fifty randomly chosen PCR products with normal SSCP pattern and PCR product of the positive control (heterozygote for the A333V mutation) were additionally sequenced. After purification with QIAquick PCR Purification kit (Qiagen), the nucleotide sequences were analyzed with DNA Sequencing Big Dye Terminator v3.0 kit on an ABIPrism 310 Genetic Analyzer (Applied Biosystems), according to the manufacturer's instructions. The data were analyzed using Sequencing Analysis software®. For effective removal of excess DyeDeoxy™ terminators from completed DNA sequencing, reaction before the analysis on the ABIPrism 310 Genetic Analyzer CENTRI‐SEP Columns were used.
RESULTS
For screening for A333V mutation in collected samples, we employed for the first time PCR‐SSCP analysis which was shown to be a relatively simple and user‐friendly procedure. Because there are no theoretical models for SSCP analysis, conditions for detecting specific mutation had to be found experimentally.
Because there are no theoretical models for conditions of SSCP analysis for detecting specific mutation they had to be found experimentally.
PCR Optimization
In this study, the blood samples were collected on Blood Stain Cards® and genomic DNA was isolated by Chelex®100 extraction method. Using this protocol, the obtained samples (∼150 μl) contained DNA in concentrations that were not measurable by standard spectrophotometric method. This is why the volume of the sample containing template DNA for PCR, as well as the concentration of the primers, had to be optimized. This step was especially important, because the DNA quantity affects sensitivity of the SSCP analysis.
It was found that PCR performed with 5 μl of the sample obtained by Chelex®100 extraction method yielded 100–250 ng of PCR products in 50 μl reaction mixture that was an adequate amount for the SSCP analysis (approximately 50 ng). The sample volumes higher than optimal (7 and 10 μl) allowed amplification of nontarget products, whereas lower (3 μl) yielded insufficient quantity of PCR products.
The PCRs with concentrations of 0.1, 0.2, 0.5, or 1 μmol/l of each primer were carried out. The results confirmed that lower concentrations of the primers (0.1 and 0.2 μmol/l) were insufficient to yield enough PCR products, whereas the highest applied concentration (1 μmol/l) resulted in unspecific products as well as primer–dimer formation. We assumed 0.5 μmol/l concentration of the primers to be optimal, because the specificity and quantity of the PCR products was considerably high. The reproducibility of the optimized PCR conditions was proved using different thermal cyclers (data not shown).
SSCP Optimization
To optimize the SSCP procedure, we tested electrophoretic separation in polyacrylamide gels of different concentrations: 6, 8, and 10% (w/v), (acrylamide:bisacrylamide (37.5:1)) at three different gel temperatures of 4, 15, and 25°C; approximately 50 ng of DNA was loaded on the gel and the electrophoresis was run at 200 V (30 mA) for 5 hr and 30 min. By electrophoresis performed in 6% polyacrylamide at 4°C, a band shift in the positive controls (heterozygous for A333V) was achieved (Fig. 2). Therefore, additional modifications of other electrophoretic conditions were not necessary.
Figure 2.
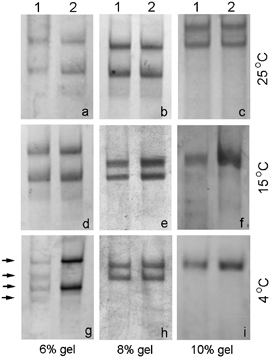
Single‐strand conformation polymorphism analysis (SSCP) of exon 11 and parts of the intronic adjacent sequences of intron 10 (−115 bp) and 11 (+101 bp) of hALG6 gene. The SSCP analysis of exon 11 and parts of intronic adjacent sequences of intron 10 (−115 bp) and 11 (+101 bp) of hALG6 gene was carried out at different electrophoretic conditions: 6, 8, and 10% polyacrylamide gel (w/v), (acrylamide:bisacrylamide (37.5:1)) at three different temperatures 4, 15, and 25°C. Approximately 50 ng of DNA was loaded on the gel and the electrophoresis was run at 200 V (30 mA) during 5 hr and 30 min. The gels were silver stained. A band shift in the positive controls (heterozygous for A333V) was achieved in 6% polyacrylamide gel at 4°C (g). The single‐strand DNA bands of interest are marked with arrows. Lines 1: positive control—heterozygous for A333V; lines 2: healthy individual.
Frequency of A333V Mutation in Croatian Population
In order to determine the frequency of the carriers of the A333V mutation in Croatian population, genomic DNA samples obtained from 600 healthy nonconsanguineous Croatian residents were analyzed. None of the examined samples carried the A333V allele, whereas heterozygous which was used as a positive control was easily detected by employed method. To confirm the accuracy of the established method, 50 randomly chosen samples obtained from healthy individuals that showed normal SSCP pattern, as well as the sample from heterozygous for A333V, were additionally sequenced. The results of the sequencing analyses confirmed those obtained by SSCP analysis (Fig. 3).
Figure 3.
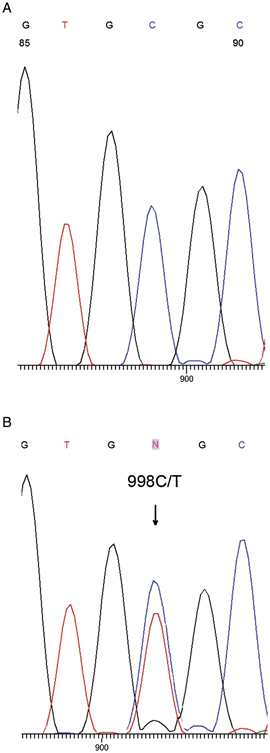
The part of hALG6 gene sequence of healthy individual (A) and heterozygote for C998T mutation (B). The position 998 in exon 11 of hALG6 gene is marked by arrow. The base T in addition to base C is present at that position in the sequence of the heterozygote for A333V mutation.
DISCUSSION
CDG‐Ic was initially described in 1998 10, 11 and more than 30 cases have been described so far, making this syndrome the second most prevalent CDG subtype. All but four (three were Indian 15 and one Arabic 21) of the reported CDG‐Ic patients were of European ancestry, whereas the most common disease‐causing mutation was shown to be C998T that results in A333V substitution.
The only method employed for screening A333V mutation until today was template‐directed primer extension with fluorescence polarization detection (FR‐TDI) 15. Although it is accurate and suitable for high‐throughput single nucleotide polymorphism genotyping 22, it requires relatively expensive instrumentation. Allelic discrimination by real‐time PCR that rapidly enters as a routine method in laboratory practice and is widely used for screening studies was not applicable for this purpose, because it was not possible to design specific primers/probes usable for the analysis of A333V mutation (according to Applied Biosystems).
This is why, in this study, we employed PCR‐SSCP analysis and set up conditions suitable for analysis of A333V mutation. This method, once set up, is accurate, simple, rapid, relatively cheap, and consequently the most widely used screening method.
Because the concentration of genomic DNA in the samples obtained by Chelex®100 extraction method from blood samples collected on Blood Stain Cards® was too low to be measurable by standard spectrophotometric method, instead of optimizing the quantity of DNA template for PCR amplification, we had to optimize the volume of the sample containing unknown DNA quantity. It was shown that PCR performed with 5 μl of the sample yielded 100–250 ng of PCR products in 50 μl of reaction mixture, a quantity sufficient for the SSCP analysis (approximately 50 ng). According to Imbach et al. 20 the initial quantity of genomic DNA for PCR (followed by sequencing analysis) was 100 ng that corresponds to the final concentration of PCR products in some of our samples. However, because of the low quantity of DNA template in the PCR mixture, we had to increase the concentration of each primer to 0.5 μmol/l that had a concentration 25 times higher than that compared with those used in the aforementioned procedure (0.02 μmol/l) 20.
To optimize SSCP analysis conditions, we first performed the electrophoreses using different gel percentage (6, 8, and 10% polyacrylamide gel (w/v), (acrylamide:bisacrylamide (37.5:1)) and gel temperature during electrophoresis (25, 15, and 4°C). Fortunately, the band shift in the positive control (heterozygous for A333V) was achieved under one of the initial conditions (6% gel, 4°C), thus further optimization was not required. The accuracy of the method was confirmed by sequencing analysis of the positive control (heterozygous for A333V) and 50 randomly chosen samples which SSCP pattern corresponded to homozygous pattern (wild‐type).
To determine the frequency of the A333V mutation in Croatian population, we analyzed a group of 600 healthy Croatian residents. Using established conditions for PCR‐SSCP analysis, we could easily differentiate homozygous (wild‐type) and heterozygous A333V individuals. None of the analyzed samples was found to be a carrier for the target mutation. Assuming that the obtained results are valid for Croatian population, the estimated incidence of homozygous A333V CDG‐Ic patients would be below 1 in 1.4×106 (calculation according to Newell et al. 15). However, to obtain the results upon which the final conclusion could be postulated, a bigger population sample has to be analyzed. Screening analysis for A333V was performed only for the United States population so far; >350 individuals of European, African, and Asian‐American origin were analyzed, and in that cohort A333V carrier was also not found 15. According to the previous studies, in half of known CDG‐Ic patients, the disease was found to be caused by homozygous form of A333V 13, 20, whereas in some cases heterozygous form of A333V is combined with other mutations in hALG6 gene 15, 23. In addition, because of relatively mild symptoms, possible absence of typical morphologic features and cerebral hypoplasia, CDG‐Ic is probably underdiagnosed 6. Furthermore, it was observed that in young age, children with CDG‐Ic are not dysmorfic and do not have neurologic involvement 16. These data suggest that CDG‐Ic should be considered in cases of unexplained enteropathy in patients with mild developmental delay and corresponding neurological symptoms.
In conclusion, we established the conditions for PCR‐SSCP analysis for detection of A333V mutation, which is suitable for primary screening, in population studies as well as for diagnosis of CDG‐Ic, especially when the initial sample volume is small or when the quantity of DNA is limited.
Acknowledgements
The authors thank Professor H.H. Freeze for providing genomic DNA from the individual who is heterozygote for A333V mutation.
REFERENCES
- 1. Freeze HH. Genetic defects in the human glycome. Nat Rev Genet 2006;7:537–551. [DOI] [PubMed] [Google Scholar]
- 2. Schachter H, Freeze HH. Glycosylation diseases: Quo vadis? Biochim Biophys Acta 2009;1792:925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eklund EA, Freeze HH. The congenital disorders of glycosylation: A multifaceted group of syndromes. NeuroRx 2006;3:254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rind N, Schmeiser V, Thiel C, et al. A severe human metabolic disease caused by deficiency of the endoplasmatic mannosyltransferase hALG11 leads to congenital disorder of glycosylation‐Ip. Hum Mol Genet 2010;19:1413–1424. [DOI] [PubMed] [Google Scholar]
- 5. Haeuptle MA, Hennet T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol‐linked oligosaccharides. Hum Mutat 2009;30:1628–1641. [DOI] [PubMed] [Google Scholar]
- 6. Grunewald S, Matthijs G, Jaeken J. Congenital disorders of glycosylation: A review. Pediatr Res 2002;52:618–624. [DOI] [PubMed] [Google Scholar]
- 7. Carchon HA, Chevigne R, Falmagne JB, Jaeken J. Diagnosis of congenital disorders of glycosylation by capillary zone electrophoresis of serum transferrin. Clin Chem 2004;50:101–111. [DOI] [PubMed] [Google Scholar]
- 8. O'Brien JF. Methods for detection of carbohydrate‐deficient glycoprotein syndromes. Semin Pediatr Neurol 2005;12:159–162. [DOI] [PubMed] [Google Scholar]
- 9. Parente F, Ah Mew N, Jaeken J, Gilfix BM. A new capillary zone electrophoresis method for the screening of congenital disorders of glycosylation (CDG). Clin Chim Acta 2010;411:64–66. [DOI] [PubMed] [Google Scholar]
- 10. Korner C, Knauer R, Holzbach U, Hanefeld F, Lehle L, von Figura K. Carbohydrate‐deficient glycoprotein syndrome type V: Deficiency of dolichyl‐P‐Glc:Man9GlcNAc2‐PP‐dolichyl glucosyltransferase. Proc Natl Acad Sci USA 1998;95:13200–13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burda P, Borsig L, de Rijk‐van Andel J, et al. A novel carbohydrate‐deficient glycoprotein syndrome characterized by a deficiency in glucosylation of the dolichol‐linked oligosaccharide. J Clin Invest 1998;102:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Imbach T, Burda P, Kuhnert P, et al. A mutation in the human ortholog of the Saccharomyces cerevisiae ALG6 gene causes carbohydrate‐deficient glycoprotein syndrome type‐Ic. Proc Natl Acad Sci USA 1999;96:6982–6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grunewald S, Imbach T, Huijben K, et al. Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N‐glycan synthesis. Ann Neurol 2000;47:776–781. [PubMed] [Google Scholar]
- 14. Marquardt T, Denecke J. Congenital disorders of glycosylation: Review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359–379. [DOI] [PubMed] [Google Scholar]
- 15. Newell JW, Seo NS, Enns GM, McCraken M, Mantovani JF, Freeze HH. Congenital disorder of glycosylation Ic in patients of Indian origin. Mol Genet Metab 2003;79:221–228. [DOI] [PubMed] [Google Scholar]
- 16. Damen G, de Klerk H, Huijmans J, den Hollander J, Sinaasappel M. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr 2004;38:282–287. [DOI] [PubMed] [Google Scholar]
- 17. Sun L, Eklund EA, Van Hove JL, Freeze HH, Thomas JA. Clinical and molecular characterization of the first adult congenital disorder of glycosylation (CDG) type Ic patient. Am J Med Genet A 2005;137:22–26. [DOI] [PubMed] [Google Scholar]
- 18. Eklund EA, Sun L, Yang SP, Pasion RM, Thorland EC, Freeze HH. Congenital disorder of glycosylation Ic due to a de novo deletion and an hALG‐6 mutation. Biochem Biophys Res Commun 2006;339:755–760. [DOI] [PubMed] [Google Scholar]
- 19. Bjursell C, Erlandson A, Nordling M, et al. PMM2 mutation spectrum, including 10 novel mutations, in a large CDG type 1A family material with a focus on Scandinavian families. Hum Mutat 2000;16:395–400. [DOI] [PubMed] [Google Scholar]
- 20. Imbach T, Grunewald S, Schenk B, et al. Multi‐allelic origin of congenital disorder of glycosylation (CDG)‐Ic. Hum Genet 2000;106:538–545. [DOI] [PubMed] [Google Scholar]
- 21. Al‐Owain M, Mohamed S, Kaya N, Zagal A, Matthijs G, Jaeken J. A novel mutation and first report of dilated cardiomyopathy in ALG6‐CDG (CDG‐Ic): A case report. Orphanet J Rare Dis 2010;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen X, Levine L, Kwok PY. Fluorescence polarization in homogeneous nucleic acid analysis. Genome Res 1999;9:492–498. [PMC free article] [PubMed] [Google Scholar]
- 23. Westphal V, Schottstadt C, Marquardt T, Freeze HH. Analysis of multiple mutations in the hALG6 gene in a patient with congenital disorder of glycosylation Ic. Mol Genet Metab 2000;70:219–223. [DOI] [PubMed] [Google Scholar]