

Abstract
Analysis of gene expression can provide important information related to prognostic or predictive factors in clinical work. Analysis of DNA or RNA isolated from formalin fixed and paraffin‐embedded tissue is generally accepted to be difficult due to degradation and crosslinking. Factors affecting mRNA quality such as formalin fixation length and autolysis period before fixation/freezing have not been systematically addressed. In this study, we analyzed the effect of formalin fixation length and autolysis period before fixation/freezing on the expression of several reference genes in formalin‐fixed paraffin‐embedded (FFPE) colorectal carcinoma cells and tissue. We also used a factorial experimental design to further analyze the interaction of both variables. It was found that mRNA levels can be reproducibly quantified, independent of the tested variables. Our findings confirm that clinical tumor specimens removed by routine surgical procedures and combined with real‐time RT‐PCR can be used for the analysis of gene expression in routine FFPE materials and provide some useful information related to the achieved tissues largely stored in histopathology departments. J. Clin. Lab. Anal. 25:166–173, 2011. © 2011 Wiley‐Liss, Inc.
Keywords: formalin fixed, paraffin embedded, colorectal carcinoma cell, Real‐time RT‐PCR, factorial experimental design
INTRODUCTION
Analysis of gene expression can provide important information related to prognostic or predictive factors in clinical experimental work. Technological progress, such as the development of sensitive real‐time PCR systems, permit rapid and specific quantification of small amounts of mRNA 1, 2. Although mRNA is relatively stable in fresh‐frozen tissues, collecting and preserving fresh‐frozen tissue samples is difficult in daily routine work. On the contrary, the widely available archives of formalin‐fixed paraffin‐embedded (FFPE) tissue represent an invaluable research resource, making it possible to perform large retrospective studies that can provide information on both prognostic factors and predictive factors such as estimates of benefit from specific therapies 3, 4, 5. In this context, DNA isolated from FFPE tissue is used in pharmacogenetic studies. Indeed, profiling of gene expression is, in addition to DNA sequence variation, an interesting tool to study inter‐individual differences in drug response. However, the reliability of analyzing mRNA isolated from FFPE tissue is subject to discussion since the stability of mRNA from this source and potentially influenced by pretreatments (such as fixation) has not been systematically studied. The dogma is that the quality of mRNA isolated from FFPE tissue is poor and not reliable for gene‐expression profiling. The reason is that the extracted RNA is believed to be extensively degraded and cross‐linked by formalin fixation 6. It is a shared opinion that several factors including the duration of fixation and autolysis before fixation/freezing (the delay time before fixation) could affect the degradation of mRNA in formalin‐fixed tissue 7, 8, 9. Although mRNA can be detected in FFPE tissue 10, the extent to which these disturbing factors affect quantitative gene expression analysis has not been systematically addressed. The purpose of our study was to determine the effect of formalin fixation length and autolysis period before fixation and their interaction on quantitative gene expression. Several colorectal carcinoma cells and tissue samples were paraffinned and embedded; meanwhile, the relative expression of several reference genes were determined after different incubation and formalin fixation periods.
MATERIAL AND METHODS
Cell Culture and Paraffin Embedding
Colorectal carcinoma cell lines HCT15, LoVo, DLD‐1, and Colo320 were grown in DMEM supplemented with 5% FCS at 37°C and 5% CO2 conditions. Cells obtained when they are totally full of T7 cell culture bottle. FFPE cells were made using Shandon Cytoblock Kit according to the manufacturer's instructions (Thermo Electron Corporation, Pittsburgh, PA). Following this protocol, cells were trypsinized and clumped to test formalin fixation and autolysis. For HCT15 and LoVo cells, the effect of the fixation time was analyzed. HCT15 cells were incubated in neutral‐buffer formalin for 0, 1, and 3 days; LoVo cells were incubated for 0, 3, and 24 hrs. A factorial experimental design for DLD‐1 and Colo320 cells were used to compare the effect of fixation length, autolysis period (delay time before fixation), and a combination of both interaction, which included nine different treatment from S1 to S9 Schematically depicted in Table 1. Colorectal carcinoma tissue kindly provided by the department of Colorectal Surgery. The experiments were approved by the Ethics Committee of China Medical University. Tumor tissue samples were obtained fresh, directly from the operating theater from two different patients suffering from colorectal carcinoma. The diagnosis of each tumor was confirmed by histological examination with supplementary immunohistochemical staining (data not shown). Each fresh tumor sample was cut into nine macroscopically similarly sized pieces and placed in a separate Eppendorf tubes. Each sample was processed as the instruction schematically depicted in Table 1.
Table 1.
Experimental Design to Test Autolysis and Formalin Fixation
| Fixation time | |||
|---|---|---|---|
| Autolysis time | 0 day | 1 day | 3 days |
| 0 h | Sample 1 | Sample 2 | Sample 3 |
| 1 h | Sample 4 | Sample 5 | Sample 6 |
| 3 h | Sample 7 | Sample 8 | Sample 9 |
Each sample processes as the following instruction: Sample 1: snap frozen in −80°C liquid nitrogen; Sample 2: immediately insert the cell pellet into 10% neutral formalin buffer for 1 day; Sample 3; immediately insert the cell pellet into 10% neutral formalin buffer for 3 days; Sample 4: after an hour insert the cell pellet into −80°C liquid nitrogen; Sample 5: after an hour insert the cell pellet into 10% neutral formalin buffer for 1 day; Sample 6: after an hour insert the cell pellet into 10% neutral formalin buffer for 3 days; Sample 7: after an hour insert the cell pellet into −80°C liquid nitrogen; Sample 8: after 3 hr insert the cell pellet into 10% neutral formalin buffer for 1 day; Sample 9: after 3 hr insert the cell pellet into 10% neutral formalin buffer for 3 days.
Deparaffinization, RNA Isolation, and cDNA Synthesis
FFPE cell blocks or colorectal tumor tissue samples were cut with a disposable blade into 20 µm sections (corresponding to 10–15 µg of cells) depending on the size of the embedded cell sample and placed in RNase‐free tubes. Deparaffination and RNA isolation was done with High Pure RNA Paraffin Kit (Roche Diagnostics, Almere, the Netherlands) according to the manufacturer's instructions. Briefly, paraffin section deparaffinized by incubation in xylol and repeated several time. Then, it was incubated with ethanol and centrifuged, and the tissue residue was air‐dried and incubated again with digestion with Proteinase K, SDS, and lysis buffer overnight at 55°C. RNA was purified by the addition of RNA extraction buffer. The aqueous phase was removed to fresh tubes, and the RNA was precipitated with an equal volume of isopropanol and washed twice in 75% ethanol, and then the pellet was air‐dried and resuspended in 10 µl of RNA storage solution. To avoid any operational mistake, the extractions were prepared in duplicate.
The amount of RNA in the samples was quantified using Nanodrop (Isogen, The Meern, the Netherlands). In addition, RNA quality was visualized on ethidiumbromide‐stained agarose gel. The total 300 ng RNA was reverse transcribed into cDNA using first‐strand synthesis kit of Invitrogen (Breda, the Netherlands). As primer we tested random hexamers and antisense PCR primer (Table 2). First, RNA reaction adding random hexamer or antisense PCR primer annealed 10 min at 70°C; Second, a total of 20 µl volume including 50 U M‐MLV‐RT RNase‐Transcriptase (Invitrogen), 20 U RNase inhibator (Invitrogen) DTT RT‐buffer, dNTP incubated with annealed RNA reaction 1 hr at 37°C.
Table 2.
Primer Sequence and Amplicon Size
| Gene name | Sequence | Amplicon size (bp) |
|---|---|---|
| β‐Actin | 5′‐ATGTGCAAGGCCGGTTC‐3′ | 265 bp |
| 5′‐GCTGGAAGAGTGCCTCAG‐3′ | ||
| β2‐Microglobulin | 5′‐TGCCGTGTGAACCATGTGA‐3′ | 98 bp |
| 5′‐CCAAATGCGGCATCTTCAA‐3′ | ||
| CPSF6 | 5′‐AAGATTGCCTTCATGGAATTGAG‐3′ | 88 bp |
| 5′‐TCGTGATCTACTATGGTCCCTCTCT‐3′ | ||
| HNRPM | 5′‐GAGGCCATGCTCCTGGG‐3′ | 80 bp |
| 5′‐TTTAGCATCTTCCATGTGAAATCG‐3′ |
Real‐Time Quantitative RT‐PCR
The cDNA was five times diluted and 4 µl was used as a template for real‐time PCR in a 20 µl PCR reaction mixture containing 12.5 µl ABI‐TaqMan Universal PCR Master Mix (Applied Biosystems, Nieuwerkerk a/d Ussel, the Netherlands). Relative quantification of samples was determined using universal SYBR green master mixture in the Taqman 7500 (Applied Biosystems). The PCR conditions were 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 10 min, and 40 cycles of 95°C for 15 sec, and 60°C for 1 min. We tested the following house‐keeping genes: β2‐microglobulin, β‐actin, Heterogenous Nuclear RibonucleoProtein M gene (HNRPM), and Cleavage and Polyadenylation Specific Factor 6 (CPSF6). We used relative quantification (RQ) to determine the change in the expression of a nucleic acid sequence (target) compared with a calibrator, after all the measurements are standardized. The relative expression of each house‐keeping gene are compared to each other in order to reflect the specific effect of fixation and autolysis. RNA for a calibration curve was included in every RT together with one negative control (without RNA) to exclude potential contamination. Each sample was analyzed in triplicate, and the average value of triplicates was used as a quantitative value.
Statistics
The data for each gene were statistically evaluated for the main effect of fixation, delay time, and their interaction using the SPSS 15.0 for Windows. Analysis of variance (ANOVA) or Kruskal–Wallis test of independent or factorial experimental design were used to evaluate the data for a single gene according to the normality and variance of homogeneity of data. All tests were two‐sided and P‐values <0.05 were considered significant.
RESULTS
RNA Integrity in Fresh and Fixed Paraffin‐Embedded Cells
The RNA quality of all samples was checked by spectrophotometer analysis. The A260/A280 value measures the ratio of nucleic acid to protein, and good quality RNA samples have the A260/A280 values in the range from 1.8 to 2.0. Figure 1A shows the yield of RNA, A260/A280, and A260/A230 in DLD‐1 and Colo320 cells following the different fixation and autolysis time. Freshly isolated cells were of better quality compared to FFPE‐isolated RNA. Of note, the yield of RNA differed from each other following the fixation and autolysis time. In addition, we isolated total RNA from LoVo cells that have been incubated in formalin and compared with RNA isolated from fresh cells to test the effect on RNA integrity. The electrophoresis analysis revealed that the RNA was degraded to a certain extent and there were no clear 28S and 18S rRNA bands in fixed cells (Fig. 1B). RNA was highly degraded after formalin fixation (S2 and S3) compared to RNA from fresh cells (S1), independent of incubation time. Degradation of RNA in the cells was comparable with the FFPE tissue control (S4: colorectal carcinoma tissue sample). For the colorectal carcinoma tissue from two patients, the yield of RNA also differed from each other following the fixation and autolysis time, and the average RNA concentration range was from 52 to 317 ng/µl (data not shown).
Figure 1.

(A) Statistic descriptive of Nanodrop Spectrophotometer measurements used as an indicator of RNA purity of DLD‐1 and COLO320 cell (Mean+Std.dev) (valid N=3). (B) RNA integrity checked by electrophoresis on 2% agarose gels stained with ethidium bromide. (S1) frozen LoVo cell sample; (S2) 3hrs fixation; (S3) 24hrs fixation; (S4) FFPE tissue control; (S5) positive control (unfrozen cells); (M) 500bp marker (Promega, Leiden, the Netherlands).
Effect of Fixation Time and Amplicon Size on Quantification of mRNA
Primers for reference genes β2‐microglobulin and β‐actin were used to generate different amplicons (β2‐microglobilin 98 bp; β‐actin 265 bp). The threshold cycle (C T value) of β2‐microglobulin was used for normalization. The averages of the normalized C T value and coefficient of variation from the different samples were calculated (Table 3). No significant difference in quality and quantity of cDNA was found between cDNA synthesized using hexamer primers or cocktail antisense PCR primers. cDNA obtained from FFPE cells showed a higher C T value than fresh cells. Concerning amplicon size, the coefficient of variation is larger for 265 bp (β‐actin) than for 98 bp (β2‐microglobilin). There was a significant difference in C T value of β2‐microglobulin among fresh, short fixation, and long fixation (K‐W test, χ2=6.525, P=0.038), There was no statistical significance in relative quantification of β‐actin against β2‐microglobulin among fresh, short fixation, and long fixation (ANOVA test, F=1.758, P=0.188).
Table 3.
Coefficient of Variation (CV) Calculated for C T Value With Different Fragment and Formalin Fixation in FFPE Tissue (Hexamer and Cocktail PCR primer)
| C T value (hexamer) | C T value (cocktail PCR primer) | ||||||
|---|---|---|---|---|---|---|---|
| Gene/amplicon | Fixation period | n | Mean (arbitrary unit) | CV (%) | n | Mean (arbitrary unit) | CV (%) |
| β2‐Microglobulin 98 bp | Fresh | 3 | 18.319 | 0.328 | 4 | 19.662 | 15.80 |
| 3 hr | 3 | 23.075 | 8.093 | 8 | 24.127 | 13.00 | |
| 3 days | 3 | 25.302 | 6.881 | 8 | 24.550 | 15.09 | |
| β‐actin 265 bp | Fresh | 3 | 17.500 | 3.543 | 4 | 18.724 | 15.18 |
| 3 hr | 3 | 24.026 | 14.360 | 8 | 22.726 | 22.31 | |
| 3 days | 3 | 28.808 | 23.187 | 8 | 25.252 | 21.87 | |
| RQ of β‐actin/β2‐microglobulin | Fresh | 3 | 1.047 | 0.382 | 4 | 1.052 | 10.31 |
| 3 hr | 3 | 0.873 | 6.758 | 8 | 1.0716 | 13.40 | |
| 3 days | 3 | 0.902 | 4.213 | 8 | 0.995 | 11.58 | |
Effect of Formalin Fixation Time on the Quantification of mRNA
Reference genes β2‐macroglobulin, HNRPM, and CPSF6 were taken to generate short amplicons (98, 88, and 86 bp respectively). The threshold cycle (C T value) of β2‐microglobulin, HNRPM, and CPSF6 from FFPE HCT cells with different fixation lengths were compared to each other. The data showed a significant difference among fresh, short, and long fixation if we only compared C T values. However, relative quantification of HNPRM and CPSF6 expression normalization for β2‐microglobulin and HNRPM normalized for CPSF6 showed no statistical difference between different incubation times (Table 4). These results were confirmed for Lovo cells (data not shown).
Table 4.
Comparison of Quantitation Results With Different Fixation in HCT15 Cell Block (Hexamer and Cocktail PCR Primer)
| C T value (hexamer) | C T value (cocktail PCR primer) | ||||||
|---|---|---|---|---|---|---|---|
| Gene and amplicon | Fixation time | Mean (arbitrary unit) | CV (%) | F and P‐value for ANOVA | Mean (arbitrary unit) | CV (%) | F and P‐value for ANOVA |
| β2‐Microglobulin 98 bp | Fresh | 19.943 | 0.645 | F=32.147 | 20.139 | 0.0199 | F=86.869 |
| 1 day | 20.773 | 1.889 | P=0.001 | 22.105 | 2.781 | P=0.000 | |
| 3 days | 22.238 | 2.049 | 24.729 | 1.675 | |||
| HNRPM 88 bp | Fresh | 18.346 | 0.679 | F=109.360 | 19.281 | 0.4634 | F=109.838 |
| 1 day | 19.968 | 2.265 | P=0.000 | 21.697 | 2.816 | P=0.000 | |
| 3 days | 21.759 | 0.647 | 23.661 | 0.4847 | |||
| CPSF6 86 bp | Fresh | 18.480 | 1.708 | F=53.268 | 18.885 | 2.436 | F=70.473 |
| 1 day | 19.822 | 1.081 | P=0.000 | 21.548 | 1.538 | P=0.000 | |
| 3 days | 21.172 | 1.893 | 23.084 | 2.187 | |||
| RQ of HNRPM/β2‐microglobulin | Fresh | 0.9199 | 0.316 | F=4.413 | 0.9574 | 0.4687 | F=4.024 |
| 1 day | 0.9553 | 4.124 | P=0.066 | 0.9815 | 0.0584 | P=0.078 | |
| 3 days | 0.9787 | 1.532 | 0.9570 | 2.1406 | |||
| RQ of CPSF6/β2‐microglobulin | Fresh | 0.9266 | 1.059 | F=2.422 | 0.9378 | 2.4169 | F=2.026 |
| 1 day | 0.9543 | 0.987 | P=0.169 | 0.9755 | 4.1915 | P=0.213 | |
| 3 days | 0.9523. | 2.786 | 0.9335 | 1.4995 | |||
| RQ of HNRPM/CPSF6 | Fresh | 1.0073 | 1.134 | F=1.520 | 0.9795 | 2.7274 | F=0.266 |
| 1 day | 0.9999 | 3.579 | P=0.292 | 0.9939 | 4.2167 | P=0.775 | |
| 3 days | 0.9731 | 2.318 | 0.9757 | 2.6265 | |||
Effect of Fixation and Autolysis Time on the Quantification of mRNA
The factorial experimental design for DLD‐1 and Colo320 cell block was not only used to investigate the main effect of fixation and autolysis period (delay time before freezing and fixation) but also used to compare the interaction of both. We tested 0, 1, and 3 hr delay before fixation. In addition we investigated the fixation times of 0, 1, and 3 days. The relative quantification of HNRPM against β2‐microglobulin was determined. We found no statistical significance in the expression of HNRPM between different fixation times, autolysis delay, and the combination of both. We confirmed these results in Colo320 cells (Fig. 2 and Table 5). The colorectal carcinoma tissue samples were treated as the same process as the previous cell block. We tested 0, 1, and 3 hr delay before fixation. In addition, we investigated the fixation times of 0, 1, and 3 days. The relative quantification of HNRPM against β2‐microglobulin was determined. As expected, no statistically significant differences in the expression of HNRPM between fixation times, autolysis delay, and the combination of both were noted. For further confirmation, Figure 3 and Table 6 show the data of colorectal carcinoma tissue from two patients.
Figure 2.
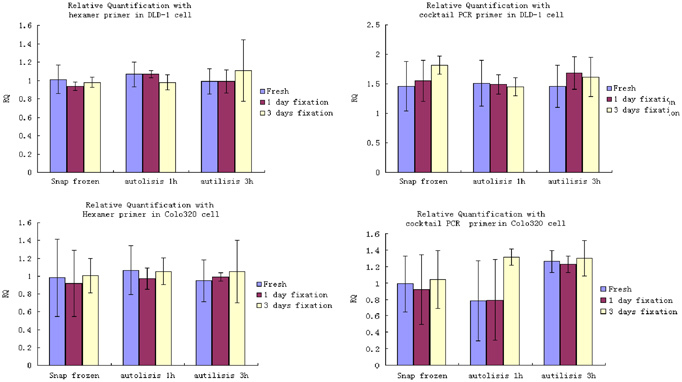
Effect of fixation, autolysis, and their interaction on relative quantification in DLD‐1 and Colo320 cells showed a factorial experimental design for DLD‐1 and Colo320 cells, the main effect of fixation, autolysis period, and their interaction on the relative quantification of HNRPM against β2‐microglobulin were all investigated. Meanwhile, Hexamer and Cocktail PCR primer were used respectively. The ANOVA results showed no statistical significance in relative quantification of HNRPM among different fixation times, autolysis delay, and the combination of both.
Table 5.
Effect of Fixation, Autolysis, and Their Interaction on Relative Quantification in DLD‐1 and Colo320 Cells
| DLD‐1 cell | Colo320 cell | |||||||
|---|---|---|---|---|---|---|---|---|
| Hexamer | PCR cocktail Primer | Hexamer | PCR cocktail Primer | |||||
| Summary of fit R 2 adjusted | 0.433 | 0.376 | 0.504 | 0.314 | ||||
| Statistic and term | F‐value | Prob | F‐value | Prob | F‐value | Prob | F‐value | Prob |
| Intercept | 1,079.31 | ≤0.0001 | 112.27 | ≤0.0001 | 1,091.86 | ≤0.0001 | 21.709 | ≤0.0001 |
| Fixation | 0.312 | 0.736 | 1.709 | 0.209 | 0.177 | 0.839 | 1.436 | 0.264 |
| Autolysis time | 1.214 | 0.320 | 0.196 | 0.824 | 0.123 | 0.885 | 2.372 | 0.122 |
| Fixation×autolysis time | 2.679 | 0.065 | 1.763 | 0.180 | 0.059 | 0.993 | 0.705 | 0.599 |
Figure 3.
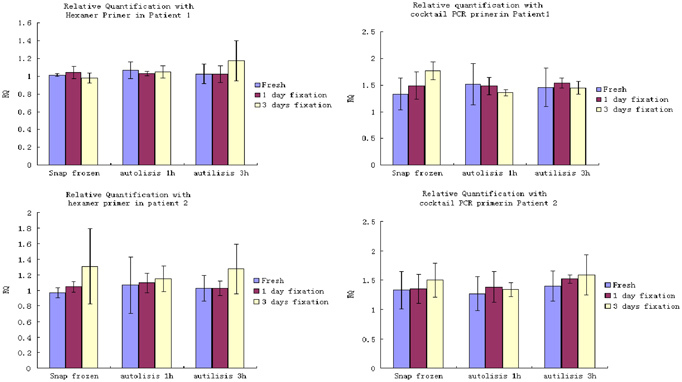
Effect of fixation, autolysis, and their interaction on relative quantification in colorectal carcinoma tissue from two patients showing a factorial experimental design for colorectal carcinoma tissue from two patients, the main effect of fixation, autolysis period, and their interaction on the relative quantification of HNRPM against β2‐microglobulin were all investigated. Meanwhile, Hexamer and Cocktail PCR primer were used respectively. The ANOVA results showed no statistical significance in relative quantification of HNRPM among different fixation times, autolysis delay, and the combination of both.
Table 6.
Effect of Fixation, Autolysis, and Their Interaction on Relative Quantification in Colorectal Carcinoma Tissue From Two Patients
| Patient 1 | Patient 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Hexamer | PCR cocktail Primer | Hexamer | PCR cocktail Primer | |||||
| Summary of fit R 2 adjusted | 0.287 | 0.264 | 0.205 | 0.184 | ||||
| Statistic and term | F‐value | Prob | F‐value | Prob | F‐value | Prob | F‐value | Prob |
| Intercept | 2874.43 | ≤0.0001 | 1050.67 | ≤0.0001 | 748.95 | ≤0.0001 | 810.91 | ≤0.0001 |
| Fixation | 0.354 | 0.709 | 0.337 | 0.719 | 0.438 | 0.652 | 0.734 | 0.494 |
| Autolysis time | 0.872 | 0.435 | 0.235 | 0.793 | 0.180 | 0.837 | 1.046 | 0.372 |
| Fixation×autolysis time | 1.122 | 0.377 | 1.331 | 0.297 | 0.848 | 0.513 | 0.127 | 0.971 |
DISCUSSION
The fixation of human tissue with formalin, and subsequent embedding in paraffin, has been a routine method of collecting and preserving surgical samples for many years. These archived materials are the most widely available specimens for retrospective genetic clinical studies. Although FFPE tissues provide a valuable source of stable RNA for gene expression analysis, RNA isolation from FFPE tissue samples remains difficult and inefficient, due to degradation of the RNA by ubiquitous RNases 11, 12. To date, the most successful method for the extraction of total RNA from FFPE tissues utilizes a proteinase K digestion before acid–phenol chloroform extraction and carrier precipitation 6, 10, 13. However, the RNA extracted from FFPE tissue is significantly degraded 10, 13. Our present study confirmed that mRNA (at least for the reference genes we studied) can be detected by using real‐time RT‐PCR in FFPE cells, even though RNA isolation from FFPE samples was degraded as was seen by gel electrophoreses. The fixation process causes cross‐linkage between nucleic acids and proteins, and covalently modifies RNA by the addition of monomethyl groups to the bases, so the molecules in FFPE sample are rigid and fragile. Formalin fixation damages the integrity of RNA and also limits the size of the cDNA product produced 14, 15. Therefore, the size of nucleic acid fragments amplified by PCR should be kept to a minimum, which is less than 120 bp. It is generally accepted that whole tissue specimens have important disadvantage when they are used for gene expression analysis. For example, whole tumor tissue is generally composed of tumor cells, as well as stromal components, inflammatory cells, vessels, and others. Tumor heterogeneity may be a source of possibly serious bias when quantifying RNA extracted from tissues. The effects of tumor heterogeneity can be limited by analyzing several different areas of a tumor. In order to overcome the problem of tissue heterogeneity, cells have to be selected for further analysis. Therefore, laser capture microdissection (LCM) has been introduced and is now generally accepted as a powerful tool to dissect morphologically identified cell populations. There are some reports to support combination LCM with RNA isolation from FFPE tissue with real‐time PCR to quantify some target gene expressions 16, 17. One important factor in quantifying gene expression study is to choose one or more reference genes to control variation due to different degradation of mRNA in FFPE tissue. In this study, we investigated several reference genes, including β2‐macroglobulin, β‐actin, HNRPM, and CPSF6. The ideal reference gene is the one that is constantly expressed in moderate levels of all cells. There are three common priming methods that can be employed for the cDNA synthesis: oligo‐dT primers, random hexamers, and gene‐specific primers. Oligo‐dT priming is not ideal for FFPE samples because of the chemical modification of the poly(A) tail of the mRNA. Random hexamer priming, although used widely for FFPE tissue samples, suffers from the fact that any RNA template, not just mRNA, can be primed. Gene‐specific priming can be used to increase the specificity of the cDNA synthesis and enhance the detection of low‐abundance RNA transcripts 18. We use both hexamer and PCR‐specific primer to synthesize cDNA. We found no significant differences between these two approaches. Several factors may influence the relative quality and quantity of RNA in FFPE tissue. One such factor that may be a problem for mRNA quantification is the length of fixation time 19, 20. Some researchers concluded that the duration of fixation should be kept between 0.5 and 2 days since long fixation times drastically reduced the number of mRNA molecules 8. Conversely, other groups concluded that prolonged fixation times up to 6 days caused no significant change in mRNA quantity. Furthermore, archived tissue blocks after 5, 10, 15, and 20 years failed to identify a clear time‐dependent loss of RNA and the variable results also obtained from such kind of tissues 8, 21. In our experiment, we tested the effect caused by the length of formalin fixation. Prolonged fixation times up to several days caused significant loss of detectable mRNA molecules; however, relative quantification data showed no differences (P=0.292 for hexamer; P=0.775 for cocktail primer), indicating that the degradation of mRNA accounts for all genes and not only for a subset of genes. Nevertheless, should tissue be fixed as soon as possible and is it necessary to prevent autolysis‐induced degradation? There are some reports about gene autolysis in mRNA level occurred within first 2 hr after which a steady state appeared. On the other hand, some researchers concluded that a delay in tissue processing with pre‐fixation times of up to a few hours did not appear to be a problematic for quantitative gene expression study 22, 23. To evaluate in vitro RNA decay, we choose a model reflecting daily routine as closely as possible. Freezing or fixing delay up to 3 hr was chosen to reflect the situation for delayed tissue processing. Although much is known about the degradation of mRNA because of the effect of autolysis and fixation, little has been published concerning the interaction effect between that. In our study, we investigated the effect of fixation and delay time before freezing and fixation, and we also compared the combination of these two variables, by calculation of the relative quantification of several reference genes. It appears crucial that the ratio between the copy numbers of different genes remained constant 24 and has not been altered by some artificial factors in the process of tissue treatment such as formalin fixation and autolysis period before fixation/freezing. We demonstrated that in our settings mRNA levels in FFPE cells could be reproduced and quantified. We realize that cell lines do not reflect tissue to test the effect of formalin fixation and autolysis, because of their cell constitution. Although logistically hard to perform, we suggest to confirm these results in tumor tissue. In conclusion, our findings do suggest that clinical tumor specimens removed by routine surgical procedures and embedded in paraffin are a justified source for the analysis of gene expression. We showed that mRNA levels in FFPE cells could be reproducibly quantified, independent of the nonstandardized fixation processes. We believe that this conclusion enables the use of FFPE tissue to provide useful information related to prognostic and predictive study of these important material mainly stored in pathology department.
References
- 1. Heid CA, Stevens J, Livak KJ, et al. Real time quantitative PCR. Genome Res 1996;6:986–994. [DOI] [PubMed] [Google Scholar]
- 2. Bustin SA. Absolute quantification of mRNA using real‐time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 2000;25:169–193. [DOI] [PubMed] [Google Scholar]
- 3. Lewis F, Maughan NJ, Smith V, et al. Unlocking the archive—Gene expression in paraffin‐embedded tissue. J Pathol 2001;195:66–71. [DOI] [PubMed] [Google Scholar]
- 4. Harbeck N, Nimmrich I, Hartmann A, et al. Multicenter study using paraffin‐embedded tumor tissue testing PITX2 DNA methylation as a marker for outcome prediction in tamoxifen‐treated, node‐negative breast cancer patients. J Clin Oncol 2008;26:5036–5042. [DOI] [PubMed] [Google Scholar]
- 5. Chang JC, Makris A, Gutierrez MC, et al. Gene expression patterns in formalin‐fixed, paraffin‐embedded core biopsies predict docetaxel chemosensitivity in breast cancer patients. Breast Cancer Res Treat 2008;108:233–240. [DOI] [PubMed] [Google Scholar]
- 6. Masuda N, Ohnishi T, Kawamoto S, et al. Analysis of chemical modification of RNA from formalin‐fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res 1999;27:4436–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene 2001;265:11–23. [DOI] [PubMed] [Google Scholar]
- 8. Godfrey TE, Kim SH, Chavira M, et al. Quantitative mRNA expression analysis from formalin‐fixed, paraffin‐embedded tissues using 5′ nuclease quantitative reverse transcription‐polymerase chain reaction. J Mol Diagn 2000;2:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abrahamsen HN, Steiniche T, Nexo E, et al. Towards quantitative mRNA analysis in paraffin‐embedded tissues using real‐time reverse transcriptase‐polymerase chain reaction: A methodological study on lymph nodes from melanoma patients. J Mol Diagn 2003;5:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jackson DP, Lewis FA, Taylor GR, et al. Tissue extraction of DNA and RNA and analysis by the polymerase chain reaction. J Clin Pathol 1990;43:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mizuno T, Nagamura H, Iwamoto KS, et al. RNA from decades‐old archival tissue blocks for retrospective studies. Diagn Mol Pathol 1998;7:202–208. [DOI] [PubMed] [Google Scholar]
- 12. Li J, Smyth P, Cahill S, et al. Improved RNA quality and TaqMan Pre‐amplification method (PreAmp) to enhance expression analysis from formalin fixed paraffin embedded (FFPE) materials. BMC Biotechnol 2008;6:8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stanta G, Schneider C. RNA extracted from paraffin‐embedded human tissues is amenable to analysis by PCR amplification. Biotechniques 1991;11:304–308. [PubMed] [Google Scholar]
- 14. Jiang YH, Davidson LA, Lupton JR, et al. A rapid RT‐PCR method for detection of intact RNA in formalin‐fixed paraffin‐embedded tissues. Nucleic Acids Res 1995;23:3071–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Specht K, Richter T, Müller U, et al. Quantitative gene expression analysis in microdissected archival tissue by real‐time RT‐PCR. J Mol Med 2000;78:B27. [PubMed] [Google Scholar]
- 16. Emmert‐Buck MR, Bonner RF, Smith PD, et al. Laser capture microdissection. Science 1996;274:998–1001. [DOI] [PubMed] [Google Scholar]
- 17. Luzzi V, Mahadevappa M, Raja R, et al. Accurate and reproducible gene expression profiles from laser capture microdissection, transcript amplification, and high density oligonucleotide microarray analysis. J Mol Diagn 2003;5:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paik S, Kim CY, Song YK, et al. Technology insight: Application of molecular techniques to formalin‐fixed paraffin‐embedded tissues from breast cancer. Nat Clin Pract Oncol 2005;2:246–254. [DOI] [PubMed] [Google Scholar]
- 19. Ben‐Ezra J, Johnson DA, Rossi J, et al. Effect of fixation on the amplification of nucleic acids from paraffin‐embedded material by the polymerase chain reaction. J Histochem Cytochem 1991;39:351–354. [DOI] [PubMed] [Google Scholar]
- 20. Bresters D, Schipper ME, Reesink HW, et al. The duration of fixation influences the yield of HCV cDNA‐PCR products from formalin‐fixed, paraffin‐embedded liver tissue. J Virol Methods 1994;48:267–272. [DOI] [PubMed] [Google Scholar]
- 21. Rupp GM, Locker J. Purification and analysis of RNA from paraffin‐embedded tissues. Biotechniques 1988;6:56–60. [PubMed] [Google Scholar]
- 22. Beelman CA, Parker R. Degradation of mRNA in eukaryotes. Cell 1995;81:179–183. [DOI] [PubMed] [Google Scholar]
- 23. Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene 2001;265:11–23. [DOI] [PubMed] [Google Scholar]
- 24. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]