

Abstract
Fluorescence, visible circular dichroism (CD), absorption, and resonance Raman spectroscopy techniques were combined to explore structural changes of ferricytochrome c upon its binding to cardiolipin-containing liposomes (20% 1,1′,1,2′-tetraoleyolcardiolipin and 1,2-deoleyol-sn-glycero-3-phosphocholine) at acidic pH (6.5). According to the earlier work of Kawaiet al. [J. Biol. Chem. 2005, 280, 34709–347171],cytochrome c binding at this pH is governed by interactions between the phosphate head groups of cardiolipin and amino acid side chains of the so-called L-site, which contains the charged residues K22, K25, K27, and potentially H26 and H33. We found that L-site binding causes a conformational transition that involves a change of the protein’s ligation and spin state. In this paper, we report spectroscopic responses to an increasing number of cardiolipin-containing liposomes at pH 6.5 in the absence and presence of NaCl. The latter was found to mostly inhibit protein binding already with 50 mM concentration. The inhibition effect can be quantitatively reproduced by applying the electrostatic theory of Heimburget al. [Biophys. J. 1995, 68, 536–546]. A comparison with corresponding spectroscopic response data obtained at pH 7.4 reveals major differences in that the latter indicates hydrophobic binding, followed by an electrostatically driven conformational change. Visible CD data suggest that structural changes in the heme pocket of liposome-bound ferricytochrome c resemble to some extent those in the denatured protein in urea at neutral and acidic pH. The measured noncoincidence between absorption and CD Soret band of cytochrome c in the presence of a large access of cardiolipin is caused by the electric field at the membrane surface. The very fact that its contribution to the internal electric field in the heme pocket is detectable by spectroscopic means suggests some penetration of the protein into membrane surface.
1. Introduction
Cytochrome c is a multifunctional heme protein found primarily in the mitochondria of cells, where it carries out an electron-transfer process that drives cellular respiration.1,2 The multifunctionality of this protein is due to its conformational flexibility in its oxidized state3,4 and native folded state. Its primary function of electron transfer between cytochrome c reductase and oxidase is carried out via the slightly solvent-exposed heme group. The heme iron has a high reduction potential owing to its methionine (M80) axial ligand.5 An alternative biological function of cytochrome c is its role in initiating apoptosis after it is released into the cytosol.6 The release from the intermembrane space of mitochondria is facilitated by a complex biochemical cascade during which the protein acts as a lipid peroxidase. It oxidizes an increasing number of anionic phospholipid cardiolipin (CL) that makes up about 20% of the lipids constituting the resting state of the inner mitochondrial membrane (IMM).7
The heme environments of native cytochrome c and classical peroxidases are significantly different. While cytochrome c contains a hexacoordinate low-spin heme iron with histidine and methionine as axial ligands, classical peroxidases like horseradish peroxidase generally adopt a pentacoordinate high-spin or quantum-mixed spin state of the heme iron.8,9 The distal environment is configured in a way that amino acid side chains of, e.g., a histidine and an arginine can stabilize intermediates like compound I via hydrogen bonding. In cytochrome c, covalent linkages of the heme to two cysteins via thioether bridges cause substantial ruffling of the heme group, which reduces the protein’s redox potential.10,11 In classical peroxidases, interactions between the heme macrocycle and the protein are solely noncovalent, which produces a mixing of macrocycle saddling and ruffling.12,13 These structural differences make it apparent that cytochrome c has to adopt a non-native structure for acquiring peroxidase activity. Multiple lines of evidence do indeed suggest that the interaction of ferricytochrome c with anionic lipid surfaces indeed induces structural changes.14−21 The anionic phospholipid CL has a high binding affinity for cytochrome c, thus making CL-containing vesicles excellent tools for studying the thermodynamic and structural aspects of cytochrome c–IMM interactions.21−24
Several binding sites on the protein have been proposed thus far in the literature involving the interaction between cytochrome c and negatively charged phospholipids. Unfortunately, the multitude of binding studies reported over the last 25 years does not provide a consistent picture regarding the physical determinants of binding processes and their dependence on external parameters such as pH and ionic strength. A detailed discussion of unresolved issues has been given in a recent review.4 Here, we confine ourselves to a brief summary of existing contradictions. Generally, the two-site binding model of Rytömaa and Kinnunen is still considered as a kind of ultima ratio for the interpretation of binding studies.25,26 On the basis of fluorescence quenching studies of ferricytochrome c binding to liposomes composed of different mixtures of anionic and zwitterionic lipids, these researchers proposed two types of binding sites termed A- and C-site. A-site binding occurs at a patch of positively charged lysine residues (K72 and K73) (Figure 1). Although the authors originally determined the binding mechanism to be electrostatic, they later reported experimental findings that suggested an irreversibility of the process. They tried to explain this discrepancy by a two-step binding process where electrostatic binding is followed by a lipid insertion into a hydrophobic pocket in the protein.25 It does not depend significantly on pH above 5. C-site binding was proposed to involve hydrogen bonding between the N52 residue (as acceptor) and a protonated phosphate head group of CL. For liposomes with physiological CL content (20% and lower), this binding process requires a pH value below 5 but data reported by Rytömaa and Kinnunen suggest that the pH threshold moves into the physiological pH region with increasing CL content of liposomes.26 Thus, it could become biologically relevant since the CL contents of the inner and outer membranes of mitochondria increase during the initial phase of the apoptotic process.7 C-site binding is not electrostatic in nature. Therefore, it cannot be inhibited by the addition of NaCl or other salts.
Figure 1.
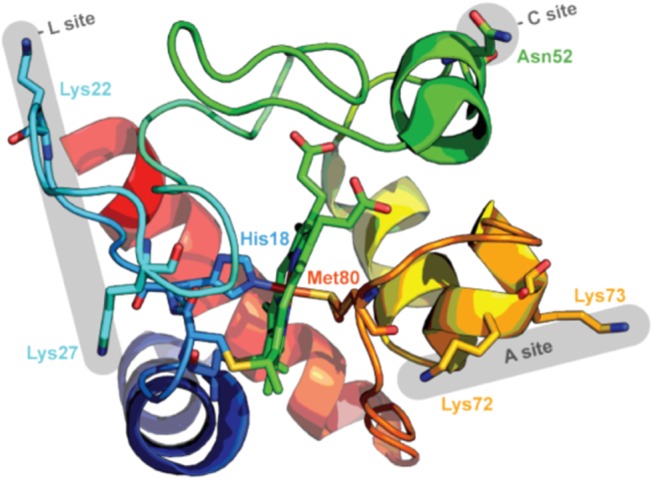
Proposed binding sites A, C, and L involved in cytochrome c–cardiolipin interactions. Taken from Soffer.27
The binding model of Rytömaa and Kinnunen has been recently augmented by a third so-called L-site proposed by Kawai et al., which is active only at pH below 7.0.24 The existence of this binding site was first deduced from the observation of pH-dependent turbidity after allowing ferricytochrome c to interact with liposomes containing 20% CL. The turbidity was related to vesicle fusion caused by the simultaneous interactions of cytochrome c with two different liposomes via its A- and L- binding sites. According to this model, the protein operates like a bidentate ligand (Figure 1). A combination of carbethoxylation (histidine) and alkylation (lysine) modifications with matrix-assisted laser desorption ionization time-of-flight experiments enabled the authors to identify the involved protein residues as K22, K25, K27, H33, and H26 (cf. Figure 1). This result led the authors to conclude that L-site binding is electrostatic in nature. The pH range of L-site binding for which experimental data indicate an effective pK value of seven overlaps with the reported pH values of the IMM (6.8–7),28 thus giving it a high physiological relevance. The latter has been corroborated by a follow-up study of Kawai et al., who observed that the respiration of cytochrome c-depleted mitoplasts can be restored by the addition of cytochrome c in a pH-dependent manner that resembles the observed L-site binding.29 While the experiments of Kawai et al. provide compelling evidence for the existence and biological relevance of L-site binding, its very existence is actually at variance with the results of Rytömaa and Kinnunen in that L-site binding was shown to increase the apparent binding constant for cytochrome c from ca. 7.5 × 106 at pH 7.4 to ca. 2.5 × 107 M–1 at pH 6.2. This variation would lead to a much more pronounced variation of binding isotherms in this pH range than those reported by the Kinnunen group.24,26 Further contradiction to the A/C-site binding model arose from conflicting observations regarding cytochrome c binding to CL-containing liposomes at neutral pH, which generally reflect binding affinities well below those reported in the above-cited studies (in the 104–105 M–1 range).20,21,30 Work of the Santucci group on liposomes with 100% CL content seems to suggest a mixture of A- and C-site binding,20 while Pandisica and Schweitzer-Stenner proposed a model that combines a nonelectrostatic first binding step with a subsequent conformational change that establishes an equilibrium between native-like and non-native-like states. This equilibrium was found to depend on the protein occupation of liposomes.21 According to their results, the addition of NaCl solely shifts the equilibrium between the two populations toward native-like state. Although their binding model is fully consistent with the results of very comprehensive fluorescence resonance energy transfer (FRET)-based structural studies of Pletneva and co-workers,19,30 the derived binding affinities and the proposed mechanism do not seem to be in agreement with the A/C-site binding model. In addition, the very possibility of the C-site binding mechanism has recently been questioned based on the observation that both phosphate groups of CL are deprotonated above pH 3.0 even for liposomes with 100% CL content.31−33
Another binding site option has been reported by O’Brien et al.34 These authors carried out a very detailed solid-state NMR investigation of interactions between encapsulated ferricytochrome c and CL that was titrated to the protein solution. Besides A- and L-site binding, they observed a mostly hydrophobic binding site termed N-site, which involves the residues F36, G37, T58, Y59, and K60.
The current study focuses on a further characterization of L-site binding. Recently, we have reported results of a spectroscopic study demonstrating that structural changes caused by this mode of binding involve the population of non-native states, where ferricytochrome c adopts a mixture of penta- and hexacoordinate high-spin states. They are clearly distinct from the non-native hexacoordinate low-spin conformations adopted at more neutral pH (7.4).35 We attributed these differences to the protonation of a second histidine ligand that replaces the native M80 at neutral pH in the non-native states. To clarify whether the L-site binding is indeed electrostatic in nature, we measured the spectroscopic response of W59 fluorescence, Soret band circular dichroism (CD), and charge-transfer band absorption as a function of CL concentration in the absence and presence of NaCl at pH 6.5. Conformational changes induced by the addition of salt are further verified by resonance Raman spectroscopy. A global analysis of the current and earlier spectroscopic response data21,36 for cytochrome c–CL interactions obtained at neutral pH is being carried out, which considers two mechanisms of how the presence of sodium cations can affect the binding processes. Model 1 just assumes that the conversion of native-like to non-native-like conformations of CL-bound ferricytochrome c is inhibited by the direct interactions of sodium cations with negatively charged CL head groups in a Langmuir-type manner.21,37 For model 2, we assume that the initial binding step is electrostatic in nature, which we describe by utilizing the electrostatic binding theory of Heimburg and Marsh.38 This theory takes into account the electrostatic contributions to the binding constant dependent on the electrostatic free-energy change upon cytochrome c binding to the anionic surface. The addition of positively charged salt ions reduces the electrostatic double-layer potential, thus decreasing the binding free energy and the number of bound proteins at high lipid concentrations.
2. Results
2.1. Spectroscopic Response Probing Cytochrome c L-Site Binding to CL-Containing Liposomes
2.1.1. W59 Fluorescence
Figure 2 (left) shows the background-corrected W59 fluorescence of horse heart ferricytochrome c in the absence of NaCl observed with the indicated effective CL concentrations at pH 6.5. Since W59 is the only tryptophan residue in horse heart cytochrome c, the protein’s fluorescence in the near-UV region is very weak in its folded state owing to fluorescence resonance energy transfer to the nearby heme group. Conformational changes that lead to a partial unfolding of the protein’s tertiary structure causes the W59 fluorescence to increase. The dominant band in the region between 320 and 380 nm (termed F-band in our earlier studies)21,36 steadily increases in intensity with CL concentration, suggesting tertiary structure changes upon binding. As also shown in Figure 2 (right), the addition of NaCl results in a significant decrease of fluorescence at high CL concentration (200 μM), which is by far more pronounced than the corresponding fluorescence quenching observed at neutral pH (7.4).21 At lower CL concentration, the residual W59 fluorescence only slightly exceeds the background scattering at the employed liposomes so that a background subtraction leads to very noisy spectra. This observation is qualitatively consistent with the expectation that L-site binding is electrostatic in nature.
Figure 2.

Left) Background-corrected fluorescence spectra of horse heart ferricytochrome c–liposome mixture recorded as a function of the indicated CL concentration (liposome concentration). The arrow highlights the spectral changes in the direction of increasing CL concentration. Starting from the solid black line, the CL concentrations are 0, 50, 75, 100, 125, 150, 175, 200, 225, and 250 μM. (Right) Background-corrected fluorescence spectra of a mixture of 5 μM ferricytochrome c with 200 μM cardiolipin in the absence of NaCl (black line) as well as in the presence of 50 mM (blue line) and 100 mM (teal line) NaCl. Experimental details are given in the Section 4 of this paper.
2.1.2. Soret Band Circular Dichroism
Figure 3 displays the changes in the Soret band CD spectra of cytochrome c upon binding to CL-containing liposomes. The indicated CL concentrations correspond to the same CL/protein ratios as the above CL concentrations used for the fluorescence measurements. Generally, the circular dichroism associated with this band is indicative of electronic interactions between the heme chromophore and the surrounding protein.39 In an ideal D4h symmetry of the porphyrin macrocycle of the heme, the underlying electronic transition of the Soret band would be 2-fold degenerate with transition dipole moments along the FeN lines. The electric field produced by the protein and vibronic perturbations lifts the degeneracy.39−41 Owing to their perpendicular orientation, the two dipole moments interact differently with the heme environment. In the case of native horse heart ferricytochrome c, this leads to a negative couplet, i.e., two overlapping signals with rotational strengths of different sign. The rotational strength of the negative signal is generally believed to be predominantly caused by interactions between transition dipole moments of the heme and the nearby F82 residue.42 Upon addition of CL-containing liposomes, the Soret band CD changes from a couplet to a positive Cotton band, indicating that the electronic environment around the heme is changing due to structural changes in the protein.20,43,44 The very pronounced positive Cotton band observed at high CL concentrations in the absence of salt is indicative of a large population of partially unfolded proteins. The addition of NaCl causes a return of the couplet, which is diagnostic of a repopulation of the native unfolded state. In line with the above fluorescence data, this observation is qualitatively consistent with the notion that L-site binding is electrostatic in nature.
Figure 3.

Soret band circular dichroism spectra of horse heart ferricytochrome c–liposome mixture recorded as a function of CL concentration (liposome concentration) in the absence (left) of NaCl as well as in the presence of 50 mM (middle) and 100 mM (right) NaCl. The arrows highlight the spectral changes in the direction of increasing CL concentration. The corresponding CL concentrations are 0, 100, 150, 200, 250, 300, 350, 400, 450, and 500 μM. Experimental details are given in the Section 4 of this paper.
The Soret band CD spectrum measured at high CL concentration exhibits a very pronounced Cotton band with a peak value of ca. 40 M–1 cm–1. The corresponding spectrum measured at pH 7.4 depicts a very similar intensity.21 At room temperature, the Soret bands in the CD spectrum of the partially unfolded alkaline states IV and V of ferricytochrome c are by far less intense (<20 M–1 cm–1).45 Upon further thermal unfolding, only the state V band reaches the level of the peak dichroism that we observed for the binding of the protein to CL-containing liposomes. This led us to conjecture that the latter represents a nearly denatured state of the protein. To check for this possibility, we measured the absorption and visible CD spectra of ferricytochrome c in neutral (pH 7.4) and acidic (pH 5.5) solutions at denaturing conditions (8 M urea). The choice of the two pH values was guided by the results of Elöve et al.,46 who showed that the refolding kinetics of cytochrome denatured by the addition of a large amount of either urea or guanidinium chloride depends on pH in the region between 4.5 and 7. They attributed this to denatured states with different heme ligands at neutral and acidic pH. At the former, the heme group is axially ligated with two histidines that produce a hexacoordinate low-spin state. At pH below 5.7, the non-native histidine ligand is replaced by water to produce a hexacoordinate high-spin state. Hence, the pH-induced conformational transition of denatured ferricytochrome c resembles very much the transition between the f and f+ states of the membrane-bound protein.
Figure 4 compares the Soret CD and absorption spectra of denatured ferricytochrome c at pH 5.5 (vide supra) with the spectra measured in the presence of 500 μM accessible CL at pH 6.5. Both absorption spectra are very similar; the Soret bands peak at the same wavelength and the total intensities are comparable if one considers the different baselines that originate from higher-lying transitions. The CD spectra, however, look slightly different. At both conditions, we observed a very intense positive Cotton band, but the respective band positions are clearly different, i.e., at 403.5 (chemically denatured) and 410 nm (CL-bound). The red shift of the latter reveals a noncoincidence between the position of the CD Cotton band and the respective absorption band, which indicates excited-state splitting between the Bx and By states and significantly different rotational strengths of the underlying transitions.39 This splitting and/or the asymmetry between rotational strengths must be much smaller in the denatured protein as they are in partially unfolded states of the protein. A similar noncoincidence was earlier observed for cytochrome–CL mixtures at pH 7.4 and was assigned to an additional electric field component produced by the phosphate groups.21 This addition to the internal electric field of the protein can change its strength at the heme group as well as its direction. We argued that a change of the latter flips the position of Bx and By. The different total intensities of the two Cotton bands also suggest that the underlying structures are slightly different in that the lower-lying transition carries some more rotational strength in the membrane-bound protein.47 This can easily be caused by slight differences between the positions of aromatic residues in the heme pocket or by different degrees of heme nonplanarity.48,49 The relationship between protein unfolding on the membrane surface and noncoincidence is demonstrated in Figures S1 and S2. The noncoincidence values are positive at low lipid concentrations (the positive maximum of the CD spectrum is blue-shifted relative to the absorption spectrum) and negative at high lipid concentrations (the positive maximum is now on the red side of the absorption spectrum). The response curve clearly displays two phases. We also measured the visible CD and absorption spectra of denatured ferricytochrome c at pH 7.4 (Figure S3). They appear slightly blue-shifted with respect to the corresponding spectra measured at acidic pH, which reflects the low-spin state of the heme iron. The noncoincidence between CD and absorption is again small. The amplitude of the Cotton band is still rather large (26 M–1 cm–1) compared to the corresponding peak values of alkaline states at room temperature,45 but lower than the corresponding value of liposome-bound ferricytochrome c at pH 7.4 (36 M–1 cm–1).36 Altogether, these results suggest that the corresponding denatured states in solution and on the surface of CL-containing membranes are very similar and that spectroscopic differences between the CD spectra reflect specific forces that membrane surface groups exert on the heme pocket.
Figure 4.
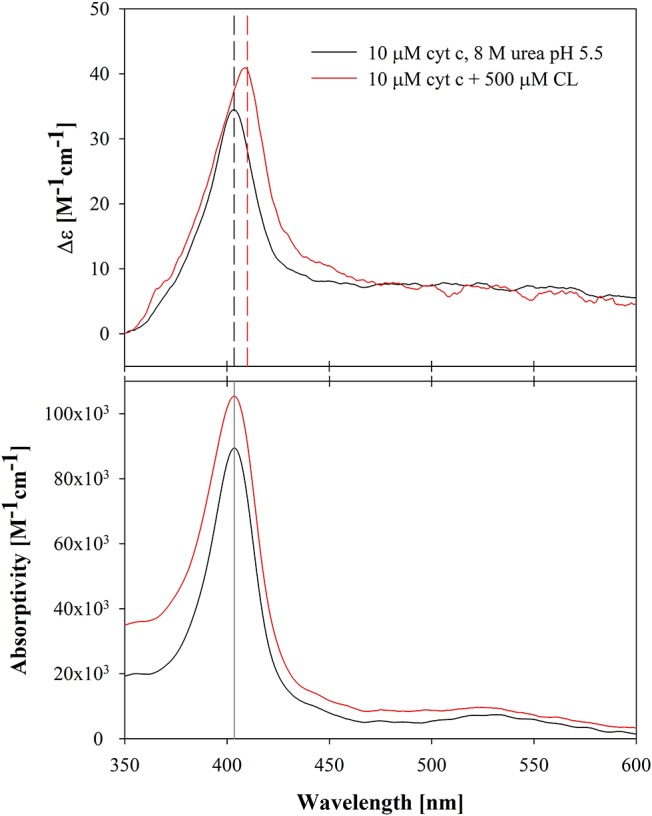
Visible CD and absorption spectra of ferricytochrome c in the presence of 8 M urea at pH 5.5 (black) and 500 μM cardiolipin 20% 1,1′,1,2′-tetraoleyolcardiolipin (TOCL)/80% 1,2-deoleyol-sn-glycero-3-phosphocholine (DOPC) liposomes (red).
2.1.3. Charge-Transfer Absorption Bands
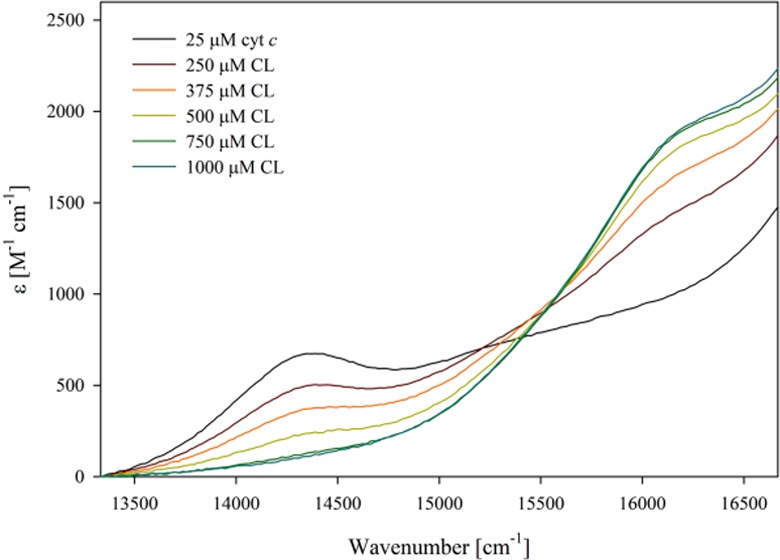
Figure 5 shows the visible absorption of ferricytochrome c in the region between 13 000 and 17 000 cm–1 (770–590 nm) measured as a function of CL concentration. Higher protein and CL concentrations were used for these measurements to ensure a good signal-to-noise ratio. The recorded spectra depict two weak absorption bands that are assignable to charge-transfer transitions. The first (CT1) appears at 14 389 cm–1 (695 nm). It is an indicator of M80 coordination of the heme iron and therefore assigned to an M80 → Fe(III) transition.50 However, the underlying changes of the electronic configurations have not yet been identified. The decrease in CT1 intensity is therefore indicative of a ligand exchange involved in the conformational change from a native-like into a partially unfolded structure, in agreement with the above fluorescence and CD data. On a qualitative level, all of these spectroscopic changes are reminiscent of the results that Pandiscia and Schweitzer-Stenner reported for cytochrome–CL binding at pH 7.4.21 The most important difference between the spectral responses obtained at pH 6.5 and 7.4 is the appearance of a second charge-transfer band (CT2) at 16 000 cm–1 (625 nm) under acidic conditions. This band is indicative of a population of high-spin cytochrome c, which could either be a pentacoordinate with an exposed heme group or a hexacoordinate with a water ligand. As reported earlier, the corresponding resonance Raman spectra reveal a mixture of both ligation states at high CL concentration, where CT2 is most intense.35 In the presence of 50 mM NaCl, the CT1 band reappears at even very high CL concentrations, while the CT2 band is no longer present in the spectrum. This observation is clearly indicative of a recovery of native-state-like conformations.
Figure 5.

Optical absorption spectra of horse heart ferricytochrome c–liposome mixture between 13 000 and 17 000 cm–1 recorded as a function of cardiolipin concentration (liposome concentration) in the absence (left) of NaCl as well as in the presence of 50 mM (middle) and 100 mM (right) NaCl. Note that in the presence of NaCl, the baseline first increases and subsequently decreases with cardiolipin concentration. Experimental details are given in the Section 4 of this paper.
2.1.4. Resonance Raman Spectroscopy
Figure 6 compares the resonance Raman spectra of horse heart cytochrome c–liposome–potassium ferrocyanide mixtures without and with NaCl (150 mM) taken with 442 nm excitation at pH 6.5. The CL concentration of 2 mM corresponds to a CL/protein ratio of 40. The presence of potassium ferrocyanide ensures that cytochrome molecules in the native state (i.e., with M80 as axial ligand) become photoreduced,51 while non-native proteins remain in their oxidized state. The spectrum recorded in the absence of NaCl is indicative of the earlier reported mixture of penta- and hexacoordinate high-spin species,35 whereas the spectrum taken in the presence of NaCl is that of a totally reduced sample. This observation suggests that the addition of NaCl practically eliminates the f+-state population.
Figure 6.
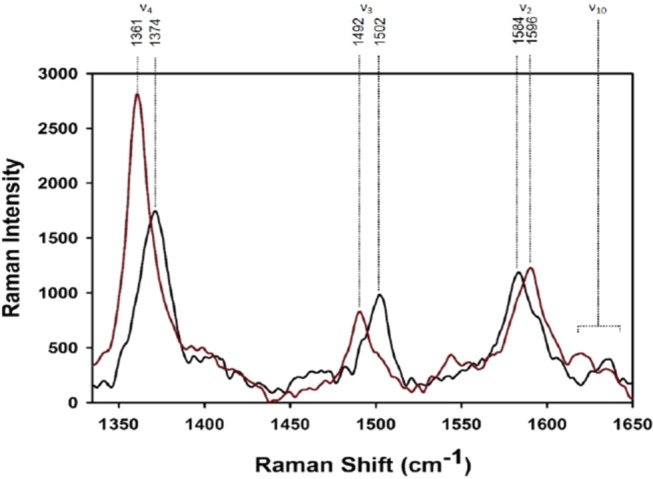
Resonance Raman spectrum of 50 μM horse heart cytochrome c and 20% TOCL/80% DOPC–liposome mixtures with 2 mM accessible cardiolipin measured in the absence (black) and presence of 150 mM NaCl (red).
2.2. Analysis of Spectral Response Data
Our data analysis can be represented by the following general reaction scheme
| R1 |
which is based on the assumption that protein binding to CL-containing liposomes produces an equilibrium between native-like and non-native partially unfolded protein populations.21,30 The former is termed nf+ (nonfluorescing) because the quantum yield of the W59 fluorescence is considered to be zero or very small. The non-native state is termed f+ (fluorescing) because one observes W59 fluorescence. The f states populated at pH 7.4 and 6.5 are different in that the corresponding heme iron adopts a hexacoordinate, low-spin state at the former51 and a mixture of penta- and hexacoordinate high-spin states at the latter.35 We can also expect some differences between the respective nf states. Therefore, we term the states populated at pH 6.5 nf+ and f+ to indicate that they become populated due to the protonation of amino acid side chains.24 Mechanistic details are being dealt with in the Section 3 below.
The spectral response curves in Figure 7 were produced after measured spectra were subjected to data analysis described in detail in the Supporting Information. We represent the manifold of the spectroscopic response data displayed in Figure 7 by a Langmuir-type function s([CL])
| 1 |
where [CL] is the concentration of free CL (not bound to cytochrome c), s0 is the spectroscopic parameter for the fully folded native protein in solution, snf+ and sf+ are the spectroscopic parameters of the membrane-bound nf+ and f+ states, respectively, K+ denotes the apparent equilibrium binding (association) constant, and Kc+ reflects the apparent equilibrium between nf+ and f+ states. It should be noted that symbols nf+ and f+ represent heterogenous ensembles of subconformations similarly to the C- and E-state distributions reported by Hong et al.19 It is very likely that there is a close correspondence between nf and C states as well as between f and E states at neutral pH. Equation 1 resembles the formalism that we used to describe cytochrome c binding at neutral pH in terms of an nf ⇆ f equilibrium, where we denoted the corresponding equilibrium constants without the superscript.21
Figure 7.

Spectral response data of visible circular dichroism (top left), the integrated intensities of CT1 (top middle), and CT2 (top right); integrated fluorescence (bottom left), polarized fluorescence Ivv (bottom middle), and Ivh (bottom right) measured as a function of cardiolipin CL concentration at pH 6.5 in the absence and presence of NaCl. The NaCl concentrations employed are indicated. The solid lines result from fits and simulations described in the manuscript. Note that the corresponding data points in the different figures were obtained over the same range of CL-to-protein ratio. The error bars of the CT1 data points at the two highest cardiolipin concentrations are asymmetric and indicate rather large errors due to uncertainties with regard to the choice of the baseline.
Equation 1 describes a classical Langmuir-type binding process followed by a conformational change, if the binding affinity K+ was independent of the protein concentration. However, as shown earlier, this cannot be assumed. The consideration of mixing entropies, protein–protein interactions, and electrostatic effects leads to CL dependence of the apparent binding affinity.38 Moreover, the equilibrium between nf+ and f+ shifts toward the latter with increasing CL concentration owing to the decrease of molecular crowding on the liposome surface. Details of how all of these effects contribute to K+ and Kc+ are described in the Section 4 of this paper.
In principle, there are two mechanisms by which the addition of sodium ions can affect cytochrome c–CL interactions. First, Na+ ions can bind to the ionic and functional groups of TOCL/DOPC liposomes, form a so-called Stern layer, and shield electric charges.37 Second, the positive charges of CL cause the sodium ions to accumulate in a layer at the membrane–water interface.37 This leads to a reduction of the membrane’s surface potential and thus of the Gibbs energy of cytochrome c binding. In an earlier paper, we provided evidence for the notion that shielding due to sodium binding reduces Kc rather than K at neutral pH. We invoked such an effect (model 1) to explain why the addition of NaCl at pH 7.4 reduces the spectroscopic amplitudes more than the midpoint positions of spectral response curves.36 However, if the initial binding is electrostatic in nature, the reduction of the membrane surface potential has to be taken into consideration (model 2).38 The algorithms used for both models are described in detail in the Section 4 of this paper to which the reader is referred for a complete overview over the free parameters used in the fits to the data in Figure 7 described below. They are also listed in Table 1.
Table 1. List of Fitting Parameters Obtained from a Global Analysis of Spectroscopic Response Data Indicating Ferricytochrome c–Cardiolipin Interactions at pH 6.5 and Various NaCl Concentrations.
| fitting parameters | obtained values |
|---|---|
| K0+ (M–1) | 5 × 10–9 ± 3 × 10–9 a |
| Kc,high+ | 20 ± 14b |
| ΔΔεnf+ (M–1 cm–1) | –8 ± 19b |
| ΔΔεf+ (M–1 cm–1) | 21 ± 80b |
| fnf+ (M–1 cm–2) | 2.2 × 105 ± 0.3 × 105 b |
| If+ | 5.94 × 103 ± 103 b |
| IVV+·f+ | 37 ± 68b |
| IVH,f+ | 23 ± 68b |
| Kmod | 0.018 ± 0.006b |
| N | 3.1 ± 0.3b |
| KNa (M–1) | 60 ± 10a |
Errors obtained manually.
Errors of fitting parameters. Note that the spectroscopic parameters for fluorescence and circular dichroism cannot be directly compared to the corresponding values for pH 7.4 because they were obtained differently.
In a first step of our analysis, however, we employed model 1 to this set of spectral response data. The spectroscopic response data measured without NaCl were subjected to a nonlinear least-squares fitting (cf. Section 4). Except for K0+, we allowed all parameters listed in Table 1 to vary. For K0+, we initially used the value reported in the earlier study of pH 7.4 binding.21 Next, we simulated the spectroscopic response data for the three employed NaCl concentrations (50, 100, and 150 mM). Differences between predicted and experimentally obtained responses were then minimized by solely varying K0+ and KNa (cf. eqs 7a and 7b in the Section 4). While we could reproduce the response data obtained in the absence of NaCl, none of the data set representing cytochrome c binding to liposomes in the presence of salt could be reproduced. Results did not improve after we allowed K0+, Kc+, and all of the spectroscopic parameters to vary. All spectroscopic response curves predicted for NaCl concentrations of 50 and 100 mM drastically underestimated the observed spectroscopic changes even for very large and physically unreasonable KNa values (up to 200 M–1). The failure of model 1, which works well for the pH 7.4 data, is not surprising since a comparison of corresponding response plots in Figures 7 (pH 6.5) and S6 (pH 7.4) clearly suggests that the addition of NaCl is by far a more efficient inhibitor at pH 6.5.
These negative results led us to conclude that model 1 alone does not account for the observed NaCl-induced changes of spectroscopic response data at pH 6.5. Therefore, we combined models 1 and 2 to reproduce the spectral response data in Figure 7. We opted for an integration of model 1 in our analysis because it makes no sense to assume that the underlying effect, namely, the stabilization of the nf state by the binding of sodium ions to the membrane surface, is suddenly absent at acidic pH. We used a KNa value of 60 M–1, which accounts well for the NaCl-induced spectral changes at pH 7.4 (cf. Supporting Information). Again, we subjected only the response data obtained in the absence of NaCl to a global nonlinear fitting procedure. To this end, we had to keep K0+ constant again. We obtained good fits to all spectroscopic response data obtained without NaCl, including the observed changes of the integrated intensity of the CT2 band. Next, we simulated the spectroscopic response data for the three employed NaCl concentrations (50, 100, and 150 mM). Differences between predicted and experimentally obtained responses were then minimized by solely varying K0+. As shown in Figure 7, this procedure yielded a very satisfactory reproduction of most of the experimental data. To fine-tune the fits to the CT1 and CT2 data, the Kmod constant obtained from the fits to the fluorescence and CD response data had to be multiplied by a factor of 1.2. This small change lies still within the statistical error of Kmod. The spectral responses predicted for the investigated NaCl concentrations of 50 and 100 mM (obtained by solely varying the common K0+ value) reproduce the respective integrated fluorescence, CD, IVV, and IVH quite well. It should be noted that the integrated CT1 intensity of the unbound protein depends on the NaCl concentration. The CT1 recovery at 50 mM NaCl is underestimated, while the agreement between prediction and experiment is satisfactory for 100 mM NaCl. The model still predicts some CT2 intensity at 50 mM NaCl and high CL concentration, which we did not observe, but it does reproduce its absence at 100 mM NaCl. In view of the limitations of the Heimburg–Marsh model discussed below, we find its capability to reproduce the influence of NaCl on spectroscopic response data very satisfactory. All parameters used for this global analysis are listed in Table 1.
One would expect that the ionic concentration in eq 10 is the sum of the NaCl concentration and the concentration of the zwitterionic N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) buffer (25 mM). However, we show in the Supporting Information (Figure S5) that the zwitterionic buffer is not contributing to the ionic strength in the same concentration-dependent manner that NaCl does. The concentration of HEPES buffer did influence the initial binding of cytochrome c to CL-containing liposomes, but the addition of NaCl affected both the initial binding and the amplitude of spectroscopic response. We found that assuming an effective ionic strength contribution of 8 mM for the buffer yields the best global fit and the most reasonable parameter values for Kc,high+. A full consideration of the ionic strength of the buffer produced very high Kc,high+ values in the range of 100. As shown in the Section 3, such values would be physically unreasonable.
To allow for a full comparison of cytochrome c–CL interactions at pH 6.5 and 7.4, we reanalyzed the pH 7.4 response data first with model 1 and subsequently with a combination of models 1 and 2. In our earlier paper, we used a more heuristic version of model 1 for our analysis, where we inferred KNa indirectly from changes of Kc,high.21 As shown in the Supporting Information, model 1 yields a very good reproduction of the pH 7.4 data set (Figure S6; the corresponding parameters are listed in Table S1). In contrast, attempts to fit these data with a combination of models 1 and 2 failed completely (Figure S7). In all cases, where we could fit the response data obtained without salt, we overestimate the inhibition of spectroscopic changes. Our analysis of the pH 7.4 and 6.5 data sets therefore led us to conclude that the underlying binding mechanisms must indeed be different, as suspected.
3. Discussion
3.1. Summary of Results
As reported earlier, a combination of fluorescence, visible circular dichroism, and absorption data suggests that the binding of ferricytochrome c to liposomes prepared with a mixture of 20% TOCL and 80% DOPC at pH 6.5 involves a conformational change of the protein in that native-like structures with a hexacoordinate low-spin heme iron are converted into non-native ones. The subensemble of non-native proteins is a mixture of conformations with penta- and hexacoordinate high-spin heme irons.35 The analysis of the data presented in this study reveal that that the binding process must be electrostatic in nature, where the initial binding step is affected by the reduction of the liposomes’ surface potential by accumulation of Na+ ions. The obtained electrostatic character of the binding process is consistent with the results of Kawai et al., who provided strong experimental evidence for the notion that the so-called L-site of the protein (Figure 1) is involved in binding processes below pH 7.24 This site encompasses the positively charged residues K22 and K27 as well as H33 in the blue foldon region of the protein. The L-site binding is clearly distinct from the binding process at neutral pH (7.4), where we found the initial binding step to be unaffected by the addition of NaCl, which instead stabilizes native- over non-native-like conformations of membrane-bound proteins. The heme iron of the latter is still hexacoordinate with a low-spin heme iron.51 An error analysis of our fit to the L-site data set is given in the Supporting Information.
3.2. Interpretation of Fitting Parameters
The effective binding affinity K+(1 + Kc+) (eqs 1, 3, and 4) depends on the CL concentration. Figure S7 shows K as a function of the CL concentration for [NaCl] = 0 and 100 mM. The displayed curves were calculated with the parameters obtained from the fits to the spectroscopic response data measured at pH 7.4 and 6.5. In the absence of NaCl, the binding affinity values are very similar at low CL concentrations (around 106 M–1), while they diverge at high CL concentrations, where the binding affinity of L-site binding becomes significantly larger (7.3 × 106 vs 1.8 × 106 M–1). The L-site values cannot directly be compared to the binding affinities reported by Kawai et al.24 because the experimental conditions are too different. For the binding of ferricytochrome c to the inner mitochondrial membrane, the observed binding affinities vary between 1.5 × 107 M–1 at pH 6.5 and 7.5 × 106 M–1 at pH 7.4.29 These values are somewhat larger than ours, while the affinity increase at acidic pH is less pronounced. In view of the different systems used in our study and from Kawai et al., we can consider the qualitative agreement between the respective binding affinities as mutually corroborating. It should be mentioned that the fitting procedure carried out in the current study yields K values for pH 7.4, which are ca. 50 times larger than those reported in our earlier paper.21
With regard to the intrinsic binding constant K0+, reference points in the literature are rare. Heimburg and Marsh obtained a value of 1.9 × 10–5 M–1 from their analysis of isotherms reflecting the binding of ferricytochrome c to dioleoyl-phosphatidylglycerol (DOPG) liposomes.38 This value is by 4 orders of magnitude larger than the value listed in Table 1. However, a closer inspection of their text led us to the conclusion that their K0 value already contained van der Waals and entropy corrections. Our earlier calculation revealed that for liposomes with 20% TOCL, these contributions increase the binding constant by nearly 3 orders of magnitude at low CL concentration. That leads us very close to the value reported by Heimburg and Marsh. The difference becomes even smaller if one considers the fact that these authors used liposomes with 100% protein binding DOPG, which can be expected to increase the entropic contributions to the effective binding free energy at low and intermediate CL concentrations.
The plots in Figure S8 clearly reveal the different influence of ions on cytochrome c binding at pH 6.5 and 7.4. At acidic pH, the addition of 100 mM NaCl decreases the effective binding constants by nearly 4 orders of magnitude. The same amount of salt has practically no effect on K at low CL concentration and neutral pH, while it decreases the affinity by a factor of 2 at high CL concentration, which reflects the decrease of the equilibrium constant Kc,high+. Thus, the present analysis confirms the rather limited influence of the ionic strength on the overall binding affinity at pH 7.4.21
The Kc,high+ value of 20 obtained for pH 6.5 is considerably larger than the corresponding value of 2.7 observed for the conformational change at 7.4. These values imply that the f state is predominantly populated at high CL concentration and acidic pH, while it accounts only for 2/3 of the bound proteins at pH 7.4. This difference is illustrated by the plots in Figure S9, which depicts the molar fractions of nf+ and f+ as a function of CL concentration as derived from the above analysis of the pH 6.5 response data.
The difference between the two Kc,high+ values can be rationalized as follows. The binding models used to fit the spectral response data clearly separate the different f states populated at the investigated pH (cf. Section 4). A reaction scheme that takes into account the coexistence of protonated and deprotonated state can be written as follows
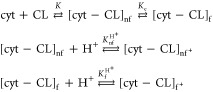 |
R2 |
The binding polynomial f[CL] for this scheme reads as follows
| 2 |
where KnfH+ = 10pKnf and Kf = 10pKf are the apparent association constants associated with the effective pK value representing protonatable residues in the nf/nf+ and f/f+ states, respectively. Note that we used K and Kc without the superscript + in R2 and eq 2 because the binding occurs in the deprotonated state. Since the effective binding affinity K is very similar for both binding processes at low CL concentration (Figure S8), we can assume that the effective increase of Kc solely reflects the difference between the Gibbs energies of nf- and f-state protonation. In this case
| 3 |
where Kc = [f]/[nf]. If one now represents the set of m protonatable groups by an effective pK value and if one further assumes an apparent pK value of 6.9 for the f state,24 one obtains an effective pKnf value of 6.1. This difference between the two pK values could be reasonably well explained by assigning them to the imidazole group of a distal histidine ligand (in the f state) and the corresponding unbound histidine in the nf state. It should be noted that both pK values reflect to some extent the higher hydronium-ion concentration close to the surface of the membrane. Alternatively, one could consider the possibility that two or more protonation processes are involved in the nf → nf+ and f → f+ transitions, which would reduce the apparent pK shift between the two conformations. Overall, this estimation reveals that the obtained difference between the Kc,high values at pH 6.5 and 7.4 makes sense from a thermodynamic point of view.
In Section 2, we emphasize that we adjusted the value for the effective ionic strength of the buffer solution (no NaCl) for the fits to spectroscopic response data in Figure 7 to avoid very large Kc values (Kc,high >100). Based on the rational presented in the preceding paragraphs, such values would be unreasonable from a thermodynamic point of view.
Some parameters which entered our calculation of the electrostatic energy should be briefly discussed. Following Heimburg and Marsh, we assumed an effective protein charge of Z = 3.8 (eqs 10 and 11d). Changes of this value in the upper or lower direction led to increasing discrepancies between simulated and experimental spectroscopic responses for nearly all NaCl concentrations investigated. Regarding the number α of CL molecules attached to membrane-bound cytochrome c, we assumed αnf+ = 2 and af+ = 4.38 A larger number for the latter makes sense since experimental evidence suggests that the terminal helices of the protein are interacting with the membrane surface in the adopted extended structure of the protein. Since our lipid mixture contains only 20% CL, both numbers implicitly require some lipid–lipid demixing to occur upon protein binding.52 The current literature does not provide a clear picture of the effective stoichiometry of cytochrome c–CL interaction; generally reported numbers vary between 1 and 4.20,38,44 The number certainly depends on the protein conformation and on the lipid/protein ratio. Interestingly, variations of αnf+ led to a substantial deterioration of the fits to the response data obtained in the presence of NaCl, which could not be compensated for by changes of K0. On the contrary, changes of αf+ between 2 and 6 did affect the simulations only mildly. Heimburg and Marsh used different α values for low (4.9) and high (11.9) NaCl concentrations for their analysis of cytochrome c binding to liposomes formed with DOPG.38
It should be finally mentioned that the electrostatic theory employed should be considered as an approximation to a more complex reality. The theory of Heimburg and Marsh assumes a homogeneous distribution of charges on the membrane surface.38 This scenario would certainly not apply to the employed TOCL/DOPC mixture if the lipids were ideally mixed. However, multiple lines of evidence suggest that cytochrome c binding causes lipid demixing and thus a clustering of CL around cytochrome c binding sites. Heimburg et al. considered lipid demixing in an extension of the theory utilized in this study.52 These authors showed that in the case of total demixing, the employed approach can still be utilized if one considers the real charge density by modifying the n value in eq 10 to account for the number of available lipids.
The simplifications embedded in our theoretical approach are also reflected by the rather large KNa value of 60 M–1, which emerged from our analysis. Literature values for sodium binding to anionic lipids are generally more than an order of magnitude lower.37 This discrepancy does certainly reflect the fact that we used the bulk concentration of sodium in eq 7b. For a more exact modeling of Na+ binding to CL, we would have to use the actual concentration in double layer of the membrane, which is certainly higher than the bulk concentration.
3.3. Comparing Proposed Binding Models and Binding Affinities
Although there is a constantly growing number of cytochrome c binding studies in the literature, a comprehensive and consistent picture of cytochrome c–anionic lipid interactions that includes the identified binding sites has still to be developed. In the following paragraphs, we compare the properties of proposed binding sites and delineate possible relationships and discrepancies. Since C-site binding is thought to be only active at very low pH for the CL content used in this study,26 we omit it from our discussion, which focuses on binding processes at mildly acidic and neutral conditions.
We first consider the classical A-site binding, which is thought to be completely electrostatic in nature. A large number of studies employing horse heart and yeast cytochrome c mutants have led to the identification of the lysine residues K72, 73, 86, and 87 as the involved amino acid residues.44,53,54 Depending on solution conditions, these residues can become involved to a different extent.54 Kinnunen and co-workers did not analyze their binding isotherms in their earlier studies, but the reported data indicate binding affinities in the range of ca. 5 × 106 M–1.26 Gorbenko et al. employed a sophisticated theoretical approach based on scaled particle theory to subject fluorescence binding isotherms to a global analysis.23 The authors assumed that the phosphate head group of CL is only single-protonated at neutral pH and becomes double-protonated below pH 6.5 for liposomes with 40% CL. For the former, they derived binding constants varying between 7.9 × 105 M–1 at high protein concentration and 4 × 107 M–1 at low protein concentration, while the double-protonated state exhibits a lower affinity (104 and 4.5 × 105 M–1). This result, while properly reflecting molecular crowding effects at high protein concentration, is counterintuitive because one would certainly expect a higher affinity for completely ionized phosphate groups if the binding was solely electrostatic in nature.
The binding constants inferred from the Kinnunen data for single-protonated CL molecules are in the same order of magnitude as the affinities obtained for L-site binding by Kawai et al.24 and in this study, but substantially larger than apparent binding constants observed from studies at neutral pH. The latter were mostly obtained by measuring spectroscopic responses caused by the addition of CL-containing liposomes (large (LUV) and small unilamellar vesicles (SUVs)) to a fixed concentration of ferricytochrome c and lie consistently in the 104–105 M–1 range.20,21,30,44,54 The reason for this discrepancy remains unclear. We wonder, however, whether it might be related to the different protocols. The binding isotherms observed by Rytömaa and Kinnunen were measured by adding an increasing number of cytochrome c to a fixed amount of liposomes.22,26 The lower binding constant, however, emerged from studies where the CL rather than the protein concentration was changed. If this explanation has some merits, it would point toward some nonequilibrium, metastable situation produced by at least one of these protocols. An alternative explanation of the above discrepancy could be that the spectroscopic studies from which the lower binding affinities were derived probe the population of f(E) states rather than the initial binding process. Such a notion is in fact supported by the results of our new global analysis of the pH 7.4 response data, which yielded effective equilibrium constant values between 106 and 2 × 106 M–1. These values exceed our earlier ones by a factor of 20.21 This issue deserves further consideration in the future.
Several lines of evidence and the very nature of the involved amino acid residues strongly suggest that binding should be electrostatic even at neutral pH. The analysis of our binding data presented in this and earlier papers are at odds with this notion. Only the equilibrium between nf and f seems to be governed by electrostatic forces. The latter notion is consistent with the results of FRET studies by Hanske et al., which revealed an ionic strength-dependent equilibrium between compact and extended states.30 The influence of salt on the amplitude of a fluorescence anisotropy change of Zn-substituted cytochrome c caused by its interaction with CL-containing liposomes is rather limited and comparable to our observations. Earlier resonance Raman studies of cytochrome c binding to DOPG liposomes by Oellerich et al. also strongly suggests that cytochrome c binding becomes nonelectrostatic at low lipid/protein ratios.17 The only way to reconcile these results with the composition and the undeniable involvement of A-site binding is actually a combination of interaction with N- and A-sites. The former was observed by O’Brien et al., and it involves the residues F36, G37, T58, W59, and K60.34 We propose that the initial binding process is governed by N-site binding, which could not be inhibited by NaCl addition. In agreement with the structural result of O’Brien et al., this binding process would cause only limited structural changes and maintain a native-like conformation with M80 as the sixth heme ligand. With increasing CL concentration, the protein unfolds due to electrostatic interactions between the protein and the lipid surface. This is in line with the mechanism proposed by Muenzner et al.55 An inhibition of the latter by screening effects (Na+ binding) or selective mutation would reduce the overall binding constant at high CL concentrations. Interestingly, the model is at least qualitatively consistent with the binding data of Rytömaa and Kinnunen,26 who showed that even in the presence of 150 mM NaCl, cytochrome c binding to CL-containing liposomes is not totally inhibited, which one would expect if the binding was entirely electrostatic.
The rather complex picture outlined above is complicated further by the fact that the above-cited studies were performed with different types of liposomes (SUVs and LUVs) and with liposomes containing different contents of anionic lipids. The influence of the latter on cytochrome c binding has been studied only sporadically. The early binding studies of Rytöman and Kinnunen indicate that the effective binding increases with CL content at pH 7, where both A- and L-site binding might overlap.26 Their data also seem to indicate that C-site binding becomes relevant in the physiological pH region for cytochrome c binding to liposomes with very high CL content. This view was modified later by Gorbenko et al., who, based on the results of their analysis of FRET binding isotherms, proposed a model that predicted a switch from binding to totally deprotonated phosphate groups for very low CL content to interactions with single-protonated phosphate groups for CL contents above 20 mol %.23 Hong et al. showed that the mole fraction of their unfolded E conformation increases with increasing CL content of liposomes until it reaches a maximum of 60% at 50 mol % CL.19 We showed in an earlier paper that at neutral pH, the intrinsic affinity Ko for liposomes with 100 mol % CL is slightly larger than the respective values obtained for 20 and 50 mol %.21 Interestingly, both K0 and Kc,high were found to decrease with increasing salt concentration for 100 mol % CL liposomes, which suggests that the first binding step has become at least to some extent electrostatic. This observation and the appearance of a CT2 band at high CL concentration seems to suggest an involvement of L-site binding. Such a scenario is indeed reasonable because it is very likely that the effective pH at the liposome surface decreases with increasing CL content. To avoid any interference from L-site binding, Elmer-Dixon and Bowler have recently used visible CD and W59 fluorescence to probe the binding of yeast iso-1-cytochrome c (wild type and mutants) to liposomes with 100 CL content at pH 8.54 They obtained effective binding affinities in the 105 M–1 range, which is somewhat lower than the corresponding range of effective binding affinities obtained at pH 7.4 with horse heart cytochrome c (3 × 105–4 × 105 M–1).21 If one assumes similar binding properties of horse heart and yeast cytochrome c at neutral and mildly alkaline pH, this difference could indeed be indicative of an L-site influence at pH 7.4. Further investigations of the influence of the CL content on cytochrome c binding and structural conversion are certainly necessary for a development of a model that accounts for all available experimental data.
Our results suggest that at the pH where L-site binding becomes active, the electrostatic driving forces involved overcome other binding mechanisms. This is why, we are able to invoke a comprehensive electrostatic binding analysis at pH 6.5.24 At this point, one may ask the question why electrostatic binding could be so pH-dependent. The answer could lie in the findings of Kawai et al., who showed that at least some lysine residues of the L binding site as well as the two histidines 26 and 33 are protonated below pH 7. This would increase the effective charge of the protein. In an attempt to simulate such an effect, we reduced the effective charge number from 3.8 to 2 and calculated the spectral response curves. This leads to a nearly total breakdown of the binding process. The apparent affinity was lowered by nearly 2 orders of magnitude. Hence, the proposed protonation surface residues at the L-site can explain the onset of electrostatic binding, in particular if one takes into consideration that the A-site residues do not directly contribute to the first binding step.
While the above reasoning explains the onset of L-site binding, it does not account for another observation. One would expect that, in principle, both L- and N/A-site bindings were operative at acidic pH, with a dominance of the former. This was also assumed by Kawai et al., who suggested that the protein might act like a gigantic bidentate ligand by interacting with two vesicles simultaneously, thus triggering vesicle fusion, giving rise to an increased turbidity.24 This binding model is actually at variance with two observations. While we also observed an increased turbidity at low CL concentration, this effect is absent at high lipid-to-protein ratios. If the bidentate binding was operative, it should be the other way around since an increasing number of liposomes would just offer more opportunities for this type of interaction. Moreover, a functioning N/A-site binding would still allow for substantial binding at high NaCl concentrations. This would have led to spectral responses very similar to those obtained at neutral pH, contrary to our observation. Therefore we conclude that for yet unknown reasons, the N/A-site binding becomes less effective at acidic pH.
Kawai et al. used the bidentate binding model to explain the onset of vesicle fusion, but we have observed the same phenomenon at neutral pH and very low CL concentration.36 In line with Oellerich et al.,17 we consider the surface pressure exerted by the high density of surface-bound proteins, which significantly destabilize the membrane. Some lines of evidence suggest that this could lead to a protein penetration into the membrane or even to the transfer into the interior space of the employed liposomes.56 Such an effect has been clearly established for cytochrome binding to gigantic unilamellar vesicles, but regarding its occurrence on LUVs and SUVs, experimental results are somewhat contradictory.55,57,58
Taken together, we propose the following reaction sequence for cytochrome c binding to 20% TOCL/80% DOPC SUVs. At neutral pH, cytochrome binds hydrophobically via the proposed N-site. With increasing CL concentration and decreasing liposome occupation by proteins, this facilitates a conformational change in which an unfolded state resembling the denatured state of ferricytochrome c is stabilized via A-site binding to the surface. This interaction is inhibited in the presence of sodium ions. With decreasing pH, the effective protein surface charge increases and allows L-site binding to become dominant. The unfolded state populated at high CL concentrations becomes stabilized by the protonation of the distal histidine ligand and its concomitant dissociation, while for yet unidentified reasons, N/A-site binding becomes less effective. Obviously, this conformational change stabilizes the unfolded/denatured state of the protein to a significant extent. The similarity between structural changes induced by the protonation of second histidine ligand of liposome-bound and denatured ferricytochrome deserves particular attention. In view of the slightly acidic environment of the inner membrane of mitochondria, the L-site binding and its inhibition by salt might be the processes that prevent conformational changes of the protein and the acquisition of peroxidase activity at normal conditions. Experiments exploring the peroxidase activity of membrane-bound cytochrome c at various conditions are underway in our laboratory.
3.4. Structural Aspects
Multiple lines of evidence suggest that upon binding, the native M80 ligand is replaced by a histidine residue,59 but it is then displaced at mildly acidic pH owing to the protonation of its imidazole group. This process is very much reminiscent of what Roder, Rousseau, and their respective co-workers observed for ferricytochrome c under denaturing conditions.46,60−62 One is therefore tempted to suggest that the acidic and neutral denatured states obtained in the presence of, e.g., urea should be very similar to the f+ and f states inferred from our data. This notion is strongly corroborated by the observation that the rotational strengths of the respective CD Soret band spectra are of comparable magnitude, which suggest very similar heme environments. The differences between the CD bands of denatured ferricytochrome c in solution and on the membrane surface can be related to a modified Stark effect caused by the electric field on the latter.
The observed noncoincidence between CD and absorption band positions deserves some further consideration. As outlined above, the noncoincidence value changes by ca. 800 cm–1 upon cytochrome c binding to the liposome surface (Figure S2). A comparison of the lipid concentration dependence of the noncoincidence effect and of the molar fractions of nf+ and f+ reveals that the change of the spectroscopic noncoincidence occurs in a CL concentration region, where f+ becomes mostly populated. Hence, the switch of the former can be clearly related to the conformational change on the membrane surface. As shown earlier, the electrical charges on the membrane surface have to produce an electric field of ca. 2 MV cm–1 to produce the change of the noncoincidence at pH 7.4.21 The noncoincidence shift obtained at pH 6.5 is slightly larger, but since the electric field change scales with the square root of the noncoincidence change, the difference does not matter. The estimated value of the electric field seems to be large compared to what one would infer from the reported electrostatic profiles (ca. 0.5–0.7 MV cm–1).63 However, if the protein penetrates the membrane in its nf/nf+ state, then it would move the heme group in close proximity to the surface charges. This interpretation is corroborated by the results of Domanov et al.,64 who analyzed fluorescence quenching experiments with a donor fluorophore in the membrane and the heme group as acceptor. They found that the protein increases its penetration into the membrane with a decreasing surface density of proteins (high CL concentrations in our case) by ca. 10 Å. This substantial change would increase the electric field in the heme pocket. If the heme group becomes positioned close to the membrane–solvent interface, then it might be subjected to electrostatic potentials of several hundred millivolts, which decay on a length scale of a few angstroms. The corresponding electric field strengths would lie in the range of several tens of MV cm–1, comparable to the intrinsic electric field of the protein.65 Therefore, the observed noncoincidence effect can be considered as reflecting the electric field probed by the heme group of a denatured protein that has somewhat penetrated into the outer membrane.
4. Materials and Methods
The methods used are the same as in the Supporting Information of our most recent publication, but are presented again here with any minor changes for the convenience of the reader.
4.1. Experimental Methods
4.1.1. Protein Preparation
Horse heart cytochrome c (Sigma-Aldrich Co., St. Louis, MO) was dissolved in 25 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) buffer. Potassium ferricyanide was added to the solution in minimal amounts as an oxidizing agent, the protein was allowed to oxidize at 5 °C for 1 h, and then the pH was adjusted to 7.0 before running the solution over a Sephades G15 column (GE Healthcare). The sample was collected and readjusted to pH 7.0. The sample was collected and readjusted to pH 7.0. The concentration and oxidation state were confirmed by scanning the Soret and Q band regions with UV/visible absorption.
4.1.2. Liposome Preparation and Dynamic Light Scattering Experiments
1,1′,1,2′-Tetraoleyolcardiolipin (TOCL) and 1,2-deoleyol-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids, Birmingham, AL) were dissolved in a 2:1 mixture of chloroform and methanol. A protocol for liposome preparation by Hanske et al. was followed.30 Rotary evaporation was used to remove the solvent at room temperature. This was followed by further drying overnight in a vacuum desiccator. The lipid film was rehydrated in a sufficient amount of 25 mM HEPES buffer to reach the desired liposome concentration. The lipid mixture was then sonicated in an ice bath at 100 W for 1 h 30 min before being centrifuged at 12 000 rpm for 50 min to remove any ultrasonicator tip contamination. The supernatant was decanted and stored overnight in a refrigerator to equilibrate. Liposomes were always stored in the refrigerator under nitrogen gas to avoid oxidation. After equilibrating, the size distribution of the liposomes was measured at room temperature in a 0.2 cm path length quartz cuvette using a Horiba Lb-500 dynamic light scattering particle size analyzer (Edison, NJ).
4.1.3. Sample Preparation
Solutions for cytochrome c–CL binding studies were made to span a series of total lipid-to-protein ratios from 0 (no liposomes added) to 200. The corresponding CL-to-protein ratios are 0–40. Samples for fluorescence measurements had a protein concentration of 5 μM; samples for CD measurements had a protein concentration of 10 μM; and samples for charge-transfer measurements had a protein concentration of 25 μM. Samples were stored in a refrigerator for about 25 min to allow equilibration before taking spectroscopic measurements. All CL concentration values refer to the outer leaflet of the membrane. We assumed that CL is equally distributed among the inner and outer leaflets of the produced liposomes.
For experiments involving the chemical denaturing of cytochrome c, ferricytochrome c was diluted in a solution of 25 mM HEPES buffer and 8 M urea to the desired concentration. The pH of these solutions was adjusted to 5.5 to ensure that the denatured protein adopts a conformation with a hexacoordinate, high-spin heme iron.
4.1.4. Circular Dichroism Measurements
Spectra were measured with a JASCO J810 spectropolarimeter purged with nitrogen gas. The quartz cell (Crystal Laboratories, Garfield, NJ) had a 0.2 cm path length. The spectra spanned 350–600 nm at a scan speed of 500 nm min–1, a data pitch of 0.5 nm, a bandwidth of 5 nm, and a response time of 1 s. A Peltier solid-state heating and cooling module held the cell at 20 °C while five spectra were collected and averaged. The spectra were all background-corrected using the JASCO spectral analysis program.
4.1.5. Fluorescence Measurements
Emission spectra were measured at room temperature in an ICL quartz cell with a path length of 1 cm using a PerkinElmer LS55 Luminescence Spectrometer. The spectra were recorded from 300 to 550 nm with an excitation wavelength of 293 nm and a scan speed of 200 nm min–1. Excitation and emission slit widths of 5 and 2.5 nm, respectively, were used.
4.1.6. UV/visible Absorption Measurements
Spectra were measured at room temperature using a 1 cm quartz cuvettes with a PerkinElmer Lambda 35 UV/visible spectrometer. Spectra were measured between 350–700 and 600–700 nm for charge-transfer measurements with an excitation slit width of 2.5 nm. Charge-transfer bands were decomposed using our fitting program Multifit with sub-bands positioned at 14 020, 14 400, and 14 700 cm–1 for CT1 and 15 467, 16 001, and 16 285 cm–1 for CT2, in agreement with previous results.66−68
4.1.7. Resonance Raman Spectroscopy
The Raman spectrum was obtained by using a Renishaw RM-1000 Ramascope with a BH-2 confocal Raman microscope and the 442 nm excitation of a HeCd laser (Kimmon) with a final laser power of approximately 7.6 mW at the sample. The spectrometer was calibrated using a silicon wafer by setting the silicon peak to 520.0 cm–1. Multiple spectra for each sample were taken (and later averaged in Multifit)16 after relocating the focal point in between subsequent spectra to reduce any effects from prolonged laser exposure.
4.2. Theoretical Background
4.2.1. CL Dependence of Equilibrium Constants
The theory presented below combines several approaches, which have been presented by different authors in earlier papers.21,52 We think that presenting them briefly in this paper serves its readability.
As indicated in Section 3, several physical effects can lead to a CL dependence of the equilibrium constants K+ and Kc+ in eq 2. First, we take into account that molecular crowding at low lipid/protein ratios (i.e., high occupancy of liposomes with proteins) and mixing entropy contributions can affect the apparent binding affinity. The van der Waals gas approach of Heimburg and Marsh takes these effects into account and leads to the following equation for K+(38)
| 4 |
where K0+ is the intrinsic binding constant for the protonated state, σp is the number of occupied CL/liposome, NL is the number of CL lipids per liposome, and Kagg+ is an aggregation constant accounting for the aggregation of proteins at surface crowding. In the limit of high CL/protein ratios, CLb is small compared to NL and approaches K0+NL
Second, we assumed that Kc+ depends on the density of bound cytochromes and thus on the CL concentration. The rationale behind that approach is that molecular crowding effects prevent the protein from unfolding at high occupancy. Hints in that direction can be inferred from the work of the Pletneva group and recent papers of Bowler and associates.19,30,44 Following our earlier approach,21 we describe the CL dependence of Kc by a sigmoidal function
| 5 |
where f([CL]) is the phase transition between two states of the membrane shown by a Hill-type equation
| 6 |
where χb is the mole fraction of bound cytochrome c, Kmod denotes a thermodynamic constant that reflects the free energy of the transition between the two phases of the membrane, and n is the Hill coefficient.
4.2.2. Electrostatic Screening
The spectral response curves measured at pH 7.4 and 6.5 are both significantly affected by the addition of NaCl. One might therefore be tempted to conclude that cytochrome c binding to CL-containing liposome interactions is governed by attractive electrostatic interactions. However, a closer look at the data does not corroborate this notion. At pH 7.4, the addition of NaCl caused the amplitudes of spectroscopic changes to decrease without being accompanied by a drastic change of the midpoint. However, at pH 6.5, the addition of salt decreases the amplitude and shifts the midpoint out of the concentration range used for our experiments.
In our earlier paper, we rationalized the influence of NaCl on the spectroscopic response curves with a screening model, where we assumed that sodium cations bind to CL-containing liposomes in Langmuir-type manner.21 Sodium ions not only bind to the phosphate groups of CL, but they can also interact with carbonyl groups and with the zwitterionic head groups of DOPC.38 Hong et al. suggested that unfolded, extended states of cytochrome c on CL-containing liposomes are stabilized by electrostatic interactions between the decoupled, terminal helices and the liposome surface.19 Cation binding can screen the surface charges and thus destabilize the unfolded states. Heuristically, we account for this effect by the following formalism
| 7a |
| 7b |
Equations 7a and 7b describe a decrease of the equilibrium constant Kc,high+ with increasing sodium concentration. The binding of sodium to the liposome surface is accounted for by a Langmuir-type equation with an apparent binding constant KNa. This screening model is termed model 1 throughout this paper.
In the second step, we consider the possibility that the initial binding step is driven by electrostatic forces (model 2), in line with the characteristics of the L-site identified by Kawai et al. To this end, we utilize the purely electrostatic double-layer theory of Heimburg and Marsh.38 In this theory, the surface potential produced by negatively charged head groups is reduced upon addition of salt owing to the accumulation of cations in layers above the membrane surface. This leads to a reduction of the electrostatic contribution to the binding free energy. The theory assumes that head group charges are continuously distributed over the membrane surface. The electrostatic contribution to the apparent binding constant is written as
| 8 |
where fvdW is the van der Waals gas term in eq 4, kB is the Boltzmann constant, and T is the temperature (in Kelvin). The electrostatic binding energy function ΔFel(σp) is shown below
| 9 |
where the first term is the surface energy with σp number of ligands, the second term is the surface energy with no ligands, and the last term is the energy of the ligand in solution. By invoking the high potential limit approximation of Jähnig,69 Heimburg and Marsh38 showed that the first derivative of the free-energy function in eq 8 can be written as follows
| 10 |
where Z is the ligand charge, α is the number of CL per bound cytochrome c, n is the number of binding sites, and c0 is the effective ionic strength of the buffer solution. The parameters Λi are written as
| 11a |
| 11b |
| 11c |
| 11d |
where ε denotes the dielectric constant of the solution, NA is the Avogardo constant, e is the elementary charge, r0 is the radius of the ligand, and κ is the Debye length, which can be calculated with
| 12 |
To utilize the above formalism for fitting experimentally obtained response data, the concentrations of unoccupied CL and unbound cytochrome c have to be calculated for each CL concentration with an iterative procedure described in the Supporting Information of our earlier paper.21
4.2.3. Fitting Procedure
For all nonlinear fits to experimentally obtained spectroscopic response data, we used the module Nlinfit of the Matlab 2018 program package. Generally, we subjected only the response data measured in the absence of NaCl to the fitting procedure and used the obtained parameter values to predict the spectral response for different NaCl concentrations. Predictions were optimized by varying the equilibrium constant K0+ (models 1 and 2) and KNa (model 1). Generally, Kc,high, Kmod, n, and the snf/sf parameters (eq 1) If,nf+, IVV,nf+, IVH,nf+, ΔΔεnf+, ΔΔεf+, εCT1,nf+, and εCT2,f+ were used as free parameters for models 1 and 2. For the fluorescence parameters of the nf state, we assumed If,nf+ = If,0, IVV,nf+ = IVV,0 and IVH,nf+ = IVH,0. All s0 values were determined experimentally. Furthermore, we assumed that εCT1,f+ and εCT2,nf+ are zero.
Acknowledgments
R.K. acknowledges the receipt of a Maryanoff fellowship for summer research at the Department of Chemistry, Drexel University.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02574.
Error analysis of our fits, reanalysis of spectroscopic response data reflecting the binding of ferricytochrome c to liposomes with 20% TOCL/80% DOPC, plots of effective equilibrium constants and mole fractions of cytochrome c conformations as a function of cardiolipin concentration, visualization of liposome binding-induced changes of the noncoincidence between absorption and CD spectra in the Soret band region, and the cardiolipin concentration dependence of CD difference spectra (PDF)
Author Present Address
† College of Medicine, The Ohio State University, 370 W 9th Avenue, Columbus, Ohio 43210, United States (D.M.).
The authors declare no competing financial interest.
Supplementary Material
References
- Alvarez-Paggi D.; Hannibal L.; Castro M. A.; Oviedo-Rouco S.; Demicheli V.; Tórtora V.; Tomasina F.; Radi R.; Murgida D. H. Multifunctional Cytochrome c Learning New Tricks from an Old Dog. Chem. Rev. 2017, 117, 13382–13460. 10.1021/acs.chemrev.7b00257. [DOI] [PubMed] [Google Scholar]
- Adman E. T. A Comparison of the Structures of Electron Transfer Proteins. Biochim. Biophys. Acta, Rev. Bioenerg. 1979, 549, 107–144. 10.1016/0304-4173(79)90012-0. [DOI] [PubMed] [Google Scholar]
- Hannibal L.; Tomasina F.; Capdevila D. A.; Demicheli V.; Tórtora V.; Alvarez-Paggi D.; Jemmerson R.; Murgida D. H.; Radi R. Alternative Conformations of Cytochrome c: Structure, Function and Detection. Biochemistry 2016, 55, 407–428. 10.1021/acs.biochem.5b01385. [DOI] [PubMed] [Google Scholar]
- Schweitzer-Stenner R. Relating the Multi-Functionality of Cytochrome to Membrane Binding and Structural Conversion. Biophys. Rev. 2018, 10, 1151–1185. 10.1007/s12551-018-0409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkstone G. G.; Cooper C. E.; Svistunenko D. A.; Wilson M. T. EPR and Optical Spectroscopy Studies of Met80X Mutants of Yeast Ferricytochrome c. Models for Intermediates in the Alkaline Transition. J. Am. Chem. Soc. 2005, 127, 92–99. 10.1021/ja045719b. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Wang X. Cytochrome C-Mediated Apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Kagan V. E.; Bayir H. A.; Belikova N. A.; Kapralov O.; Tyurina Y. Y.; Tyurin V. A.; Jian J.; Stoyanovsky D. A.; Wipf P. M.; Kochanek P. M.; et al. Cytochrome c/Cardiolipin Relations in Mitochondria: A Kiss of Death. Free Radicals Biol. Med. 2009, 46, 1439–1453. 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajhede M.; Schuller D. J.; Heriksen A.; Smith A. T.; Poulos T. L. Crystal Structure Determination of Classical Horseradish Peroxidase at 2.15 Å Resolution. Nat. Struct. Biol. 1997, 4, 1032–1038. 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- Huang Q.; Szigeti V.; Fidy J.; Schweitzer-Stenner R. Structural Disorder of Native Horseradish Peroxidase Probed by Resonance Raman and Low Temperature Optical Absorption Spectroscopy. J. Phys. Chem. B 2003, 107, 2822–2830. 10.1021/jp026935e. [DOI] [Google Scholar]
- Jentzen W.; Ma M.-G.; Shelnutt J. A. Conservation of the Conformation of the Porphyrin Macrocycle in Hemoproteins. Biophys. J. 1998, 74, 753–763. 10.1016/S0006-3495(98)74000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel L. V.; Ye T.; Bowman S. E. J.; Levin B. D.; Hahn M. A.; Russell B. S.; Elliott S. J.; Bren K. L. Heme Attachment Motif Mobility Tunes Cytochrome c Redox Potential. Biochemistry 2007, 46, 11753–11760. 10.1021/bi701177j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q.; Schweitzer-Stenner R. Non-Planar Heme Deformations and Excited State Displacements in Horseradish Peroxidase Detected by Resonance Raman Spectroscopy at Soret Excitation. J. Raman Spectrosc. 2005, 36, 363–375. 10.1002/jrs.1326. [DOI] [PubMed] [Google Scholar]
- Howes B. D.; Schiødt C. B.; Welinder K. G.; Marzocchi M. P.; Ma J.-G.; Zhang J.; Shelnutt J. A.; Smulevich G. The Quantum Mixed-Spin Heme State of Bareley Peroxidase C: A Paradigm for Class III Peroxidases. Biophys. J. 1999, 77, 478–492. 10.1016/S0006-3495(99)76905-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J. S.; Kon H.; Levin I. W. Low-Temperature Electron Paramagnetic Resonance Study of the Ferricytochrome c-Cardiolipin Complex. Biochemistry 1987, 26, 2312–2314. 10.1021/bi00382a036. [DOI] [PubMed] [Google Scholar]
- Heimburg T.; Hildebrandt P.; Marsh D. Cytochrome c-Lipid Interactions Studied by Resonance Raman and 31P NMR Spectroscopy. Correlation between the Conformational Change of the Protein and the Lipid Bilayer. Biochemistry 1991, 30, 9084–9089. 10.1021/bi00101a025. [DOI] [PubMed] [Google Scholar]
- Hildebrandt P.; Stockburger M. Cytochrome c at Charged Interfaces: 1. Conformational and Redox Equilibria at the Electrode/Electrolyte Interface Probed by Surface Enhanced Resonance Raman Spectroscopy. Biochemistry 1989, 28, 6710–6721. 10.1021/bi00442a026. [DOI] [PubMed] [Google Scholar]
- Oellerich S.; Lecomte S.; Paternostre M.; Heimburg T.; Hildebrandt P. Peripheral and Integral Binding of Cytochrome c to Phospholipids Vesicles. J. Phys. Chem. B 2004, 108, 3871–3878. 10.1021/jp036799t. [DOI] [Google Scholar]
- Kapralov A. A.; Kurnikov I. V.; Vlasova I. I.; Belikova N. A.; Tyurin V. A.; Basova L. V.; Zhao Q.; Tyurina Y. Y.; Jiang J.; Bayir H.; et al. The Hierachy of Structural Transitions Induced in Cytochrome c by Anionic Phospholipids Determines Its Peroxidase Activation and Selective Peroxidation during Apoptosis in Cells. Biochemistry 2007, 46, 14232–14244. 10.1021/bi701237b. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Muenzner J.; Grimm S. K.; Pletneva E. V. Origin of the Conformational Heterogeneity of Cardiolipin-Bound Cytochrome C. J. Am. Chem. Soc. 2012, 134, 18713–18723. 10.1021/ja307426k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi F.; Fiorucci L.; Patriarca A.; Lauceri R.; Ferri T.; Coletta M.; Santucci R. Insights into Cytochrome C-Cardiolipin Interaction. Role Played by Ionic Strength. Biochemistry 2008, 47, 6928–6935. 10.1021/bi800048v. [DOI] [PubMed] [Google Scholar]
- Pandiscia L. A.; Schweitzer-Stenner R. Coexistence of Native-Like and Non-Native Cytochrome c on Anionic Lipsomes with Different Cardiolipin Content. J. Phys. Chem. B 2015, 119, 12846–12859. 10.1021/acs.jpcb.5b07328. [DOI] [PubMed] [Google Scholar]
- Rytömaa M.; Mustonen P.; Kinnunen P. K. J. Reversible, Nonionic and pH-Dependent Association of Cytochrome c with Cardiolipin- Phosphatidylcholine Liposomes. J. Biol. Chem. 1992, 267, 22243–22248. [PubMed] [Google Scholar]
- Gorbenko G. P.; Molotkovsky J. G.; Kinnunen P. K. J. Cytochrome c Interaction with Cardiolipin/Phosphatidylcholine Model Membranes: Effect of Cardiolipin Protonation. Biophys. J. 2006, 90, 4093–4103. 10.1529/biophysj.105.080150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai C.; Prado F. M.; Nunes G. L. C.; Di Mascio P.; Carmona-Ribeiro A. M.; Nantes I. L. pH-Dependent Interaction of Cytochrome c with Mitochondrial Mimetic Membranes: The Role of an Array of Positively Charged Amino Acid Residues. J. Biol. Chem. 2005, 280, 34709–34717. 10.1074/jbc.M412532200. [DOI] [PubMed] [Google Scholar]
- Rytömaa M.; Kinnunen P. K. J. Reversibility of the Binding of Cytochrome c to Liposomes. J. Biol. Chem. 1995, 270, 3197–3202. 10.1074/jbc.270.7.3197. [DOI] [PubMed] [Google Scholar]
- Rytömaa M.; Kinnunen P. K. J. Evidence for Two Distinct Acidic Phospholipid-Binding Sites in Cytochrome C. J. Biol. Chem. 1994, 269, 1770–1774. [PubMed] [Google Scholar]
- Soffer J.The Folded, Partially Folded Misfolded, and Unfolded Conformations of Cytochrome c Probed by Optical Spectroscopy; Drexel University, 2013. [Google Scholar]
- Porcellli A. M.; Ghelli A.; Zanna C.; Pinto P.; Rizzulo R.; Rugollo M. pH Difference across the Outer Mitochondrial Membrane Measured with a Green Fluorescent Protein Mutant. Biochem. Biophys. Res. Commun. 2005, 326, 799–804. 10.1016/j.bbrc.2004.11.105. [DOI] [PubMed] [Google Scholar]
- Kawai C.; Pessoto F. S.; Rodrigues T.; Mugnol K. C. U.; Tórtora V.; Castro L.; Milícchio V. A.; Tersariol I. L. S.; Di Mascio P.; Radi R.; et al. pH-Sensitive Binding of Cytochrome c to the Inner Mitochondrial Membrane. Implications for the Participation of the Protein in Cell Respiration and Apoptosis. Biochemistry 2009, 48, 8335–8342. 10.1021/bi9006463. [DOI] [PubMed] [Google Scholar]
- Hanske J.; Toffey J. R.; Morenz A. M.; Bonilla A. J.; Schiavoni K. H.; Pletneva E. V. Conformational Properties of Cardiolipin-Bound Cytochrome C. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 125–130. 10.1073/pnas.1112312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyshka D.; Pandiscia L. A.; Schweitzer-Stenner R. Cardiolipin Containing Liposomes Are Fully Ionized at Physiological pH. An FT-IR Study of Phosphate Group Ionization. Vib. Sprectrosc. 2014, 75, 86–92. 10.1016/j.vibspec.2014.10.003. [DOI] [Google Scholar]
- Sathappa M.; Alder N. N. The Ionization Properties of Cardiolipin and Its Variants in Model Bilayers. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1362–1372. 10.1016/j.bbamem.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijman E. E.; Swim L. A.; Garber Z. T.; Tyurina Y. Y.; Bayir H.; Kagan V. R. Magic Angle Spinning 31NMR Spectroscopy Reveals Two Essentially Identical Ionization States for the Cardiolipin Phosphates in Phospholipid Liposomes. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 61–68. 10.1016/j.bbamem.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien E. S.; Nucci N. V.; Fuglestad B.; Tommos C.; Wand A. J. Defining the Apoptopic Trigger: The Interaction of Cytochrome c and Cardiolipin. J. Biol. Chem. 2015, 290, 30879–30887. 10.1074/jbc.m115.689406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milorey B.; Malyshka D.; Schweitzer-Stenner R. pH Dependence of Ferricytochrome c Conformational Transitions during Binding to Cardiolipin Membranes: Evidence for Histidine as the Distal Ligand at Neutral pH. J. Phys. Chem. Lett. 2017, 8, 1993–1998. 10.1021/acs.jpclett.7b00597. [DOI] [PubMed] [Google Scholar]
- Pandiscia L. A.; Schweitzer-Stenner R. Coexistence of Native-like and Non-Native Partially Unfolded Ferricytochrome c on the Surface of Cardiolipin-Containing Liposomes. J. Phys. Chem. B. 2015, 119, 1334–1349. 10.1021/jp5104752. [DOI] [PubMed] [Google Scholar]
- Khomich D. A.; Nesterenko A. M.; Kostritskii A. Y.; Kondinskaia D. A.; Ermakov Y.; Gurtovenko A. A. Independent Adsorption of Monovaletn Cations and Cationic Polymers at PE/PG Lipid Membranes. J. Phys.: Conf. Ser. 2017, 794, 012010 10.1088/1742-6596/794/1/012010. [DOI] [Google Scholar]
- Heimburg T.; Marsh D. Protein Surface-Distribution and Protein-Protein Interactions in the Binding of Peripheral Proteins to Charged Lipid Membranes. Biophys. J. 1995, 68, 536–546. 10.1016/S0006-3495(95)80215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer-Stenner R. The Internal Electric Field in Cytochrome C Explored by Visible Electronic Circular Dichroism Spectroscopy. J. Phys. Chem. B 2008, 112, 10358–10366. 10.1021/jp802495q. [DOI] [PubMed] [Google Scholar]
- Manas E. S.; Wright W. W.; Sharp K. A.; Friedrich J.; Vanderkooi J. M. The Influence of Protein Environment on the Low Temperature Electronic Spectroscopy of Zn-Substituted Cytochrome C. J. Phys. Chem. B 2000, 104, 6932–6941. 10.1021/jp0005975. [DOI] [Google Scholar]
- Rasnik I.; Sharp K. A.; Fee J. A.; Vanderkooi J. M. Spectral Analysis of Cytochrome c: Effect of Heme Conformation, Axial Ligand, Peripheral Substituents, and Local Electric Fields. J. Phys. Chem. B 2001, 105, 282–286. 10.1021/jp002656k. [DOI] [Google Scholar]
- Pielak G. J.; Oikawa K.; Mauk A. G.; Smith M.; Kay C. M. Elimination of the Negative Soret Cotton Effect of Cytochrome c by Replacement of the Invariant Phenylalanine Using Site-Directed Mutagenesis. J. Am. Chem. Soc. 1986, 108, 2724–2727. 10.1021/ja00270a035. [DOI] [Google Scholar]
- Pandiscia L. A.; Schweitzer-Stenner R. Salt as a Catalyst in the Mitochondria: Returning Cytochrome c to Its Native State after It Misfolds on the Surface of Cardiolipin Containing Membranes. Chem. Commun. 2014, 50, 3674–3676. 10.1039/C3CC48709A. [DOI] [PubMed] [Google Scholar]
- Elmer-Dixon M. M.; Bowler B. E. Site A-Dediated Partial Unfolding of Cytochrome c on Cardiolipin Vesicles Os Species-Dependent and Does Not Require Lys72. Biochemistry 2017, 56, 4830–4839. 10.1021/acs.biochem.7b00694. [DOI] [PubMed] [Google Scholar]
- Hagarman A.; Duitch L.; Schweitzer-Stenner R. The Conformational Manifold of Ferricytochromec Explored by Visible and Far-UV Electronic Circular Dichroism Spectroscopy. Biochemistry 2008, 47, 9667–9677. 10.1021/bi800729w. [DOI] [PubMed] [Google Scholar]
- Elöve G. A.; Bhuyan A. K.; Roder H. Kinetic Mechanism of Cytochrome c Folding: Involvement of the Heme and Its Ligands. Biochemistry 1994, 33, 6925–6935. 10.1021/bi00188a023. [DOI] [PubMed] [Google Scholar]
- Hsu M. C.; Woody R. W. The Origin of the Heme Cotton Effects in Myoglobin and Hemoglobin. J. Am. Chem. Soc. 1971, 93, 3515–3525. 10.1021/ja00743a036. [DOI] [PubMed] [Google Scholar]
- Kiefl C.; Sreerama N.; Haddad R.; Sun L.; Jentzen W.; Lu Y.; Qiu Y.; Shelnutt J. A.; Woody R. W. Heme Distortions in Sperm-Whale Carbonmonoxy Myoglobin: Correlations between Rotational Strengths and Heme Distortions in MD-Generated Structures. J. Am. Chem. Soc. 2002, 124, 3385–3394. 10.1021/ja011961w. [DOI] [PubMed] [Google Scholar]
- Woody R. W.; Pescitelli G. The Role of Heme Chirality in the Circular Dichroism of Heme Proteins. Z. Naturforsch. 2014, 69, 313–325. 10.5560/zna.2014-0004. [DOI] [Google Scholar]
- McKnight J.; Cheesman M. R.; Thomson A. J.; Miles J. S.; Munro A. W. Identification of Charge Transfer Transitions in the Optical Spectrum of Low-Spin Ferrocytochrome P-450 Bacillus Megaterium. Eur. J. Biochem. 1993, 213, 683–687. 10.1111/j.1432-1033.1993.tb17808.x. [DOI] [PubMed] [Google Scholar]
- Malyshka D.; Schweitzer-Stenner R. Ferrocyanide-Mediated Photoreduction of Ferricytochrome C Utilized to Selectively Probe Non-Native Conformations Induced by Binding to Cardiolipin-Containing Liposomes. Chem. – Eur. J. 2017, 23, 1151–1156. 10.1002/chem.201604992. [DOI] [PubMed] [Google Scholar]
- Heimburg T.; Angerstein A.; Marsh D. Binding of Peripheral Proteins to Mixed Lipid Membranes: Effect of Lipid Demixing upon Binding. Biophys. J. 1999, 76, 2575–2586. 10.1016/S0006-3495(99)77410-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi F.; Howes B. D.; Droghetti E.; Polticelli F.; Piro M. C.; Di Pierro D.; Fiorucci L.; Coletta M.; Smulevich G.; Santucci R. Role of Lysines in the Cytochrome c - Cardiolipin Interactions. Biochemistry 2013, 52, 4578–4588. 10.1021/bi400324c. [DOI] [PubMed] [Google Scholar]
- Elmer-Dixon M. M.; Bowler B. E. Electrostatic Constituents of the Interaction of Cardiolipin with Site A of Cytochrome C. Biochemistry 2018, 57, 5683–5695. 10.1021/acs.biochem.8b00704. [DOI] [PubMed] [Google Scholar]
- Muenzner J.; Toffey J. R.; Hong Y.; Pletneva E. V. Becoming a Peroxidase: Cardiolipin-Induced Unfolding of Cytochrome C. J. Phys. Chem. B 2013, 117, 12878–12886. 10.1021/jp402104r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales P. A.; Bergstrom C. L.; Geerts N.; Groves J. T.; Vanderlick T. K. Single Vesicle Observations of the Cardiolipin-Cytochrome c Interaction: Introduction of Membrane Morphology Changes. Langmuir 2011, 27, 6107–6115. 10.1021/la104924c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A.; Hoop C. L.; DeLucia M.; Kodali R.; Kagan V. E.; Ahn J.; van der Wei P. C. A. Structural Changes and Proapoptopic Peroxidase Activity of Cardiolipin-Bound Mitochondrial Cytochrome C. Biophys. J. 2015, 109, 1873–1884. 10.1016/j.bpj.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusova V. M.; Gorbenko G. P.; Molotkovsky J. G.; Kinnunen P. K. J. Cytochrome c-Lipid Interactions: New Insight from Resonance Energy Transfer. Biophys. J. 2010, 99, 1754–1763. 10.1016/j.bpj.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi F.; Milazzo L.; Howes B. D.; Piro M. C.; Florucci L.; Polticelli F.; Ascenzi P.; Coletta M.; Smulevich G.; Santucci R. The Key Role Played by Charge in the Interaction of Cytochrome c with Cardiolipin. J. Biol. Inorg. Chem. 2017, 22, 19–29. 10.1007/s00775-016-1404-5. [DOI] [PubMed] [Google Scholar]
- Colón W.; Elöve G. A.; Walken L. P.; Sherman F.; Roder H. Side Chain Packing of the N- and C-Terminal Helices Plays a Critical Role in the Kinetics of Cytochrome c Folding. Biochemistry 1996, 35, 5538–5549. 10.1021/bi960052u. [DOI] [PubMed] [Google Scholar]
- Colón W.; Roder H. Kinetic Intermediates in the Formation of the Cytochrome c Molten Globule. Nat. Struct. Biol. 1996, 3, 1019–1025. 10.1038/nsb1296-1019. [DOI] [PubMed] [Google Scholar]
- Yeh S.-R.; Takahashi S.; Fan B.; Rousseau D. L. Ligand Exchange during Cytochrome c Folding. Nat. Struct. Mol. Biol. 1997, 4, 51–56. 10.1038/nsb0197-51. [DOI] [PubMed] [Google Scholar]
- Cevc G. Membrane Electrostatics. Biochim. Biophys. Acta, Rev. Biomembr. 1990, 1031, 311–382. 10.1016/0304-4157(90)90015-5. [DOI] [PubMed] [Google Scholar]
- Domanov Y. A.; Molotkovsky J. G.; Gorbenko G. P. Coverage-Dependent Changes of Cytochrome c Transverse Location in Phospholipid Membranes Revealed by FRET. Biochim. Biophys. Acta, Biomembr. 2005, 1716, 49–58. 10.1016/j.bbamem.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Manas E. S.; Vaderkooi J. M.; Sharp K. A. The Effects of Protein Environment on the Low Temperature Electronic Spectroscopy of Cytochrome c and Microperoxidase-11. J. Phys. Chem. B 1999, 103, 6334–6348. 10.1021/jp9908552. [DOI] [Google Scholar]
- Dragomir I.; Hagarman A.; Wallace C.; Schweitzer-Stenner R. Optical Band Splitting and Electronic Perturbations of the Heme Chromophore in Cytochrome c at Room Temperature Probed by Visible Electronic Circular Dichroism Spectroscopy. Biophys. J. 2007, 92, 989–998. 10.1529/biophysj.106.095976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbaro D.; Hagarman A.; Soffer J.; Schweitzer-Stenner R. The pH Dependence of the 695 nm Charge Transfer Band Reveals the Population of an Intermediate State of the Alkaline Transition of Ferricytochrome c at Low Ion Concentrations. Biochemistry 2009, 48, 2990–2996. 10.1021/bi802208f. [DOI] [PubMed] [Google Scholar]
- Verbaro D.; Hagarman A.; Kohli A.; Schweitzer-Stenner R. Microperoxidase 11: A Model System for Porphyrin Networks and Heme-Protein Interactions. J. Biol. Inorg. Chem. 2009, 14, 1289–1300. 10.1007/s00775-009-0574-9. [DOI] [PubMed] [Google Scholar]
- Jähnig F. Electrostatic Free Energy and Shift of the Phase Transition from Charged Lipid Membranes. Biophys. Chem. 1976, 4, 309–318. 10.1016/0301-4622(76)80012-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.