

Abstract
Vinblastine (VLB) is an antimitotic drug that binds to the vinca site of tubulin. The molecule possesses a high molecular weight and a complex chemical structure with many possibilities of metabolization. Despite advances in drug discovery research in reducing drug toxicity, the cause and mechanism of VLB-induced adverse drug reactions (ADRs) remains poorly understood. VLB is metabolized to at least 35 known metabolites, which have been identified and collected in this present work. This study also explores how VLB metabolites affect nausea-associated receptors such as muscarinic, dopaminergic, and histaminic. The metabolites have stronger binding interactions than acetylcholine (ACh) for muscarinic M1, M4, and M5 receptors and demonstrate similar binding profiles to that of the natural substrate, ACh. The affinities of VLB metabolites to dopaminergic and histaminic receptors, their absorption, distribution, metabolism, excretion, toxicity properties, and the superiority of VLB to ACh for binding to M5R, indicate their potential to trigger activation of nausea-associated receptors during chemotherapy with VLB. It has been shown that metabolite 20-hydroxy-VLB (metabolite 10) demonstrates a stronger binding affinity to the vinca site of tubulin than VLB; however, they have similar modes of action. VLB and metabolite 10 have similar gastric solubility (FaSSGF), intestinal solubility (FeSSIF), and log P values. Metabolite 10 has a more acceptable pharmacokinetic profile than VLB, a better gastric and intestinal solubility. Furthermore, metabolite 10 was found to be less bound to plasma proteins than VLB. These are desired and essential features for effective drug bioavailability. Metabolite 10 is not a substrate of CYP2D6 and thus is less likely to cause drug–drug interactions and ADRs compared to its parent drug. The hydroxyl group added upon metabolism of VLB suggests that it can also be a reasonable starting compound for designing the next generation of antimitotic drugs to overcome P-glycoprotein-mediated multidrug resistance, which is often observed with vinca alkaloids.
1. Introduction
1.1. Vinblastine
Vinblastine (VLB) or vincaleukoblastine is an anticancer drug that was initially isolated from the alkaloids of Madagascar periwinkle (Catharanthus roseus) plant in 1960.1 It acts by inhibiting the polymerization of the microtubules (MTs) because it binds to the tubulin heterodimer, preventing the formation of the mitotic spindle, and consequently the cell proliferation process.2 VLB is effective against many types of cancers such as renal cell carcinoma,3 Hodgkin’s lymphoma,4 and small cell cancer such as lung, breast, and colon cancers.5
Anticancer drugs are known to cause many adverse drug reactions (ADRs) during chemotherapy.6 It has been more than 50 years since VLB was approved, but the ADRs caused by the high drug toxicity remain poorly understood. The most common ADRs of VLB are nausea, vomiting, diarrhea, alopecia, myelosuppression, and anemia.7
VLB is known to be highly distributed throughout the body. It reaches a maximum plasma concentration (Cmax) and time (Tmax) of 7.95 μg/mL and 0.08 h, respectively.4 The slow excretion comes from the high percentage of drug bound to plasma proteins, which is above 90% for VLB.8 Because of the high metabolism rate by the hepatobiliary system, the concentration of VLB is found 50–100 times higher in the bile than in blood samples.9
VLB has a molecular weight (MW) of ∼813 g/mol and a chemical formula of C46H60N4O9, and thus it is considered a large and complex structure. Catharantine (CT) of VLB (labeled C1′–C26′) has a 9-membered azacyclononane ring, which contains a protonated and charged nitrogen atom at position 6′, a hydroxyl and methyl group at C4′, an ester group at C18′, and an indole ring. Vindoline moiety (VD) of VLB (labeled N1–C30) is the major alkaloid from the periwinkle plant.7a It also has an indole ring that contains an N-methyl at position 1 and a methoxy moiety at C16. It contains an ester and a hydroxyl group at C3, as well as an ester and an ethyl group at C4 and C5, respectively. According to the crystal structure of VLB, the molecule exists in dicationic form whereas the N6′ of CT and the N9 of VD are both protonated and positively charged10 (Figure 1A).
Figure 1.

Chemical structures of (A) VLB and (B) vinorelbine.
1.2. Metabolites of VLB
VLB is likely to form a variety of metabolites due to its molecular complexity and hence leads to several ADRs. The majority of the experiments regarding the identification of metabolites of VLB are performed in vivo, but they lack the chemical elucidation of the possible compounds formed during the VLB metabolism. The three recent studies have proposed a possible fragmentation of VLB. Twenty chemical structures were indicated as metabolites of VLB and elucidated in vitro utilizing the mass spectrophotometry technique11 (Figure 2).
Figure 2.

Thirty-five identified metabolites of VLB.
Moreover, an in vivo study of the metabolites of vinorelbine, an analogue of VLB is also included in this study.12 The chemical structure of vinorelbine only differs from VLB in the CT moiety, which lacks the hydroxyl group observed in position C20′ of VLB (Figure 1B).
A metabolite of vinorelbine, which undergoes oxidation and cyclization in the VD was also previously elucidated by Elmarakby et al. as a possible metabolite of VLB.13 This suggested that vinorelbine and VLB both go through the same metabolic reactions. There are 35 known metabolites of VLB identified to date. VLB mainly undergoes hydroxylation, demethylation, and hydrolysis metabolic reactions. The MW of VLB metabolites varies from 353.44 to 828.99 g/mol; however, only a minority of them have their MW increased (higher than the MW of VLB) due to aromatic hydroxylation.11,12 The VD moiety of VLB is more susceptible to metabolic reactions than its CT due to a high number of active functional groups prone to metabolism (Figure 2 and Table 1).
Table 1. Metabolic Reactions, Site of Reaction, and Molecular Weight of VLB Metabolites, CT, and VD.
| metabolite | location | metabolic reaction | MW (g/mol) | refs |
|---|---|---|---|---|
| MTB1 | C4 (VD) | ester hydrolysis | 770.95 | (12) |
| MTB2 | C3 (VD) | deprotonation, oxidation, cyclization | 810.97 | |
| MTB3 | C6 (VD) | olefin hydroxylation | 828.99 | |
| MTB4 | C7 (VD) | olefin hydroxylation | 828.99 | |
| MTB5 | C16 (VD) | O-demethylation | 798.96 | |
| MTB6 | N1 (VD) | N-demethylation | 798.96 | |
| MTB7 | C12′ (CT) | aromatic hydroxylation | 828.99 | |
| MTB8 | C13′ (CT) | aromatic hydroxylation | 828.99 | |
| MTB9 | C17 (VD) | hydroxylation | 828.99 | |
| MTB10 | C20 (VD) | hydroxylation | 828.99 | |
| MTB11 | N6′ (CT) | N-oxidation | 827.98 | |
| MTB12 | C16 (VD) | O-demethylation | 756.93 | |
| MTB13 | N1 (VD) | N-demethylation | 756.93 | |
| MTB14 | C16 (VD) | O-demethylation | 796.95 | |
| MTB15 | N1 (VD) | N-demethylation | 796.95 | |
| MTB16 | C3 (VD) | dehydration | 794.98 | (11) |
| MTB17 | C4′ (CT) | dehydration | 794.98 | |
| MTB18 | C3 (VD) | hydrolysis of ester | 752.94 | |
| MTB19 | C4 (VD) | hydrolysis of ester | 752.94 | |
| MTB20 | C18′ (CT) | no characterized | 664.81 | |
| MTB21 | (VD) | retro Diels–Alder | 651.86 | |
| MTB22 | C18′ (CT) | hydrolysis/dehydration | 354.44 | |
| MTB23 | C18′ (CT) | hydrolysis/dehydration | 353.44 | |
| MTB24 | C4′ (CT) | dehydration | 734.92 | |
| MTB25 | C4′ (CT) | dehydration | 734.92 | |
| MTB26 | C10 (VD) | no characterized | 526.6 | |
| MTB27 | C10 (VD) | no characterized | 524.67 | |
| MTB28 | (CT) | dehydration | 664.81 | |
| MTB29 | (VD) | no characterized | 598.71 | |
| MTB30 | (VD) | oxidation | 692.89 | |
| MTB31 | (VD) | no characterized | 544.70 | |
| MTB32 | (VD) | no characterized | 542.69 | |
| MTB33 | C4′ (CT) | dehydration | 675.88 | |
| MTB34 | C18′ (CT) | hydrolysis of ester | 484.65 | |
| MTB35 | C4′ (CT) | dehydration | 466.64 |
1.3. Nausea-Associated Receptors
Chemotherapy-induced nausea and vomiting (CINV) is the most common and unpleasant ADR during cancer treatment.14 CINV does not only impact the quality of life of a cancer patient but it can also lead to disease complications, such as dehydration and malnutrition.14a There are prophylactic interventions to avoid or attenuate CINV such as antiemetic drugs (e.g., olanzapine and ondansetron) or a combination of other nausea-associated receptor antagonists.14a,15 However, CINV still remains a concern for the healthcare system and cancer patients.
Nausea is stimulated by a variety of mechanisms in our body.6b The chemoreceptor trigger zone and the vestibular system are vulnerable areas found outside the blood brain barrier (BBB) that can trigger nausea.16 Other mechanisms such as those in the gastrointestinal (GI) tract and the cerebral cortex can also stimulate the vomiting center and cause emesis.16 We have previously investigated the main neurotransmitters involved in nausea and identified some of the main receptors whose activation is associated with nausea.6b
Dopamine, histamine, acetylcholine (ACh), and serotonin are the main neurotransmitters known to play important roles in emesis.6b These molecules bind specifically to nausea-associated receptors, dopaminergic (D1, D2, D3, D4, and D5), histaminic (H1, H2, H3, and H4), muscarinic (M1, M2, M3, M4, and M5), and serotoninergic receptors, respectively. Moreover, cannabinoid, corticosteroid, neurokinin-1, gabaminergic, and opioid receptors are also included in the nausea pathway.6b,16,17 These, along with the serotoninergic receptors, will be elaborated elsewhere.
1.3.1. Dopaminergic Receptors
Dopaminergic receptor subtypes, D1, D2, D3, D4, and D5, share a high homology with distinct biological features. They are composed of seven-transmembrane domains and their molecular mechanisms involve regulation of cyclic adenosine monophosphate, which triggers a variety of biological signaling. Antagonists of dopaminergic receptors have been extensively studied for the treatment of nausea and psychological disorders.18 Osinski et al. have shown that the activation of D2R and D3R triggers the emesis process in ferrets treated with specific receptor agonists.19
1.3.2. Histaminic Receptors
The natural substrate of histaminic receptors subtypes, H1, H2, H3, and H4, is histamine. It plays an important role in regulating the mucosa immunity, motility, neurotransmission, and visceral nociception of the GI tract.20
Sullivant et al. have shown that the presence of subtypes H1, H2, and H3 is more highly expressed than the H4 subtype throughout the GI tract of dogs with canine inflammatory bowel disease.20c This is in agreement with previous research performed in humans that demonstrates a similar distribution of histaminic receptors in the GI tract.21 In particular, H1R is extensively distributed in the smooth muscle and gastric mucosa, and H3R is mainly found at the myenteric plexus, a major nerve supply also involved in motility.20c,21
A case report of 2008 shows that cyclizine, a second generation of the H1R antagonists, is effective in managing nausea induced by chemotherapy.22 Studies of H1R antagonists have also demonstrated that this class of drugs is effective in postoperative nausea and vomiting as well.23
1.3.3. Muscarinic Receptors
The identified muscarinic receptor subtypes, M1, M2, M3, M4, and M5, are also expressed in the parasympathetic system. They regulate function of the GI tract and endocrine and cardiac systems.24
The endogenous substrate responsible for the activation of the muscarinic receptors, ACh, exerts physiological effects in hormonal and neuronal pathways. As ACh is found both in the vestibular and GI systems, antagonists of the natural ligand are useful drugs to treat nausea induced by different mechanisms. Anticholinergic agents such as scopolamine, a nonspecific antagonist of muscarinic receptors, have been extensively studied for the relief of nausea caused by chemotherapy and/or postoperative situations.25 More specifically, M1R and M5R antagonists have been reported as effective drug targets during nausea induced by cancer treatment.26
1.4. Tubulin
MTs are part of the cytoskeleton and are structurally formed by 13–16 protofilaments of tubulin, which are aligned side-by-side with a head-to-tail layout forming hollow cylindrical MTs. Highly dynamic in nature, MTs denote the mitotic spindle of eukaryotic cells and are strongly involved in cellular proliferation.2b
The cellular division promoted by the MTs has made this structure an attractive target for anticancer drug design, more specifically by targeting the tubulin heterodimer. The structure of tubulin is composed of two distinct subunits of proteins known as α- and β-tubulin that forms a tubulin heterodimer.27
VLB targets the MTs by inhibiting the polymerization of the protofilaments impairing the mitotic spindle of cells.28 We have previously studied the binding interactions of VLB with the αβ-tubulin heterodimer by means of molecular dynamics simulation.2b In addition, we investigated how carbon nanotubes function as a drug delivery option for VLB in order to enhance target specificity and decrease ADRs.29
The presented work aims to identify the mode of action of VLB metabolites against MT and looks into their role as nausea-triggering elements.
2. Results and Discussion
VLB and its metabolites were docked into the nausea-associated receptors D2, H1, H3, M1, M4, M5, and tubulin using the molecular docking method. The binding energy values and main binding interactions of VLB and its metabolites with the target sites were compared to the known binding interactions of the endogenous substrates for each receptor (dopamine, histamine, and ACh), in order to evaluate their likelihood of inducing nausea during chemotherapy. Moreover, the pharmacokinetics (PK) properties of VLB and its metabolites were also calculated.
2.1. Dopaminergic D2R
The natural substrate of D2R, dopamine, interacts with residues Phe361 and Val115 through hydrophobic contacts. Additionally, the hydroxyl group of dopamine makes H-bonds with residue Ser193 (1.9 and 1.8 Å), and the amine group makes an H-bond and ionic interactions with residue Asp114 (1.8 Å). Dopamine has a binding energy of −12.89 kJ/mol when it interacts with D2R (Figure 3B and Table S1).
Figure 3.
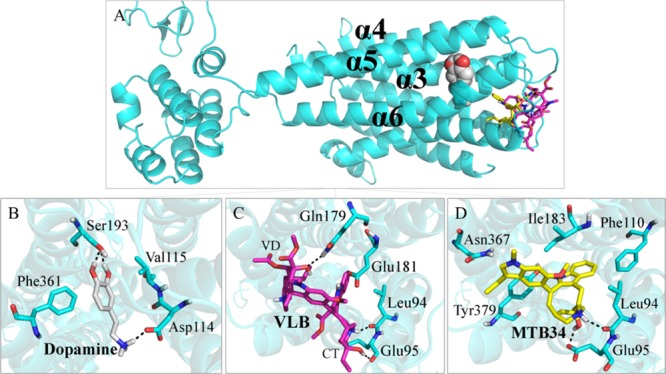
Main binding interactions at the dopaminergic receptor D2. (A) Pose views of overlaid dopamine (spheres), VLB, and metabolite 34 docked into D2R (sticks). Binding interactions of (B) dopamine; (C) VLB; and (D) metabolite 34. Hydrogen bonds are shown in dashed lines.
The binding energy of VLB when docked into D2R is higher than that of dopamine (−2.37 kJ/mol vs −12.89 kJ/mol). VLB makes H-bonds with residues Glu181, Gln179, Glu95, and with the main chain of Leu94. Because of the binding affinity of VLB (−2.37 kJ/mol) for D2R and lack of involvement with residues that interact with the natural substrate, it is suggested that the anticancer drug would not compete with dopamine for D2R active site (Figure 3C and Table S1).
The majority of metabolites of VLB would not play a role in the binding site of D2R due to their binding energies being weaker than that of dopamine. However, metabolite 34 interacts more strongly than dopamine in the D2R binding site (−17.58 kJ/mol vs −12.89 kJ/mol). The protonated nitrogen atom of the azacyclononane ring as well as the hydroxyl group establishes H-bonds with the binding site of D2R through residues Glu95 and Leu94. Metabolite 34 makes hydrophobic interactions with residues Asn367, Ile183, Phe110, and Tyr379. Despite the lack of biological studies on the VLB metabolites and the urge for both in vitro and in vivo experiments, the present in silico results suggest that metabolite 34 could compete with dopamine for binding to D2R (Figure 3D and Table S1).
2.2. Histaminic H1R
VLB, its metabolites, and histamine were docked into the known histamine binding site in H1R. The docking results of histamine, VLB, and its metabolites demonstrate that only three metabolites (metabolites 22, 23, and 35) have stronger binding energy than the natural substrate, among which metabolite 22 has the lowest binding energy, −18.10 kJ/mol (Table S2).
The imidazole ring of histamine makes H-bonds with residues Asp178 (2.2 Å) and the protonated amine makes ionic interactions and H-bonds with Asp107 (1.9 and 2.3 Å) within the binding site of H1R. Moreover, histamine also interacts with residues Ile454 and Tyr431 through hydrophobic contacts with a binding energy of −14.40 kJ/mol for H1R (Figure 4B and Table S2).
Figure 4.
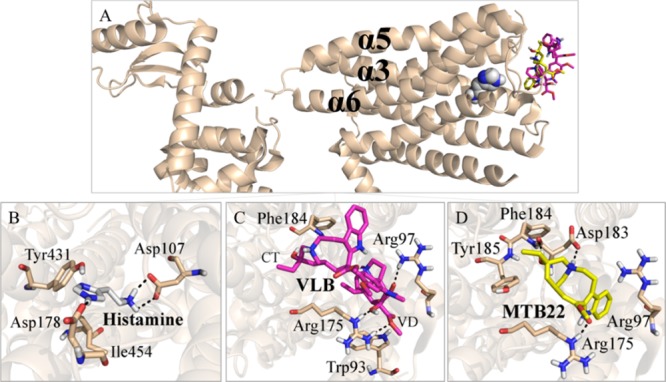
Main binding interactions at the histaminic receptor H1. (A) Pose view of histamine (spheres), VLB, and metabolite 22 (sticks) docked into H1R. Binding interactions of (B) histamine; (C) VLB; and (D) metabolite 22. Hydrogen bonds are shown in dashed lines.
The binding energy of VLB in the substrate binding site of H1R is −0.17 kJ/mol. VLB makes H-bonds with residues Arg175, Arg97, and Trp93, in which both methyl groups of the ester groups and the hydroxyl group are involved. Although VLB is able to interact with H1R residues, it is less likely to play any effect on the receptor’s function. This is because VLB does not interact with the same residues involved in the binding of the natural substrate (histamine) and possesses a binding energy of only −0.17 kJ/mol (Figure 4C and Table S2).
The metabolites of VLB that are most likely to display a significant role in the functionality of H1R are metabolite 22, metabolite 23, and metabolite 35 due to their binding energies (Table S2).
Metabolite 22 (binding energy of −18.10 kJ/mol) interacts with residues from helices α5 and α6 of H1R, such as Asp183. Hydrogen bonds are observed between the metabolite and Asp183 (2.0 and 1.9 Å), Arg175 (1.7 Å), and hydrophobic interactions with Phe184, Tyr185, and Arg97. Therefore, metabolite 22 competes with histamine for the binding site of H1R due to its binding energy (−18.10 kJ/mol vs −14.40 kJ/mol). Thus, it is suggested that metabolite 22 may account for nausea during chemotherapy with VLB through interaction with H1R (Figure 4D and Table S2).
2.3. Histaminic H3R
Docking of histamine into its binding site in the H3 receptor has a calculated binding energy of −13.42 kJ/mol, and its imidazole ring makes an H-bond with residue Asp114 (2.1 Å) and the charged amine with Ser121 and Asp80 at a distance of 2.1 and 1.8 Å, respectively. Moreover, hydrophobic interactions are seen among the aliphatic chain of histamine and residue Trp371 (Figure 5B and Table S3).
Figure 5.
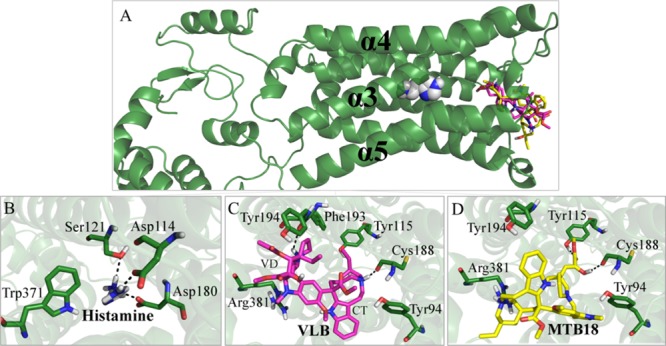
Main binding interactions at the histaminic receptor H3. (A) Pose view of histamine (spheres), VLB, and metabolite 18 (sticks) docked into H3R. Binding interactions of (B) histamine; (C) VLB; and (D) metabolite 18. Hydrogen bonds are shown in dashed lines.
In the same binding site, VLB binds with an energy of −13.78 kJ/mol, with its azacyclononane ring having an H-bond with Cys188 (1.8 Å). Residues Tyr194 (through an H-bond), Phe193, Tyr115, Arg381, and Tyr94 (via vdW forces) participate in the interactions with VLB. VLB and histamine have very close binding energies (−13.78 kJ/mol vs −13.42 kJ/mol), and thus the anticancer drug could compete with the natural substrate for the binding site of H1R (Figure 5C and Table S3).
Metabolite 18 has the lowest binding energy among the metabolites of VLB (−19.67 kJ/mol vs −13.42 kJ/mol of VLB). Similar to VLB, metabolite 18 is located at the entrance of the binding pocket (Figure 5A and Table S3).
Metabolite 18 makes H-bonds with Tyr115, Cys188, and Arg381, whereas residue Tyr115 binds to the carbonyl group of ester, and the main chain of Cys188 interacts with the hydroxyl group of VD and with Tyr94 and Tyr194 by vdW forces. Therefore, because of the binding energy of metabolite 18 (−19.67 kJ/mol) and its occupation in the same site as the substrate, this metabolite would compete with histamine for the binding pocket of H3R regardless of its interactions with the residues in proximity to the residues in the histamine network (Figure 5D and Table S3).
2.4. Muscarinic M1R
ACh binds to M1R with a total energy of −4.79 kJ/mol resulting from H-bonds with Tyr404 and Tyr106, within distances of 2.2 and 2.4 Å, respectively. Hydrophobic interactions are seen among the N-trimethyl group of ACh and residues Asp105 and Tyr381 (Figure 6B and Table S4).
Figure 6.
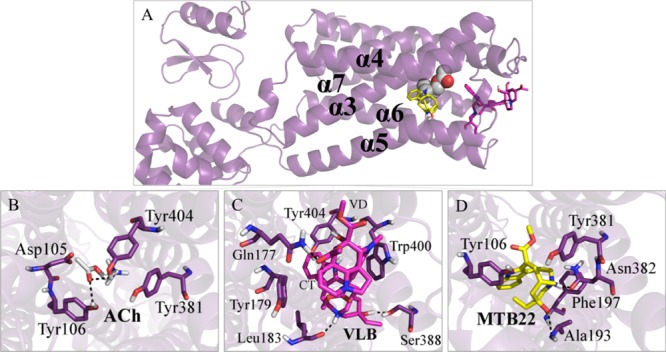
Main binding interactions at the muscarinic receptor M1. (A) Pose view of ACh (spheres), VLB, and metabolite 22 (sticks) docked into M1R. Binding interactions of (B) ACh; (C) VLB; and (D) metabolite 22. Hydrogen bonds are shown in dashed lines.
VLB, which has a lower binding affinity for M1R than ACh (−3.77 kJ/mol vs −4.79 kJ/mol, respectively), possesses H-bonds to Leu183 and Ser388 through its CT moiety. Gln177 interacts with the ester group at C4 of VLB’s VD moiety and with Trp400, Tyr404, and Tyr179 via hydrophobic interactions. VLB shares Tyr404 with ACh interaction subsite that is reinforced by the vdW force (Figure 6C and Table S4).
The metabolites of VLB that would play a major role in the activation of M1R are metabolites 22 and 23 due to their binding energies (−18.10 and −16.86 kJ/mol, respectively) and their similar interaction profiles to the ACh.
Metabolite 22 has the lowest binding energy in the ACh binding site of M1R with the estimated magnitude of −18.24 kJ/mol. The binding energy of metabolite 22 is more than 3 times stronger than the binding energy of ACh (−4.79 kJ/mol) (Table S4).
The binding energy of metabolite 22 results from the H-bond with Asn382 within 1.6 Å distance and hydrophobic interactions with Ala193, Tyr381, Tyr106, and Phe197. Similar to ACh, metabolite 22 interacts with residues Tyr106 and Tyr381. Metabolite 22 has shown to be potentially a better agonist of M1R than ACh due to its stronger binding (−18.24 kJ/mol vs −4.79 kJ/mol). It shows as well a binding profile similar to that of ACh (i.e., interacts with Tyr106 and Tyr381). Thus, it could account for nausea during chemotherapy with VLB through muscarinic receptor activation. This suggests that further biological studies are required for supplementing these findings (Figure 6D and Table S4).
Tyr106 and Tyr404 are involved in the binding of ACh and metabolite 23. An H-bond is observed among residue Tyr106 and the ester group of metabolite 23, within a distance of 1.7 Å. This interaction is 0.7 Å shorter than that of ACh with Tyr106 (2.4 Å). Moreover, Tyr404 interacts with the protonated nitrogen of indole ring within a distance of 1.6, 0.6 Å shorter than that with ACh (2.2 Å). Metabolite 23 also makes H-bonds with Leu183 and Ile180, thus metabolite 23 with a interaction profile similar to the ACh, and a higher binding affinity could contribute in triggering nausea through activation of M1R (Figure S1).
The metabolites other than 22 and 23 also bind to the binding site of M1R; however, they do not demonstrate any significant difference with ACh in terms of their binding energy nor do they share the same residues with the ACh interaction network. This implies that they are less likely to compete with ACh or interfere largely with the ACh-M1R natural function. Nevertheless, metabolites 22 and 23 have significant binding affinities for M1R and share similar binding interactions to those of ACh (Tyr106, Tyr404 and Tyr381) and hence could induce nausea during treatment with VLB.
2.5. Muscarinic M4R
ACh binds to M4R with −7.65 kJ/mol of binding energy through hydrophobic contacts and H-bonds with Tyr439, Tyr113, and Asp112. The carbonyl of the ester group of ACh makes an H-bond with Tyr439 (3.0 Å) and Tyr113 (2.2 Å) (Figure 7B and Table S5).
Figure 7.
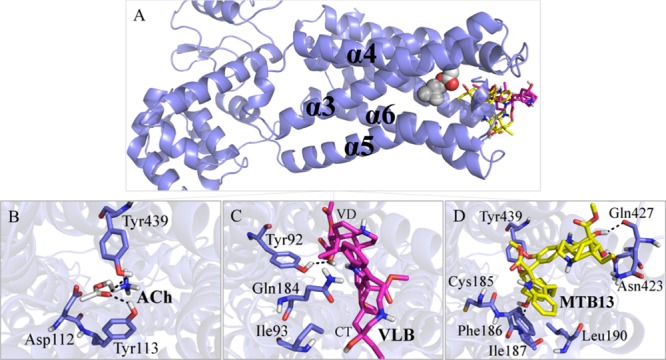
Main binding interactions at the muscarinic receptor M4. (A) Pose view of ACh (spheres), VLB, and metabolite 13 (sticks) docked into M4R. Binding interactions of (B) ACh; (C) VLB; and (D) Metabolite 13. Hydrogen bonds are shown in dashed lines.
VLB has a binding energy of −3.90 kJ/mol when it interacts with Tyr92 through an H-bond and with Ile93 and Gln184 by vdW forces. However, it has less affinity for the receptor compared with ACh (−3.77 kJ/mol vs −7.65 kJ/mol). This could prevent VLB from playing any major role in ACh’s natural effect by binding to the M4R. However, some of its metabolites have significant binding affinities for M4R (Figure 7C and Table S5).
Metabolite 13 has the lowest binding energy among the metabolites, which is nearly more than twice than that of ACh (−20.65 kJ/mol vs −7.65 kJ/mol). The affinity of metabolite 13 results from its H-bonds with Ile187, Gln427, and Asn423, and the hydrophobic interactions of its ethyl group with Tyr439, an amino acid shared with ACh binding network, as well as vdW interactions with Phe186, Leu190, and Cys185. Thus, it could potentially contribute to the onset of nausea (Figure 7D and Table S5).
Other metabolites with better binding affinity than ACh that display interactions similar to that of ACh are metabolite 19, metabolite 18, metabolite 34, metabolite 22, metabolite 23, and metabolite 10, and thus they could trigger the onset of nausea during chemotherapy with VLB (Table S5).
Many of the VLB metabolites have better binding affinity for M4R than ACh; however, their interaction profiles are different from that of ACh. This may result in a different induced effect during the signaling process. Metabolite 19 binds to M4R with a binding strength of −18.63 kJ/mol. This is mainly due to hydrophobic interactions and an H-bond with Tyr439, which is also involved with ACh binding. Tyr439 interacts with metabolite 19 within a distance of 2.2, 0.8 Å shorter than ACh (3.0 Å). In addition, hydrophobic contacts are seen with Trp435 and Cys185. Hence, metabolite 19 could also contribute to triggering nausea, similar to metabolite 18 (−17.19 kJ/mol), metabolite 23 (−16.63 kJ/mol), metabolite 34 (−17.10 kJ/mol), metabolite 22 (−16.71 kJ/mol), or even metabolite 10, 20-hydroxy-VLB (−10.16 kJ/mol) (Figure 8 and Table S5).
Figure 8.
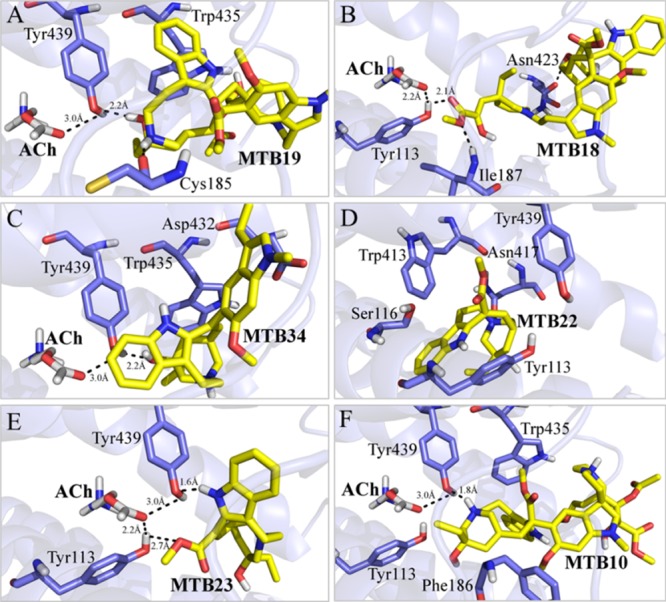
Binding interactions of VLB metabolites docked into M4R. (A) Metabolite 19; (B) metabolite 18; (C) metabolite 34; (D) metabolite 22; (E) metabolite 23; and (F) metabolite 10. ACh is shown in white sticks and hydrogen bonds in dashed lines.
2.6. Muscarinic M5R
ACh binds to M5R via Tyr480, Tyr110, and Tyr457, while its carbonyl group makes H-bonds to Tyr480 and Tyr110 within respective distances of 2.6 and 2.2 Å. It has hydrophobic interactions with Tyr457 as well as Asp109 (Figure 9B and Table S6).
Figure 9.
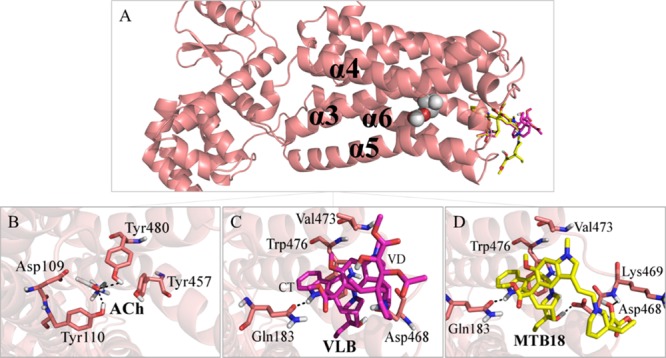
Main binding interactions at the muscarinic receptor M5. (A) Pose view of ACh (spheres), VLB, and metabolite 18 (sticks) docked into M5R. Binding interactions of (B) ACh; (C) VLB; and (D) metabolite 18. Hydrogen bonds are shown in dashed lines.
VLB with a binding energy of −9.28 kJ/mol for M5R has ∼4 kJ/mol lower than that of ACh (−4.51 kJ/mol), which stems from VLB’s H-bonds with Asp468 and Gln183. The VLB hydroxyl group at C4′ and the protonated nitrogen at N9 both make H-bonds with Asp468, within respective 1.6 and 1.9 Å distances. The positively charged protonated nitrogen in the azacyclononane ring of VLB (N6′) electrostatically interacts with the negative charge of Asp468, while Gln183 interacts with the carbonyl ester group through an H-bond (2.2 Å). Hydrophobic interactions of Asp468, Gln183, Trp476, and Val473 with the azacyclononane ring and ester methyl groups of CT moiety of VLB also contribute to the total binding strength of the ligand. Although VLB does not bind to the same residues involved with ACh in M5R, it occupies the same binding site with nearly twice the strength of ACh (−9.28 kJ/mol vs −4.51 kJ/mol) (Figure 9C and Table S6).
Similar to muscarinic M1 and M4 receptors, many of VLB metabolites, such as metabolite 18 and metabolite 23, bind to M5R more strongly than ACh and often interact with the same residues binding to those involved with ACh (e.g., Tyr480 and Tyr110) (Tables S4–S6).
The calculated binding energy of metabolite 18 is −18.68 kJ/mol when docked into the binding site of M5R. It interacts with residues Asp468, Lys469, and Gln183 through H-bonds. The amide group of Gln183 interacts with the carbonyl ester (2.0 Å) and with the indole ring (1.7 Å) of metabolite 18. It has hydrophobic interactions with Val473 and Trp476, as well. Thus, metabolite 18 could trigger the onset of nausea during chemotherapy with VLB (Figure 9D and Table S6).
The interactions of metabolite 23 to the binding site of M5R are over 3 times stronger than that of ACh (−14.11 and −4.51 kJ/mol). The hydroxyl group makes H-bonds within 2.5 Å radiuses of Tyr110, which is 0.3 Å farther than the ACh (2.5 Å vs 2.2 Å). However, it has an extra H-bond with residue Thr193 (2.2 Å), implying that metabolite 23 is capable of inducing nausea through activation of M5R (Figure 10A and Table S6).
Figure 10.
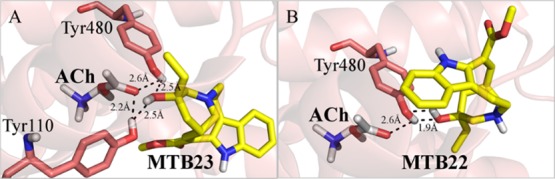
Binding interactions of (A) metabolite 23 and (B) metabolite 23 docked into M5R. ACh is shown in white sticks and hydrogen bonds in dashed lines.
Tyr480, which makes an H-bond with the endogenous substrate ACh (2.6 Å), makes H-bond with the hydroxyl group of metabolite 22 within a distance of 1.9 Å, which is 0.7 Å shorter than the one seen with the substrate (1.9 Å vs 2.6 Å). Metabolite 22 binds over 3 times more strongly than ACh (−18.41 kJ/mol) to M5R. Metabolite 22 binds to the same residues involved with ACh in the binding pocket of M5R and similar to metabolite 23 could trigger the onset of nausea (Figure 10B).
2.7. Binding to Tubulin
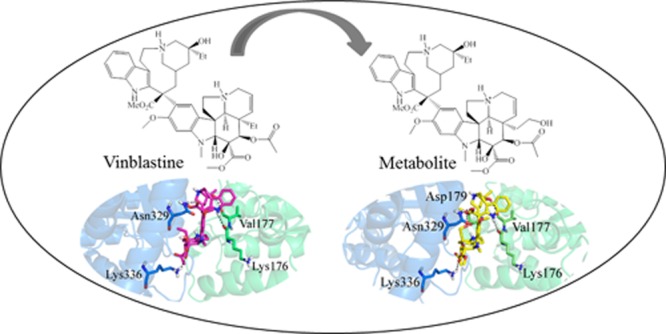
The virtual screening of the ligand library against tubulin vinca site resulted in a binding solution with an energy of −11.23 kJ/mol for VLB. Its conformation adaption shows the major role of CT moiety in VLB binding and agrees with the previously published data in this regard.2b,30 VLB makes H-bonds with Asn329 and Lys336 of α-tubulin along with Lys176 and Val177 of β-tubulin (Tables 2, S7 and Figure S2).
Table 2. Residues That Are Majorly Involved in the Binding of VLB into the Tubulin Heterodimera.
| subunit | hydrogen bonds | hydrophobic interactions |
|---|---|---|
| α-tubulin | Asn329, Lys336 | Leu248, Ile332 Thr349, Gly350, Phe351, Val353 |
| β-tubulin | Lys176, Val177 | Ser178, Asp179, Tyr210, Pro222 |
Residues underlined bind via H-bonds with the CT moiety of VLB, whereas those in italic interact with the vindoline moiety of VLB.
More specifically, the protonated N16′ interacts with the carbonyl group of the main chain of Lys176 within a distance of 2.3 Å; the hydroxyl group at C4′ interacts with the main chain of Val177 within a distance of 1.9 Å; the carbonyl of ester at C18′ interacts with the amide group of Asn329 (2.1 Å); and the carbonyl group at C3 interacts with Lys336 within a distance of 2.3 Å (Figure 11B and Table 3).
Figure 11.
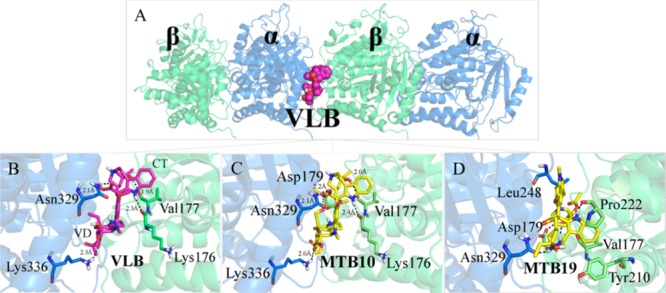
Main binding interactions at the vinca site of tubulin. (A) Structure of α- and β-tubulin coupled with docked VLB (spheres), colored in blue and green, respectively. Binding interactions of (B) VLB, (C) metabolite 10 and, (D) metabolite 19. Hydrogen bonds are shown in dashed lines.
Table 3. Distances between Heteroatoms of the Vinca Site of Tubulin and Those of VLB, Metabolites 8, 10, and 11 Involved in Hydrogen Bonds.
| distance
among hydrogen bond donor (HBD) and hydrogen
bond acceptor (HBA) atoms (Å) |
|||||
|---|---|---|---|---|---|
| subunit | H-bonds interactions | VLB | MTB8 | MTB10 | MTB11 |
| CT | N16′ + Lys176 | 2.3 | 2.2 | 2.4 | 2.3 |
| OH + Val177 | 1.9 | 2.0 | 2.0 | 1.7 | |
| VD | ester C4 + Asn329 | 2.1 | 2.0 | 2.1 | 1.9 |
| ester C3 + Lys336 | 2.3 | 2.4 | 2.6 | 2.5 | |
| ΔGbinding (kJ/mol) | –11.23 | –12.18 | –13.39 | –14.24 | |
Most of the VLB metabolites bind to the heterodimer of tubulin with the binding strength ranging from −21.90 kJ/mol of metabolite 19 to −0.45 kJ/mol of metabolite 4 (Table S7).
That could be due to the fact that the three rings of VD which are opened after metabolism provides metabolite 19 more conformational flexibility to fit into the deep tubulin binding site. Although the metabolite does not bind to all of the exact residues interacting with VLB, it makes a shorter H-bond with Asn329 (α-tubulin) than VLB2b (1.6 Å vs 2.1 Å). Furthermore, it binds more strongly than the parent compound, VLB (−21.90 kJ/mol vs −11.23 kJ/mol). Metabolite 19 makes H-bonds with β-tubulin amino acids, including Asp179 and Pro222, which are also involved in VLB binding. Moreover, hydrophobic interactions with Ser178, Val177, Tyr210 (β-tubulin), and Leu248 (α-tubulin), also observed with VLB, contribute to the metabolite binding (Figure 11D).
Metabolites that have adapted conformation similar to that of VLB in the vinca site of tubulin are metabolite 8, metabolite 10, and metabolite 11. These compounds have additional moieties in their structure that enhance their binding affinity for tubulin. Metabolite 11 has undergone an N-oxidation reaction at N6′ during VLB metabolism. Metabolite 10 is hydroxylated at position C20 of VD and metabolite 8 has undergone aromatic hydroxylation at C13′ of CT. Besides adapting conformation similar to that of VLB at the tubulin heterodimer binding site, metabolite 8, metabolite 10, and metabolite 11 have stronger binding energy (−12.18, −13.39, and −14.24 kJ/mol, respectively) than VLB (−11.23 kJ/mol). As expected, these metabolites bind to the same residues that interact with the parent drug through H-bonds (Lys176, Val177, Asn329, and Lys336). The strength of these interactions and high binding energy are due to the close proximity of atoms involved in H-bonds (Figure 2, Tables 3 and S7).
Studies have investigated possible analogues of VLB that display stronger target selectivity and hence higher cytotoxicity. Chemical modifications on the CT of VLB (CT) at the C20′ ethyl substituent, such as the addition of hydrophobic groups, have shown significant increased potency toward destabilization of tubulin.30c In a lesser extent, the C5 ethyl substituent at the VD of VLB, an important chemical group for biological activity of VLB, has also been chemically modified.31 Va et al. have analyzed different compounds and their cytotoxic potency against cancer cell lines (i.e., colon cancer HCT116) mainly by modifying the C5 ethyl moiety. However, the chemical bond among C6 and C7 of VD was also modified to either a double or single bond. In a C6–C7 single bond version, the addition of CH2OH group at C5 position (referring C19 of VD) has reduced the biological activity by 10-fold (compound 56)31 (Figure 1).
As compared to VLB, metabolite 10 has one extra H-bond with Asp179. The hydroxyl group added at C5 of VD (attached to C20) shares its electron deficiency with the side chain of Asp179 of β-tubulin within a distance of 2.2 Å. The corresponding heteroatoms of Lys336, Asn329, Val177, and Lys176 are farther from metabolite 10 than they are from VLB. However, an extra H-bond between the hydroxyl group at C5 of metabolite 10 and Asp179 strengthens the interaction of the metabolite with tubulin. Thus, metabolite 10 is a stronger binder than VLB (−13.39 kJ/mol vs −11.23 kJ/mol) while it displays the same binding profile (Figure 11C and Table S7).
2.8. In Silico Prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity Properties of VLB and Its Metabolites
2.8.1. Solubility, log P, and Human Jejunal Effective Permeability
The gastric solubility (FaSSGF) of VLB is higher than the intestinal solubility in the fasted state (FaSSIF) and fed state (FeSSIF) (4.61 mg/mL vs 4.61 × 10–2 and 3.12 × 10–2 mg/mL, respectively) (Table 4).
Table 4. Solubility of VLB and Its Metabolites in Fed and in Fasted Gastric/Intestinal Fluids, log P, and Their Human Jejunal Effective Permeability (Peff).
| drug & metabolites | solubility in simulated fasted stated gastric fluid FaSSGF (mg/mL) | solubility in simulated fasted stated intestinal fluid FaSSIF (mg/mL) | solubility in simulated fed stated intestinal fluid FeSSIF (mg/mL) | log P | human jejunal effective permeability (cm/s × 104) |
|---|---|---|---|---|---|
| VLB | 4.61 | 0.05 | 0.03 | 3.95 | 0.58 |
| MTB1 | 12.89 | 0.14 | 0.03 | 3.61 | 0.53 |
| MTB2 | 4.19 | 0.01 | 0.03 | 4.57 | 0.57 |
| MTB3 | 3.94 | 0.11 | 0.03 | 3.26 | 0.41 |
| MTB4 | 3.91 | 0.10 | 0.03 | 3.31 | 0.43 |
| MTB5 | 5.06 | 0.14 | 0.03 | 3.48 | 0.48 |
| MTB6 | 0.43 | 0.05 | 0.07 | 3.73 | 0.47 |
| MTB7 | 3.80 | 0.08 | 0.05 | 3.56 | 0.46 |
| MTB8 | 3.78 | 0.12 | 0.05 | 3.60 | 0.46 |
| MTB9 | 4.16 | 0.07 | 0.03 | 3.81 | 0.60 |
| MTB10 | 4.65 | 0.11 | 0.03 | 2.99 | 0.43 |
| MTB11 | 77.43 | 0.70 | 0.08 | 1.62 | 0.21 |
| MTB12 | 12.76 | 0.40 | 0.04 | 3.12 | 0.43 |
| MTB13 | 2.69 | 0.17 | 0.07 | 3.36 | 0.42 |
| MTB14 | 4.88 | 0.03 | 0.03 | 4.09 | 0.46 |
| MTB15 | 0.31 | 0.02 | 0.06 | 4.34 | 0.45 |
| MTB16 | 2.16 | 0.02 | 0.03 | 4.71 | 0.84 |
| MTB17 | 2.46 | 0.01 | 0.02 | 5.00 | 0.95 |
| MTB18 | 30.79 | 0.04 | 0.00 | 3.99 | 0.30 |
| MTB19 | 35.07 | 0.15 | 0.00 | 3.71 | 0.30 |
| MTB20 | 217.19 | 0.29 | 0.30 | 0.77 | 0.24 |
| MTB21 | 5.79 | 0.05 | 0.00 | 3.96 | 0.27 |
| MTB22 | 3.12 | 0.90 | 0.63 | 2.40 | 1.99 |
| MTB23 | 6.44 | 3.70 | 0.57 | 0.22 | 0.36 |
| MTB24 | 22.01 | 0.05 | 0.00 | 4.50 | 0.35 |
| MTB25 | 26.62 | 0.07 | 0.00 | 4.50 | 0.38 |
| MTB26 | 0.03 | 0.01 | 0.07 | 6.41 | 3.67 |
| MTB27 | 2.56 | 0.11 | 0.07 | 3.93 | 0.43 |
| MTB28 | 183.45 | 0.15 | 0.24 | 1.45 | 0.27 |
| MTB29 | 0.02 | 0.01 | 0.02 | 5.11 | 1.08 |
| MTB30 | 39.10 | 0.05 | 0.00 | 3.92 | 0.25 |
| MTB31 | 0.34 | 0.07 | 0.08 | 4.93 | 1.44 |
| MTB32 | 5.02 | 0.23 | 0.08 | 3.09 | 0.35 |
| MTB33 | 21.88 | 0.03 | 0.00 | 4.70 | 0.30 |
| MTB34 | 0.48 | 0.04 | 0.09 | 5.69 | 2.22 |
| MTB35 | 0.07 | 0.01 | 0.08 | 7.04 | 4.73 |
The calculated amount for human jejunal effective permeability (Peff) of VLB is 0.58 cm/s × 104. In vivo studies by Ogihara et al.32 have demonstrated that VLB has an intestinal permeability of less than 1.0 × 10–6 cm/s. Compounds with a Peff lower than 0.5 cm/s × 104 have low permeability.33 VLB has a Peff of slightly higher than 0.5 cm/s × 104 (∼0.58 cm/s × 104), and thus it is considered to be poorly absorbed by the intestines as it is administered by IV route (Table 4).
The predicted absorption, distribution, metabolism, excretion, toxicity (ADMET) properties of VLB compares to two of the Lipinski’s rule of five (RO5) criteria, as it has a log P 3.95 and three HBD groups within the criteria suggested by Lipinski (log P and number of HBD groups both <5).6a,34 Despite log P value of VLB indicating its suitable drug likeness (log P < 5.0) according to the RO5, it is poorly absorbed via the oral route as mentioned before. This is due to its extensive binding to plasma proteins and to P-glycoprotein (P-gp) transporter, which are contributing factors to poor oral bioavailability. In this case, a drug requires to first unbind from any plasma proteins prior to reaching its target. Once it binds to the cell target, it effluxes the cell by P-gp.35 Therefore, the poor oral bioavailability of VLB may be due to its gastric and intestinal solubility, Peff and affinity for both P-gp and plasma proteins. Moreover, VLB has an MW of ∼813 g/mol, which ∼300 g/mol exceeds the acceptable magnitude according to the RO5, and thus influences the low likelihood of oral drug bioavailability6a,34 (Table 4).
Metabolite 23 has 3.70 mg/mL intestinal solubility in the fasted state (FaSSIF). Having the highest intestinal solubility among the metabolites (3.70 mg/mL vs 0.01 mg/mL), it is likely to be reabsorbed into the blood circulation system. However, considering its log P value of 0.22 and Peff through the small intestine (0.36 cm/s × 104), metabolite 23 is probably not reabsorbed into the portal circulation in the liver. This is because compounds with a Peff less than 0.5 cm/s × 104 and a low log P (<1) have low permeability and are too hydrophilic to cross the cellular membrane33 (Table 4).
Very hydrophilic compounds with poor membrane permeability (less than 0.5 cm/s × 10–4) are more likely to be excreted through the kidneys or remain in the intestines rather than to be reabsorbed in the blood system.35 Thus, it is expected that metabolite 23 remains in the intestines or is excreted through the renal route. If the former is the case, metabolite 23 could interact with H1R, M1R, M4R, and M5R as these receptors are found throughout the body.36 In addition, metabolite 23 binds to these receptors with stronger affinities, with respective values of −15.13, −16.86, −16.63, and −14.11 kJ/mol, than the corresponding natural substrates histamine and ACh (−14.40, −4.79, −7.65, and −4.51 kJ/mol, respectively). Thus, according to the predicted ADME properties for metabolite 23 and its likelihood of interaction with nausea-associated receptors, the metabolite could cause nausea symptoms during chemotherapy with VLB (Tables 4, S2, S4–S6).
Moreover, metabolite 26, metabolite 29, metabolite 31, metabolite 34, and metabolite 35 all have poor intestinal solubility (values close to 0 mg/mL) in the fed or fasted states (gastric and intestinal). They have high log P (>5.0) and high Peff (>0.5 cm/s × 104) ranging from 4.93 to 7.04 and 1.08 to 4.73 cm/s × 104 (Table 4).
The three parameters, solubility, log P, and Peff, could be used for assessing whether a drug could reach to the blood circulation. For instance, there is a higher possibility for metabolites 26, 29, 31, 34, and 35 than metabolite 23 to cross the intestinal membranes and to be reabsorbed into the blood system according to their Peff and log P values. If these metabolites are reabsorbed into the blood system, they could account for further off-target interactions with proteins or receptors and cause ADRs. In addition, metabolite 22 and metabolite 34 have a high Peff, 1.99 and 2.22 cm/s × 104, compared to the Peff calculated for the other metabolites, and a high likelihood to cross the BBB for their ability to cross the membrane. These physicochemical properties facilitate their promiscuous off-target binding. In addition, metabolite 22 and metabolite 34 display affinities for all nausea-associated receptors studied in this work (H1R, H3R, D2R, M1R, M4R, and M5R) and thus have increased potential of causing ADRs (Table 4).
2.8.2. Volume of Distribution, Plasma Proteins, log D, and BBB
The first possible interaction of VLB and the blood protein human serum albumin (HSA) was demonstrated in vitro in 1973,37 showing that VLB is 75% bound to the plasma proteins. Despite affinity for blood proteins, the biological effects of VLB are increased compared to other anticancer agents with biological effects diminished in the presence of albumin.37 Pandya et al. demonstrated that VLB binds more weakly to HSA than to its target tubulin (−7.6 kJ/mol vs −10.0 kJ/mol).38 These are in agreement with the in silico results that shows VLB is 89.22% bound to plasma proteins due to its hydrophilic character and affinity for these proteins, meaning that only ∼10.78% of the drug is in the free form and available to interact with its target tubulin. Therefore, the in vitro data support the in silico results demonstrating that although VLB is highly bound to plasma proteins, it still displays desired biological effects (Table 5).
Table 5. Volume of Distribution, Percentage of Unbound Plasma Proteins, log D, and log BBB Coefficient Partition of VLB and Its Metabolites.
| drug & metabolites | volume of distribution Vd (L/kg) | % of unbound to plasma proteins | log D | log BBB partition coefficient |
|---|---|---|---|---|
| VLB | 14.02 | 10.78 | 3.37 | –0.95 |
| MTB1 | 11.25 | 12.23 | 2.96 | –0.89 |
| MTB2 | 17.60 | 7.13 | 4.08 | –0.77 |
| MTB3 | 17.65 | 15.80 | 2.76 | –1.26 |
| MTB4 | 17.26 | 15.06 | 2.82 | –1.40 |
| MTB5 | 12.21 | 13.56 | 2.91 | –1.09 |
| MTB6 | 5.55 | 11.91 | 3.14 | –0.87 |
| MTB7 | 2.78 | 13.13 | 3.04 | –1.16 |
| MTB8 | 2.80 | 13.27 | 3.04 | –1.08 |
| MTB9 | 13.59 | 11.35 | 3.28 | –0.90 |
| MTB10 | 12.43 | 18.83 | 2.43 | –1.29 |
| MTB11 | 0.46 | 46.06 | –1.71 | –1.43 |
| MTB12 | 9.74 | 15.71 | 2.48 | –1.04 |
| MTB13 | 4.34 | 13.81 | 2.70 | –0.82 |
| MTB14 | 15.81 | 9.09 | 3.61 | –0.93 |
| MTB15 | 6.58 | 8.02 | 3.83 | –0.66 |
| MTB16 | 18.81 | 5.90 | 4.18 | –0.59 |
| MTB17 | 13.57 | 3.00 | 4.80 | –0.80 |
| MTB18 | 0.49 | 11.93 | 3.25 | –1.13 |
| MTB19 | 0.36 | 16.81 | 1.04 | –1.20 |
| MTB20 | 0.72 | 55.70 | 0.72 | –1.10 |
| MTB21 | 0.46 | 13.86 | 0.78 | –0.30 |
| MTB22 | 1.67 | 15.90 | 1.89 | –0.25 |
| MTB23 | 0.59 | 77.22 | –0.85 | –0.79 |
| MTB24 | 0.42 | 10.45 | 2.02 | –1.02 |
| MTB25 | 0.38 | 9.99 | 2.16 | –1.07 |
| MTB26 | 1.53 | 0.46 | 6.28 | 0.16 |
| MTB27 | 0.49 | 9.46 | 1.31 | –0.29 |
| MTB28 | 2.95 | 45.32 | 1.39 | –0.81 |
| MTB29 | 1.47 | 1.12 | 4.93 | –0.45 |
| MTB30 | 1.18 | 15.40 | 0.80 | –0.66 |
| MTB31 | 1.58 | 2.20 | 4.22 | –0.10 |
| MTB32 | 0.57 | 16.59 | 0.86 | –0.68 |
| MTB33 | 0.96 | 9.09 | 1.62 | –0.51 |
| MTB34 | 1.43 | 1.06 | 5.13 | 0.17 |
| MTB35 | 1.22 | 0.60 | 6.79 | 0.43 |
The calculated Vd value of VLB is 13.41 L/kg, indicating its high potential of distribution throughout the body. This is in agreement with previous research stating that vinca alkaloids have significant Vd.8a,39 It could explain the long half-life of 35 h of VLB compared to other anticancer drugs4 as it is dispersed throughout the body and thus takes a long time to be excreted (Table 5).
Metabolite 11 and metabolite 20 are found in high amounts in the body according to their predicted Vd values of 0.46 and 0.72 L/kg, respectively. However, neither of these metabolites displays a significant interaction with any of the nausea-associated receptors in this study. Therefore, more research needs to be done to evaluate other possible interactions during their clearance to identify other off-targets receptors (Table 5).
Metabolite 17, metabolite 26, metabolite 29, metabolite 31, metabolite 34, and metabolite 35 have high affinities for the plasma proteins (<3% unbound). Even though metabolite 17 does not have an affinity for M5R (−4.34 kJ/mol vs −4.51 kJ/mol for ACh), all of the aforementioned metabolites have stronger binding energies than ACh for M5R (−11.07, −12.40, −12.08, −17.93, and −16.06 kJ/mol vs −4.51 kJ/mol). It is useful to compare the binding affinity of these metabolites against the muscarinic receptors and plasma proteins as the former receptors are also present in the blood circulation. Therefore, more research is required to investigate the binding profile of them in respect to the plasma proteins (Table S6).
Drugs capable of crossing the BBB must be lipophilic enough to be absorbed by the cell membranes and enter into the brain.35 The low likelihood of VLB to cross the BBB is calculated to be log −0.95. VLB has a partial hydrophilic character with charged atoms such as the protonated nitrogen atoms at N6′ and N9 that make its entrance through the endothelial cells of the central nervous system difficult.40 Studies have shown that the likelihood of drugs crossing the BBB is also related to their affinity for P-gp, either as substrates or inhibitors of the efflux transporter.41 P-gp removes drugs out of the endothelial cells into the blood vessels, thus hindering them to cross the BBB. As will be discussed next, the in silico results show that VLB is both a substrate and inhibitor of P-gp. An in vivo study suggests that the poor permeability of VLB into the BBB is linked to the P-gp-mediated efflux in the brain, which limits the capacity of VLB to enter the brain.41a Therefore, previous research supports the in silico data found for VLB that it is not likely to cross the BBB (Table 5).
2.8.3. P-gp and Human Ether-à-go-go-Related Gene
VLB and its metabolites are all transported by P-gp, whereas a majority of them can also inhibit this protein according to the in silico results. The metabolites transported out of the cell by P-gp are metabolite 11, metabolite 20, metabolite 22, metabolite 23, metabolite 27, metabolite 28, and metabolite 32. Horio et al. have explained that VLB is actively transported by P-gp in Hodgkin’s disease cells (KB cells) with a transport affinity of 2 μM for P-gp.42 Posterior studies have shown that VLB is also an inhibitor and inducer of P-gp.3,39,43 Thus, VLB is transported out of the cell by P-gp and inhibit the functionality of this protein and/or induce its activity to transport other xenobiotics. According to the in silico results, not only VLB but also its metabolites are transported by P-gp and can inhibit the P-gp protein.
Metabolite 22 and metabolite 23 have a high probability to cross the BBB (−0.25 and −0.79) and are transported by P-gp. If drugs capable of crossing the BBB are taken in concomitance with VLB, metabolites 22 and 23 could potentially compete with them for interaction with P-gp in the brain. If metabolite 22 and metabolite 23 are better binders to P-gp than the concomitant drugs, an increase of blood concentration of either metabolite or of the concomitant drugs in the brain would be expected. Therefore, as these metabolites are transported out of the cell membrane due to their P-gp affinity, the chances of off-target events and thus drug–drug interactions (DDI) induced by these compounds are increased within the brain environment (Table 5).
The human ether-à-go-go-related gene (hERG) receptor, a potassium channel receptor responsible for the action potential of myocardiocytes, is also an important enzyme in PK studies due to its cardiotoxicity induced by a drug inhibitory effect.44 Compounds with an inhibitory concentration (IC50) less than or equal to 10 μM have an affinity for the hERG potassium channel and thus block the receptor.33 In agreement with previous research,45 the in silico predicted ADMET data show that VLB does not interact with hERG receptor, but some of its metabolites such as metabolite 19, metabolite 21, metabolite 27, metabolite 30, metabolite 31, metabolite 32, metabolite 34, and metabolite 35 have an affinity for the receptor. Their predicted inhibitory concentrations (pIC50) for hERG inhibition are 6.25, 6.07, 6.09, 6.03, 6.18, 6.02, 6.88, and 6.71 mol/L, respectively. Therefore, the metabolites blocking hERG channel could cause postchemotherapy cardiovascular complications. Previous research has shown that tubulin-binding drugs have a probability of inducing cardiotoxicity during cancer treatment through damage of the endothelial cells of the myocardium; however, this mechanism is not yet fully known.46 Thus, further research is required to evaluate detailed mechanism of triggering cardiotoxic events during the clearance of VLB.
2.8.4. Cytochrome P450 Complex
Similar to ondansetron, metabolite 22, metabolite 23, metabolite 34, and metabolite 35 are all metabolized by CYP1A2. Thus, it is expected that the binding competition among the antiemetic drug and VLB or its metabolites for CYP1A2 could determine their differences in plasma concentrations as these drugs are usually used in concomitance during chemotherapy.47
The predicted ADMET properties show that metabolite 2, metabolite 3, metabolite 15, metabolite 16, metabolite 17, and metabolite 34 are treated by CYP2C8 enzyme. In addition, metabolite 2, metabolite 15, metabolite 17, metabolite 20, metabolite 24, metabolite 25, metabolite 26, and metabolite 27 are able to inhibit CYP2C9, whereas only metabolite 35 is metabolized by CYP2C9.
The in silico results have shown that VLB and almost all of its metabolites can inhibit CYP2D6, but not all of them are metabolized by this enzyme (NB). VLB and its metabolites which are treated by CYP2D6 might have DDI with inhibitors of this enzyme, such as quinidine, cimetidine, and ritonavir.48 The concomitant intake of compounds that are metabolized by or inhibit CYP2D6 can potentially lead to an increase on the plasma concentration level of VLB. This is due to the lower metabolism rate of VLB triggered by enzymatic competition of CYP2D6. For instance, if VLB is taken along with the antiarrhythmic agent quinidine, an inhibitor of CYP2D6, the plasma concentrations of VLB are increased because CYP2D6 is inhibited by quinidine and thus can cause drug toxicity49 (Table S8).
Moreover, VLB can also cause DDI with drugs that are metabolized by CYP2D6.50 Ondansetron is an antiemetic agent often administered during cancer treatment.51 Because it is metabolized by CYPD2652 similarly to VLB, ondansetron would compete with VLB for metabolism with CYP2D6 and would therefore be likely to cause DDI because one of the two compounds would have its plasma concentrations increased.
CYP3A4 plays the major role in the metabolism of VLB and its metabolites.53 Some VLB metabolites are metabolized by both CYP3A4 and CYP2D6, such as metabolite 1, metabolite 2, metabolite 5, metabolite 7, metabolite 13, and metabolite 14. As these metabolites could potentially bind to CYP3A4 similar to VLB, they could interfere with the metabolism of the drugs binding to CYP3A4 when another drug is taken in concomitance (Table 6 and S8).
Table 6. Kinetics and Intrinsic Parameters of VLB and Its Metabolites for CYP3A4a.
| drug & metabolites | kinetic Michaelis–Menten (Km) | kinetic Michaelis–Menten (Vmax) | Intrinsic clearance (CLint) constants |
|---|---|---|---|
| VLB | 19.9 | 25.2 | 299.1 |
| MTB1 | 17.5 | 13.2 | 183.6 |
| MTB2 | 26.3 | 24.8 | 345.0 |
| MTB3 | 17.3 | 25.0 | 109.5 |
| MTB4 | 14.8 | 26.7 | 82.1 |
| MTB5 | 21.6 | 25.0 | 134.7 |
| MTB6 | 21.8 | 23.1 | 239.7 |
| MTB7 | 17.8 | 11.2 | 144.8 |
| MTB8 | 18.9 | 12.9 | 154.8 |
| MTB9 | 16.8 | 9.3 | 333.2 |
| MTB10 | 17.6 | 27.7 | 140.0 |
| MTB11 | 64.5 | 7.2 | 139.4 |
| MTB12 | 19.4 | 13.3 | 75.2 |
| MTB13 | 19.5 | 15.8 | 146.9 |
| MTB14 | 30.0 | 25.3 | 156.9 |
| MTB15 | 28.9 | 21.3 | 282.1 |
| MTB16 | 32.8 | 76.8 | 327.3 |
| MTB17 | 6.6 | 12.7 | 1107.9 |
| MTB18 | 13.2 | 2.3 | 1871.7 |
| MTB19 | 21.3 | 3.7 | 993.2 |
| MTB20 | 28.7 | 6.4 | 183.2 |
| MTB21 | 17.4 | 2.8 | 1781.9 |
| MTB22 | 192.5 | 36.4 | 11.0 |
| MTB23 | 326.2 | 3.8 | 19.5 |
| MTB24 | 23.3 | 1.8 | 1614.7 |
| MTB25 | 22.4 | 2.2 | 1952.1 |
| MTB26 | 6.2 | 2.5 | 556.5 |
| MTB27 | 67.0 | 0.7 | 503.9 |
| MTB28 | 25.8 | 4.1 | 498.5 |
| MTB29 | 6.3 | 3.1 | 149.2 |
| MTB30 | 22.0 | 3.5 | 3507.8 |
| MTB31 | 31.1 | 10.1 | 93.4 |
| MTB32 | 72.8 | 1.4 | 187.9 |
| MTB33 | 23.4 | 2.0 | 6361.0 |
| MTB34 | 28.5 | 8.1 | 132.3 |
| MTB35 | 11.4 | 1.8 | 592.5 |
Km (μM), Vmax (nmol/min–1/nmol–1), CLint (μL/min/mg HLM protein), NB: non substrate.
According to the in silico predictions, the metabolism of VLB and all of its metabolites can be treated by CYP3A4 as well; however, they also inhibit CYP3A4. This is in agreement with the in vitro PK study of VLB, which has shown that VLB is both a substrate39,54 and an inhibitor of CYP3A4,55 as it is observed for its metabolites. Levêque and Jehl have revised the PK of the main vinca alkaloids used to treat cancer and shown that VLB is highly metabolized by CYP3A4 in human liver microsomes.39 In addition, Zhou-Pan et al. have demonstrated that the Km and Vmax constants of VLB for CYP4A4 are 6.82 ± 0.27 μM and 0.64 ± 0.06 nmol/min/mg P450, respectively.54 The inhibitory effect of CYP3A4 caused by VLB was investigated by Baumhäkel et al., who have shown that the IC50 of VLB was 20 and 44 μmol/L in two human liver microsome samples, respectively, compared with the plasma concentrations of antineoplastic drugs taken during chemotherapy.55 Both inhibition of, or metabolism by CYP3A4 are important factors to explain treatment failures commonly seen with VLB as well as ADRs caused by DDI when co-administered with other drugs (Table 6).
VLB has a Michaelis–Menten constant (Km) of 19.9 μM and a maximum metabolic rate (Vmax) of 25.2 nmol/min–1/nmol–1 P450 for CYP3A4. VLB has a much higher Km for CYP2D6 (1443.0 μM) than for CYP3A4 (19.9 μM), which indicates a significant affinity for the latter enzyme (Table 6 and S8).
2.8.5. Phase II Metabolism–Glucuronidation Reactions
VLB and its metabolites were screened against uridine 5′-diphosphate-glucuronosyltransferase (UGT), an enzyme responsible for catalyzing glucuronidation reaction during phase II drug metabolism.56 In particular, the UGT isoforms UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15 were experimentally tested for VLB and its metabolites in this work. The in silico data show that VLB is not catalyzed by any of the UGT enzymes. This indicates that VLB is not conjugated through glucuronidation metabolism. This correlates with Owellen et al. that demonstrated that VLB undergoes neither glucuronide nor sulfate formation.57 However, some of the metabolites undergo glucuronidation reactions with enzymes UGT1A3 (metabolite 9, metabolite 26, metabolite 27 and metabolite 32), UGT1A8 (metabolite 19), and UGT2B7 (metabolite 22 and metabolite 23) while the remaining metabolites do not undergo glucuronidation (Table 7).
Table 7. Phase II of Metabolites of VLB with Isoenzymes UGT1A3, UGT1A8, and UGT2B7.
| qualitative model of glucuronidation by UGT1A3 | qualitative model of glucuronidation by UGT1A8 | qualitative model of glucuronidation by UGT2B7 |
|---|---|---|
| MTB9 (45%) | MTB19 | MTB22 (61%) |
| MTB26 (49%) | MTB23 (65%) | |
| MTB27 (49%) | ||
| MTB32 (47%) |
3. Conclusions
VLB and its metabolites were docked into nausea-associated receptors D2, H1, H3, M1, M4, and M5 by means of molecular docking and their binding interactions were compared to those of dopamine, histamine, and ACh. Furthermore, the ADMET properties of VLB and its metabolites were predicted through in silico experiments. The results have shown that VLB plays a role in the M5R, and it might compete with histamine for the binding site of H3R. VLB and histamine share most of the residues in their nearest interaction subsites, which include Tyr194, Cys188, Tyr115, Tyr194, and Arg381. Thus, it is probable that VLB competes closely with histamine for occupying the binding site of H3R. The metabolites of VLB bind to all of the nausea-associated receptors used in this study and thus are able to trigger emesis during chemotherapy with VLB. The molecular docking has shown that the metabolites of VLB have binding affinities mainly for the muscarinic receptors, with binding interactions similar to that of ACh.
Because metabolite 22 and metabolite 23 have both binding affinities stronger than that of ACh and share the same binding profile as that of ACh in the binding pocket of M1R, they could potentially involve the onset of nausea through M1R activation (Table S4).
The metabolites of VLB also display binding affinities for M4R and thus are involved with nausea during chemotherapy with VLB through this additional receptor. Metabolite 13 has a binding energy of −20.65 kJ/mol, which is more than twice that of ACh (−7.65 kJ/mol). Although it does not interact with any of the residues directly interacting with ACh, it can compete with the natural substrate for stronger interactions with M4R. Moreover, metabolite 23 and metabolite 10 both have the same type of interactions of ACh and with the same residues in M4R and have shown the potential for initiating nausea through activation of M4R, as well.
In respect to M5R, metabolite 18, metabolite 22, and metabolite 23 have better binding affinities for the binding site of the receptor than the substrate. Metabolite 22 and metabolite 23 make H-bonds and hydrophobic interactions with all of the residues involved with ACh and both have higher affinities for M5R than ACh (−18.40 and −14.11 kJ/mol, respectively, vs −4.51 kJ/mol for ACh). Therefore, they can compete with ACh and affect the function of M5R during chemotherapy with VLB, causing the onset of nausea.
The ADMET results for VLB and metabolites have shown that almost all of them potentially inhibit CYP2D6 and CYP3A4. They could be treated by CYP3A4 and transported out of the cell by P-gp as well. This could explain the DDI seen with VLB when a variety of drugs are taken in concomitance. In addition, drugs that are metabolized by, inhibit CYP2C8 and/or CYP2C9, and are taken concurrently with VLB also account for DDI during chemotherapy with VLB. This is because the metabolites of VLB can also be metabolized by or inhibit these two isoenzymes, and thus an enzymatic competition is likely to occur.
Among the VLB metabolites, metabolite 23 interacts the most with nausea-associated receptors. Its PK properties are important for evaluation of how this metabolite behaves inside the body. It has an intestinal solubility (FaSSIF) of 3.70 mg/mL and an affinity for plasma proteins of 22.8%, thus indicating its availability for off-targets binding. In addition, it has a Vd of 0.59 L/kg, showing that it would not be scattered out in the tissues. It is noteworthy to point out that metabolite 23 has low MW (<400 g/mol) nearly less than half of VLB. Although metabolite 23 has a low predicted Peff (0.36 cm/s × 104), it is known that the less a molecule is bound to the plasma proteins, and the smaller it is, the easier it can traverse the cell membrane and be considered active to interact with protein-receptors.35 As it was shown earlier, metabolite 23 has affinity for all nausea-associated receptors in this study, thus supporting the idea that metabolite 23 can induce the onset of nausea during chemotherapy with VLB.
Moreover, the calculated data demonstrate that only a few metabolites of VLB undergo phase II metabolism through UGT. It is noteworthy that metabolite 22 and metabolite 23, which display strong binding affinities and binding profile similar to the substrates for all nausea-associated receptors, can potentially undergo glucuronidation through catalysis of UGT2B7. Therefore, their effect within the body must be investigated as glucuronidation products may cause toxicity. In addition, a few metabolites of VLB block the hERG receptor involved in cardiotoxic events and can cross the BBB, thus raising attention for further investigation on the effects of these metabolites in different organ tissues.
The results suggest that the metabolites formed during excretion of VLB are likely to be involved in the onset of nausea during chemotherapy with the vinca alkaloid, potentially through activation of muscarinic receptors.
In respect with VLB’s target, VLB binds to tubulin with a binding energy of −11.23 kJ/mol, whereas the majority of its metabolites demonstrate better binding energy than the parent drug, where metabolite 19 has the lowest (best) binding energy. Despite the interaction of metabolite 19 with Asn329 (α-tubulin) similar to VLB, it adapts conformation nearly similar to VLB in the vinca site of tubulin. It interacts with more residues of β-tubulin than of α-tubulin compared to VLB,29 and thus it induces different conformation to tubulin upon binding. Further in vitro studies are required to evaluate whether it induces the same antimitotic effects as VLB. This study is worth developing because the metabolite possesses ∼65 g/mol less MW than VLB and the study could be conducted to designing low MW, less chemically complex VLB analogues, potentially with less ADRs and improved properties that would meet the requirements in Lipinski’s RO5.6a
Metabolite 8 and metabolite 11 have ADMET profiles similar to that of VLB. However, metabolite 10 has a stronger binding energy than VLB for tubulin (−13.39 kJ/mol vs −11.23 kJ/mol). It has better PK properties and would be potentially more tolerable than VLB. Metabolite 10 is unique because it possesses a similar binding profile of VLB in the tubulin, however, with more acceptable PK profile than VLB. It has a better gastric and intestinal solubility and is calculated to be less bound to plasma proteins than VLB. VLB and metabolite 10 share a similar gastric solubility (FaSSGF) 4.65 mg/mL versus 4.61 mg/mL, intestinal solubility (FeSSIF) (2.95 × 10–2 mg/mL vs 3.12 × 10–2 mg/mL), and log P (2.99 vs 3.95), which are all essential drug features for desired drug bioavailability. Moreover, it is less bound to plasma proteins than VLB (81.2% vs 89.2%), thus more available throughout the body.
Metabolite 10, known as 20-hydroxy-VLB, is not a substrate of CYP2D6 as is VLB and thus is less likely to cause DDI and ADRs than its parent drug. The in silico ADMET results show that metabolite 10 does not undergo glucuronidation, and hence it has a phase II metabolism similar to that of VLB. Moreover, its hydroxyl group added upon metabolism of VLB suggests that it can also be a candidate as the starting compound to design the next generation of antimitotic drugs to overcome P-gp-mediated multidrug resistance often observed with vinca alkaloids.58 This would lead to lower dosage requirements and thus less aggressive chemotherapy, providing a more target-selective treatment with diminished ADRs for cancer patients.
4. Materials and Method
4.1. Ligand–Protein Binding
A library consisting of the structures of VLB and its 35 known metabolites was built up using SYBYL-X 2.1.1, a commercial molecular modeling and simulation package developed by Certara. Metabolites were minimized stepwise using Pullman atomic charges and Tripos force field starting from 1.0 to 0.001 kJ/mol energy gradient each through 1000 iterations. In addition, the chemical structures of the natural substrates histamine, dopamine, and ACh were set to correct protonation states for molecular docking. The natural endogenous substrates as well as the library of VLB and metabolites were docked into the binding pocket of each nausea-associated receptor H1, H3, D2, M1, M4, and M5. Each individual in silico experiment was carried out using FlexX program embedded in the LeadIT software package (version 2.1.8). We have previously performed studies on identifying nausea-associated receptors and simulated their structure in the absence of crystallographic data by means of homology modeling. The method and the verification of the structure qualities of H3R, D2R, M1R, M4R, and M5R studied here have been described elsewhere.6b Briefly, the structures of H3, D2 and M1, M4, and M5 receptors were determined by means of homology modeling using the Phyre Protein Homology Recognition Engine.59 D2R was simulated based on the beta-2-adrenergic receptor (PDB entry 2HR1, for additional details see Flynn et al.,6b), and the muscarinic receptors were based on the structure of the human M2 muscarinic ACh receptor as template all with 100% prediction confidence. The templates were used to model the target proteins using the SWISS-MODEL workspace.60 After removing atomic clashes in the simulated structure by energy minimization, the quality of the structures was assessed using PROCHECK61 and confirmed based on various factors including the high percentage of the residues in the core regions of the Ramachandran plot.6b
The crystal structure of H1R was retrieved from the Protein Data Bank (PDB) entry 3RZE,20b in which water molecules were removed, side chains and atom charges were calculated, and protonation states were adjusted for energy minimization with AMBER7 FF99 force field.
The binding pocket of H1R can accommodate two different charge states of the agonist histamine, dicationic with a protonated imidazole ring and monocationic with a neutral imidazole ring.62 All endogenous substrates of the nausea receptors (i.e., histamine, dopamine, and ACh)6b have the amine group in the protonated state and are positively charged.62,63
4.1.1. H1R
The binding pocket of H1R (PDB 3RZE),20b composed of hydrophobic residues, is formed by three α-helices: α3 (residues 96–130), α5 (188–216), and α6 (408–441).6b,20b The crystal structure used for H1R contains the antagonist doxepin that was used as a reference ligand for the docking experiments into H1R. A spherical region with a radius of 20 Å surrounding doxepin was chosen as the target site for molecular docking of histamine, VLB, and its metabolites.
4.1.2. H3R
The binding pocket of H3R (UniProtKB Q9Y5N1) is also formed by hydrophobic interactions that involves helices α3, α4, and α5, which respectively comprise residues 104–137, 141–178, and 196–225.6b,20a,64 A spherical region with a radius of 20 Å around residue Asp114 located at α3 was selected as the target site for docking VLB, its metabolites, and histamine into the binding site of the homology model of H3R.
4.1.3. D2R
The binding pocket of D2R (UniProtKB P14416) is located between helices α3 (residues 104–137), α4 (151–171), α5 (187–220), and α6 (336–369). Asp114 is directly involved in H-bonding with the endogenous substrate dopamine as well as with known dopaminergic antagonists (e.g., clozapine) leading to receptor activation and inactivation, respectively.6b,65 A spherical region with a radius of 20 Å around Asp114 was defined as the target site for docking the metabolites, VLB, and dopamine into D2R. However, choosing the central point for the space encompassing the docking region technically does not have to be based on the role of the central amino acid but rather the residue’s coordination, such that the specified sphere around it should cover all the residues that potentially could involve with a ligand binding.
4.1.4. M1R
The hydrophobic pocket located at helices α2, α6, and α7 is involved in the binding of ACh and antagonists into M1R (UniProtKB P11229) whereas α3, α4, α5, α6, and α7 interact with the anticholinergic agents. To define the binding site of M1R, a spherical region with a radius of 20 Å around residue Asp105 was chosen as the target site for docking VLB, its metabolites, and ACh. Asp105 is involved in the binding of agonists and antagonists of M1R through electrostatic interactions.6b,66
4.1.5. M4R
For M4R (UniProtKB P08173), a spherical region with a radius of 20 Å around residue Tyr113 was defined as the target site for docking VLB, its metabolites, and ACh.66b,67
4.1.6. M5R
The binding site of M5R (UniProtKB P08912) is located between α2 (residues 62–93), α3 (99–133), α4 (142–166), α5 (189–215), α6 (435–467), and α7 (472–495). A spherical region with a radius of 20 Å surrounding residue Asp109 was chosen as the target site for docking the natural substrate, VLB, and its metabolites. Asp109 is involved in the agonist and antagonist interactions within M5R.6b,68
4.1.7. Tubulin
The crystal structure of tubulin was retrieved from PDB with entry code of 4EB6.69 Water molecules were removed, side chains with missing residues were completed, and atom charges were set as appropriate. Protonation states were adjusted prior to energy minimization with AMBER7 FF99 force field. The crystal structure used in this study (4EB6) is originated from Ovis aries species (sheep). However, the amino acid sequence is more than 90% similar to the human sequence. The residues known to be involved in the binding of VLB share 100% sequence identity with Homo sapiens.28,69 The structure of tubulin crystalized with VLB2b,69 shows that the drug binds to the interface of β- and α-tubulin. Thus, a spherical region with a radius of 10 Å surrounding the reference ligand was selected as the target site for docking.
4.2. In Silico Prediction of ADMET Properties
In the absence of in vitro and in vivo data, ADMET Predictor (version 7.2),33 a software package developed by Simulation Plus, was utilized for predicting the ADMET properties of the VLB metabolites according to their chemical structure under pH 7.4. The software provided in silico data for over 140 PK parameters such as the gastric and intestinal solubility, log P, and the likelihood of drugs crossing the BBB. In addition, it calculated the affinity for a diverse number of proteins/enzymes or receptors involved in drug effectiveness and DDI, such as P-gp, plasma proteins, hERG channel, as well as phases I and II of drug metabolism. This was performed by assessing the likelihood of compounds binding to CYP450 enzymes and glucuronosyltransferases (UGT).
Our research group has recently investigated the ADMET properties of tamoxifen and its metabolites providing satisfactory results which are in agreement with previous in vivo and in vitro research.6b In addition, many other research groups have demonstrated in-depth characterization of the ADMET profiles of promising anti-cancer agents,70 including PK properties of drug metabolites and their roles in drug toxicity,71 for which ADMET predictor was utilized as an in silico modeling tool for prediction of the PK of VLB and its metabolites.
Glossary
Abbreviations
- VLB
vinblastine
- ADMET
absorption, distribution, metabolism, excretion, toxicity
- ADRs
adverse drug reactions
- VD
vindoline moiety of VLB
- CT
catharantine moiety of VLB
- P-gp
P-glycoprotein
- PK
pharmacokinetics
- hERG
human ether-à-go-go-related gene channel
- CINV
chemotherapy-induced nausea and vomiting
- DDI
drug–drug interactions
- GI
gastrointestinal
- MW
molecular weight
- ACh
acetylcholine
- Vd
volume of distribution
- Peff
human jejunal effective permeability
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00652.
Binding interactions of metabolite 23 docked into the binding site of M1R; binding interactions of VLB docked into the vinca site of tubulin; binding energies of VLB, its metabolites, and dopamine docked into D2R; binding energies of VLB, its metabolites, and histamine docked into H1R; binding energies of VLB, its metabolites, and histamine docked into H3R; binding energies of VLB, its metabolites, and ACh docked into M1R; binding energies of VLB, its metabolites, and ACh docked into M4R; binding energies of VLB, its metabolites, and ACh docked into M5R; binding energy of VLB and its metabolites docked into the vinca site of tubulin; and kinetic and intrinsic parameters (Km, Vmax, and CLint) of VLB and its metabolites for CYP2D6 (PDF)
Author Contributions
The manuscript was written through contributions of the authors. The authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Johnson I. S.; Armstrong J. G.; Gorman M.; Burnett J. P. Jr. The Vinca Alkaloids: A New Class of Oncolytic Agents. Cancer Res. 1963, 23, 1390–1427. [PubMed] [Google Scholar]
- a Moudi M.; Go R.; Yien C. Y.; Nazre M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235. [PMC free article] [PubMed] [Google Scholar]; b Li Z.; Alisaraie L. Microtubules dual chemo and thermo-responsive depolymerization. Proteins 2015, 83, 970–981. 10.1002/prot.24793. [DOI] [PubMed] [Google Scholar]
- Long Q.-Z.; Zhou M.; Liu X.-G.; Du Y.-F.; Fan J.-H.; Li X.; He D.-L. Interaction of CCN1 with αvβ3 integrin induces P-glycoprotein and confers vinblastine resistance in renal cell carcinoma cells. Anticancer Drugs 2013, 24, 810–817. 10.1097/cad.0b013e328363046d. [DOI] [PubMed] [Google Scholar]
- Malik M. Z.; Ahmad M.; Muahammad S.; Aamir M. N. Pharmacokinetic evaluation of anticancer drugs in Hodgkin’s lymphoma patients after their simultaneous administration. Pak. J. Pharm. Sci. 2016, 29, 2079–2082. [PubMed] [Google Scholar]
- Auyeung K. K. W.; Law P. C.; Ko J. K. S. Combined therapeutic effects of vinblastine and Astragalus saponins in human colon cancer cells and tumor xenograft via inhibition of tumor growth and proangiogenic factors. Nutr. Cancer 2014, 66, 662–674. 10.1080/01635581.2014.894093. [DOI] [PubMed] [Google Scholar]
- a Chagas C. M.; Moss S.; Alisaraie L. Drug metabolites and their effects on the development of adverse reactions: Revisiting Lipinski’s Rule of Five. Int. J. Pharm. 2018, 549, 133–149. 10.1016/j.ijpharm.2018.07.046. [DOI] [PubMed] [Google Scholar]; b Flynn M.; Heale K. A.; Alisaraie L. Mechanism of Off-Target Interactions and Toxicity of Tamoxifen and Its Metabolites. Chem. Res. Toxicol. 2017, 30, 1492–1507. 10.1021/acs.chemrestox.7b00112. [DOI] [PubMed] [Google Scholar]
- a Zhou X.-J.; Rahmani R. Preclinical and clinical pharmacology of vinca alkaloids. Drugs 1992, 44, 1–16. 10.2165/00003495-199200444-00002. [DOI] [PubMed] [Google Scholar]; Cancer Care Ontario —Vinblastine, 2016; [Google Scholar]; c Magee M.Clinical Guide to Antineoplastic Therapy—A Chemotherapy Handbook, 3rd ed.; Oncology Nursing Society, 2014. [Google Scholar]
- a BC Cancer Agency . Cancer Drug Manual—Vinblastine, 2015;; b Pfizer . Product Monography—Vinblastine Sulfate Injection, 2017. [Google Scholar]
- Zhou X. J.; Martin M.; Placidi M.; Cano J. P.; Rahmani R. In vivo and in vitro pharmacokinetics and metabolism of vincaalkaloids in rat II. Vinblastine and Vincristine. Eur. J. Drug Metab. Pharmacokinet. 1990, 15, 323–332. 10.1007/bf03190222. [DOI] [PubMed] [Google Scholar]
- Bau R.; Jin K. K. Crystal structure of vinblastine. J. Chem. Soc., Perkin Trans. 1 2000, 2079–2082. 10.1039/b001855o. [DOI] [Google Scholar]
- a Béni Z.; Háda V.; Dubrovay Z.; Szántay C. Jr. Structure elucidation of indole–indoline type alkaloids: a retrospective account from the point of view of current NMR and MS technology. J. Pharm. Biomed. Anal. 2012, 69, 106–124. 10.1016/j.jpba.2012.02.015. [DOI] [PubMed] [Google Scholar]; b Háda V.; Dubrovay Z.; Lakó-Futó Á.; Galambos J.; Gulyás Z.; Aranyi A.; Szántay C. Jr. NMR and mass spectrometric characterization of vinblastine, vincristine and some new related impurities-Part II. J. Pharm. Biomed. Anal. 2013, 84, 309–322. 10.1016/j.jpba.2012.09.008. [DOI] [PubMed] [Google Scholar]
- de Graeve J.; van Heugen J. C.; Zorza G.; Fahy J.; Puozzo C. Metabolism pathway of vinorelbine (Navelbine) in human: characterisation of the metabolites by HPLC-MS/MS. J. Pharm. Biomed. Anal. 2008, 47, 47–58. 10.1016/j.jpba.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Elmarakby S. A.; Duffel M. W.; Rosazza J. P. N. In vitro metabolic transformations of vinblastine: oxidations catalyzed by human ceruloplasmin. J. Med. Chem. 1989, 32, 2158–2162. 10.1021/jm00129a023. [DOI] [PubMed] [Google Scholar]
- a Shankar A.; Roy S.; Malik A.; Julka P.; Rath G. Prevention of Chemotherapy-Induced Nausea and Vomiting in Cancer Patients. Asian Pac. J. Cancer Prev. 2015, 16, 6207–6213. 10.7314/apjcp.2015.16.15.6207. [DOI] [PubMed] [Google Scholar]; b Beusterien K.; Grinspan J.; Kuchuk I.; Mazzarello S.; Dent S.; Gertler S.; Bouganim N.; Vandermeer L.; Clemons M. Use of conjoint analysis to assess breast cancer patient preferences for chemotherapy side effects. Oncologist 2014, 19, 127–134. 10.1634/theoncologist.2013-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Holt K. Common side effects and interactions of colorectal cancer therapeutic agents. J. Pract. Nurs. 2011, 61, 7–20. [PubMed] [Google Scholar]; d Kuchuk I.; Bouganim N.; Beusterien K.; Grinspan J.; Vandermeer L.; Gertler S.; Dent S. F.; Song X.; Segal R.; Mazzarello S.; Crawley F.; Dranitsaris G.; Clemons M. Preference weights for chemotherapy side effects from the perspective of women with breast cancer. Breast Canc. Res. Treat. 2013, 142, 101–107. 10.1007/s10549-013-2727-3. [DOI] [PubMed] [Google Scholar]
- Yoodee J.; Permsuwan U.; Nimworapan M. Efficacy and safety of olanzapine for the prevention of chemotherapy-induced nausea and vomiting: A systematic review and meta-analysis. Crit. Rev. Oncol.-Hematol. 2017, 112, 113–125. 10.1016/j.critrevonc.2017.02.017. [DOI] [PubMed] [Google Scholar]
- O’Brien C. Nausea and vomiting. Can. Fam. Physician 2008, 54, 861–863. [PMC free article] [PubMed] [Google Scholar]
- Lohr L. Chemotherapy-induced nausea and vomiting. Cancer J. 2008, 14, 85–93. 10.1097/ppo.0b013e31816a0f07. [DOI] [PubMed] [Google Scholar]
- Salmas R. E.; Yurtsever M.; Stein M.; Durdagi S. Modeling and protein engineering studies of active and inactive states of human dopamine D2 receptor (D2R) and investigation of drug/receptor interactions. Mol. Divers. 2015, 19, 321–332. 10.1007/s11030-015-9569-3. [DOI] [PubMed] [Google Scholar]
- Osinski M.; Uchic M.; Seifert T.; Shaughnessy T.; Miller L.; Nakane M.; Cox B.; Brioni J.; Moreland R. Dopamine D2, but not D4, receptor agonists are emetogenic in ferrets. Pharmacol. Biochem. Behav. 2005, 81, 211–219. 10.1016/j.pbb.2005.03.012. [DOI] [PubMed] [Google Scholar]
- a Ishikawa M.; Watanabe T.; Kudo T.; Yokoyama F.; Yamauchi M.; Kato K.; Kakui N.; Sato Y. Investigation of the histamine H3 receptor binding site. Design and synthesis of hybrid agonists with a lipophilic side chain. J. Med. Chem. 2010, 53, 6445–6456. 10.1021/jm100643t. [DOI] [PubMed] [Google Scholar]; b Shimamura T.; Shiroishi M.; Weyand S.; Tsujimoto H.; Winter G.; Katritch V.; Abagyan R.; Cherezov V.; Liu W.; Han G. W.; Kobayashi T.; Stevens R. C.; Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sullivant A.; Mackin A.; Pharr T.; Cooley J.; Wills R.; Archer T. Identification of histamine receptors in the canine gastrointestinal tract. Vet. Immunol. Immunopathol. 2016, 182, 29–36. 10.1016/j.vetimm.2016.09.010. [DOI] [PubMed] [Google Scholar]
- Thurmond R. L. Histamine inflammation. Preface. Adv. Exp. Med. Biol. 2010, 709, vii–viii. [PubMed] [Google Scholar]
- Bailey F.; Davies A. The misuse/abuse of antihistamine antiemetic medication (cyclizine) by cancer patients. Palliat. Med. 2008, 22, 869–871. 10.1177/0269216308094337. [DOI] [PubMed] [Google Scholar]
- Doenicke A. W.; Hoernecke R.; Celik I. Premedication with H1 and H2 blocking agents reduces the incidence of postoperative nausea and vomiting. Inflamm. Res. 2004, 53, S154–S158. 10.1007/s00011-004-0367-0. [DOI] [PubMed] [Google Scholar]
- Ishii M.; Kurachi Y. Muscarinic acetylcholine receptors. Curr. Pharm. Des. 2006, 12, 3573–3581. 10.2174/138161206778522056. [DOI] [PubMed] [Google Scholar]
- Lochner M.; Thompson A. J. The muscarinic antagonists scopolamine and atropine are competitive antagonists at 5-HT 3 receptors. Neuropharmacology 2016, 108, 220–228. 10.1016/j.neuropharm.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhong W.; Picca A. J.; Lee A. S.; Darmani N. A. Ca2+ signaling and emesis: Recent progress and new perspectives. Auton. Neurosci. 2017, 202, 18–27. 10.1016/j.autneu.2016.07.006. [DOI] [PubMed] [Google Scholar]; b Golding J. F.; Stott J. R. R. Comparison of the effects of a selective muscarinic receptor antagonist and hyoscine (scopolamine) on motion sickness, skin conductance and heart rate. Br. J. Clin. Pharmacol. 1997, 43, 633–637. 10.1046/j.1365-2125.1997.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brouhard G. J.; Rice L. M. The contribution of αβ-tubulin curvature to microtubule dynamics. J. Cell Biol. 2014, 207, 323–334. 10.1083/jcb.201407095. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stanton R. A.; Gernert K. M.; Nettles J. H.; Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. 10.1002/med.20242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B.; Wang C.; Ravelli R. B. G.; Roussi F.; Steinmetz M. O.; Curmi P. A.; Sobel A.; Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- Li Z.; Tozer T.; Alisaraie L. Molecular dynamics studies for optimization of noncovalent loading of vinblastine on single-walled carbon nanotube. J. Phys. Chem. A 2016, 120, 4061–4070. 10.1021/acs.jpcc.5b10646. [DOI] [Google Scholar]
- a Rashid A.; Kuppa A.; Kunwar A.; Panda D. Thalidomide (5HPP-33) suppresses microtubule dynamics and depolymerizes the microtubule network by binding at the vinblastine binding site on tubulin. Biochemistry 2015, 54, 2149–2159. 10.1021/bi501429j. [DOI] [PubMed] [Google Scholar]; b Chi S.; Xie W.; Zhang J.; Xu S. Theoretical insight into the structural mechanism for the binding of vinblastine with tubulin. J. Biomol. Struct. Dyn. 2015, 33, 2234–2254. 10.1080/07391102.2014.999256. [DOI] [PubMed] [Google Scholar]; c Allemann O.; Cross R. M.; Brütsch M. M.; Radakovic A.; Boger D. L. Key analogs of a uniquely potent synthetic vinblastine that contain modifications of the C20′ ethyl substituent. Bioorg. Med. Chem. Lett. 2017, 27, 3055–3059. 10.1016/j.bmcl.2017.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Va P.; Campbell E. L.; Robertson W. M.; Boger D. L. Total Synthesis and Evaluation of a Key Series of C5-Substituted Vinblastine Derivatives. J. Am. Chem. Soc. 2010, 132, 8489–8495. 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogihara T.; Kamiya M.; Ozawa M.; Fujita T.; Yamamoto A.; Yamashita S.; Ohnishi S.; Isomura Y. What kinds of substrates show P-glycoprotein-dependent intestinal absorption? Comparison of verapamil with vinblastine. Drug Metab. Pharmacokinet. 2006, 21, 238–244. 10.2133/dmpk.21.238. [DOI] [PubMed] [Google Scholar]
- Predictor A.ADMET Predictor User Manual, 7.2; March 13, 2017.
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3-25. 1. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Graham L. P.Introduction to Medicinal Chemistry, 5th ed.; Oxford University Press: Oxford, 2013. [Google Scholar]
- Haga T. Molecular properties of muscarinic acetylcholine receptors. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 226–256. 10.2183/pjab.89.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donigian D. W.; Owellen R. J. Interaction of vinblastine, vincristine and colchicine with serum proteins. Biochem. Pharmacol. 1973, 22, 2113–2119. 10.1016/0006-2952(73)90110-x. [DOI] [PubMed] [Google Scholar]
- Pandya P.; Agarwal L. K.; Gupta N.; Pal S. Molecular recognition pattern of cytotoxic alkaloid vinblastine with multiple targets. J. Mol. Graph. Model. 2014, 54, 1–9. 10.1016/j.jmgm.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Levêque D.; Jehl F. Molecular pharmacokinetics of catharanthus (vinca) alkaloids. J. Clin. Pharmacol. 2007, 47, 579–588. 10.1177/0091270007299430. [DOI] [PubMed] [Google Scholar]
- Daneman R.; Prat A. The blood-brain barrier. Cold Spring Harbor Perspect. Biol. 2015, 7, a020412. 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cisternino S.; Rousselle C.; Debray M.; Scherrmann J. M. In vivo saturation of the transport of vinblastine and colchicine by P-glycoprotein at the rat blood-brain barrier. Pharm. Res. 2003, 20, 1607–1611. 10.1023/a:1026187301648. [DOI] [PubMed] [Google Scholar]; b Drion N.; Lemaire M.; Lefauconnier J. M.; Scherrmann J. M. Role of P-glycoprotein in the blood-brain transport of colchicine and vinblastine. J Neurochem 1996, 67, 1688–1693. 10.1046/j.1471-4159.1996.67041688.x. [DOI] [PubMed] [Google Scholar]
- Horio M.; Gottesman M. M.; Pastan I. ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 3580–3584. 10.1073/pnas.85.10.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sharom F. J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. 10.1042/bse0500161. [DOI] [PubMed] [Google Scholar]; b Silva R.; Vilas-Boas V.; Carmo H.; Dinis-Oliveira R. J.; Carvalho F.; de Lourdes Bastos M.; Remião F. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. 10.1016/j.pharmthera.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Diaz G. J.; Daniell K.; Leitza S. T.; Martin R. L.; Su Z.; McDermott J. S.; Cox B. F.; Gintant G. A. The [3H]dofetilide binding assay is a predictive screening tool for hERG blockade and proarrhythmia: Comparison of intact cell and membrane preparations and effects of altering [K+]o. J. Pharmacol. Toxicol. Methods 2004, 50, 187–199. 10.1016/j.vascn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- DrugBank . Vinblastine https://www.drugbank.ca/drugs/DB00570 (accessed March 25, 2017).
- Mikaelian I.; Buness A.; de Vera-Mudry M.-C.; Kanwal C.; Coluccio D.; Rasmussen E.; Char H. W.; Carvajal V.; Hilton H.; Funk J.; Hoflack J.-C.; Fielden M.; Herting F.; Dunn M.; Suter-Dick L. Primary endothelial damage is the mechanism of cardiotoxicity of tubulin-binding drugs. Toxicol. Sci. 2010, 117, 144–151. 10.1093/toxsci/kfq189. [DOI] [PubMed] [Google Scholar]
- Legha S. S.; Hodges C.; Ring S. Efficacy of ondansetron against nausea and vomiting caused by dacarbazine-containing chemotherapy. Cancer 1992, 70, 2018–2020. . [DOI] [PubMed] [Google Scholar]
- Chandrani G. Drug Metabolism & Pharmacokinetics in Drug Discovery: A Primer for Bioanalytical Chemists, Part I. Curr. Sep. 2000, 19, 17. [Google Scholar]
- Medscape . Drug Interactions, 2017. [Google Scholar]
- Wang B.; Yang L.-P.; Zhang X.-Z.; Huang S.-Q.; Bartlam M.; Zhou S.-F. New insights into the structural characteristics and functional relevance of the human cytochrome P450 2D6 enzyme. Drug Metab. Rev. 2009, 41, 573–643. 10.1080/03602530903118729. [DOI] [PubMed] [Google Scholar]
- Wenzell C. M.; Berger M. J.; Blazer M. A.; Crawford B. S.; Griffith N. L.; Wesolowski R.; Lustberg M. B.; Phillips G. S.; Ramaswamy B.; Mrozek E.; Flynn J. M.; Shapiro C. L.; Layman R. M. Pilot study on the efficacy of an ondansetron- versus palonosetron-containing antiemetic regimen prior to highly emetogenic chemotherapy. Support. Care Cancer 2013, 21, 2845–2851. 10.1007/s00520-013-1865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef A. S.; Parkman H. P.; Nagar S. Domperidone interacts with pioglitazone but not with ondansetron via common CYP metabolismin vitro. Xenobiotica 2014, 44, 792–803. 10.3109/00498254.2014.899406. [DOI] [PubMed] [Google Scholar]
- Zanger U. M.; Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Zhou-Pan X. R.; Seree E.; Zhou X. J.; Placidi M.; Maurel P.; Barra Y.; Rahmani R. Involvement of human liver cytochrome P450 3A in vinblastine metabolism: drug interactions. Cancer Res. 1993, 53, 5121–5126. [PubMed] [Google Scholar]
- Baumhäkel M.; Kasel D.; Rao-Schymanski R. A.; Böcker R.; Beckurts K. T.; Zaigler M.; Barthold D.; Fuhr U. Screening for inhibitory effects of antineoplastic agents on CYP3A4 in human liver microsomes. Int. J. Clin. Pharmacol. Ther. 2001, 39, 517–528. 10.5414/cpp39517. [DOI] [PubMed] [Google Scholar]
- Sorich M. J.; McKinnon R. A.; Miners J. O.; Smith P. A. The Importance of Local Chemical Structure for Chemical Metabolism by Human Uridine 5′-Diphosphate–Glucuronosyltransferase. J. Chem. Inf. Model. 2006, 46, 2692–2697. 10.1021/ci600248e. [DOI] [PubMed] [Google Scholar]
- Owellen R. J.; Hartke C. A.; Hains F. O. Pharmacokinetics and metabolism of vinblastine in humans. Cancer Res. 1977, 37, 2597–2602. [PubMed] [Google Scholar]
- Szakács G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery 2006, 5, 219–234. 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Kelley L. A.; Sternberg M. J. E. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Bordoli L.; Kiefer F.; Arnold K.; Benkert P.; Battey J.; Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2008, 4, 1–13. 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/s0021889892009944. [DOI] [Google Scholar]
- Ratnala V. R. P.; Kiihne S. R.; Buda F.; Leurs R.; de Groot H. J. M.; DeGrip W. J. Solid-state NMR evidence for a protonation switch in the binding pocket of the H1 receptor upon binding of the agonist histamine. J. Am. Chem. Soc. 2007, 129, 867–872. 10.1021/ja0652262. [DOI] [PubMed] [Google Scholar]
- a Lichman B. R.; Gershater M. C.; Lamming E. D.; Pesnot T.; Sula A.; Keep N. H.; Hailes H. C.; Ward J. M. “Dopamine-first” mechanism enables the rational engineering of the norcoclaurine synthase aldehyde activity profile. FEBS J. 2015, 282, 1137–1151. 10.1111/febs.13208. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schyman P.; Lai W.; Chen H.; Wang Y.; Shaik S. The directive of the protein: how does cytochrome P450 select the mechanism of dopamine formation?. J. Am. Chem. Soc. 2011, 133, 7977–7984. 10.1021/ja201665x. [DOI] [PubMed] [Google Scholar]; c Lioe H.; Barlow C. K.; O’Hair R. A. J. How does acetylcholine lose trimethylamine? A density functional theory study of four competing mechanisms. J. Am. Soc. Mass Spectrom. 2009, 20, 238–246. 10.1016/j.jasms.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Uveges A. J.; Kowal D.; Zhang Y.; Spangler T. B.; Dunlop J.; Semus S.; Jones P. G. The role of transmembrane helix 5 in agonist binding to the human H3 receptor. J. Pharmacol. Exp. Ther. 2002, 301, 451–458. 10.1124/jpet.301.2.451. [DOI] [PubMed] [Google Scholar]
- a Durdagi S.; Salmas R. E.; Stein M.; Yurtsever M.; Seeman P. Binding Interactions of Dopamine and Apomorphine in D2High and D2Low States of Human Dopamine D2 Receptor Using Computational and Experimental Techniques. ACS Chem. Neurosci. 2016, 7, 185–195. 10.1021/acschemneuro.5b00271. [DOI] [PubMed] [Google Scholar]; b Kalani M. Y. S.; Vaidehi N.; Hall S. E.; Trabanino R. J.; Freddolino P. L.; Kalani M. A.; Floriano W. B.; Kam V. W. T.; Goddard W. A. 3rd The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 3815–3820. 10.1073/pnas.0400100101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Platania C. B. M.; Salomone S.; Leggio G. M.; Drago F.; Bucolo C. Homology modeling of dopamine D2 and D3 receptors: molecular dynamics refinement and docking evaluation. PLoS One 2012, 7, e44316 10.1371/journal.pone.0044316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Peng J. Y.-c.; Vaidehi N.; Hall S. E.; Goddard W. A. 3rd The predicted 3D structures of the human M1 muscarinic acetylcholine receptor with agonist or antagonist bound. ChemMedChem 2006, 1, 878–890. 10.1002/cmdc.200600047. [DOI] [PubMed] [Google Scholar]; b Thal D. M.; Sun B.; Feng D.; Nawaratne V.; Leach K.; Felder C. C.; Bures M. G.; Evans D. A.; Weis W. I.; Bachhawat P.; Kobilka T. S.; Sexton P. M.; Kobilka B. K.; Christopoulos A. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. 10.1038/nature17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K.; Davey A. E.; Felder C. C.; Sexton P. M.; Christopoulos A. The role of transmembrane domain 3 in the actions of orthosteric, allosteric, and atypical agonists of the M4 muscarinic acetylcholine receptor. Mol. Pharmacol. 2011, 79, 855–865. 10.1124/mol.111.070938. [DOI] [PubMed] [Google Scholar]
- Huang X.; Zheng G.; Zhan C.-G. Microscopic binding of M5 muscarinic acetylcholine receptor with antagonists by homology modeling, molecular docking, and molecular dynamics simulation. J. Phys. Chem. B 2012, 116, 532–541. 10.1021/jp210579b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranaivoson F. M.; Gigant B.; Berritt S.; Joullie M.; Knossow M. Structural plasticity of tubulin assembly probed by vinca-domain ligands. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 927–934. 10.1107/s0907444912017143. [DOI] [PubMed] [Google Scholar]
- a El-Saadi M. W.; Williams-Hart T.; Salvatore B. A.; Mahdavian E. Use of in-silico assays to characterize the ADMET profile and identify potential therapeutic targets of fusarochromanone, a novel anti-cancer agent. In Silico Pharmacol. 2015, 3, 6. 10.1186/s40203-015-0010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Uba A. I.; Yelekçi K. Pharmacophore-based virtual screening for identification of potential selective inhibitors of human histone deacetylase 6. Comput. Biol. Chem. 2018, 77, 318–330. 10.1016/j.compbiolchem.2018.10.016. [DOI] [PubMed] [Google Scholar]; c Vaidhyanathan S.; Wang X.; Crison J.; Varia S.; Gao J. Z. H.; Saxena A.; Good D. Bioequivalence Comparison of Pediatric Dasatinib Formulations and Elucidation of Absorption Mechanisms Through Integrated PBPK Modeling. J. Pharm. Sci. 2019, 108, 741–749. 10.1016/j.xphs.2018.11.005. [DOI] [PubMed] [Google Scholar]; d Majcher U.; Klejborowska G.; Kaik M.; Maj E.; Wietrzyk J.; Moshari M.; Preto J.; Tuszynski J. A.; Huczynski A. Synthesis and Biological Evaluation of Novel Triple-Modified Colchicine Derivatives as Potent Tubulin-Targeting Anticancer Agents. Cells 2018, 7, 216. 10.3390/cells7110216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S. S.; Chavan B. B.; Kushwah B. S.; Yerra N. V.; Mukesh S.; Sangamwar A. T.; Thaota J. R.; Talluri M. V. N. K. In vitroand in vivo investigation of metabolic fate of riociguat by HPLC-Q-TOF/MS/MS and in silico evaluation of the metabolites by ADMET predictor. J. Pharm. Biomed. Anal. 2019, 164, 326–336. 10.1016/j.jpba.2018.10.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.