

Abstract
With more than a third of the genome encoding for metal-containing biomolecules, the in silico prediction of how metal ions bind to proteins is crucial in chemistry, biology, and medicine. To date, algorithms for metal-binding site prediction are mainly based on sequence analysis. Those methods have reached enough quality to predict the correct region of the protein and the coordinating residues involved in metal-binding, but they do not provide three-dimensional (3D) models. On the contrary, the prediction of accurate 3D models for protein–metal adducts by structural bioinformatics and molecular modeling techniques is still a challenge. Here, we present an update of our multipurpose molecular modeling suite, GaudiMM, to locate metal-binding sites in proteins. The approach is benchmarked on 105 X-ray structures with resolution lower than 2.0 Å. Results predict the correct binding site of the metal in the biological scaffold for all the entries in the data set. Generated 3D models of the protein–metal coordination complexes reach root-mean-square deviation values under 1.0 Å between calculated and experimental structures. The whole process is purely based on finding poses that satisfy metal-derived geometrical rules without needing sequence or fine electronic inputs. Additional post-optimizations, including receptor flexibility, have been tested and suggest that more extensive searches, required when the host structures present a low level of pre-organization, are also possible. With this new update, GaudiMM is now able to look for metal-binding sites in biological scaffolds and clearly shows how explicitly considering the geometric particularities of the first coordination sphere of the metal in a docking process provides excellent results.
Introduction
With more than 30% of the genome encoding for metal-containing biomolecules, the study of the interactions between metal ions and proteins bares a fundamental role in biology, pharmacy and medicine.1 Metals provide life with unique structural and catalytic properties for transport, storage, or enzymatic functions. Humans have taken advantage of this complementarity and now apply it in many fields like therapy (metallodrugs) and diagnosis (biosensors) of many diseases ranging from cancer to ulcer.2
Experimental techniques like X-ray crystallography or NMR spectroscopy can provide accurate or partial three-dimensional (3D) information of the metal-bound structure of a protein, but when those data are unreachable, the determination of the region where a metal ion could bind must be addressed computationally.3 A complete simulation of the metal-binding process should take into account: (1) the vast conformational space that must be explored to detect metal-binding sites in a protein; (2) the intrinsic properties of the first coordination sphere of the metal during binding, i.e., the directionality of the metal–ligand interactions or the possible changes in the number of chemical groups bound to metal. Nowadays, the panoply of molecular modeling methods struggles for this kind of predictions.
To the best of our knowledge, the available methods for predicting metal-binding sites are based on sequence analysis or structural motif analogies between the query and well-characterized metal-binding in proteins.4 Those relying on sequence homology tend to be limited to a specific subset of metal elements and protein residues, e.g., MetalDetector v2.05 considers transition metals and Cys or His residues; MetalPredator6 is focused on iron–sulfur proteins; SeqCHED Server7 supports Zn, Fe, Ni, Cu, Co, Mn, Mg, and Ca, and Cys, His, Glu, or Asp residues; and ZincFinder8 is available for Zn–proteins only. Structural motif analogies have been used by software such as mFASD,9 FINDSITE-metal,10 MIB,11 TEMSP,12 and FEATURE metal scanning,13 the latter two limited to Zn; however, they are restricted to a reduced subset of all possible metal elements.
To design a metal-binding predictor with a broader scope, we thought of using a strategy inspired by protein–ligand dockings, since these are designed to explore wide conformational spaces and provide fast predictions of ligand-binding modes to proteic receptors. Although docking is mainly focused on small organic species,14 in principle, the simplified force fields make the modifications of energetic terms (scoring functions) more approachable than in Molecular Mechanics, which opens some opportunities for metal implementations. However, attempts to predict metal-binding sites with protein–ligand dockings have only reached partial success.3c,15 Some stand on pure electrostatic models, which, despite their step forward, fail to respect coordination rules (e.g., providing the metal with an octahedral environment). Others, like those we recently developed inside the GOLD framework, stand on introducing some directionality to the vacant sites of the metal by explicitly simulating coordination bonds but are still restricted in terms of first coordination sphere description.16 Indeed, the user needs to define how the metal–protein interaction should take place in terms of angle and direction.
Here, we present a completely different approach to the problem. It is a new molecular descriptor (or objective) for our GaudiMM platform,25 which reproduces coordination geometries of metal moieties. Unlike existing strategies, this objective does not rely on structural templates extracted from large protein data sets but on pure coordination rules (mainly geometrical) that are structure agnostic. A benchmark of 105 structures shows that the algorithm can find the correct binding site in 100% of the entries of the data set and generate 3D models with root-mean-square deviation (RMSD) values under 1.0 Å from the crystallographic pose. The best solutions are always in the top 5 results, according to the coordination score detailed below. Post-analysis including protein flexibility maintains an excellent trend.
Computational Section
Data Set
To validate the new GaudiMM objective,25 a data set of 105 high-quality X-ray structures was built. All were selected via the MetalPDB web server17 and downloaded from the Protein Data Bank (PDB).18 They feature a bare metal cation bound to one or more side chains of a proteic host. Whereas the objective supports any arbitrary geometry that can be provided as a set of origin-centered vectors, the data set is focused on octahedral or derived geometries (i.e., square pyramidal or T-shaped) having at least one vacant coordination position (to consider possible water occupancy). Tetrahedral geometries are easier to model: a single angle can be used for all four vertices. Our chosen subset includes nontrivial coordination geometries commonly found in biological scaffolds. The vacancies could be already present in the original crystallographic structure or correspond to nonproteic ligands (e.g., water or carbonate), which are removed before running the calculation. The rationale behind this decision is to develop a general method able to find any potential coordination sites with a minimum number of donors and a compatible orientation according to the chosen geometry. Labile ligands like water molecules are, therefore, considered implicitly. Two additional criteria were taken into account to select the entries: (1) it must be a high-resolution structure (<2.0 Å when possible) and (2) no co-factors are bound to the metal. The resulting data set includes a wide range of biologically relevant metals (Mg, Ca, Mn, Fe, Co, Ni, Cu, Zn) and donor types (NHis, –SCys, –OSer, –OTyr, –OOCAsp, –OOCGlu, SMet, OCamide, –Namide, etc.; see Table S1 of the Supporting Information for further details).
Setup
All structures of the data set were prepared using a PyChimera19 script with the following instructions: (1) remove water and other nonproteic molecules present in the PDB structure, (2) add hydrogen atoms with UCSF Chimera v1.1114a (“addh” command), and (3) extract the coordinates of the bare metal ions and proteins in new, separate Mol2 files. It must be highlighted that for the largest proteins only one subunit chain, the one containing the binding site, was chosen for the simulation.
Calculation Protocol
GaudiMM provides a modular optimization framework for molecular modeling in which multiple competing evaluation criteria can be set up simultaneously. In GaudiMM, each iteration of the optimization is divided in two stages: exploration and evaluation. In the exploration stage, genes generate new possible solutions by assigning different values to the structural properties. In the evaluation stage, one or more objectives compute a score for each of those candidate solutions.
The GaudiMM genes used for this benchmark were (1) a molecule gene for the isolated protein structure, (2) another molecule gene for the isolated metal ion, and (3) a search gene instructed to move the metal ion within 20 Å of the crystallographic binding site position. This search radius covers, on average, ca. 55% of the volume of the selected proteins.
The objectives to be minimized were (1) the evaluation of clashes (interatomic unfavorable contacts) and (2) the coordination evaluator set to identify any possible binding site of the ion, with at least three donors within 3.5 Å of the metal, compatible with an octahedral geometry. The possible donor atom types considered by the coordination objective were Npl (trigonal planar sp2 nitrogen with a lone pair, e.g., His), O3 (tetrahedral sp3 oxygen, neutral with two lone pairs or negatively charged with three lone pairs, e.g., R–OH/R–O– of Tyr, Ser, or Thr), O2 (trigonal sp2 oxygen with two lone pairs, e.g., ketone group of the backbone), O2– (trigonal sp2 negatively charged oxygen in a carboxylic group, e.g., COO– of Asp or Glu), and S3 (tetrahedral sp3 sulfur, neutral with two lone pairs and negatively charged with 3, e.g., R–S–R/R–S– of Met or Cys). All calculations were performed with 150 generations and an initial population size of 100 individuals. The complete input configuration used in our benchmark can be found in the Supporting Information. The bare ions, extracted from the crystallographic structure, were re-docked to reproduce the experimental binding sites without any kind of geometrical constrains or energy restrains. The protein was treated as a rigid body. The GaudiMM solutions were analyzed by means of GaudiView,25,20 a UCSF Chimera extension developed to browse through docking solutions coming from GOLD, GaudiMM and others.
Coordination Objective
The coordination objective discovers sets of donor residues able to coordinate a metal ion (bare in this study, but it could be complexed by some ligands, if desired). Based on an early work,21 the method evaluates the immediate surroundings of different metal ion positions, both chemically and geometrically, as generated by the exploration stage of the algorithm. For each candidate, this GaudiMM objective first analyzes the presence of suitable donor atom types within a user-defined distance (3.5 Å by default) from the metal ion; if the number of atoms found is high enough to fulfill the minimum number of atoms requested (three, at least), a series of geometrical aspects are also calculated.
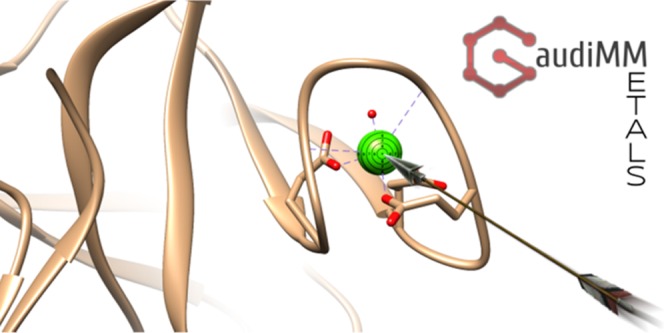
First, a RMSD calculation between the ideal polyhedron and the one obtained from the candidate’s coordination sphere is performed with a 3D rigid implementation of the Coherent Point Drift (CPD) algorithm.22 This term tells if the positions of the potential coordinating ligand atoms are adequate: a value of zero will report a perfect overlap of the atoms with the chosen coordination polyhedron. However, the directionality of the neighbor bonds is equally important. To account for that, for each donor found, the angles and dihedrals formed by (1) the metal center (probe), (2) the donor atom (donor), (3) its immediate neighbor (1st_neighbor), and (4) a nonterminal neighbor of 1st_neighbor (2nd_neighbor) are computed and the absolute sines of the difference between the found angles and the expected ideal values (as calculated out of the ideal atom positions suggested by the chimera.bondGeom module) are summed (see Figure 1). Thus, a sum equal to zero would correspond to an ideal directionality. Since the CPD algorithm scales the ideal polyhedron to fit the candidate vertices, the hypothetical coordination bonds can end up being accidentally larger or shorter than expected. We add one more term to control this error by computing the ideal distance deviation as the absolute difference between the ideal element–element distance reported by the chimera.bondGeom routine and the calculated probe–donor distance. It must be noted that no assumptions are made on the coordinating structure: the algorithm simply looks for suitable coordinating atoms arranged in a particular geometry; the atoms can belong to protein residues, but this does not constitute a requirement.
Figure 1.

Given a metal ion M, potential donors (D) are searched in its surroundings. If the distance M–D is smaller than the r threshold for more than three potential donors, the RMSD between the donor atom positions and the chosen ideal polyhedron (an octahedron in this figure) is computed. If the test is successful and D is bound to another atom (1st_neighbor), the M–D–1st_neighbor angle α is computed. If 1st_neighbor has another nonterminal neighbor (2nd_neighbor), the M–D–1st_neighbor–2nd_neighbor dihedral angle θ is also computed. The absolute sines of the differences between the calculated angles and their ideal counterparts (according to chimera.bondGeom routines) are then calculated. Since the polyhedron matching procedure is scalable, the absolute difference between the M–D distances and the expected ideal coordination distances for those elements are also computed. All the obtained terms are then summed together, and the sum should be zero for an ideal coordination geometry.
Finally, the total score (Coord_Fitness) is calculated by summing the RMSD, distance deviation, and directionality terms. Smaller values will correspond to better geometries (closer to the ideal polyhedron). A more detailed description of the algorithm, along with discarded alternative strategies, can be found in the Supporting Information (Figure S1).
Results and Discussion
The capabilities to predict metal-binding sites with our upgraded GaudiMM with coordination-aware docking features were evaluated considering the Coord_Fitness value and computing the RMSD of each docked metal ion versus the crystallographic coordinates. The binding site was considered fully reproduced if the Coord_Fitness value is below 5.0 and the obtained RMSD is below 1.0 Å. In the docking benchmarks reported in literature, the RMSD threshold applied to identify a successful solution usually ranges from 2.0 to 3.0 Å.3c,14b,23 In our case, working with a single atom, it is more appropriate to consider a smaller value. In combination with the Coord_Fitness score, this criterion can help avoid false-positive geometries. Steric clashes, measured as the volumetric overlap of the van der Waals spheres of close atoms by the contacts objective, were always below 3.0 Å3.
A detailed analysis of the GaudiMM solutions for the entire data set is summarized in Table 1, which shows that the crystallographic binding sites are reproduced with a success rate of 100% with a RMSD ≤ 1.0 Å by structures contained in the top 5 results. The mean of the Coord_Fitness score is close to 4.0 units. The mean RMSD was 0.519 Å, with an associated standard deviation value of 0.175 Å, highlighting an error distribution very close to the mean of the set. In Figure 2, as an example, a comparison between three simulated structures is reported.
Table 1. Summary of RMSD between the Experimental and the Predicted Binding Sites (First Column) and Coord_Fitness Distribution of the Data Set.
Value reported in Å.
RMSD computed via UCSF Chimera.
Value calculated by GaudiMM using eq S1 in the Supporting Information.
Figure 2.

Comparison between the GaudiMM solution (in orange) and the original X-ray diffraction (XRD) structure (in dots surface) for the PDB structures (a) 1EJJ, (b) 414A, and (c) 2Y12.
Although for each structure, the solution with the best value of the coordination objective (lowest Coord_Fitness score) correctly predicts the metal binding site, only in 86.7% of the cases the same solution presented the best RMSD value of the run. The polyhedron models considered in our objective are likely too ideal when it comes to cases where the metal geometry is skewed or lacks one or more coordination vertices. Also, the ideal distances and angles parametrized in chimera.bondGeom are measured for generic element atom types and do not account for partial charges, polarizabilities, etc. Since some ligands were removed prior to the calculation (e.g., water or carbonate ions, see Computational Section), deviations from the ideal polyhedra are expected in these systems and can explain why low-RMSD solutions can exhibit relatively high Coord_Fitness scores (see Table 1). Even so, the proposed geometric terms are enough in most situations: they still reach a 100% prediction power within the top 5.
To conclude this part of the study, our updated version of GaudiMM with metal coordination prediction capabilities provides the correct binding site of the ions with an average success rate ranging from ca. 87 to 100% (best RMSD value in top 1 or top 5 scores, respectively), even if not all the coordination positions are occupied.
Dealing with Receptor Flexibility and Rearrangement
The previous benchmark successfully reproduced the coordination geometries of the metal ions after exploring a rigid protein structure. Even if the possibility of vacant sites of the metal is considered, it could be argued that the calculation benefitted from the existing structural pre-organization of the protein. In this second assessment, we studied the performance of the coordination objective when considering full side-chain flexibility. Since this obviously implies tremendously wider computational time, we limited the test to a subset corresponding to the highest coordinated entry for each metal, for a total of eight structures from the complete data set (1AVW, 1AX1, 1B8C, 1B71, 1FX7, 1J5Y, 1XJS, and 3AWS). Moreover, we slightly changed the search strategy to save the computational time and avoid possible bias (i.e., starting the search too close to the X-ray structure). To do so, we applied an automated, structure-agnostic strategy that first locates potential metal-binding sites by probing the protein space for accessible regions in which the center of mass has β-carbon of three or more potential coordinating amino acids (Glu, His, Asp, etc.) within a threshold distance of 3.5 Å. The resulting script, called multisite.py, and the accompanying documentation are included in the Supporting Information.
The three best ranked metal-binding sites predicted with the multisite.py script were consequently processed with GaudiMM exploring the rotameric states of the surrounding amino acids (using the Dunbrack rotamer database24 included in the rotamers gene) and using two objectives: clashes and coordination. It is important to highlight that (1) from the three best ranked metal centers, the crystallographic one was systematically identified, showing that the multisite.py script could already be used in a standalone manner and (2) prior to the rotameric exploration, the orientation of all residues of the potential binding sites were randomly modified to avoid any possible bias in the pre-organization of the receptor.
The results, detailed in Table 2, successfully found 87.5% of the metal centers of this reduced data set with RMSD ≤ 1.2 Å. In 85.7% of the correctly found sites, three coordinating donors were correctly described, with a mean RMSD of 0.79 ± 0.34 Å.
Table 2. Summary of Reproduced Binding Sites, Donors, and RMSD Values Obtained after the Flexible Side Chain Docking Simulation.
| structure (PDB) | donorsXRD | rotamers | donorscalcd | RMSDa |
|---|---|---|---|---|
| 1AVW (Ca) | 3(−OOCAsp); 2(OCamide) | Glu70, Glu80, Glu77, Val75, Asn72 | –OOC(Glu80); OCamide(Val75) | 0.556 |
| 1AX1 (Mn) | 2(−OOCAsp); (−OOCGlu); (NHis) | Asp129, Asp136, Glu127, His142 | (−OOC)Asp129; (−OOC)Asp136; (−OOC)Glu127; (N)His142 | 1.238 |
| 1B8C (Mg) | 4(−OOCAsp); (OCamide) | Asp90, Asp92, Asp94, Ile96, Asp101 | (−OOC)Asp90; (−OOC)Asp92; (−OOC)Asp101; (OCamide)Lys96 | 0.792 |
| 1B71 (Fe) | 3(−OOCGlu); (NHis) | Glu94, Glu128, Glu53, His131 | (−OOC)Glu94; (−OOC)Glu128; (N)His131 | 0.268 |
| 1FX7 (Co) | 2(−OOCGlu); 2(NHis); (OCNGln) | His79, Glu83, His98, Glu172, Gln175 | (N)His79; (−OOC)Glu83; (N)His98; (−OOC)Glu172 | 0.889 |
| 1J5Y (Ni) | (−OOCGlu); 3(NHis)b | His148, His79, His146, Glu87 | c | c |
| 1XJS (Zn) | 3(−SCys); (−OOCAsp)b | Cys66, Cys128, Cys41, Asp43 | (−S)Cys128; (−S)Cys41; (−OOC)Asp43b | 1.023 |
| 3AWS (Cu) | (−OOCAsp); (NHis); (OCamide); (−Namide) | His82, Glu67, His68 | (−OOC)Glu67; (N)His82 | 0.391 |
| success rate | 87.5% |
RMSD value computed via UCSF Chimera, reported in Å.
Bidentate coordination of the carboxylate group.
The correct binding site was not identified during the calculation.
The high success rate of this reduced data set indicates that the coordination objective in our metal docking approach has an excellent predictive power even for explorations accounting for the flexibility of the receptor. The slightly worse result with respect to the rigid receptor exercise is likely related to the wider search space dimensionality of considering side-chain flexibility. Improving the exploration algorithm could potentially improve this success rate, and we are now working at possible updates. We mainly aim to implement a smarter search procedure and a heuristics-guided side-chain exploration gene.
Conclusions
The in silico prediction of the binding sites of metal ions in proteic scaffolds offers many possible applications at the chemical biology interface, like decoding the molecular processes that allowed life to take advantage of the inorganic realm or generating alternative metal-binding sites in proteins to provide them with novel functions or activities (e.g., artificial enzymes). Despite its interest, standard molecular modeling approaches fail to generate ab initio prediction of 3D models for metal-binding poses. Conceptually, the multiobjective genetic algorithm at the core of GaudiMM presents a suitable environment to advance along this line: each important descriptor involved in metal–protein interactions could be encoded as an objective. This work presents a new objective designed to reproduce geometric features without explicitly resorting to fine electronic details and offers an encouraging degree of predictiveness. We believe that this approach provides computational chemists and molecular modelers with a novel tool to handle metal ions in their computational workflows.
Acknowledgments
J.-D.M., G.S., and J.R.-G.P. are thankful for the support given by the Spanish grant CTQ2017-87889-P and the Generalitat de Catalunya grant 2017SGR1323. Support of COST Action CM1306 is kindly acknowledged. G.S. thanks the Universitat Autònoma de Barcelona for its support to his Ph.D. E.G. thank Fondazione di Sardegna (project FdS15Garribba) and FFABR 2017 “Fondo per il finanziamento delle attività base di ricerca” for the financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03457.
Data set specification (Table S1); coordination objective flowchart including supporting eqs S1 and S2 (Figure S1); strategies for increasing performance, RMSD deviations and Coord_Fitness values (Tables S2 and S3), RMSD distribution of the GaudiMM benchmarks (Table S4), and supporting eqs S3 and S4; benchmark input details; Multisite.py script (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Rehder D.Bioinorganic Chemistry; Oxford University Press: Oxford, 2014. [Google Scholar]
- a Guo Z.; Sadler P. J. Metals in Medicine. Angew. Chem., Int. Ed. 1999, 38, 1512–1537. 10.1002/(SICI)1521-3773(19990601)38:113.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; b Farrell N.Metal Complexes as Drugs and Chemotherapeutic Agents. In Comprehensive Coordination Chemistry II; McCleverty J. A.; Meyer T. J., Eds.; Pergamon: Oxford, 2003; Vol. 9, pp 809–840. [Google Scholar]; c Metallotherapeutic Drugs and Metal-Based Diagnostic Agents. The Use of Metals in Medicine; Gielen M.; Tiekink E. R. T., Eds.; John Wiley & Sons Ltd: Chichester, 2005. [Google Scholar]; d Thompson K. H.Medicinal Inorganic Chemistry: An Introduction. In Encyclopedia of Inorganic and Bioinorganic Chemistry; King R. B., Ed.; John Wiley & Sons, Ltd: Chichester, 2011. [Google Scholar]; e Interrelations between Essential Metal Ions and Human Diseases; Sigel A.; Sigel H.; Sigel R. K. O., Eds.; Springer Science+Business Media: Dordrecht, 2013. [Google Scholar]; f Mjos K. D.; Orvig C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. 10.1021/cr400460s. [DOI] [PubMed] [Google Scholar]; g Medici S.; Peana M.; Nurchi V. M.; Lachowicz J. I.; Crisponi G.; Zoroddu M. A. Noble metals in medicine: Latest advances. Coord. Chem. Rev. 2015, 284, 329–350. 10.1016/j.ccr.2014.08.002. [DOI] [Google Scholar]; h Jones C.; Thornback J.. Medicinal Applications of Coordination Chemistry; The Royal Society of Chemistry: Cambridge, 2007. [Google Scholar]; i Dabrowiak J. C.Metals in Medicine; John Wiley & Sons, Ltd, 2009. [Google Scholar]; j Bioinorganic Medicinal Chemistry; Alessio E., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA, 2011. [Google Scholar]
- a Farid R.; Day T.; Friesner R. A.; Pearlstein R. A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. 10.1016/j.bmc.2005.12.032. [DOI] [PubMed] [Google Scholar]; b Rarey M.; Kramer B.; Lengauer T.; Klebe G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]; c Jain A. N. Scoring Functions for Protein-Ligand Docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. 10.2174/138920306778559395. [DOI] [PubMed] [Google Scholar]
- Akcapinar G. B.; Sezerman O. U. Computational approaches for de novo design and redesign of metal-binding sites on proteins. Biosci. Rep. 2017, 37, BSR20160179 10.1042/BSR20160179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerini A.; Lippi M.; Frasconi P. MetalDetector v2.0: predicting the geometry of metal binding sites from protein sequence. Nucleic Acids Res. 2011, 39, W288–W292. 10.1093/nar/gkr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valasatava Y.; Rosato A.; Banci L.; Andreini C. MetalPredator: a web server to predict iron–sulfur cluster binding proteomes. Bioinformatics 2016, 32, 2850–2852. 10.1093/bioinformatics/btw238. [DOI] [PubMed] [Google Scholar]
- Levy R.; Edelman M.; Sobolev V. Prediction of 3D metal binding sites from translated gene sequences based on remote-homology templates. Proteins 2009, 76, 365–374. 10.1002/prot.22352. [DOI] [PubMed] [Google Scholar]
- Passerini A.; Andreini C.; Menchetti S.; Rosato A.; Frasconi P. Predicting zinc binding at the proteome level. BMC Bioinf. 2007, 8, 39 10.1186/1471-2105-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.; Liang Z.; Teng M.; Niu L. mFASD: a structure-based algorithm for discriminating different types of metal-binding sites. Bioinformatics 2015, 31, 1938–1944. 10.1093/bioinformatics/btv044. [DOI] [PubMed] [Google Scholar]
- Brylinski M.; Skolnick J. FINDSITE-metal: Integrating evolutionary information and machine learning for structure-based metal binding site prediction at the proteome level. Proteins 2011, 79, 735–751. 10.1002/prot.22913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-F.; Cheng C.-W.; Shih C.-S.; Hwang J.-K.; Yu C.-S.; Lu C.-H. MIB: Metal Ion-Binding Site Prediction and Docking Server. J. Chem. Inf. Model. 2016, 56, 2287–2291. 10.1021/acs.jcim.6b00407. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Xu M.; Liang Z.; Ding B.; Niu L.; Liu H.; Teng M. Structure-based de novo prediction of zinc-binding sites in proteins of unknown function. Bioinformatics 2011, 27, 1262–1268. 10.1093/bioinformatics/btr133. [DOI] [PubMed] [Google Scholar]
- Ebert J. C.; Altman R. B. Robust recognition of zinc binding sites in proteins. Protein Sci. 2008, 17, 54–65. 10.1110/ps.073138508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yuriev E.; Holien J.; Ramsland P. A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. 10.1002/jmr.2471. [DOI] [PubMed] [Google Scholar]; b Warren G. L.; Andrews C. W.; Capelli A.-M.; Clarke B.; LaLonde J.; Lambert M. H.; Lindvall M.; Nevins N.; Semus S. F.; Senger S.; Tedesco G.; Wall I. D.; Woolven J. M.; Peishoff C. E.; Head M. S. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]; c Guedes I. A.; de Magalhães C. S.; Dardenne L. E. Receptor–ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. 10.1007/s12551-013-0130-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Glen R. C. A fast empirical method for the calculation of molecular polarizability. J. Comput.-Aided Mol. Des. 1994, 8, 457–466. 10.1007/BF00125380. [DOI] [PubMed] [Google Scholar]; c Scholz C.; Knorr S.; Hamacher K.; Schmidt B. DOCKTITE—A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment. J. Chem. Inf. Model. 2015, 55, 398–406. 10.1021/ci500681r. [DOI] [PubMed] [Google Scholar]
- a Sciortino G.; Rodríguez-Guerra Pedregal J.; Lledós A.; Garribba E.; Maréchal J.-D. Prediction of the interaction of metallic moieties with proteins: an update for protein-ligand docking techniques. J. Comput. Chem. 2018, 39, 42–51. 10.1002/jcc.25080. [DOI] [PubMed] [Google Scholar]; b Sciortino G.; Garribba E.; Maréchal J.-D. Validation and Applications of Protein–Ligand Docking Approaches Improved for Metalloligands with Multiple Vacant Sites. Inorg. Chem. 2019, 58, 294–306. 10.1021/acs.inorgchem.8b02374. [DOI] [PubMed] [Google Scholar]; c Sanna D.; Ugone V.; Sciortino G.; Buglyo P.; Bihari Z.; Parajdi-Losonczi P. L.; Garribba E. VIVO complexes with antibacterial quinolone ligands and their interaction with serum proteins. Dalton Trans. 2018, 47, 2164–2182. 10.1039/C7DT04216G. [DOI] [PubMed] [Google Scholar]; d Sciortino G.; Sanna D.; Ugone V.; Micera G.; Lledós A.; Maréchal J.-D.; Garribba E. Elucidation of Binding Site and Chiral Specificity of Oxidovanadium Drugs with Lysozyme through Theoretical Calculations. Inorg. Chem. 2017, 56, 12938–12951. 10.1021/acs.inorgchem.7b01732. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Guerra Pedregal J.; Sciortino G.; Guasp J.; Municoy M.; Maréchal J.-D. GaudiMM: A modular multi-objective platform for molecular modeling. J. Comput. Chem. 2017, 38, 2118–2126. 10.1002/jcc.24847. [DOI] [PubMed] [Google Scholar]
- Andreini C.; Cavallaro G.; Lorenzini S.; Rosato A. MetalPDB: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2012, 41, D312–D319. 10.1093/nar/gks1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burley S. K.; Berman H. M.; Christie C.; Duarte J. M.; Feng Z.; Westbrook J.; Young J.; Zardecki C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018, 27, 316–330. 10.1002/pro.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rose P. W.; Prlić A.; Altunkaya A.; Bi C.; Bradley A. R.; Christie C. H.; Costanzo L. D.; Duarte J. M.; Dutta S.; Feng Z.; Green R. K.; Goodsell D. S.; Hudson B.; Kalro T.; Lowe R.; Peisach E.; Randle C.; Rose A. S.; Shao C.; Tao Y.-P.; Valasatava Y.; Voigt M.; Westbrook J. D.; Woo J.; Yang H.; Young J. Y.; Zardecki C.; Berman H. M.; Burley S. K. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. 10.1093/nar/gkw1000. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Guerra Pedregal J.; Maréchal J.-D. PyChimera: use UCSF Chimera modules in any Python 2.7 project. Bioinformatics 2018, 34, 1784–1785. 10.1093/bioinformatics/bty021. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Guerra J.Insilichem/gaudiview: Pre-alpha public release; Zenodo, 2017.
- Mujika J. I.; Rodríguez-Guerra Pedregal J.; Lopez X.; Ugalde J. M.; Rodríguez-Santiago L.; Sodupe M.; Maréchal J.-D. Elucidating the 3D structures of Al(III)–Aβ complexes: a template free strategy based on the pre-organization hypothesis. Chem. Sci. 2017, 8, 5041–5049. 10.1039/C7SC01296A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myronenko A.; Song X. Point Set Registration: Coherent Point Drift. IEEE Trans. Pattern Anal. Mach. Intell. 2010, 32, 2262–2275. 10.1109/TPAMI.2010.46. [DOI] [PubMed] [Google Scholar]
- a Ajay N. J. Scoring Functions for Protein-Ligand Docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. 10.2174/138920306778559395. [DOI] [PubMed] [Google Scholar]; b Hevener K. E.; Zhao W.; Ball D. M.; Babaoglu K.; Qi J.; White S. W.; Lee R. E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. 10.1021/ci800293n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huang S.-Y.; Grinter S. Z.; Zou X. Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. 10.1039/c0cp00151a. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bianco G.; Forli S.; Goodsell D. S.; Olson A. J. Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25, 295–301. 10.1002/pro.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack R. L.; Karplus M. Backbone-dependent Rotamer Library for Proteins Application to Side-chain Prediction. J. Mol. Biol. 1993, 230, 543–574. 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.