

Abstract
End-stage renal disease afflicts ∼750,000 Americans and claims >100,000 lives annually in the United States. Kidney transplantation is associated with longest survival and least cost but is limited by scarcity of donor organs. The balance of patients are treated with dialysis, a cumbersome, morbid, and expensive procedure. Each hemodialysis treatment consumes in excess of 160 liters of water and anchors the patient to a machine for 12–15 h per week. Cultured tubule cells can reduce the obligate fluid requirements of a bioengineered artificial kidney by concentrating wastes and reabsorbing filtered salt and water. Primary tubule epithelial cells rapidly dedifferentiate in culture and form a flattened epithelium lacking the brush border essential to apicobasal transport. We hypothesized that substrate mechanical properties have a strong influence on differentiation in primary cell culture. We cultured primary renal tubule cells on polyacrylamide hydrogels of varying elasticity and measured expression of key transporter proteins essential to renal tubule cell function. Primary tubule cells cultured on soft substrates for extended periods showed increased expression of key transporters characteristic of differentiated proximal tubule cells. These data support the hypothesis that scaffold elasticity is a critical factor in cell culture, and, unexpectedly, that prolonged culture of primary cells was essential to observing this difference.
Impact Statement
Successful clinical tissue engineering requires functional fidelity of the cultured cell to its in vivo counterpart, but this has been elusive in renal tissue engineering. Typically, renal proximal tubule cells in culture have a flattened morphology and do not express key transporters essential to their function. In this article, we show for the first time that in vitro substrate mechanical properties dictate differentiation of cultured renal proximal tubule cells. Remarkably, this effect was only discernable after 4 weeks in culture, longer than usually reported for this cell type. These results demonstrate a new tunable parameter to optimize cell differentiation in renal tissue engineering.
Keywords: renal tubule cell, matrix elasticity, NHE3
Introduction
Irreversible kidney failure affects ∼700,000 Americans and 2 million people worldwide. Our group has focused on biohybrid artificial organs as a technology solution to the epidemic of end-stage renal disease. Hemodialysis requires large volumes of water for dialysate or heavy sorbent cartridges, both of which pose a fundamental barrier to wearable or implantable therapies. Renal proximal tubule cells use chemical energy to transport large volumes of salt and water while they maintain a barrier to uremic toxins both known and unknown. Substitution of chemical energy for electrically driven roller pumps is fundamentally enabling for implanted therapies.
A strategy combining high-efficiency filters with a bioreactor of cultured renal epithelial cells offers the promise of clinically useful solute clearance with obligate fluid losses small enough to be managed by oral intake. Proximal tubule cells are responsible for 50–70% of salt and water reabsorption in the tubule, accomplish additional metabolic tasks, and grow readily in culture. Because of these features, proximal tubule cells may be the best initial choice for use in a bioartificial kidney.
Renal tubule cells grown in artificial culture incompletely copy the in vivo phenotype. Renal proximal tubule cells show a flattened morphology with an attenuated brush border when grown in culture.1,2 The architecture of the cell is essential to function. It is generally accepted that the elaborate apical brush border of proximal tubule cells is essential for cell function; similarly invaginations of the basolateral membrane place sodium–potassium ATPase in proximity to mitochondrial sources of ATP. However, in vitro renal tubule cells rapidly lose the morphologic features characteristic of their in vivo counterparts. We sought to establish cell culture conditions that restored differentiated morphology to renal tubule cells in vitro.
Characteristics of the extracellular matrix (ECM) in vivo have emerged as critical to cellular proliferation, differentiation, and fibrosis.3–9 In vitro, matrix elasticity appears to direct stem cell differentiation to osteogenic, myogenic, and neurogenic fates.2,10,11 Matrix elasticity influences cytokine signaling to mature cells by regulating ligand availability and may thereby perpetuate fibrotic responses to injury.12 We hypothesized that primary renal tubule cells might vary appearance and function when cultured on substrates with elasticities similar to healthy tissue. To test that hypothesis, we compared cell morphology and expression of transport and barrier genes between primary renal tubule cells grown on hydrogels with elasticities of 0.5–1 kPa and cells grown on hydrogels with elasticities of 40–50 kPa. Substrate stiffness values were chosen to represent physiological (0.5–1 kPa) and pathological stiffnesses (40–50 kPa). Physiological stiffness is based on equilibrium compressive modulus of decellularized porcine cortex (unpublished data under review) and acellular kidney tissue (glomerular basement membrane).13 Pathological kidney ECM stiffness in chronic kidney disease is unknown; however, it is likely significantly higher stiffness than that seen under normal physiological conditions. The stiffnesses chosen are similar to those used in a number of in vitro studies in multiple cell types, including kidney epithelial cells.14,15
Methods
Cell culture
Human renal epithelial cells (HRECs) (Innovative Biotherapies, Inc., Ann Arbor, MI) were cultured at 37°C in a humidified 5% CO2 atmosphere. Cells were maintained in a 50/50 mix of DMEM (Sigma D5030) and Ham's F12 (US Biological N8542-12) medium supplemented with 10 mL/L insulin, transferrin, and selenium (ITS-Gibco 100X), 5.5 mM glucose (Sigma), 2 mM sodium pyruvate (Gibco), 2 mM l-glutamine (Gibco), 5 mM HEPES (pH 7.4) (Gibco), 0.7 μg/L triiodothyronine (T3) (Sigma), 25 ng/mL prostaglandin E1 (#P7527; Sigma), 25 ng/mL hydrocortisone (#H0888, Sigma), 50 μM ascorbate 2-phosphate (#013-19641; Wako), 10 μg/L recombinant human epidermal growth factor (Invitrogen), and 2 mL/L Normosin (InvivoGen). Transforming growth factor β (TGF-β) type II receptor conditionally deficient murine proximal tubule cells were the generous gift of Dr. Leslie Gewin.16 Murine cells were maintained in DMEM/F-12 medium with supplements. For most hydrogel experiments, 100k low passage (P1–P4) human primary renal tubule epithelial cells were seeded per well, on either soft (0.5–1.0 kPa) or stiff (40–50 kPa) hydrogels in six-well plates. Soft (1.0 kPa) and stiff (40 kPa) hydrogels were cast in the principal investigator's laboratory, whereas the 0.5 and 50 kPa hydrogels were purchased commercially.
Scaffold construction
Scaffolds for this study were initially cast from polyacrylamide (PA) in the principal investigator's laboratory (0.5, 1.0, 10, and 40 kPa, Figs. 1, 6, and 7), then later purchased commercially (0.5 and 50 kPa) (Softwell Easy Coat, Matrigen Life Technologies, Brea, CA). PA gels were prepared as described in Tse and Engler.14 In brief, various amounts of acrylamide and bis-acrylamide were mixed in water to achieve specified stiffness. After polymerization between an aminosilanated coverslip and a chlorosilanated glass slide, the gels were incubated in Sulfo-SANPAH (Sigma, Saint Louis, MO) and exposed to UV light with a wavelength of 360 nm for 30 min. After thorough rinsing with 0.5 M HEPES buffer pH 8.5, the gels were then stored in phosphate buffered saline (PBS) at 4°C until used. Before cell seeding, functionalized hydrogel surfaces were cross-linked with protein for cell attachment by incubating with a 10 μg/mL solution of basement membrane matrix proteins (Matrigel, BD Biosciences) in PBS for 1–3 h at room temperature. The elastic modulus of the PA gels was measured using an Enduratech measurement system (Enduratec Electroforce 3100 testing system; Bose, Eden Prairie, MN) to confirm that the method produced stiffnesses very close to the expected range. Measurements were performed in a manner similar to Barnes et al.17 The system was fitted with a 50 g load cell with 1.5 mm radius spherical indentation tip. For preparation of hydrogels from cross-linked basement membrane matrix proteins, Matrigel was diluted to 6 mg/mL in the presence or absence of 0.05% glutaraldehyde (GLA; Sigma). About 1 mL of Matrigel was loaded into each Costar Transwell insert (Corning Incorporated–Life Sciences, Oneonta, NY) to form a thick layer of gel. For gels containing GLA, 50 mM NH4Cl was used to wash out the excess GLA before cells were plated on top as described.18 Softwell and Softslip Easy Coat hydrogels were purchased from Matrigen Life Technologies (Brea, CA) and stored at 4°C until needed. Before cell seeding, Matrigen hydrogels were incubated with a 10 μg/mL solution of reduced growth factor basement membrane proteins (Geltrex, Invitrogen) in PBS for 1–3 h at room temperature to cross-link the functionalized surfaces with protein for cell attachment. Hydrogels were rinsed once in sterile PBS after protein coating, then trypsinized cells in standard HREC medium were seeded and allowed to attach at 37°C.
FIG. 1.

Primary human renal tubule cell morphology varies as a function of substrate elasticity. Blue, DAPI, green, ZO-1. On softer substrates (A, 0.5 kPa, 1.0 kPa; B: Matrigel), renal tubule cells formed rounded colonies. On stiffer substrates (A: 10 kPa, 40 kPa, and B: Matrigel crosslinked with glutaraldehyde “Matrigel+GLA”), renal tubule cells formed more spread confluent monolayers. Scale bars = 20 μm. DAPI, 4′,6-diamidino-2-phenylindole.
FIG. 6.

Effect of TGF-β on morphology of cultured renal tubule cells. Smad2 phosphorylation was more pronounced in cells grown on gels with modulus of 10 and 40 kPa than in those grown on more elastic gels (A). Cells cultured on 1 kPa substrates formed rounded colonies of cells, unless TGF-β was added to culture medium, in which case cells formed more spread out confluent sheets (B; blue: DAPI; red: acetylated tubulin). Similarly, cells grown on 40 kPa substrates formed flat sheets, unless an inhibitor of TGF-β, SB431542 10 μM (Sigma) as added to culture medium, in which case cells formed rounded colonies (A). Murine renal proximal tubule cells also showed a stiffness-dependent culture morphology, but deletion of the TGF-β receptor 2 on those cells blocked the spreading effect of increased stiffness (C). The deletion of TGF-β receptor 2 in these murine proximal tubule cells was sufficient to block Smad2 phosphorylation, indicating that the deletion was successful (D). TGF, transforming growth factor.
FIG. 7.

Effect of substrate elasticity on TGF-β receptor and TGF-β expression by cultured renal tubule cells. TGF-β and TGF-β receptor expression levels were determined by qPCR in primary HREC cultured on soft (0.5 kPa) and stiff (50 kPa) hydrogels. TGF-B1R1 and R2 mRNA increased with increasing substrate stiffness from 0.5 to 40 kPa (A). However, TGF-β mRNA did not vary significantly as a function of substrate elasticity (B). The gene expression level was shown as the relative fold comparison with that from cells on 0.5 kPa gel, which was set as 1. *p < 0.05, **p < 0.01. The results were calculated from three independent experiments.
TGF-β signaling
TGF-β1 10 ng/mL (Invitrogen) and SB431542 10 μM (Sigma) of indicated concentrations were applied to subconfluent cells 24 h after cells had been plated. TGF-β1 was added to cells in a serum-free condition. SB431542, an inhibitor of TGF-β receptor 1 was added to medium 24 h before TGF-β1 treatment. Cells were cultured for 7–10 days with medium change on alternate days before analysis. For longer (4 week) experiments on soft (0.5 kPa) and stiff (50 kPa) hydrogels (Fig. 8), HRECs were seeded at 100k/well on six-well plates (Matrigen) and maintained for 10 days postconfluence with standard complete medium. After 10 days, medium was supplemented with either TGF-β1 (10 ng/mL) or SB431542 (10 μM) for the remaining 2.5 weeks, with fresh reagents added with every medium change at 2–3-day intervals.
FIG. 8.
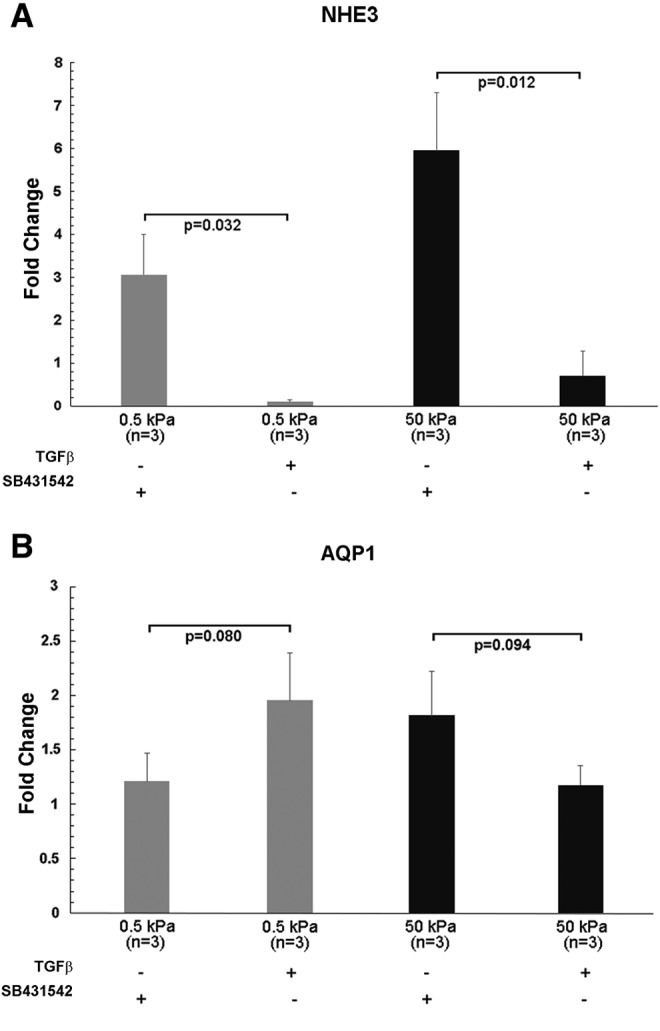
Effect of exogenous TGF-β on NHE3 and aquaporin-1 (AQP1) expression in cultured primary renal proximal tubule cells on soft and stiff substrates. Expression levels of SLC9A3 (NHE3) (A) and AQP1 (B) were determined by qPCR in primary HREC cultured on soft (0.5 kPa) and stiff (50 kPa) hydrogels for 4 weeks. Confluent cells in duplicate wells were cultured for 10 days on soft (0.5 kPa) and stiff (50 kPa) hydrogels, then treated with TGF-β (10 ng/mL) or treated with the TGF-β inhibitor SB431542 (10 μM) for 2.5 weeks before harvest. Fold change differences were normalized relative to GAPDH levels, and results from replicate wells were pooled for analysis.
RNA isolation and real-time PCR
Total RNA was isolated using the Micro or Mini RNeasy kits (Qiagen). RNA quality was determined by measuring absorbance at 260 and 280 nm on a Nanodrop One Spectrometer (Thermo Scientific). First-strand cDNA was synthesized from total RNA using the iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer's instructions. Real-time PCR was performed on triplicate samples using SsoAdvanced Universal SYBR Green Supermix and a Bio-Rad CFX96 Real-Time PCR System. Data were normalized to human GAPDH mRNA levels as an endogenous control. Relative expression (RE) levels are expressed relative to static control using the ΔΔCt formula (RE = 2 − [(CT(gene, test sample) − CT(GAPDH, test sample)) − (CT(gene, static sample) − CT(GAPDH, static sample))]), in which CT is the threshold cycle number. Bio-Rad CFX Manager Software version 3.1 was used to determine CT numbers, RE levels, and fold change differences. DNA sequences used for synthesizing PCR primers (shown in Supplementary Table S1 were from the PrimerBank database and oligonucleotides were synthesized by the Vanderbilt University Molecular Biology Core. Fold change differences were determined relative to GAPDH levels and normalized to a specified time point within the gene study data set.
Western blotting
Cells were rinsed once with PBS, then lysed in sodium dodecyl sulfate lysis buffer with protease inhibitor cocktail (Sigma) and phosphatase inhibitors (Roche) and gently scraped from hydrogels or tissue culture plates, and collected on ice. Cell suspensions were sheared by passing through a 27 guage needle, and centrifuged for 10 min at 10,000 g. Supernatants were collected and protein was quantified using bicinchoninic acid assays (Pierce). Equal amounts of protein were separated on 4–20% Mini-PROTEAN TGX Precast Gels (Bio-Rad) and transferred to polyvinylidene difluoride membranes. Samples were blocked in 5% milk and probed with primary antibody overnight at 4°C. Membranes were washed 3 × in Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated in goat antirabbit (#W401B, Promega) or goat antimouse horseradish peroxidase (#7076P2; Cell Signaling) secondary antibodies (1:2000) for 1 h at room temperature. Membranes were washed 3 × in TBST and developed with Immobilon chemiluminescence substrate (Millipore) and visualized on a ChemiDoc MP Imaging System (Bio-Rad) or by exposure to X-ray film. Antibodies used were AQP1 (#AB2219) 1:1000 (Millipore), Claudin 2 (#12H12) 1:1000 (Invitrogen), GAPDH (#14C10) 1:5000, phosphorylated Smad2 (#3101) 1:1000 (Cell Signaling), and NHE3 (#sc-16103), 1:1000, (Santa Cruz).
Immunofluorescence staining and imaging
Cells were fixed with 4% paraformaldehyde at room temperature and stained as described previously.19 After 5 min permeabilization with 0.2% Triton-X-100 in PBS followed by 1 h blocking in 10% normal goat serum, proteins of interest were probed with primary antibodies to ZO-1 (#61–7300) (Invitrogen, Eugene, Oregon), acetylated-α-tubulin (#32–2700) (Zymed, San Francisco, CA), NHE3 (#sc-16103) (Santa Cruz Biotechnology, Dallas, TX) at 1:200 to 1:300 dilutions, and visualized with Alexa Fluor 555 or 488 secondary antibodies (Invitrogen) at 1:200 to 1:2000 dilutions. Cells stained for NHE3 were also stained to visualize F-actin by adding Alexa Fluor 488 phalloidin (#A12379; Invitrogen) to the secondary antibody solution at a concentration of 25 U/mL. Both primary and secondary antibodies were diluted in 5% goat serum in PBS with 0.2% Triton-X-100. Primary antibodies were incubated overnight at 4°C; secondary antibodies were incubated for 45 min at room temperature. Excess unbound antibody was removed by washing with PBS. DAPI was present in the mounting medium (Vector Laboratory, Burlingame, CA). To reduce background when staining cells on Matrigel, 0.1 M glycine in PBS was added to quench antigen after fixation; in addition, a second blocking with 10% goat serum was applied before incubation with secondary antibody. NHE3 staining was imaged on an Olympus FV-1000 inverted confocal microscope. All other fluorescent images were captured on Nikon Microphot FXA or Zeiss Axioskop 2 microscopes paired with Jenoptik ProgRes digital cameras.
Proliferation assay
Primary HRECs were seeded on both 0.5 and 50 kPa six-well hydrogels at a density of 25k per well and allowed to grow for 5 days. Proliferation rates were determined using the Click-iT EdU Proliferation Assay kit (Invitrogen, Eugene, Oregon). The deoxynucleotide analogue EdU was added to the tissue culture medium for 16 h before fixation with 4% paraformaldehyde. The incorporation of EdU into the DNA of cells was visualized by conjugation to an Alexa Fluor 555 picolyl azide. Slides were mounted with mounting medium containing DAPI (Vector Laboratory, Burlingame, CA) and images were captured with a Zeiss Axioskop 2 fluorescent microscope paired with a Jenoptik ProgRes digital camera. Subconfluent areas of similar cell densities were chosen for sampling, and number of cells was counted using ImageJ software.
Statistical analysis
Experiments were designed to test both the reproducibility of multiple biological replicates performed in parallel and also repeated at different times to ensure that differences were not the result of variations related to cell culture conditions or changes in hydrogels or other reagents that may arise during storage. Technical repeats were performed for all real-time PCR by measuring each sample in triplicate, and cDNA synthesis and real-time analysis were repeated a minimum of two times for each RNA sample. Reported number of experimental replicates represent repeated individual experiments or a mixture of repeated individual experiments and parallel replicates, unless otherwise indicated. Results in which cells attached poorly or were detaching from hydrogels were excluded from analysis. Poor quality RNA samples with a ratio of absorbance at 260 and 280 nm <2.0, or concentrations <50 ng/mL, were excluded from analysis. Student's t-test (two-tailed, two sample unequal variance) was used for statistical analysis of qPCR fold change results and p-values <0.05 were considered significant.
Results
Proximal tubule cell morphology varies with substrate elasticity
Kidney epithelial cells have been grown in vitro for >60 years for applications ranging from viral culture to bioartificial organs and microphysiologic systems for drug toxicity assessment.20–23 Although expression of kidney-specific transporters can be observed in vitro, renal tubule cells in 2D and 3D culture lack fully elaborated brush borders and may not express a complete complement of transporters.2,1,15 Apicobasal fluid transport independent of hydrostatic pressure gradients has to our knowledge only been observed in fresh intact tubules.24 Multiple investigators have reported enhanced expression of various kidney-specific proteins when tubule cells are cultured in three-dimensional hydrogels. We first tested whether substrate elasticity had an effect on primary cell proliferation and monolayer formation, as reported by Beamish.25 When seeded at identical densities, human primary renal tubule cells tended to form rounded colonies on 0.5 and 1.0 kPa PA gels, and more flattened confluent monolayers on 10 and 40 kPa gels (Fig. 1A). On both stiff and soft substrates, cells polarized as shown by localization of ZO-1 (green) to cell–cell junctions.
It is not clear whether cells on the stiffer substrates underwent more divisions and doublings as cells grown on soft gels tended to form cysts and tubule-like structures that were easily dislodged during medium changes. To address this question, we determined the proliferation rates of primary human renal cells seeded on 0.5 and 50 kPa gels by measuring the incorporation of EdU into the DNA of proliferating cells during a 16 h period. This assay avoids the possibility of dislodging cells during medium changes, as it is performed by adding the EdU reagent directly to undisturbed medium, followed by fixation immediately after the 16 h incubation period. Our results show that primary human renal cells on 0.5 kPa gels proliferate ∼40% slower than cells on 50 kPa gels (Fig. 2).
FIG. 2.

Proliferation rates of primary human renal tubule cells on soft and stiff substrates. (A) Relative proliferation rates based on EdU incorporation for 16 h of subconfluent primary human renal tubule cells on soft (0.5 kPa) and stiff (50 kPa) substrates. (B) Representative fluorescent images showing proliferating cells (red, Alexa Fluor 555 picolyl azide conjugated to incorporated EdU) and total nuclei (blue, DAPI). Subconfluent areas of similar cell densities were chosen for sampling, and number of cells was counted to determine percentages of proliferating cells using ImageJ software. Cells on stiffer substrates appear to be proliferating more rapidly, based on great number of cells incorporating a nucleotide analogue.
The tendency to form flat sheets on stiffer substrates and rounded colonies on more elastic matrices was also observed on Matrigel (Fig. 1B). Matrigel (Young's modulus ∼0.1–1 kPa) gels were cast on tissue culture plates and left in original state or crosslinked with GLA to increase matrix stiffness.18 On native Matrigel, primary renal tubule cells formed rounded colonies similar to those seen on highly elastic PA gels. On crosslinked Matrigel, renal proximal tubule cells formed flat sheets similar to the results on 10 and 40 kPa PA gels, supporting the hypothesis that tendency to form rounded colonies versus spreading sheets is governed by substrate mechanical properties. We also examined whether cell morphology is related to the amount of ECM crosslinked to the hydrogel surfaces. Laminin staining was measured by immunofluorescence on 0.5 and 50 kPa gels prepared for cell culture with Geltrex as mentioned. Occasional gels showed extremely low laminin staining, but the intensity of laminin staining was not different between 0.5 and 50 kPa gels (data not shown).
Transporter expression was increased on soft substrates compared with stiff substrates
Chen noted that stiff substrate elasticity was associated with epithelial to mesenchymal transition in primary murine renal tubule cells.18 From our observation that cells form isolated clusters and spheroids on soft gels, but confluent sheets on stiffer gels, we then asked whether there were functional consequences of the choice of substrate. We elected to look at some of the major functional proteins found in proximal tubules that are critical for building a functional bioreactor that can actively and selectively transport solutes from the luminal to the basal surface. These proteins include the sodium–proton antiporter 3 (NHE3) and the aquaporin-1 (AQP1) water channel. At 10 days postseeding, mRNA levels for SLC9A3 (NHE3) were not different between soft and stiff substrates. However, at 4 weeks postseeding, SLC9A3 transcripts were ∼10-fold higher on 0.5 kPa scaffolds than on 50 kPa scaffolds, and nearly undetectable on plastic (Fig. 3A). NHE3 protein levels by Western blot were barely detectable at 10 days postseeding. At 4 weeks, NHE3 expression was clearly detectable on the 0.5 kPa scaffold, but only barely detectable on the 50 kPa scaffold (Fig. 3B). Immunohistochemistry of cultures of primary human renal tubule cells grown on 0.5 and 50 kPa PA gels showed increased NHE3 (red stain) in cells on the more elastic substrates compared with stiffer substrates (Fig. 3C). On soft substrates, the NHE3 staining appeared more diffusely distributed through the cytoplasm and at the cell membranes as well. This contrasted with the punctate staining in the middle of the cells, on stiff gels. AQP1 (Fig. 4A) and aquaporin-1 protein (Fig. 4B) were increased on 0.5 kPa gels compared with 50 kPa gels. Unlike NHE3, AQP1 expression levels were similar at the 10 day and 4 week time points (Fig. 4A, B). Interestingly, the levels of AQP1 mRNA were significantly higher on plastic than on 50 kPa gels at the 10 day time point, but this difference was not significant at 4 weeks (Fig. 4A).
FIG. 3.

NHE3 expression varies with substrate stiffness. (A) Expression levels of SLC9A3 (NHE3) were determined by qPCR in primary HREC cultured on soft (0.5 kPa) and stiff (50 kPa) hydrogels. Cells were harvested at short (10 days) and long (4 weeks) time points. (B) NHE3 protein levels were determined by Western blot analysis. Protein was isolated from HREC on soft and stiff hydrogels, cultured in parallel with cells used for qPCR analysis. Twenty microgram of total protein was loaded per lane, and blots were probed with anti-NHE3 antibody (sc-16103 @ 1:1000). GAPDH was used as a loading control. (C) NHE3 (sc-16103 primary @ 1:200 with Alexa Fluor 555 secondary, red) and F-actin (Alexa Fluor 488 Phalloidin, green) staining of HRECs cultured on soft (0.5 kPa) and stiff (50 kPa) hydrogels for 4 weeks. Inset images show samples of the same images at double magnification. HREC, human renal epithelial cell.
FIG. 4.

Aquaporin expression varies with substrate stiffness. (A): Expression levels of aquaporin-1 (AQP1) were determined by qPCR in primary HREC cultured on soft (0.5 kPa) and stiff (50 kPa) hydrogels. Cells were harvested at short (10 days) and long (4 weeks) time points. (B) Aquaporin-1 protein levels were determined by Western blot analysis. Protein was isolated from HREC on soft and stiff hydrogels, cultured in parallel with cells used for qPCR analysis. Twenty micrograms of total protein was loaded per lane, and blots were probed with anti-AQP1 antibody (Millipore AB2219 @ 1:1000). GAPDH was used as a loading control.
In addition to NHE3 and aquaporin-1, we measured mRNA levels for megalin (LRP2), gamma-glutamyl transferase (GGT1), as well as for the α3 subunit of sodium–potassium ATPase, the basolateral active sodium–potassium exchanger (ATP1A3) (Fig. 5). Na-K ATPase mRNA increased over time but was not significantly different between plastic, 50 and 0.5 kPa gels. GGT1 mRNA also increased over time, and was significantly increased in cells grown on hydrogels compared with cells grown on plastic. Megalin mRNA levels were significantly higher on plastic at 10 days than at 4 weeks. mRNA levels for megalin were consistently higher on soft scaffolds than on stiff scaffolds, and similar between 10 days and 4 weeks, but the differences did not reach significance (Fig. 5C).
FIG. 5.
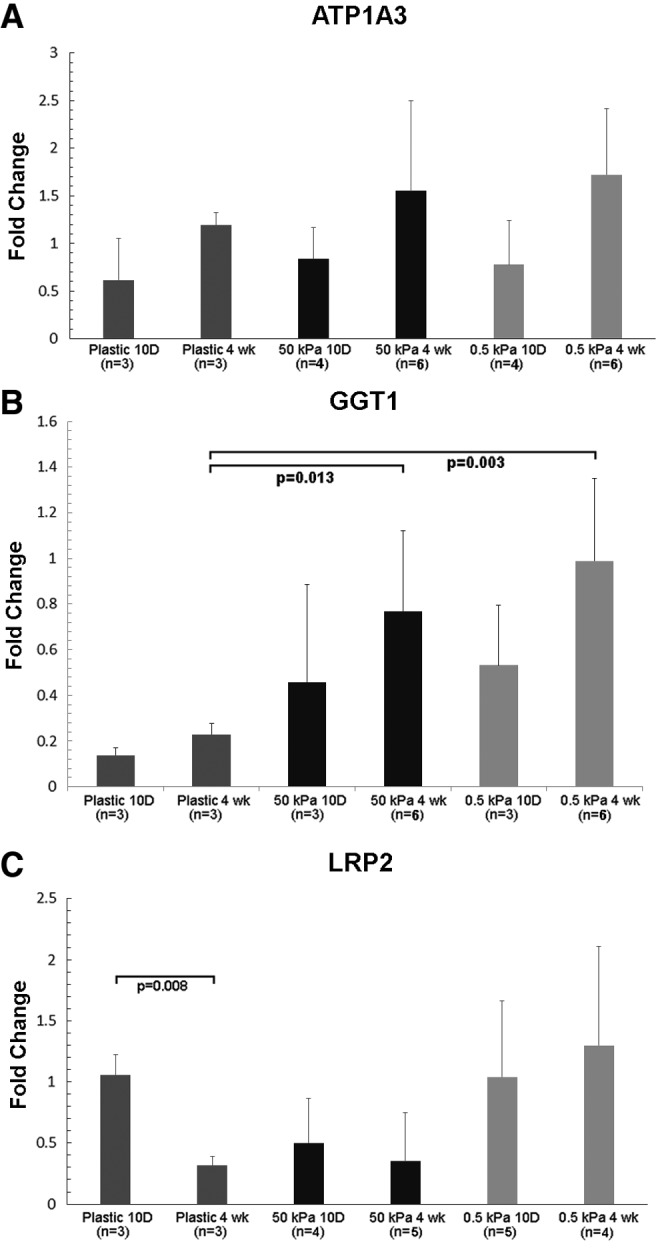
Expression of ATP1A3, gamma-glutamyl transferase, and megalin as a function of culture time and substrate stiffness. Expression levels of ATP1A3 (A), gamma-glutamyl transferase (GGT1) (B), and megalin (LRP2) (C) were determined by qPCR in primary HREC cultured on soft (0.5 kPa) and stiff (50 kPa) hydrogels. Cells were harvested at short (10 day) and long (4 weeks) time points.
TGF-β signaling mediates elasticity-driven differentiation
It has been suggested that the effects of substrate stiffness on differentiation may be mediated by growth factors such as TGF-β.18,26 We, therefore, sought evidence for differences in TGF-β signaling on soft and stiff substrates by examining a downstream mediator, Smad2 phosphorylation. Indeed, Smad2 phosphorylation was evident in cells cultured on 10 and 40 kPa gels, but not on 0.5 and 1.0 kPa gels (Fig. 6A). To further examine whether TGF-β signaling governed differentiation of cultured renal proximal tubule cells, we incubated cells on 1 kPa scaffolds with and without TGF-β. Adding TGF-β to the culture medium resulted in a more spread and flattened phenotype (Fig. 6B). Conversely, we incubated cells on stiffer (40 kPa) scaffolds with and without an inhibitor of the TGF-β receptor 1, SB431542. The inhibitor resulted in rounded clusters of cells similar to those cultured on 1 kPa scaffolds (Fig. 6B). To further confirm the role of TGF-β, we cultured murine proximal tubule cells in which the TGF-β receptor 2 was conditionally deleted. Renal tubule cells with intact TGF-βR2 showed identical morphologic response to substrate stiffness as primary human cells, but TGF-βR2-deficient murine cells formed rounded colonies on both soft and stiff substrates (Fig. 6C). We confirmed the conditional deletion of TGF-β receptor 2 in these experiments as Smad2 phosphorylation was detected after the addition of exogenous TGF-β only in murine cells with an intact TGF-β receptor 2 (Fig. 6D).
We then went on to ask whether the effect of substrate stiffness on TGF-β signaling was mediated by increased expression of TGF-β, or its receptors. Unexpectedly, the effect of substrate elasticity appeared to be mediated by increased expression of TGF-β receptors in cells grown on stiffer substrates, rather than expression of TGF-β itself (Fig. 7). mRNA levels for TGF-β receptors 1 and 2 were significantly higher on stiff versus soft substrates (Fig. 7A), whereas mRNA levels for TGF-β isoforms 1–3 did not vary significantly between cells grown on soft versus stiff substrates (Fig. 7B).
Finally, we examined the effect of TGF-β on gene expression of NHE3 and AQP1 by culturing primary renal tubule cells on soft and stiff substrates in the presence of exogenous TGF-β or the TGF-βR1 inhibitor SB431542. The addition of TGF-β essentially abolished transcription of NHE3 on 0.5 kPa substrates, whereas NHE3 mRNA levels were markedly higher on 50 kPa substrates in the presence of TGF-βR1 inhibitor (Fig. 8A). In contrast, in the same cells, AQP1 mRNA levels were not significantly different in the presence of TGF-β or inhibitor, on 0.5 and 50 kPa substrates (Fig. 8B). The reduction in NHE3 transcript levels in the presence of TGF-β confirms that TGF-β signaling not only mediates changes in the morphology of cell colonies but also expression of some genes essential to proximal tubule cell function. The increased expression of NHE3 in the presence of SB431542 on 50 kPa substrates appears to be related to time in culture and duration of treatment, as the effect was not observed in cells harvested after treatment for only 3 days (data not shown). These results strongly suggest that although TGF-β signaling is crucial to primary proximal tubule cell differentiation, TGF-β may have different effects on expression of individual membrane proteins, rather than a global switch between differentiation and dedifferentiation.
Discussion
The role of scaffold mechanics in governing mammalian cell differentiation is incompletely understood. For some time it has been known that pluripotent stem cell lineage commitment can be dictated, in part, by mechanical characteristics of the scaffold.11 Interruption of mechanotransduction pathways from integrin through focal adhesions to the actin cytoskeleton can block stiffness-guided lineage commitment, suggesting that stiffness, rather than some other factor, such as nutrient and waste diffusion through matrix, is the cue for differentiation.27,28 The role of substrate mechanical characteristics in directing the fate of mature differentiated cells is less well understood but potentially of much more immediate clinical significance.
In that regard, Chen et al. explored matrix cues for cell differentiation by comparing tubule cell growth on soft and stiff polyacrylamide matrices and Matrigel–collagen gel mixtures.18 Similar to our findings, cell morphology varied with matrix stiffness. Addition of TGF-β to medium also caused increased spreading of cells, as we observed. However, the short duration of cell culture Chen reported (2–7 days) may have influenced their ability to detect positive differences in expression between different matrix types. NHE3 message was detectable on Day 0 (the day of harvest) for primary mouse tubule cells, but was undetectable thereafter.
Weber described a three-dimensional construct composed of type 1 collagen gel coated with type IV collagen.22 Although there was no direct comparison regarding substrate elasticity, the collagen gel probably had a bulk modulus in the mid-to-low kilopascal range. Weber reported many functional readouts of differentiated tubule cell function similar to those reported by Humes.1 Hoppensack cultured primary human renal tubule cells and HK-2 cells on porcine small intestine submucosa (SIS), on polystyrene plates, and on collagen gels.2 Remarkably, the primary human cells on the SIS formed a cuboidal monolayer with a rich and evident brush border in some patches, a differentiation marker rarely observed in prior reports of cultured primary tubule cells. Taken together, these observations provide strong support for the hypothesis that scaffold elasticity can permit or prevent the optimal function of adult differentiated epithelial cells.
The data presented here are largely consistent with the prior work and extend the insight further. We showed that transmembrane proteins integral to the functional identity of proximal tubule cells are indeed expressed by cultured primary cells, both at the transcript and protein levels. The mechanism by which substrate elasticity exerts control over protein expression in these cells remains incompletely defined. The morphology of cell colonies on elastic substrates was clearly affected by TGF-β signaling, as competitive inhibition and additional TGF-β appeared to be able to override the effect of elasticity. It may be difficult to interpret our results through the accepted understanding of TGF-β interactions with scaffolds through latent transforming growth factor β binding proteins (LTBPs).29 The morphologies described in Figure 1 were evident on purely synthetic substrates as well as on native and crosslinked Matrigel. The PA scaffold with relatively sparsely bound surface fibronectin may provide specific binding partners for LTBP, particularly LTBP-1 that is highly expressed in kidney and binds fibronectin. The morphology differences are evident within days of seeding, making unlikely that the proximal tubule cells have deposited new matrix ligands for TGF-β binding partners. The role of LTBPs in guiding phenotype of renal tubule cells in culture seems a promising area for further investigation.
Our ultimate goal is a bioreactor of renal proximal tubule cells with quantitative fidelity to the in vivo counterpart regarding transport and barrier function in a manufacturable format. Flat sheet culture is advantageous in many ways, not least of which are direct optical observation of cells, simplified collection of apical and basolateral conditioned media, and ease of instrumentation for measurement of transepithelial electrical resistance.30 However, the scaffold stiffness that promotes uniform monolayer culture of proximal tubule cells also appears not to foster complete differentiation of these cells. Conversely, the soft scaffolds that in our hands were associated with expression of NHE3, aquaporin-1, and other proximal tubule transport proteins also appear to lead to rounded, cyst-, and tubule-like colonies of cells that are poorly suited to manufacturing and monitoring. A tradeoff between manufacturability and utility may prove to be a major challenge to organ-on-a-chip projects.
Supplementary Material
Acknowledgments
Drs. Fissell and Love are supported by U01EB021214 from the National Institutes of Health and a gift from the Wildwood Foundation. Dr. Roy is supported by U01EB021214 and U01EB025136. Dr. Zent is supported by R01DK069921 from the National Institutes of Health and VAI01BX002198 from the Department of Veterans Affairs. Dr. Gewin is supported by DK108968 from the National Institutes of Health. Dr. Harris is supported by i01BX000320 from the Department of Veterans Affairs and by T32DK007569, R01DK051265, R01DK062794, R01DK095785, R24DK103067, and P30DK114809 from the National Institutes of Health. Dr. Ferrell is supported by NIH grants DK092357 and DK110399 and an American Society of Nephrology Carl W. Gottschalk Research Scholar Grant. Confocal microscopy was performed through the use of the Vanderbilt Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637, and EY08126).
Disclosure Statement
No competing financial interests exist.
Supplementary Material
References
- 1. Humes H.D., Mackay S.M., Funke A.J., and Buffington D.A. Tissue engineering of a bioartificial renal tubule assist device: in vitro transport and metabolic characteristics. Kidney Int 55, 2502, 1999. [DOI] [PubMed] [Google Scholar]
- 2. Hoppensack A., Kazanecki C.C., Colter D., et al. A human in vitro model that mimics the renal proximal tubule. Tissue Eng C Methods 20, 599, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shin J.-W., and Mooney D.J. Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc Nat Acad Sci U S A 113, 12126, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schrader J., Gordon-Walker T.T., Aucott R.L., et al. Matrix stiffness modulates proliferation, chemotherapeutic reponse, and dormancy in hepatocellular carcinoma cells. Hepatology 53, 1192, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown A.C., Fiore V.F., Sulchek T.A., and Barker T.H. Physical and chemical microenvironmental cues orthogonally control the degree and duration of fibrosis-associated epithelial-to-mesenchymal transitions. J Pathol 229, 25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wells R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 47, 1394, 2008. [DOI] [PubMed] [Google Scholar]
- 7. Wells R.G. Tissue Mechanics and Fibrosis. Biochim Biophys Acta 1832, 884, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu F., Mih J.D., Shea B.S., et al. Feedback amplification of fibrosis through matrix stiffness and COX-2 suppresion. J Cell Biol 190, 693, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Szeto S.G., Narimatsu M., Lu M., et al. YAP/TAZ are mechanoregulators of TGF-β-Smad Signaling in Renal Fibrogenesis. J Am Soc Nephrol 27, 3117, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang C., DelRio F.W., Ma H., et al. Spatially patterned matrix elasticity directs stem cell fate. Proc Natl Acad Sci U S A 113, E4439, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engler A.J., Sen S., Sweeney H.L., and Discher D.E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677, 2006. [DOI] [PubMed] [Google Scholar]
- 12. Giacomini M.M., Travis M.A., Kudo M., and Sheppard D. Epithelial cells utilize cortical actin/myosin to activate latent TGF-beta through integrin alpha (v)beta(6)-dependent physical force. Exp Cell Res 318, 716, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Embry A.E., Mohammadi H., Niu X., et al. Biochemical and Cellular Determinants of Renal Glomerular Elasticity. PLoS One 11, e0167924, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tse J.R., and Engler A.J. Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol Chapter 10, unit 10.16, 2010. [DOI] [PubMed] [Google Scholar]
- 15. Chevtchik N.V., Mihajlovic M., Fedecostante M., et al. A bioartificial kidney device with polarized secretion of immune modulators. J Tissue Eng Regen Med 12, 1670, 2018. [DOI] [PubMed] [Google Scholar]
- 16. Nlandu Khodo S., Neelisetty S., Woodbury L., et al. Deleting the TGF-beta receptor in proximal tubules impairs HGF signaling. Am J Physiol Ren Physiol 310, F499, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barnes S.L., Lyshchik A., Washington M.K., Gore J.C., and Miga M.I. Development of a mechanical testing assay for fibrotic murine liver. Med Phys 34, 4439, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen W.C., Lin H.H., and Tang M.J. Regulation of proximal tubular cell differentiation and proliferation in primary culture by matrix stiffness and ECM components. Am J Physiol Ren Physiol 307, F695, 2014. [DOI] [PubMed] [Google Scholar]
- 19. Ao M., Brewer B.M., Yang L., et al. Stretching fibroblasts remodels fibronectin and alters cancer cell migration. Sci Rep 5, 8334, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morann G.L., and Melnick J.L. Poliomyelitis virus in tissue culture. VI. Use of kidney epithelium grown on glass. Proc Soc Exp Biol Med 84, 558, 1953. [DOI] [PubMed] [Google Scholar]
- 21. Secker P.F., Luks L., Schlichenmaier N., and Dietrich D.R. RPTEC/TERT1 cells form highly differentiated tubules when cultured in a 3D matrix. Altex 35, 223, 2018. [DOI] [PubMed] [Google Scholar]
- 22. Weber E.J., Chapron A., Chapron B.D., et al. Development of a microphysiological model of human kidney proximal tubule function. Kidney Int 90, 627, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Humes H.D., Krauss J.C., Cieslinski D.A., and Funke A.J. Tubulogenesis from isolated single cells of adult mammalian kidney: clonal analysis with a recombinant retrovirus. Am J Physiol 271, F42, 1996. [DOI] [PubMed] [Google Scholar]
- 24. Giebisch G., Klose R.M., Malnic G., Sullivan W.J., and Windhager E.E. Sodium movement across single perfused proximal tubules of rat kidneys. J Gen Physiol 47, 1175, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beamish J.A., Chen E., and Putnam A.J. Engineered extracellular matrices with controlled mechanics modulate renal proximal tubular cell epithelialization. PLoS One 12, e0181085, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wells R.G., and Discher D.E. Matrix elasticity, cytoskeletal tension, and TGF-beta: the insoluble and soluble meet. Sci Signal 1, 13, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lv H., Li L., Sun M., et al. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res Ther 6, 103, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McMurray R.J., Dalby M.J., and Tsimbouri P.M. Using biomaterials to study stem cell mechanotransduction, growth and differentiation. J Tissue Eng Regen Med 9, 528, 2015. [DOI] [PubMed] [Google Scholar]
- 29. Robertson I.B., Horiguchi M., Zilberberg L., Dabovic B., Hadjiolova K., and Rifkin D.B. Latent TGF-beta-binding proteins. Matrix Biol 47, 44, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferrell N., Desai R.R., Fleischman A.J., Roy S., Humes H.D., and Fissell W.H. A microfluidic bioreactor with integrated transepithelial electrical resistance (TEER) measurement electrodes for evaluation of renal epithelial cells. Biotechnol Bioeng 107, 707, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.