

Abstract
Aims: Metabolic syndrome is associated with metabolic heart disease (MHD) that is characterized by left ventricular (LV) hypertrophy, interstitial fibrosis, contractile dysfunction, and mitochondrial dysfunction. Overexpression of catalase in mitochondria (transgenic expression of catalase targeted to the mitochondria [mCAT]) prevents the structural and functional features of MHD caused by a high-fat, high-sucrose (HFHS) diet for ≥4 months. However, it is unclear whether the effect of mCAT is due to prevention of reactive oxygen species (ROS)-mediated cardiac remodeling, a direct effect on mitochondrial function, or both. To address this question, we measured myocardial function and energetics in mice, with or without mCAT, after 1 month of HFHS, before the development of cardiac structural remodeling.
Results: HFHS diet for 1 month had no effect on body weight, heart weight, LV structure, myocyte size, or interstitial fibrosis. Isolated cardiac mitochondria from HFHS-fed mice produced 2.2- to 3.8-fold more H2O2, and 16%–29% less adenosine triphosphate (ATP). In isolated beating hearts from HFHS-fed mice, [phosphocreatine (PCr)] and the free energy available for ATP hydrolysis (ΔG∼ATP) were decreased, and they failed to increase with work demands. Overexpression of mCAT normalized ROS and ATP production in isolated mitochondria, and it corrected myocardial [PCr] and ΔG∼ATP in the beating heart.
Innovation: This is the first demonstration that in MHD, mitochondrial ROS mediate energetic dysfunction that is sufficient to impair contractile function.
Conclusion: ROS produced and acting in the mitochondria impair myocardial energetics, leading to slowed relaxation and decreased contractile reserve. These effects precede structural remodeling and are corrected by mCAT, indicating that ROS-mediated energetic impairment, per se, is sufficient to cause contractile dysfunction in MHD.
Keywords: metabolism, ROS, catalase, mitochondria, contractile function, obesity, metabolic syndrome
Introduction
Metabolic syndrome, a cluster of obesity-related metabolic abnormalities, afflicts more than one third of Americans. A consequence of metabolic syndrome is metabolic heart disease (MHD), which is characterized by left ventricular (LV) hypertrophy (LVH), cardiac myocyte hypertrophy, interstitial fibrosis, diastolic dysfunction, and impaired myocardial energetics (1, 6, 9, 20). The typical findings of MHD are present in mice that develop metabolic syndrome caused by consuming a high-fat, high-sucrose (HFHS) diet for 4 or more months (20, 28, 32). A central pathophysiologic feature in these mice is increased oxidative stress in the myocardium, as evidenced by increased levels of 4-hydroxynonenal (HNE) and oxidation of multiple protein targets including sarcoplasmic reticulum Ca2+ ATPase (SERCA) (28) and mitochondrial complex II (32, 33). Another central feature in these mice is mitochondrial dysfunction (32), which is associated with decreased energy (adenosine triphosphate [ATP]) production and impaired contractile function in the beating heart (20).
Innovation
We show that increased mitochondrial reactive oxygen species (ROS) play a critical role in the early manifestations of metabolic heart disease (MHD), including energetic deficiency and impaired contractile response to increased work demand. The energetic abnormality is sufficient to cause contractile dysfunction and is corrected by transgenic expression of catalase targeted to the mitochondria, indicating that mitochondrial ROS play a critical role in MHD. These findings suggest that interventions to correct mitochondrial ROS may be of value in the prevention and treatment of MHD.
Oxidative stress in HFHS-fed mice is associated with increased production of reactive oxygen species (ROS) by mitochondria (32, 33), the pathophysiologic importance of which is supported by the demonstration that transgenic expression of catalase targeted to the mitochondria (mCAT) prevents the development of the cardiac phenotype at 4 months on an HFHS diet—with amelioration of LVH, fibrosis, and diastolic dysfunction (33). In these studies, the improvements in cardiac structure and function were associated with correction of maximal ATP production in isolated cardiac mitochondria (33). Likewise, in skeletal muscle from mice fed a high-fat diet, mCAT expression improved mitochondrial function and decreased ischemic necrosis (30). Although these observations strongly implicate oxidative stress in the pathophysiology of MHD in the HFHS-fed mouse, the effects of mCAT on cardiac energetics measured under more physiologic conditions in the beating heart are not known, nor is the relationship between energetics and cardiac structural and/or functional remodeling. Of note, all of the prior observations on mitochondrial dysfunction and cardiac energetics in the HFHS-fed mouse were made after 4 or more months on diet, at a time when LVH, myocyte hypertrophy, and increased interstitial fibrosis are present. By increasing the distance between myocytes and decreasing the ratio of capillaries to myocytes, myocyte hypertrophy and fibrosis may impair myocardial energetics by increasing the diffusion barriers for oxygen and nutrients in the heart (3, 5, 10, 11, 15, 22, 31, 37). Therefore, it is not clear whether the adverse effects of ROS on myocardial energetics are due, on the one hand, to the consequences of structural remodeling (i.e., LVH and/or fibrosis) and/or are a direct consequence of the effects of ROS on mitochondrial function.
We previously studied the effect of mCAT expression in HFHS-fed mice after 4 months on diet, a time when both myocyte hypertrophy and fibrosis are present. It remains unclear whether mitochondrial ROS and impaired energetics precede or follow the development of LVH and fibrosis, or, for that matter, whether the beneficial effects of mCAT are associated with improvements in energetics and contractile function. In preliminary studies, we found increased mitochondrial ROS production after only 1 month on an HFHS diet, a time point that precedes the development of LVH, interstitial fibrosis, and diastolic dysfunction (28). Therefore, in this study, we capitalize on this observation to elucidate the relationship between mitochondrial ROS and energetic dysfunction by studying mice with or without mCAT overexpression that are fed an HFHS diet for 1 month and measuring the effects on energetics and contractile function in the beating heart.
Results
General characteristics and echocardiography
The mice were studied after 1 month of either the HFHS or control diet (CD), at which time the average age was 13 ± 1 weeks. As previously shown, 4 months on the HFHS diet leads to a marked body weight gain that is associated with LVH and diastolic dysfunction (32, 33). In contrast, in this study, HFHS feeding for 1 month did not affect body weight or LVH. Likewise, as assessed by echocardiography, HFHS feeding for 1 month had no effect on resting LV systolic or diastolic dimensions, wall thickness, or any measure of diastolic function, including myocardial peak early diastolic velocity (Em) (Table 1). By comparison, mice fed the HFHS diet for 4 months exhibited marked weight gain, increased wall thickness, and impaired resting diastolic function including Em (Table 1).
Table 1.
Body Weight and Echocardiographic Data
| 1 Month | 4 Months | |||
|---|---|---|---|---|
| Control diet | HFHS diet | Control diet | HFHS diet | |
| BW (g) | 29.3 ± 0.6 | 32.4 ± 0.2 | 36.1 ± 1.3 | 48.2 ± 1.6a |
| TWTh (mm) | 1.50 ± 0.01 | 1.53 ± 0.02 | 1.53 ± 0.02 | 1.80 ± 0.05a |
| EDD (mm) | 2.83 ± 0.02 | 2.93 ± 0.04 | 2.93 ± 0.04 | 2.98 ± 0.04 |
| ESD (mm) | 1.13 ± 0.02 | 1.15 ± 0.02 | 1.15 ± 0.02 | 1.23 ± 0.02 |
| LVFS (%) | 59.9 ± 0.4 | 60.4 ± 0.4 | 60.5 ± 0.3 | 59.9 ± 0.3 |
| IVRT (ms) | 17.2 ± 0.9 | 18.6 ± 0.8 | 17.2 ± 0.8 | 23.5 ± 1.1a |
| DT (ms) | 18.1 ± 2.1 | 18.4 ± 1.4 | 18.9 ± 0.9 | 22.9 ± 0.3a |
| E/A ratio | 1.77 ± 0.01 | 1.67 ± 0.06 | 1.71 ± 0.07 | 1.42 ± 0.01a |
| Em (mm/s) | 25.9 ± 0.6 | 23.7 ± 0.8 | 28.0 ± 1.8 | 19.8 ± 0.7a |
N = 4. Values are mean ± SEM.
p < 0.05 versus control diet.
BW, body weight; DT, deceleration time; E/A, the ratio of early-to-late diastolic mitral inflow velocity; EDD, end-diastolic dimension; Em, myocardial peak early diastolic velocity; ESD, end-systolic dimension; HFHS, high fat, high sucrose; IVRT, isovolumic relaxation time; LVFS, left ventricular fractional shortening; SEM, standard error of the mean; TWTh, total wall thickness.
By histology, the degree of interstitial fibrosis was similar between HFHS and CD groups after 1 month on diet, but it was increased in the HFHS group after 4 months on diet (Fig. 1A, B). Likewise, myocyte cross-sectional area was not affected by HFHS (vs. CD) diet for 1 month, but it was increased after 4 months on HFHS diet (Fig. 1C, D).
FIG. 1.

HFHS causes fibrosis and cardiomyocyte hypertrophy after 4 months but not after 1 month on the diet. (A, B) Representative photomicrographs of Picro Sirius Red staining for cardiac fibrosis and quantification of cardiac fibrosis as measured by National Institutes of Health ImageJ software after (A) 1 month (mo.) and after (B) 4 months (mos.) on CD or HFHS diet. (C, D) Photomicrographs of left ventricular tissue sections stained by hematoxylin and eosin with quantification of myocyte cross-sectional area as measured by National Institutes of Health ImageJ after (C) 1 month and after (D) 4 months on CD or HFHS diet. Values are mean ± SEM; n = 3–4. *p < 0.05 versus CD-fed mice. CD, control diet; HFHS, high fat, high sucrose; SEM, standard error of the mean. Color images are available online.
Effects of HFHS diet for 1 month on H2O2 production and maximal ATP synthesis rate in isolated cardiac mitochondria
In mitochondria isolated from hearts of wild type (WT) mice fed an HFHS (vs. CD) diet for 1 month, H2O2 production was increased with either complex I (pyruvate+malate) or complex II (succinate+rotenone) substrates (Fig. 2B, D). Likewise, in mitochondria from HFHS-fed WT mice, the maximal rate of ATP synthesis was decreased for both complex I and II substrates (Fig. 2A, C). ROS produced in mitochondria can oxidatively modify components of the electron transport chain (ETC) (4), such as complex II, which is inhibited by oxidation (33). We assessed complex II function by measuring the reduction of ubiquinone to ubiquinol. In WT mice, HFHS feeding decreased complex II activity and this decrease was prevented in mCAT mice (Fig. 3). Taken together, these findings in isolated mitochondria are qualitatively and quantitatively similar to the changes we previously observed after 4 months on the HFHS diet (33), and they demonstrate that this diet affects isolated mitochondrial ROS and ATP production after only 1 month, a time that precedes the changes in cardiac structure and resting diastolic function that are evident after 4 months.
FIG. 2.

Cardiac mitochondrial ATP synthesis rate and H2O2 production in HFHS-fed mice is protected by mitochondrial catalase. Cardiac mitochondrial complex I and II substrate-driven ATP synthesis rates are reduced in HFHS-fed WT but not mCAT mice. (A) Complex I substrate-driven ATP synthesis rate (5 mM pyruvate +5 mM malate); (B) Complex II substrate-driven ATP synthesis rate (5 mM succinate +2 μM rotenone). Values are mean ± SEM; n = 4–5. Increased cardiac mitochondrial H2O2 production in HFHS-fed mice is prevented by mCAT. The mitochondrial H2O2 production rate is increased in cardiac mitochondria from WT but not mCAT mice. (C) H2O2 production rate with a complex I substrate (5 mM pyruvate +5 mM malate); (D) H2O2 production rate with a complex II substrate (5 mM succinate) and inhibition of reverse electron transport (2 μM rotenone). Values are mean ± SEM; n = 4–5. ATP, adenosine triphosphate; mCAT, transgenic expression of catalase targeted to the mitochondria; WT, wild type.
FIG. 3.

Mitochondrial catalase prevents inhibition of the Complex II. In WT mice, complex II activity assessed by the reduction of ubiquinone to ubiquinol is decreased by HFHS feeding and in mCAT mice, the HFHS-induced decrease in complex II activity is prevented (n = 4, p < 0.05).
Impaired contractile response to increased work in mice fed HFHS diet for 1 month
Although, as noted earlier, the mice fed an HFHS diet for 1 month had no structural or functional abnormalities evident under resting conditions, the decreased maximal ATP production in isolated mitochondria suggested that there might be limited ATP supply for contractile function under conditions of increased work demand (20). To assess this possibility, LV function and energetics were evaluated simultaneously in isolated beating hearts during a high work demand protocol. At the beginning of the experiments, the volume of the LV balloon was set for an LV end-diastolic pressure (LVEDP) of 8–9 mmHg at the lowest workload (pacing = 450 beats per minute [bpm], perfusate [Ca2+] = 2 mM). Work demand was then increased in a stepwise manner by increasing the [Ca2+] in the perfusate from 2 to 4 mM and/or accelerating the pacing rate from 450 to 600 bpm. Although LVEDP was, by design, similar in all experimental groups under baseline conditions, its increase at each incremental level of work demand was higher in HFHS hearts compared with CD hearts (Fig. 4A). At the highest work level, LVEDP was 21.5 ± 2 mmHg in hearts from HFHS-fed mice versus 13.2 ± 2.1 mmHg in hearts from CD-fed mice (p < 0.01). End-systolic pressure (Fig. 4B), developed pressure (DevP) (Fig. 4C), and the rate × pressure product (RPP) (Fig. 4D) increased as expected with increasing work demand in the CD group; whereas HFHS hearts showed a markedly blunted response, consistent with a decrease in cardiac contractile reserve.
FIG. 4.

Mitochondrial catalase prevents contractile dysfunction in isolated perfused HFHS hearts. LV contractile function was assessed at four levels of progressively increasing myocardial work demand. Hearts were paced at HR 450 bpm at baseline, and paced at 600 bpm and/or Ca2+ was raised (4 mM) to increase work demand. With increased work demand, the LVEDP increased more in hearts from HFHS-fed (vs. CD-fed) mice (A). Mitochondrially expressed catalase prevented the increase: LVEDP in the mCAT HFHS group remained similar to CD. ESP (B), DevP (C), and the RPP (D) failed to increase appropriately in HFHS during high work demand and increased normally in mCAT HFHS hearts. For all panels, data are shown as mean ± SEM; open circles = CD-fed mice; filled circles = HFHS-fed mice; triangles = mCAT HFHS mice; n = 8 in each group; *p < 0.05 versus CD-fed mice. bpm, beats per minute; DevP, developed pressure; ESP, end-systolic pressure; HR, heart rate; LV, left ventricle; LVEDP, left ventricular end-diastolic pressure; RPP, rate × pressure product.
Decreased cardiac high-energy phosphate concentrations in mice fed an HFHS diet for 1 month
Simultaneously with the hemodynamic assessment, we used 31P nuclear magnetic resonance (NMR) spectroscopy to measure the high-energy phosphate concentrations in the beating hearts. The ATP concentration was not different between the HFHS- and CD-hearts at baseline or during all stages of the high workload protocol (Fig. 5A). In CD hearts, the concentration of [phosphocreatine, PCr], as well as PCr/ATP, declined progressively throughout the protocol (Fig. 5B, C). In HFHS hearts, as compared to CD hearts, [PCr] and the PCr/ATP ratio were lower throughout the protocol (Fig. 5B, C). Total [Cr] measured from frozen tissue was similar in CD and HFHS groups (Supplementary Fig. S1A). Intracellular pH (Supplementary Fig. S1B) was similar for CD and HFHS hearts at all work levels. Consistent with decreased [PCr] and PCr/ATP, inorganic phosphate (Supplementary Fig. S1C) and cytosolic free [adenosine diphosphate, ADP], calculated as per Equation (1), were elevated in HFHS hearts at all levels of work demand (Fig. 5D).
FIG. 5.

Energetic dysfunction in isolated perfused hearts from HFHS-fed mice is prevented by mCAT. Myocardial high-energy phosphates were measured concurrently with hemodynamic measurements (Fig. 2), using 31P NMR spectroscopy. ATP concentrations ([ATP]) were similar in all groups at all work demands (A). The concentration of PCr (B) and the PCr/ATP ratio (C) were lower in HFHS hearts at baseline and at all workloads, reflecting an impairment in energy reserve. Calculated cytosolic free [ADP] was higher in HFHS- versus CD-fed hearts at baseline and all increased workloads (D). The energy available for chemical work in ATP is expressed by the free energy of ATP hydrolysis (ΔG∼ATP). As ΔGATP is a negative number due to the exergonic nature of ATP hydrolysis, the absolute value is depicted for the sake of clarity. ⌈ΔG∼ATP ⌈was lower in HFHS-fed hearts at baseline and at all stages of work demand (E). The lack of energy reserve in the HFHS group is even better visualized when ⌈ΔG∼ATP ⌈ is plotted against actual work performed, RPP (F). Mitochondrially expressed catalase prevented the energetic dysfunction in HFHS (B–F) In (E, F), the dotted horizontal line at a |ΔG∼ATP| of 52 kJ/mol is the value required for normal function of sarcoplasmic reticulum calcium ATP-ase. For all panels, data are shown as mean ± SEM; open circles = CD-fed mice; filled circles = HFHS-fed mice; triangles = mCAT HFHS mice; n = 8 in each group; and *p < 0.05 versus CD-fed mice. ADP, adenosine diphosphate; NMR, nuclear magnetic resonance; PCr, phosphocreatine.
Decreased free energy of ATP hydrolysis (ΔG∼ATP) in mice fed a HFHS diet for 1 month
The free energy of ATP hydrolysis (|ΔGATP |, expressed as the absolute value for the sake of clarity), was lower in the HFHS group at baseline and throughout the incremental work demand protocol (Fig. 5E). Of note, at increased levels of work demand |ΔGATP| fell below 52 kJ/mol in HFHS hearts, the level of free energy required for normal function of SERCA (14, 26, 34). The defects in energetics and contractile function in HFHS hearts are particularly striking when |ΔGATP | is plotted against work performed, as reflected by the RPP (Fig. 5F). In HFHS hearts, the expression of mCAT shifted the |ΔGATP |/RPP relationship upward and rightward, indicating normalization of energetic and contractile reserve.
Mitochondrial catalase prevents the abnormalities in mitochondrial function, contractile reserve, and energetics in mice fed an HFHS diet for 1 month
The observed increase in ROS production in isolated mitochondria raised the possibility that mitochondrial ROS mediate some or all of the observed functional abnormalities in contractile reserve and cardiac energetics after 1 month on diet. In isolated cardiac mitochondria, the HFHS-induced increase in H2O2 production with complex I or II substrates was abolished in mCAT mice (Fig. 2B, D). Likewise, the HFHS diet-induced decrease in maximal ATP synthesis rates for complex I or II substrates was prevented in mCAT mice (Fig. 2A, C).
Overexpression of mCAT prevented all of the abnormalities in LV function in isolated beating hearts, including elevation of the LV end-diastolic filling pressure, and the decreases in end-systolic pressure, developed pressure, and RPP over the range of work demands tested (Fig. 4). In parallel, overexpression of mCAT prevented the resting and work-related abnormalities in high-energy phosphates (PCr, ATP, PCr/ATP, and ADP) (Fig. 5A–D), and likewise, the decrease in |ΔGATP | was corrected to values similar to CD-fed mice at all work levels (Fig. 5E).
Discussion
There are three major new findings in this study. First, we found that energetics in the beating heart and maximal ATP production in isolated mitochondria are impaired after only 1 month on the HFHS diet, at a time that precedes cardiac structural remodeling (i.e., myocyte hypertrophy, myocardial hypertrophy, interstitial fibrosis). Second, we show that both impaired beating heart energetics and decreased mitochondrial ATP production after 1 month on diet are prevented by mCAT, indicating that they are mediated by ROS produced and/or acting in the mitochondria. Third, we show that after 1 month on diet the abnormalities in energetics are associated with impaired relaxation and decreased contractile reserve in the beating heart, which are prevented in mCAT mice, thus supporting the functional significance of the energetic impairment. These findings indicate that ROS produced and/or acting in the mitochondria cause impairment in energetic function that is sufficient to cause abnormalities in myocardial relaxation and contraction with increased work demands.
Impaired energetics, isolated mitochondrial function, and contractile reserve after 1 month on HFHS diet
A significant new finding of this study is that, after only 1 month on the HFHS diet, myocardial high-energy phosphates assessed in the beating heart were depressed with decreases in [PCr] and the [PCr]/[ATP] ratio at all work levels. These abnormalities were quantitatively similar to those we previously observed after 4 months on diet (20). Likewise, |ΔG∼ATP| was depressed after 1 month on diet—to levels that are sufficient to limit the activity of SERCA, which is exquisitely sensitive to ΔG∼ATP depletion (14, 26, 34). Consistent with the critical role of SERCA-mediated SR calcium reuptake in contraction and relaxation, energetic impairment in the beating heart was associated with both an inability to increase systolic work (i.e., decreased contractile reserve) and an elevation of end-diastolic pressures (i.e., impaired relaxation) at increased work demand. These abnormalities in contractile function support the functional significance of the observed energetic limitation.
Since 1 month of HFHS diet did not induce cardiac structural remodeling with cardiac myocyte hypertrophy or interstitial fibrosis, the observed effects on energetics and contractile function cannot be attributed to changes in myocyte and or interstitial properties. It is noteworthy that the abnormalities in isolated mitochondria function (a decrease in the maximal production of ATP and decreased activity of complex I and II in isolated mitochondria) paralleled the perturbations in energetic and contractile function in the beating heart. Complex II dysfunction is particularly interesting given our prior observations that a decrease in functional capacity of complex II, but not complex I, is fully reversible ex vivo by a cysteine-reducing agent DTT, after 4 months (33) and even 8 months on the HFHS diet (32) We have also demonstrated that complex II function is important beyond mitochondrial ETC and ROS production: Complex II also regulates insulin homeostasis (24), which could be of critical importance in obesity-mediated cardiac and metabolic disease. That said, we provide evidence that maximal ATP generation is decreased and ROS production is increased for both complex I and complex II substrates, suggesting that complex II is unlikely to be the only dysfunctional component of the ETC during HFHS diet.
Mitochondrial ROS mediate the early (1 month) impairments in energetics and contractile function
A second major finding in this study is that mCAT prevented the phenotype observed in mice on HFHS diet for 1 month, including (i) excessive mitochondrial H2O2 production, (ii) decreased mitochondrial maximal ATP synthesis, (iii) decreased contractile reserve with increased work demand, (iv) decreased high-energy phosphate concentrations, and (v) decreased |ΔGATP |. Since, as shown, the energetic and mitochondrial abnormalities occurred before the appearance of structural remodeling, they cannot be ascribed to the consequences of myocardial hypertrophy and/or fibrosis. It has been proposed that concentric LVH causes microvascular rarefaction and fibrosis (3, 10, 22, 31, 37), resulting in longer oxygen diffusion distances and oxygen demand/supply discrepancy (3, 5, 11, 15). Our observations suggest that after 1 month on HFHS diet there is energetic impairment sufficient to cause abnormalities of relaxation and contraction in response to work demands. This energetic impairment is dependent on mitochondrial ROS, but it is independent of the effects of ROS on the myocardial structure. It is possible, however, that after 4 or more months on the HFHS diet structural remodeling also contributes to impaired energetics and contractile function. For example, impaired LV filling, as measured by Doppler echocardiography in resting mice, is present after 4 months on the HFHS diet, but not after 1 month, and therefore potentially reflects the effects of structural remodeling on resting LV function.
Prior studies of the role of mitochondrial ROS in MHD
We previously showed that mCAT normalizes ROS production and preserves maximal ATP producing capacity in isolated cardiac mitochondria from mice fed an HFHS diet for 4 months (33). In these mice, HFHS-induced LVH and interstitial fibrosis were prevented, as was resting diastolic dysfunction, as assessed by echocardiography. Although these observations implicate mitochondrial ROS in the pathophysiology of structural remodeling and diastolic dysfunction in MHD, they do not clarify the relative roles of structural remodeling versus energetic impairment in causing the characteristic abnormalities in myocardial function, nor do they address the relationship between cardiac structural abnormalities and the energetic impairment. Since ROS can mediate both myocardial hypertrophy and interstitial fibrosis (7, 18, 25, 27, 29), mCAT may prevent the adverse effects of structural changes on myocardial function by several mechanisms. For example, ROS-mediated myocyte hypertrophy and interstitial fibrosis might cause a direct physical limitation to function, or they might lead to microvascular rarefication that limits the diffusion of oxygen and nutrients (3, 10, 22, 37). Alternatively, independent of structural remodeling, ROS might cause “metabolic remodeling or reprogramming” of the heart by affecting the expression and/or function of proteins involved in energy production, thereby leading to contractile dysfunction. In this regard, we have shown that HFHS feeding causes oxidative modifications of mitochondrial proteins that could lead to energetic dysfunction (4, 32, 33). Although we cannot exclude the possibility that there is also an extra-mitochondrial source of ROS, and likewise, the mitochondrial effects of extra-mitochondrial ROS could be mitigated by mCAT, our results strongly indicate that excessive ROS are deleterious once they reach mitochondria.
Summary and conclusions
These findings support the thesis that mitochondrial ROS cause mitochondrial dysfunction that is sufficient to impair the energetic response to increased work demands, thereby leading to abnormalities in systolic and diastolic LV function. These effects of ROS may be mediated by a variety of mechanisms, including changes in the expression and/or function of proteins involved in energy production. Reprogramming of metabolic genes has been shown in other forms of heart failure (17), and we have shown that HFHS feeding is associated with oxidative modifications of proteins involved in energy production (4, 33, 36). Future studies should extend these findings to other models of heart disease to further elucidate the temporal relationships and causative roles of mechanisms for the energetic impairment that we have observed after 1 month on HFHS diet. The source of the ROS in HFHS-fed mice likewise remains to be determined. It is possible that it reflects the leakage of electrons from electron transport and/or inefficient glucose oxidation with increased anaplerotic pyruvate carboxylation that depletes NADPH and glutathione pools (16). Our findings suggest that interventions that decrease the production of excess ROS by mitochondria and/or target antioxidants to the mitochondria may be of therapeutic value in MHD.
Materials and Methods
Experimental animals
mCAT overexpressing and WT male C57BL/6J mice were started on an ad libitum dietary protocol at the age of 8 weeks. The mice were fed either a CD (product No. D09071703, 10% kcal lard, 0% sucrose; Research Diets) or an HFHS diet (product No. D09071702; 58% kcal lard, 13% kcal sucrose; Research Diets) for 1 or 4 months. The custom-formulated CD matched the HFHS diet for all micronutrients (20). The protocol was approved by the Institutional Animal Care and Use Committee of the Boston University School of Medicine.
Two-dimensional and M-mode echocardiography
Measurements of LV geometry and function were performed in non-anesthetized mice by using an Acuson Sequoia C-256 echocardiograph equipped with a 15-MHz linear transducer, as described (28). Briefly, the two-dimensional images were acquired in the parasternal short-axis view. M-mode of the mid-ventricle was acquired at the level of the papillary muscles to measure anterior and posterior wall thickness, and LV end-diastolic and end-systolic dimensions.
Doppler echocardiography
Transmitral and tissue Doppler echocardiography were performed by using a VisualSonics Vevo 770 high-resolution imaging system (Toronto, Canada) equipped with a 30-MHz RMV-707B transducer (28). Briefly, isoflurane by a facemask (concentration of 2.5% for induction and then 1.5% for maintenance) was used for anesthesia. An apical four-chamber view was used to record the mitral pulsed Doppler flow parameters: early (E) and late (A) peak mitral inflow velocities, E/A ratio, deceleration time of early filing, and isovolumetric relaxation time. A parasternal short-axis view was used to record tissue Doppler to measure myocardial peak early diastolic velocity (Em). All Doppler measurements were recorded for an average of 13 ± 1 cardiac cycles, with at least five consecutive cardiac cycles selected for the average as per the American Society of Echocardiography guidelines (23). Vevo 770 Analytic software was used for the offline data analysis.
Histology
The mice were sacrificed at the end of the study. LV samples were fixed in 10% buffered formalin, embedded with paraffin, and sectioned. Myocyte cross-sectional area and fibrosis were measured as previously described (28). Briefly, after staining with hematoxylin and eosin, the sections were examined under a light microscope (BX 40; Olympus). We measured 60 myocytes from 5 random fields from each of 4 sections per animal. NIH ImageJ software was used to quantify myocyte cross-sectional area. Picrosirius Red Stain Kit (Polysciences, Warrington, PA) staining and light microscopy (BX 40; Olympus) were used to assess fibrosis.
Mitochondrial isolation
We isolated cardiac mitochondria as previously described (33). Isolation was performed at 4°C. Briefly, tissues were rinsed in 100 mM KCl, 5 mM EGTA, and 5 mM HEPES buffer at pH 7.0; subsequently homogenized in 2 mL of HEPES 5 mM, EDTA 1 mM, and sucrose 0.25 M at pH 7.4 (HES buffer) by using a Teflon-on-glass electric homogenizer; and centrifuged for 10 min at 500 g. After collection of the supernatant, another centrifugation was performed at 9000 g for 15 min. The pellet containing mitochondria was resuspended in 100 μL of HES buffer with 0.3% of fatty acid-free bovine serum albumin (BSA). BCA (Pierce) was used to quantify protein with subtraction of the value of HES-BSA buffer alone.
Mitochondrial H2O2 production
Mitochondrial H2O2 production was assessed by using the Amplex Ultra Red-Horseradish peroxidase method (Invitrogen) as previously described (33). All measurements were performed at 25°C. Briefly, 10 μg mitochondria diluted in 50 μL reaction buffer (125 mM KCl, 10 mM HEPES, 5 mM MgCl2, 2 mM K2HPO4, pH 7.44) was used to measure complex I (pyruvate/malate, 5 mM) or complex II (succinate, 5 mM) driven H2O2 production ± the complex I inhibitor rotenone (2 μM). H2O2 production was assessed fluoroscopically after the addition of 50 μL of reaction buffer (horseradish peroxidase+Amplex Ultra Red). Fluorescence was followed for 20 min (excitation wavelength 545 nm, emission wavelength 590 nm). The slope of the fluorescence increase represents the rate of H2O2 production. The results are plotted as pmoles/min/mg protein.
Maximal ATP production in isolated mitochondria
A luciferin/luciferase-based ATP Bioluminescence Assay Kit CLS II (Roche) was used with minor modifications, as previously described (33). Briefly, 10 μg of heart mitochondria suspended in 75 μL buffer A (125 KCl, 10 mM Hepes, 5 mM MgCl2 and 2 mM K2HPO4, pH 7.44) was used to measure complex I (pyruvate/malate, 5 mM final) and complex II (succinate, 5 mM final) driven ATP synthesis. Succinate-driven ATP generation was assessed in the presence of complex I inhibitor rotenone (2 μM) to avoid the reverse electron transfer, as previously published (32, 33). The measurements were repeated with inhibitors of respiratory complexes, including rotenone (Complex I) and oligomycin (Complex IV), to assess the rates of non-mitochondrial ATP production. Measurement with mitochondria alone determined the assay background. Sample measurements were started simultaneously by adding 75 μL of luciferin/luciferase buffer containing ADP for a 0.5 mM final concentration. To assess the rate of ATP production, we measured the ATP-dependent luciferase chemiluminescence and its initial slope after background subtraction to determine the rate of ATP synthesis. The slopes were converted to nmol/min/mg protein after standard curve calibration with the ATP, as provided in the kit.
Concurrent assessment of LV contractile function and energetics by 31P NMR spectroscopy
An isolated retrograde-perfused Langendorff heart preparation was used to assess LV contractile function (21). Briefly, heparin (100 U intraperitoneal [IP]) and sodium pentobarbital (150 mg/kg IP) were administered; the chest was opened; the heart was excised, cannulated through the aorta, and perfused at a constant pressure of 80 mmHg with phosphate-free KH buffer containing NaCl 118, NaHCO3 25, KCl 5.3, CaCl2 2, MgSO4 1.2, EDTA 0.5, glucose 10, pyruvate 0.5 equilibrated with 95% O2, and 5% CO2 (pH 7.4). A custom-built balloon filled with water was positioned in the LV to continuously measure LV pressure. The balloon matched the size of the LV cavity and after 20-min stabilization its volume was adjusted to yield an LVEDP of 8–9 mmHg. The balloon volume was held constant for the rest of the protocol. LV developed pressure (DevP) was calculated as DevP = systolic pressure − LVEDP. Hearts were placed in a 10-mm glass NMRtube and positioned in a 9.4-Tesla superconducting magnet where they were maintained at 37°C throughout the protocol. The LV workload was manipulated by increasing the concentration of CaCl2 in the perfusate from 2 to 4 mM and/or increasing the rate of pacing from 450 to 600 bpm (19, 20). Work performed was estimated as the RPP (RPP = DevP × heart rate [HR]) (19).
31P NMR spectra were obtained by using a Varian VNMRS spectrometer (161.4 MHz, 9.4 Tesla), as previously described (19, 20, 26). Two spectra were acquired during 16 min at each level of work demand. A single 31P NMR spectrum was signal-averaged from 208 free induction decay signals over 8 min. An external standard (phenylphosphonic acid) was placed in a capillary positioned within the NMR tube close to the isolated heart to allow measurement of absolute concentrations of high-energy phosphates (12, 26). Saturation correction factors for PCr, inorganic phosphate (Pi), and ATP were determined from fully relaxed spectra (recycle time, 20 s). Intracellular pH was determined from differences in chemical shift of intracellular Pi relative to PCr (20, 26). At the end of the perfusion protocol, all hearts were freeze-clamped while still beating and stored at −80°C for total creatine concentration measurements (13).
Determination of the free energy of ATP hydrolysis (ΔG∼ATP)
In addition to the intracellular pH, high-energy phosphate, and intracellular Pi concentrations as acquired by NMR spectroscopy, cytosolic creatine concentration is needed to calculate ΔG∼ATP. Thus, thin-layer chromatography was performed to determine the total creatine concentration [Crtotal] in the frozen samples (13). The myocardial water content was determined after drying at 100°C for 24 h. The cytosolic creatine concentration ([Cr]) was expressed as [Cr] = [Crtotal] − [PCr], as assessed by 31P NMR. Free [ADP] cannot be measured by 31P NMR directly (2). Thus, free [ADP] was calculated by using Equation (1), with the assumption that creatine kinase was at equilibrium and Keq = 1.66 × 109 M−1 (35):
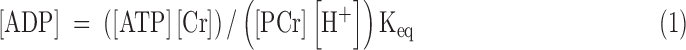 |
The free energy of ATP hydrolysis (ΔG∼ATP) was calculated by Equation (2), where ΔG0 (−30.5 kJ/mol) is the value of ΔG∼ATP under standard conditions of molarity, temperature, pH and [Mg2+], R = 8.314 J/mol · K, and T = 310 K (35).
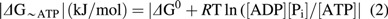
Given that ATP hydrolysis is an exergonic reaction, the value of ΔG∼ATP is always negative. In this article, we use the absolute value of ΔG∼ATP, |ΔG∼ATP| for the sake of clarity.
Statistical analysis
We present the results as mean ± standard error of the mean. Unpaired t-tests, Mann–Whitney non-parametric tests, or two-way or repeated-measures analysis of variance were used for group comparisons, as appropriate. We used GraphPad Prism software for the statistical analysis. p Value <0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of Health: HL-064750 (W.S.C.), EB 014414 (J.A.B.), NIDDK R01 DK103750 (M.M.B.), and the NHLBI-sponsored Boston University Cardiovascular Proteomics Center (Contract No. N01-HV-28178, W.S.C.); and by grants from the American Heart Association: “Grant in Aid” 16GRNT27660006 (M.M.B.), Fellow-to-Faculty Award 15FTF25890062 (I.L.), and Postdoctoral Fellowship 14POST20490003 (A.L.S.). Dr. Sverdlov was funded by the CJ Martin Fellowship (APP1037603) from the NHMRC (Australia); Future Leader Fellowship and Vanguard grant (Award IDs 101918 and 101038) from Heart Foundation of Australia; and RACP/Foundation for High Blood Pressure (Australia) Grant.
Abbreviations Used
- |ΔG∼ATP|
absolute value of free energy of ATP hydrolysis
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- bpm
beats per minute
- BSA
bovine serum albumin
- CD
control diet
- Em
myocardial peak early diastolic velocity
- ETC
electron transport chain
- HFHS
high fat, high sucrose
- IP
intraperitoneal
- LV
left ventricle
- LVEDP
left ventricular end-diastolic pressure
- LVH
left ventricular hypertrophy
- mCAT
transgenic expression of catalase targeted to the mitochondria
- MHD
metabolic heart disease
- NMR
nuclear magnetic resonance
- PCr
phosphocreatine
- ROS
reactive oxygen species
- RPP
rate × pressure product
- SERCA
sarcoplasmic reticulum Ca2+ ATPase
- WT
wild type
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
References
- 1. Ayalon N, Gopal DM, Mooney DM, Simonetti JS, Grossman JR, Dwivedi A, Donohue C, Perez AJ, Downing J, Gokce N, Miller EJ, Liang C-SS, Apovian CM, Colucci WS, and Ho JE. Preclinical left ventricular diastolic dysfunction in metabolic syndrome. Am J Cardiol 114: 838–842, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balaban RS. The application of nuclear magnetic resonance to the study of cellular physiology. Am J Physiol 246: C10–C19, 1985 [DOI] [PubMed] [Google Scholar]
- 3. Batra S. and Rakusan K. Geometry of capillary networks in hypertrophied rat heart. Microvasc Res 40: 29–40, 1991 [DOI] [PubMed] [Google Scholar]
- 4. Behring JB, Kumar V, Whelan SA, Chauhan P, Siwik DA, Costello CE, Colucci WS, Cohen RA, McComb ME, and Bachschmid MM. Does reversible cysteine oxidation link the Western diet to cardiac dysfunction? FASEB J 28: 1975–1987, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Boer RA, Pinto YM, and Van Veldhuisen DJ. The imbalance between oxygen demand and supply as a potential mechanism in the pathophysiology of heart failure: the role of microvascular growth and abnormalities. Microcirculation 10: 113–126, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Bugger H. and Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 114: 195–210, 2008 [DOI] [PubMed] [Google Scholar]
- 7. Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, and Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119: 2789–2797, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. The reference has been deleted
- 9. Fuentes L de las, Brown AL, Mathews SJ, Waggoner AD, Soto PF, Gropler RJ, and Davila-Roman VG. Metabolic syndrome is associated with abnormal left ventricular diastolic function independent of left ventricular mass. Eur Heart J 28: 553–559, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, and Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension 47: 887–893, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jameel MN. and Zhang J. Myocardial energetics in left ventricular hypertrophy. Curr Cardiol Rev 5: 243–250, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jansen MA, Shen H, Zhang L, Wolkowicz PE, and Balschi JA. Energy requirements for the Na+ gradient in the oxygenated isolated heart: effect of changing the free energy of ATP hydrolysis. Am J Physiol Heart Circ Physiol 285: H2437–H2445, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Kammermeier H. Microassay extracts of free and total of creatine. Anal Biochem 345: 341–345, 1973 [DOI] [PubMed] [Google Scholar]
- 14. Kammermeier H. High energy phosphate of the myocardium: concentration versus free energy change. Basic Res Cardiol 82: 31–36, 1987 [DOI] [PubMed] [Google Scholar]
- 15. Kayar S. and Weiss H. Diffusion distances, total capillary length volume in pressure-overload myocardial and mitochondrial hypertrophy. J Mol Cell Cardiol 24: 1155–1166, 1992 [DOI] [PubMed] [Google Scholar]
- 16. Lahey R, Carley AN, Wang X, Glass CE, Accola K, Silvestry SC, O'Donnell JM, and Lewandowski ED. Enhanced redox state and efficiency of glucose oxidation with miR based suppression of maladaptive NADPH-dependent malic enzyme 1 expression in hypertrophied hearts. Circ Res 122: 836–845, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, and Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart a multisystems approach. Circ Heart Fail 7: 1022–1031, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li G, Chen Y, Saari JT, and Kang YJ. Catalase-overexpressing transgenic mouse heart is resistant to ischemia- reperfusion injury. Am J Physiol 273: H1090–H1095, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, and Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 112: 2339–2346, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Luptak I, Sverdlov AL, Panagia M, Qin F, Pimentel DR, Croteau D, Siwik DA, Ingwall JS, Bachschmid MM, Balschi JA, and Colucci WS. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. J Mol Cell Cardiol 116: 106–114, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luptak I, Yan J, Cui L, Jain M, Liao R, and Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation 116: 901–909, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, and Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 131: 550–559, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagueh SF, Smiseth OA, Appleton CP, Byrd BF, Dokainish H, Edvardsen T, Flachskampf FA, Gillebert TC, Klein AL, Lancellotti P, Marino P, Oh JK, Popescu BA, and Waggoner AD. Recommendations for the evaluation of left ventricular diastolic function by echocardiography: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 29: 277–314, 2016 [DOI] [PubMed] [Google Scholar]
- 24. Ngo DTM, Sverdlov AL, Hess DT, Karki S, Macartney-Coxson D, Colucci WS, Carmine B, Farb MG, Stubbs RS, Colucci WS, and Gokce N. Oxidative modifications of mitochondrial complex II are associated with insulin resistance of visceral fat in obesity. Am J Physiol Endocrinol Metab 316: E168–E177, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pimentel DR, Amin JK, Xiao L, Miller T, Viereck J, Oliver-Krasinski J, Baliga R, Wang J, Siwik DA, Singh K, Pagano P, Colucci WS, and Sawyer DB. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res 89: 453–460, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Pinz I, Tian R, Belke D, Swanson E, Dillmann W, and Ingwall JS. Compromised myocardial energetics in hypertrophied mouse hearts diminish the beneficial effect of overexpressing SERCA2a. J Biol Chem 286: 10163–10168, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qin F, Lennon-Edwards S, Lancel S, Biolo A, Siwik DA, Pimentel DR, Dorn GW, Kang YJ, and Colucci WS. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q-overexpressing transgenic mice. Circ Heart Fail 3: 306–313, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin F, Siwik DA, Luptak I, Hou X, Wang L, Higuchi A, Weisbrod RM, Ouchi N, Tu VH, Calamaras TD, Miller EJ, Verbeuren TJ, Walsh K, Cohen RA, and Colucci WS. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation 125: 1757–1764, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qin F, Siwik DA, Pimentel DR, Morgan RJ, Biolo A, Tu VH, Kang YJ, Cohen RA, and Colucci WS. Cytosolic H2O2 mediates hypertrophy, apoptosis, and decreased SERCA activity in mice with chronic hemodynamic overload. Am J Physiol Heart Circ Physiol 306: H1453–H1463, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryan TE, Schmidt CA, Green TD, Spangenburg EE, Neufer PD, and McClung JM. Targeted expression of catalase to mitochondria protects against ischemic myopathy in high-fat diet-fed mice. Diabetes 65: 2553–2568, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sabbah HN, Sharov VG, Lesch M, and Goldstein S. Progression of heart failure: a role for interstitial fibrosis. In: Cellular Interactions in Cardiac Pathophysiology, edited by Slezák J. and Ziegelhöffer A. Boston, MA: Springer US, 1995, pp. 29–34 [DOI] [PubMed] [Google Scholar]
- 32. Sverdlov AL, Elezaby A, Behring JB, Bachschmid MM, Luptak I, Tu VH, Siwik DA, Miller EJ, Liesa M, Shirihai OS, Pimentel DR, Cohen RA, and Colucci WS. High fat, high sucrose diet causes cardiac mitochondrial dysfunction due in part to oxidative post-translational modification of mitochondrial complex II. J Mol Cell Cardiol 78: 165–173, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sverdlov AL, Elezaby A, Qin F, Behring JB, Luptak I, Calamaras TD, Siwik DA, Miller EJ, Liesa M, Shirihai OS, Pimentel DR, Cohen RA, Bachschmid MM, and Colucci WS. Mitochondrial reactive oxygen species mediate cardiac structural, functional, and mitochondrial consequences of diet-induced metabolic heart disease. J Am Heart Assoc 5: 1–13, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian R, Halow JM, Meyer M, Dillmann WH, Figueredo VM, Ingwall JS, and Camacho SA. Thermodynamic limitation for Ca2+ handling contributes to decreased contractile reserve in rat hearts. Am J Physiol 275: H2064–H2071, 1998 [DOI] [PubMed] [Google Scholar]
- 35. Tian R. and Ingwall JS. Energetic basis for reduced contractile reserve in isolated rat hearts. Am J Physiol 270: H1207–H1216, 1996 [DOI] [PubMed] [Google Scholar]
- 36. Wang S-BB, Foster DB, Rucker J, O'Rourke B, Kass DA, and Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res 109: 750–757, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weber KT, Clark WA, Janicki JS, and Shroff SG. Physiologic versus pathologic hypertrophy and the pressure-overloaded myocardium. J Cardiovasc Pharmacol 10: S37–S50, 1987 [PubMed] [Google Scholar]
- 38. The reference has been deleted
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.