

Abstract

The usefulness of dried Dowex H+ cation-exchange resin with or without sodium iodide (NaI) as a catalyst system for different kinds of esterifications using carboxylic acids and alcohols as starting materials has been systematically investigated. The Dowex H+/NaI approach is very effective, generally high yielding, energy-efficient, and nontoxic, and the Dowex H+ resin is reusable. Since the whole procedure from start to product isolation is also very simple, these features make the method environmentally friendly. The method is regioselective, and its potential for separation of valuable carboxylic acids like resin acids from mixtures containing other kinds of carboxylic acids has been demonstrated. Examples for green and straightforward esterification of highly important natural amino acids are also presented.
Introduction
The German chemist, Leopold Gmelin was perhaps the first person to coin the term “ester” probably as a contraction of the German Essigäther, “acetic ether”.1 In those days, esters were also called oxy-acid ethers. Generally, esters are compounds derived from acid (organic or inorganic) and alcohol.2 Triglycerides, i.e., esters of fatty acids and glycerol, are one of the main classes of lipids and important compounds in biology since animal fats and vegetable oils are mainly triglycerides.3 Polyesters are very important compounds found naturally, e.g., in the cuticle of plants to protect leaves, and their synthetic counterparts are the plastics used in bottles, resins, clothing, etc.4,5 Many esters have a pleasant “sweet” odor, and they occur naturally in fruits and plants: isoamyl acetate (pear, banana), ethyl hexanoate (pineapple), octyl acetate (orange), and methyl butyrate (apple).6,7 Esters are also used as solvents by the chemical industry, e.g., ethyl acetate is a common solvent with an annual global production of over 3.5 million tons in 2015.8 DNA and RNA structures contain phosphodiester bonds in their structures. In fact, it would not be an exaggeration to state that esters are crucial for life on Earth.9
There are numerous methods for the preparation of esters published in the literature. The most common and best known is the Fischer esterification, which is a reversible acid-catalyzed condensation reaction (see Scheme 1), but there are many irreversible reactions catalyzed by one or more catalysts, such as the Mitsunobu [activation by triphenylphosphine (PPh3) and diethyl azodicarboxylate] and Steglich [activation by carbodiimides like dicyclohexylcarbodiimide and dimethylaminopyridine] esterifications.10 Even though the literature is replete with methods for esterifications, novel approaches have been reported recently. Here are only a few examples: synthesis of ethyl lactate,11 aryl carboxylates,12 vinyl ether-containing esters,13 and lipase-catalyzed alkyl valerates.14 In addition, the use of triphenylphosphine oxide and oxalyl chloride [(COCl)2] coupling reagents to achieve esterification at room temperature has been reported as a novel method.15
Scheme 1. Esterification Methods.

Generally, perhaps one of the easiest ways to prepare esters is the use of alcohol and a more reactive carboxylic anhydride or halogenide instead of acids (see Scheme 1). These reactions are simple, high yielding, and mostly performed without any special catalysts; however, in most cases, a hydrogen halogenide (commonly HCl gas) scavenger is needed if carboxylic acid halogenide (most common is carboxylic acid chloride) is used.10 From the point of view of the environment, the best way to prepare esters would be the case presented also in Scheme 1, which is pretty close to the method we present in this paper. The use of only carboxylic acids and alcohols as starting materials instead of anhydrides or halogenides would be the ideal “green” procedure because, as generally known, acids and alcohols are so ubiquitous that they can be considered as “natural” compounds. Carboxylic acid anhydrides and halogenides (mostly chlorides) need to be prepared chemically (condensation of carboxylic acids to anhydrides or halogenation to halogenide) and that requires energy, catalyst(s), halogen source (like thionyl chloride, SOCl2), which is usually toxic, solvents, etc., i.e., these are not environmentally friendly compounds.10 Thus, we have several fundamental problems to be resolved, such as global warming and plastic exposure; micro- and nanoplastics can be found almost everywhere around the planet.16,17 It is evident that we need improved and more energy-efficient and environmentally friendly methods to prepare chemical compounds even if they are prepared on a small scale, according to the motto “great oaks from little acorns grow”.
In 2015, we published a “proof-of-concept” paper entitled, “A powerful tool for acid-catalyzed organic addition and substitution reactions”, where we presented how extremely useful “tool” oven-dried Dowex H+ cation-exchange resin with NaI is.18 By adopting the Dowex H+/NaI approach, one can prepare compounds like tertiary iodides, ethers, esters (also using R–CN as the acyl source and dimethylformamide as the formate source), biodiesel from used cooking oil (transesterification process), straight substitution of R–OH to R–I, cyclic ethers can be opened to useful building blocks,19 iodine can be added to a compound with a triple bond to form an iodinated double bond, etc. (see refs18, 19). As mentioned, the method used in those papers is based on the use of oven-dried Dowex H+ cation-exchange resin with NaX (X = I or Br). It is known that Dowex-type H+ cation-exchange resins can be used as solid catalysts in esterification reactions;20−22 however, as far as we are aware, there are very few publications where the oven-dried Dowex H+ resin has been used by itself for esterifications, and no reports where it has been used with NaI. Here, we will present systematically how extremely useful and effective the dried Dowex H+/NaI approach is for esterification reactions and why the method can be considered as a green chemistry tool/method. In this paper, we will present several examples of how dried Dowex H+ can be used effectively with or (in some cases) without NaI in the synthesis of very sterically hindered esters using only carboxylic acids and alcohols as starting materials under relatively mild conditions, and in addition, examples of regioselective esterifications will be presented. In most of the cases, the isolation of the produced esters was very simple with no further purifications needed. Examples of the esterification of highly important natural amino acids will also be presented.
It can be proposed that Dowex H+ cation-exchange resin is commonly used in all chemistry laboratories throughout the world, and therefore, it is widely available. The background describing how we initially discovered its potential for use in various chemical reactions can be found elsewhere.18 Briefly, Dowex H+ needs to be dried before use (at 120 °C in oven for around 18–20 h; see Figure 1), which makes it far more effective than undried resin (moisture content of regular Dowex H+ cation-exchange resin may be over 50%). In fact, the best way to make it as effective as possible is first to treat Dowex H+ with 2 M HCl, then wash several times with distilled water, and finally dry it (more detailed procedure can be found in Experimental Section).
Figure 1.

Dowex H+ cation-exchange resin before (left) and after (right) drying in oven at 120 °C overnight.
Results and Discussion
In Table 1, we have collected 18 examples for the preparation of esters starting from carboxylic acids, and in Table 2, we have presented six examples of acylation of selected alcohols by using dried Dowex H+ cation-exchange resin with or, in some cases (seven examples), without dried NaI. Tables 1 and 2 also describe the reaction conditions used (temperature and reaction time); detailed reaction conditions and isolation procedures can be found in Experimental Section.
Table 1. Esterification of Selected Carboxylic Acids by Dried Dowex H+/NaI Approach.

In the table, only the reaction time and temperature have been highlighted; detailed experimental procedures and conditions can be found in Experimental Section.
Isolated yields.
Conversion according to 1H NMR spectrum in parentheses.
Without NaI.
Product co-evaporation with MeOH was observed; r.t. = room temperature, n.d. = not determined, n.i. = not isolated.
Table 2. Acylation of Selected Alcohols by the Dried Dowex H+/NaI Approach.

In the table, only the reaction time and temperature have been highlighted; detailed experimental procedures and conditions can be found in Experimental Section.
Isolated yields.
Conversion according to 1H NMR spectrum in parentheses; r.t. = room temperature, n.d. = not determined.
Without NaI.
One of the most interesting starting materials for esterification experiments presented here was tropic acid (1), which is a precursor of a very important natural compound atropine (48; see Figure 2). Atropine (48) can be extracted from some plants like Atropa belladonna; it is an antagonist of the muscarinic cholinergic receptors, so it has a broad range of medical applications. Atropine (48) is also used as an antidote for poisoning from nerve gases (such as sarin and VX) and organophosphate insecticides.23 In addition to being an important precursor of atropine (48), tropic acid (1) is an attractive compound having a sterically hindered secondary carboxylic acid group and a hydroxyl group (even though this is a primary one, it has a bulky phenyl and carboxylic acid group in the β-carbon) in the same molecule. Four different derivatives of tropic acid (1) were prepared: two esters (22 and 23; see Table 1) and two acylated compounds (41 and 42; see Table 2).
Figure 2.
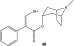
Chemical structure of atropine (48).
Interestingly, according to SciFinder search, only eight examples of ways to prepare tropic acid methyl ester (22) from tropic acid (1) have been reported with poor yields (<20%) or yields not even reported at all, and most of the esterifications were performed by basic Fischer esterification catalyzed by sulfuric acid.24 There is one paper that describes the synthesis of tropic acid isopropyl ester (23) by chlorination of carboxylic acid group using thionyl chloride; however, yield was not mentioned.25 As can be seen in Table 1, by using our method, the isolated yields of tropic acid methyl (22) and isopropyl (23) esters were 83 and 72%, respectively, much higher than in the earlier reports. Only three examples to prepare acetylated tropic acid (41) from tropic acid (1) were found in the literature, according to SciFinder search, all utilizing acetyl chloride.26 The isobutyl derivative (42) was totally unknown. By adopting the Dowex H+/NaI approach with tropic acid (1) and acetic or isobutyric acid, 99 and 44% isolated yields (99 and 88% conversions; see Table 2) were achieved for compounds 41 and 42, respectively.
The usefulness of the Dowex H+/NaI method for esterifications of benzoic acid (2) and its 4-hydroxy (3) and 2-hydroxy (4, salicylic acid) derivatives was also tested. Methyl benzoate (24) was produced with a high isolated yield (82%) by refluxing 2 for 24 h in MeOH. 4-Hydroxybenzoic acid ethyl ester (25) was isolated with a reasonable yield (51%); however, to achieve 91% conversion of 3 to 25, the reaction was refluxed for 72 h in EtOH. Only 50% conversion of salicylic acid (4) to its ethyl ester (26) was observed under the same conditions as for the synthesis of 25. One can clearly discern the inactivation effect of the presence of a hydroxy group in the 2-position versus 4-position of the benzene ring for the synthesis of compounds 25 and 26 because the conversions were 91 and 50%, respectively, under the same conditions.
We describe many examples of organic acids with different kinds of functional groups (5–10) to emphasize how effectively they can be esterified to compounds 27–32 by the Dowex H+/NaI approach under very mild reaction conditions, mostly by stirring at room temperature for 2–4 h [compound 32, which is an ethyl ester, not a methyl like the other compounds (27–31), needed stirring at 65 °C for 4 h to achieve 99% conversion]. Methyl hex-5-ynoate (28) was stirred for 24 h to achieve 100% conversion (75% isolated yield); however, the synthesis was performed without NaI, so it was not unexpected that the time to reach 100% conversion would be longer. The reason why we tested the synthesis of 28 without NaI is the possibility of an addition reaction to the triple bond, which we have reported elsewhere.18 This seems to happen even at room temperature, i.e., we observed this side reaction according to the 1H NMR spectrum in synthesis experiments to produce 28 with NaI after stirring for 24 h. Full 100% conversion of 6 to 28 can be achieved also with a catalytic amount of NaI (0.1 equiv) at room temperature with 4 h stirring; however, due to the slightly different isolation procedure, the isolated yield was 66% instead of 75%, with the procedure lacking NaI. We were not able to isolate compound 29 because of the previously observed co-evaporation with MeOH in the isolation procedure. It was also quite difficult to isolate compound 30 for the same reasons; however, we were able to obtain a 40% yield.
Malic acid (11) was chosen to be one of the starting materials in our esterification experiments because it is a dicarboxylic acid and it has two different kinds of carboxylic acid groups, primary and secondary. The dimethyl ester of malic acid (33) was effectively synthesized by stirring at room temperature for 24 h.
Amino acids are very important compounds for all life on Earth, but in addition, their structures were very interesting for the esterification experiments presented here because of α-amino groups and different side chains.27 We selected four different kinds of amino acids (as their hydrochloride salts, 12–15) for esterification experiments with the Dowex H+/NaI approach. According to a SciFinder search, by far the most commonly reported method to prepare compounds 34–37 has been the use of thionyl chloride to synthesize the acid chloride, followed by the addition of methanol, which is not an environmentally friendly approach. Surprisingly, very little attention has been paid to preparing amino acid methyl esters (34–37) from the corresponding amino acid hydrochlorides (12–15), and only 17 examples were found in the literature. We were able to prepare compounds 34–37 from 12–15 by using only dried Dowex H+ as the catalyst (see reaction conditions in Table 1). The isolation of the products was extremely straightforward: only filtration and evaporation were needed and high yields (70–82%) were acquired to compounds 34–37 (see detailed procedures in Experimental Section). The method reported here is a new, green, and straightforward way to prepare amino acid esters 34–37, and most probably it can be applied to other amino acids as well.
There were several reasons why citric acid (16) and iso-citric acid trisodium salt hydrate (17) were selected as starting materials for the esterification experiments with the Dowex H+/NaI method: (1) they are tricarboxylic acids; citric acid (16) has two primary carboxylic acid groups and one tertiary carboxylic acid group, and iso-citric acid trisodium salt hydrate (17) has one primary and two secondary carboxylic acid groups; (2) both are very important compounds and part of the citric acid cycle28 to release stored energy by all aerobic organisms; (3) citric acid triethyl ester (38) is an important precursor in the synthesis of phosphocitric acid, which is a naturally occurring biomolecule found in cells;29 (4) iso-citric acid triethyl ester (39) will become an important precursor for the synthesis of phospho-iso-citric acid, which has not yet been reported in the literature and it is not commercially available. The triethyl ester of citric acid (38) was prepared and isolated with quantitative yield (97% isolated yield; see Table 1) by refluxing compound 16 for 24 h in EtOH. The isolated yield for triethyl ester of iso-citric acid (39) was rather low (29%); however, the procedure was very straightforward because we could use directly the commercially available salt form of isocitrate (17) with no need to first convert the salt to the acid. Actually, for our needs, the 29% yield was more than sufficient, so the optimization of the procedure did not seem necessary. It should be noted that, according to the 1H NMR spectrum measured from the sample taken in the reaction mixture of 39, we were able to detect a mixture of triethyl ester (39) and diethyl esters of iso-citric acid. The presence of these other compounds and the isolation procedure of 39 (see Experimental Section) were the main reasons for the low isolated yield (29%).
As mentioned earlier, in Table 2, we have collected six examples of acylation of selected compounds by using the method reported here. Compounds 41 and 42 have been discussed earlier in the text, and compound 40 is an example of the esterification of benzyl alcohol using only dried Dowex H+ without NaI at 80 °C for 24 h. Under those conditions, a high isolated yield (85%) was achieved.
Mandelic acid (19) is an important compound, e.g., used as a precursor, in the pharmaceutical and cosmetic industries.30 It has a hindered secondary hydroxyl group, which made it a valuable compound for our esterification experiments. In the literature, only two chemical methods to prepare acetylated mandelic acid (43) from 19 and acetic acid were found, catalyzed by zinc chloride31 or silica sulfuric acid32 with 72 and 92% reported yields, respectively. In the case when silica sulfuric acid was used as the catalyst, neither the experimental procedure nor the isolation procedure were reported, also NMR characterization data were missing. In our experiment, the acetylated mandelic acid (43) was produced at 90 °C after a 24 h reaction time with a rather good isolated yield (55%; 81% conversion was observed). This is a good alternative to these previously reported methods and demonstrates well that the Dowex H+/NaI approach is effective and useful also when highly steric ester structures need to be prepared using only alcohols and carboxylic acids as starting materials.
In the preparation of octyl acetate (44) and but-3-en-1-yl acetate (45), we decided to test how effective the Dowex H+/NaI method would be if only 2 equiv of acetic acid (compared to alcohols 20 and 21) were used in the reactions with no solvent and which temperature and reaction time would be optimal for good yields. Surprisingly, at 55 °C with a reaction time of 2 h, 97% conversion of octanol (20) to octyl acetate (44) was observed and a high isolated yield (83%) was achieved. After the reaction at the same 55 °C temperature for 18 h reaction time, but-3-en-1-yl acetate (45) was isolated with a reasonable yield (52%). In the isolation of 45, we did not use any solvents, so the synthesis and isolation of 45 can be considered a very green procedure. According to a SciFinder search, there are only 18 references that are related to the synthesis of 45 from 21 using acetic acid as the acetylation reagent, 16 of which are patents. Perhaps the closest method to ours was catalyzed by sulfuric acid under the following conditions: 10 equiv acetic acid, 60 °C, 1 h; 72% isolated yield was reported.33 But-3-en-1-yl acetate (45) is a very important precursor in the synthesis of pheromones.33
According to the results reported in Tables 1 and 2, we decided to conduct some experiments to clarify the selectivity of the Dowex H+/NaI method in esterification reactions. Mixtures (1:1) of phenol with octanol or isopropanol or cyclohexanol were reacted with 10 equiv of acetic acid under the conditions reported in Scheme 2. All aliphatic alcohols were converted (100%) to their acetates over phenol. We also tested the combination of phenol, octanol, and cyclohexanol (1:1:1) under the same conditions at room temperature to determine if there was any selectivity of octanol over cyclohexanol; however, both were converted to their acetates (>95%).
Scheme 2. Selective Acetylation of Studied Alcohols (R = Octyl, Pri, Cyclohexyl) in the Presence of Phenol.

Selective synthesis of citric acid dimethyl ester (46) starting from citric acid (16) with 60% isolated yield was obtained (see Scheme 3). The whole synthesis procedure with isolation was rather straightforward, as shown in Experimental Section. There are only a few reports in the literature to prepare compound 46: one method is based on boric or boronic acid catalyzation,34 and the other utilizes a Fischer esterification procedure and sulfuric acid catalyzation with 32% isolated yield;35 another paper reported a 35% yield,36 although with rather complicated isolation procedures compared to our method. We also conducted a straightforward test with benzoic acid (2) and phenylacetic acid (5) in the same pot; after 1.5 h stirring at room temperature, the conversion of 5 to 27 was observed to be 100% and benzoic acid did not react at all; so we separated and isolated both compounds (see Scheme 4 and the detailed procedure in Experimental Section). This is not a surprise since the same kind of experiment has been described in the literature but with Amberlyst 15 as the catalyst.37 Nonetheless, this is an example demonstrating that it is possible to use the Dowex H+/NaI method for esterification of less hindered carboxylic acids and separate them and then proceed with esterification of more hindered carboxylic acids like benzoic acid (2) or its derivatives at elevated temperatures (see Table 1; preparation of compounds 24–26).
Scheme 3. Selective Synthesis of Citric Acid Dimethyl Ester.

Scheme 4. Selective Esterification of Phenylacetic Acid (5) over Benzoic Acid (2).

For the reasons mentioned earlier, dl-malic acid (11) was considered an interesting compound for selective esterification experiments. Malic acid is an important compound in biochemistry, produced by all living organisms, and it is part of the earlier mentioned citric acid cycle;28 it also confers the sour taste to fruits.38 Only one report for the synthesis of malic acid mono methyl ester (47) was found in the literature using enzymatic hydrolysis of dimethyl malic acid39 (33), and therefore, this is the first chemical synthesis of 47 reported so far in the literature. The best and most adequate isolated yield (45%) was achieved when 500 mg of 11 was stirred with 500 mg of dried Dowex H+ resin without NaI for 2 h at room temperature (see Scheme 5). The final product 47 could be readily separated from mixture containing diester 33 by crystallization (detailed procedure can be found in Experimental Section).
Scheme 5. Selective Synthesis of Malic Acid Monomethyl Ester.

The effect of NaI in the reactions can be clearly seen in the results collected in Table 3. The effect is much greater when preparing more steric esters (e.g., 42) compared to their nonsteric counterparts (e.g., 44). In addition, in the case of methyl benzoate (24), the use of 0.5 equiv of NaI instead 0.1 equiv gave better conversion, 92 and 83%, respectively. This was not the case with compound 42; these results can be seen in Table 4 and will be discussed later. As a comparison, sulfuric acid (H2SO4) catalysis to prepare compounds 24 and 44 was tested and achieved conversions were rather similar to those with esterifications with the Dowex H+/NaI method.
Table 3. Effect of NaI on Product Conversion in Selected Reactions.

Dried Dowex H+ with 0.5 equiv dried NaI.
Only dried Dowex H+.
Dried Dowex with 0.1 equiv dried NaI.
H2SO4-catalyzed.
Table 4. Effect of the Amount of Dried NaI Used in the Synthesis of the Highly Steric Compound 42.
| amount of NaI (equiv) | conversiona (%) |
|---|---|
| 0 | 19 |
| 0.1 | 74 |
| 0.2 | 66 |
| 0.5 | 25 |
50 °C, 24 h.
We also tested the effect of the amount of NaI used in the synthesis of the highly steric ester 42; the results are collected in Table 4. The best conversion (74%) was achieved with 0.1 equiv of NaI; more NaI or its absence gave lower conversions. In fact, the amounts of NaI used in the esterification reactions reported in this paper have not been optimized, and according to the above result, it might be reasonable to test the optimal amount of NaI on a case-by-case basis to achieve the best conversion.
Last but not least, we decided to test how effective and selective the Dowex H+/NaI method would be in the separation of valuable resin acids (50; see the general structure in Figure 3) from a real tall oil sample.40 Tall oil is a commercially valuable byproduct of softwood (pine or spruce) from pulp production with more than 1.6 M tonnes being produced annually. The major components of tall oil are unsaturated fatty acids and the more valuable resin acids (50), which are used in many industrial and consumer products. On an industrial scale, fatty acid components of the tall oil are separated from resin acids by high vacuum distillation.41 However, the complete separation of the components by distillation methods is difficult and not energy-efficient. Tall oil esterification by the method described here, followed by extraction, leads to quantitative separation of fatty acid methyl esters (see Scheme 6). The tertiary resin acids (50) remain intact in our method and are recovered from the process at a high yield (see procedure in Experimental Section).
Figure 3.
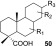
General structure of resin acids (50).
Scheme 6. Separation of Valuable Resin Acids (50, Part of the Diterpene Structure Omitted) in Mixture Containing Fatty Acids (49).

The exact reaction mechanism is unclear, but the most probable mechanism for unhindered esters follows the traditional proton-catalyzed esterification mechanism. In this case, the H+ form solid resin is an excellent proton donor and carbonyl oxygen (C=O) is protonated, the nucleophilic alcohol attacks the positively charged carbon, and water is eliminated yielding the ester (e.g., see Table 3 product 24). In the case of hindered esters (see Table 4), the mechanism is the formation of R–I from R–OH and NaI in the presence of H+ from Dowex and/or the formation of HI, which is a better proton donor than Dowex since 19 and 74% conversions were obtained without and with 0.1 equiv of NaI, respectively. Generally, substitution of alcohol group to iodine requires either activation of OH group or elevated temperatures.18,42 In the previous paper,18 we have demonstrated and discussed the formation of HI based on an addition reaction to the double bond. At least under elevated temperatures, the R–I-based mechanism might be possible.
Conclusions
The study clearly and unambiguously highlights the straightforward nature of these kinds of synthetic procedures, and usually products can be isolated without any further purifications (except two compounds, 42 and 43, which required column chromatography purifications) using only carboxylic acids and alcohols as starting materials. Our method can be compared to the basic Fisher esterification, e.g., as catalyzed by sulfuric acid; however, the product isolation by using our method seems to be simpler than in Fischer esterifications (e.g., isolation of 34–37) and no side reactions were observed under the conditions used; with sulfuric acid, side reactions are possible at least with unsaturated starting materials.18 Generally, our conditions were mild, of course some elevated temperatures were needed in reactions when very steric esters like 42 and 43 were prepared or starting materials had unreactive or steric groups like 2 and 16. We have demonstrated that the dried Dowex H+/NaI approach can be used for selective esterifications and there is clear evidence that NaI reduces the reaction energy needed in the esterification reactions, and in particular, it improves conversions when steric esters need to prepared; however, the amount of NaI may need to be determined on a test case-by-case basis to achieve the best results, as shown in Table 4. In the experiments of the esterification of amino acids 12–15, which were the last products we made, we showed that a sufficient amount of dried Dowex H+ was only 50 mg (for diacid 35, more, i.e., 75 mg was needed) vs 100 mg of starting material (0.5:1 mass ratio) and the corresponding esters (34–37) were synthesized without NaI and isolated so simply that it could be used for industrial purposes (see procedures in Experimental Section) with high yields (70–82%). We have also demonstrated how extremely useful the dried Dowex H+/NaI method is in the separation and isolation of highly valuable resin acids (50) from the mixture of acids (see Scheme 6). The reusability of Dowex H+ resin was tested for the synthesis of 27 and 28. In both cases, Dowex H+ resin was collected and reused two times (used for three times total), leading to ca. 100% conversion in all three times according to the 1H spectra (see the detailed procedure in Experimental Section).
And finally, in a nutshell, the presented method/procedure is extremely simple, very effective, and nontoxic, and Dowex H+ resin is regenerable,18 reusable, and energy-efficient, which make it a green approach.
Experimental Section
General
1H and 13C NMR spectra were recorded on a 600 MHz spectrometer operating at 600.2 and 150.9 MHz, respectively. The solvent residual peak was used as a standard for 1H and 13C measurements in CDCl3 and CD3OD (7.26 or 77.16 ppm for CDCl3 and 3.31 or 49.00 ppm for CD3OD, respectively),43 in D2O 4.79 ppm in 1H measurements and added CD3OD in the 13C measurements (49.00 ppm). The nJHH couplings were calculated from the proton spectra, and all J values are given in Hz. Appropriate two-dimensional NMR measurements were performed to confirm structures when needed. Mass spectra were recorded on a quadrupole time-of-flight mass spectrometer using electrospray ionization (ESI) with positive ionization mode for totally novel compound 42 and also for compounds 23, 39, and 47 (compounds for which chemical syntheses were not found in the literature). The purity of the products was determined from 1H spectrum and was ≥95% unless stated otherwise. All starting materials (1–21) and the Dowex H+ ion-exchange resin used in the study were commercially available.
Example of the Preparation of Dried Dowex H+ Ion-Exchange Resin
Dowex ion-exchange resin (50Wx8 hydrogen form, 100–200 mesh; 25 g) was stirred for 0.5 h in 2 M HCl solution (50 mL) at room temperature before it was filtered, and washed with distilled H2O until the filtrate pH was neutral. Finally, Dowex was dried in an oven for 18–20 h at 120 °C and stored in a closed bottle.
Preparation of 3-Hydroxy-2-phenylpropanoic Acid Methyl Ester (22)
Tropic acid (100 mg, 0.60 mmol), dried NaI (18 mg, 0.12 mmol, 0.2 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 200 mg), and dried MeOH (1 mL) were stirred overnight (around 18 h) at room temperature before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in dichloromethane (DCM) (5 mL), washed with 10% Na2S2O3 (sodium thiosulfate, 2–3 mL), dried over MgSO4, and evaporated to dryness in vacuo. 3-Hydroxy-2-phenylpropanoic acid methyl ester (90 mg, 83%) was obtained as a colorless syrup. 1H NMR (CDCl3): δ 7.37–7.32 (m, 2H), 7.32–7.28 (m, 1H), 7.28–7.25 (m, 1H), 4.16–4.11 (m, 1H), 3.88–3.84 (m, 1H), 3.84–3.80 (m, 1H), 3.71 (s, 3H), 2.28 (br, 1H, −OH). 13C NMR (CDCl3): δ 173.8, 135.7, 129.0 (2C), 128.3 (2C), 127.9, 64.7, 54.0, 52.4.
Preparation of 3-Hydroxy-2-phenylpropanoic Acid Isopropyl Ester (23)
Tropic acid (100 mg, 0.60 mmol), dried NaI (45 mg, 0.30 mmol, 0.5 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 300 mg), and 2-propanol (1.5 mL) were refluxed for 24 h before Dowex was collected after filtration and washed with 2-propanol, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in DCM (5 mL), washed with 10% Na2S2O3 (sodium thiosulfate, 2–3 mL), dried over MgSO4, and evaporated to dryness in vacuo. 3-Hydroxy-2-phenylpropanoic acid isopropyl ester (90 mg, 72%) was obtained as a colorless syrup. 1H NMR (CDCl3): δ 7.35–7.31 (m, 2H), 7.30–7.24 (m, 3H), 5.08 (h, 1H, 3JHH = 6.3), 4.13–4.07 (m, 1H), 3.83–3.78 (m, 2H), 2.32 (t, 1H, −OH, 3JHH = 6.4), 1.25 (d, 3H, 3JHH = 6.3), 1.14 (d, 3H, 3JHH = 6.3). 13C NMR (CDCl3): δ 172.9, 136.0, 128.9 (2C), 128.3 (2C), 127.7, 68.7, 64.9, 54.3, 21.9, 21.6. MS (ESI+) calcd for C12H16O3 [M + H]+ 209.1172, found: 209.1175. NMR spectra in Supporting Information p. S2.
Preparation of Methyl Benzoate (24)
Benzoic acid (100 mg, 0.82 mmol), dried NaI (61 mg, 0.41 mmol, 0.5 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 400 mg), and dried MeOH (3 mL) were refluxed for 24 h before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. Methyl benzoate (91 mg, 82%) was obtained as a colorless oil. 1H NMR (CDCl3): δ 8.06–8.03 (m, 2H), 7.58–7.53 (m, 1H), 7.46–7.42 (m, 2H), 3.92 (s, 3H). 13C NMR (CDCl3): δ 167.3, 133.0, 130.3, 129.7 (2C), 128.5 (2C), 52.2.
Preparation of Ethyl 4-Hydroxybenzoate (25)
4-Hydroxybenzoic acid (200 mg, 1.45 mmol), dried NaI (110 mg, 0.73 mmol, 0.5 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 1.0 g), and 94% (A) EtOH (8 mL) were refluxed for around 72 h before Dowex was collected after filtration and washed with EtOH, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in DCM (5 mL), washed twice with saturated NaHCO3 (3 mL), dried over MgSO4, and evaporated to dryness in vacuo. Ethyl 4-hydroxybenzoate (122 mg, 51%) was obtained as a white powder. 1H NMR (CD3OD): δ 7.88–7.85 (m, 2H), 6.83–6.80 (m, 2H), 4.31 (q, 2H, 3JHH = 7.1), 3.92 (t, 3H, 3JHH = 7.1). 13C NMR (CD3OD): δ 168.3, 163.6, 132.7 (2C), 122.5, 116.1 (2C), 61.7, 14.7.
Preparation of Methyl 2-Phenylacetate (27)
Phenylacetic acid (100 mg, 0.73 mmol), dried NaI (55 mg, 0.34 mmol, ca. 0.5 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 300 mg), and dried MeOH (3 mL) were stirred for 2 h at r.t. before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in DCM (8 mL), washed with 10% Na2S2O3 (sodium thiosulfate, 2–3 mL) and saturated NaHCO3 (2–3 mL), dried over MgSO4, and evaporated to dryness in vacuo. Methyl 2-phenylacetate (69 mg, 63%) was obtained as a colorless liquid. 1H NMR (CDCl3): δ 7.36–7.31 (m, 2H), 7.30–7.25 (m, 3H), 3.69 (s, 3H), 3.63 (s, 2H). 13C NMR (CDCl3): δ 172.2, 134.1, 129.4 (2C), 128.7 (2C), 127.2, 52.2, 41.3.
Preparation of 5-Hexynoic Acid Methyl Ester (28)
5-Hexynoic acid (300 mg, 2.68 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 250 mg), and MeOH (3 mL) were stirred for 24 h at room temperature before Dowex was collected after filtration and washed with a small portion of diethyl ether before the solvents were removed in vacuo. 5-Hexynoic acid methyl ester (254 mg, 75%) was obtained as a colorless liquid. 1H NMR (CDCl3): δ 3.68 (s, 3H), 2.46 (t, 2H, 3JHH = 7.4), 2.26 (t + d, 2H, 3JHH = 7.0, 4JHH = 2.7), 1.96 (t, 1H, 3JHH = 2.7), 1.85 (qv, 2H, 3JHH = 7.2). 13C NMR (CDCl3): δ 173.7, 83.4, 69.2, 51.7, 32.8, 23.7, 18.0.
Preparation of Hexanoic Acid Methyl Ester (30)
Hexanoic acid (300 mg, 2.58 mmol), dried NaI (39 mg, 0.26 mmol, 0.1 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 250 mg), and MeOH (3 mL) were stirred for 4 h at room temperature before Dowex was collected after filtration and MeOH removed in vacuo. The residue was dissolved in diethyl ether (5 mL), washed with 10% Na2S2O3 (sodium thiosulfate, 0.5 mL), dried over MgSO4, and diethyl ether was removed in vacuo. Methyl hexanoate (133 mg, 40%) was obtained as a colorless liquid. 1H NMR (CDCl3): δ 3.66 (s, 3H), 2.29 (t, 2H, 3JHH = 7.5), 1.62 (qv, 2H, 3JHH = 7.5), 1.35–1.26 (m, 4H), 0.89 (t, 3H). 13C NMR (CDCl3): δ 174.5, 51.6, 34.2, 31.5, 24.8, 22.4, 14.0.
Preparation of 10-Undecenoic Acid Methyl Ester (31)
10-Undecenoic acid (300 mg, 1.63 mmol), dried NaI (24 mg, 0.16 mmol, 0.1 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 200 mg), and MeOH (3 mL) were stirred for 4 h at room temperature before Dowex was collected after filtration and washed with a small portion of MeOH, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in diethyl ether (4 mL), washed with 10% Na2S2O3 (sodium thiosulfate, 0.5 mL), dried over MgSO4, and diethyl ether was removed in vacuo. 10-Undecenoic acid methyl ester (249 mg, 77%) was obtained as a colorless oil. 1H NMR (CDCl3): δ 5.84–5.77 (m, 1H), 5.01–4.90 (m, 2H), 3.66 (s, 3H), 2.30 (t, 2H, 3JHH = 7.5), 2.06–2.00 (m, 2H), 1.65–1.58 (m, 2H), 1.40–1.33 (m, 2H), 1.32–1.26 (m, 8H). 13C NMR (CDCl3): δ 174.5, 139.3, 114.3, 51.6, 34.2, 33.9, 29.41, 29.37, 29.26, 29.19, 29.0, 25.1.
Preparation of 11-Bromoundecanoic Acid Ethyl Ester (32)
11-Bromoundecanoic acid (10 g, 37.7 mmol), dried NaI (280 mg, 1.87 mmol, 0.05 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 1 g), and absolute EtOH (35 mL) were stirred for 4 h at 65 °C before Dowex was collected after filtration and washed with EtOH, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in DCM (25 mL), washed with 10% Na2S2O3 (sodium thiosulfate, 5 mL), and dried over MgSO4 before DCM was removed in vacuo. 11-Bromoundecanoic acid ethyl ester (10.9 g, 99%) was obtained as a brown oil. 1H NMR (CDCl3): δ 4.11 (q, 2H, 3JHH = 7.1), 3.39 (t, 2H, 3JHH = 6.9), 2.77 (t, 2H, 3JHH = 7.5), 1.87–1.81 (m, 2H), 1.65–1.56 (m, 2H), 1.45–1.37 (m, 2H), 1.33–1.25 (m, 10H), 1.24 (t, 3H, 3JHH = 7.1). 13C NMR (CDCl3): δ 174.0, 60.3, 34.5, 34.1, 32.9, 29.5, 29.4, 29.3, 29.2, 28.9, 28.3, 25.1, 14.4.
Preparation of Dimethyl 2-Hydroxysuccinate (33)
dl-Malic acid (100 mg, 0.75 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 200 mg), and dried MeOH (3 mL) were stirred for 24 h at room temperature before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. Dimethyl 2-hydroxysuccinate (92 mg, 76%) was obtained as a colorless syrup. 1H NMR (CDCl3): δ 4.52–4.49 (m, 1H), 3.82 (s, 3H), 3.72 (s, 3H), 2.90–2.85 (m, 1H), 2.83–2.78 (m, 1H). 13C NMR (CDCl3): δ 173.9, 171.1, 67.4, 53.0, 52.2, 38.6.
Preparation of Methyl 2-Aminoacetate Hydrochloride (34)
Glycine hydrochloride (100 mg, 0.90 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 50 mg), and MeOH (3 mL) were stirred for 24 h at 55 °C before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. Methyl 2-aminoacetate hydrochloride (92 mg, 82%) was obtained as a white solid. 1H NMR (CD3OD): 3.85 (s, 2H), 3.84 (s, 3H). 13C NMR (CD3OD): δ 168.7, 53.7, 41.2.
Preparation of (S)-Dimethyl 2-Aminosuccinate Hydrochloride (35)
l-Aspartic acid hydrochloride (100 mg, 0.59 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 75 mg), and MeOH (3 mL) were stirred for 24 h at 55 °C before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. (S)-Dimethyl 2-aminosuccinate hydrochloride (81 mg, 70%) was obtained as a colorless solid. 1H NMR (CD3OD): δ 4.41 (t, 1H, 3JHH = 5.5), 3.85 (s, 3H), 3.76 (s, 3H), 3.08 (d, 2H, 3JHH = 5.5). 13C NMR (CDCl3): δ 171.4, 169.6, 54.0, 53.0, 50.4, 34.8.
Preparation of (S)-Methyl 2-Amino-3-(4-hydroxyphenyl)propanoate Hydrochloride (36)
l-Tyrosine hydrochloride (100 mg, 0.46 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 50 mg), and MeOH (3 mL) were stirred for 24 h at 55 °C before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. (S)-Methyl 2-amino-3-(4-hydroxyphenyl)propanoate hydrochloride (80 mg, 75%) was obtained as a white solid. 1H NMR (CD3OD): δ 7.09–7.04 (m, 2H), 6.80–6.76 (m, 2H), 4.26–4.22 (m, 1H), 3.81 (s, 3H), 3.19–3.14 (m, 1H), 3.10–3.04 (m, 1H). 13C NMR (CDCl3): δ 170.6, 158.4, 131.5 (2C), 125.6, 116.9 (2C), 55.4, 53.6, 36.6.
Preparation of (R)-Methyl 2-Amino-3-mercaptopropanoate Hydrochloride (37)
l-Cysteine hydrochloride (100 mg, 0.63 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 50 mg), and MeOH (3 mL) were stirred for 24 h at 55 °C before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. (R)-Methyl 2-amino-3-mercaptopropanoate hydrochloride (83 mg, 76%) was obtained as a white solid. 1H NMR (CD3OD): δ 4.34 (t, 1H, 3JHH = 5.1), 3.87 (s, 3H), 3.09 (d, 2H, 3JHH = 5.1). 13C NMR (CDCl3): δ 167.7, 54.3, 52.5, 23.8.
Preparation of Triethyl 2-Hydroxypropane-1,2,3-tricarboxylate (Triethyl Citrate) (38)
Citric acid (200 mg, 1.04 mmol), dried NaI (234 mg, 1.56 mmol, 1.5 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 1.2 g), and abs. EtOH (10 mL) were refluxed for 24 h before Dowex was collected after filtration and washed with EtOH, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in DCM (10 mL), washed with 10% Na2S2O3 (sodium thiosulfate, 2–7 mL), dried over MgSO4, and evaporated to dryness in vacuo. Triethyl citrate (278 mg, 97%) was obtained as a colorless oil. 1H NMR (CDCl3): 4.28 (q, 2H, 3JHH = 7.2), 4.14 (q, 4H, 3JHH = 7.2), 2.90–2.86 (m, 2H), 2.80–2.76 (m, 2H), 1.30 (t, 3H, 3JHH = 7.2), 1.25 (t, 6H, 3JHH = 7.2). 13C NMR (CDCl3) δ 173.5, 169.9 (2C), 73.4, 62.5, 61.1 (2C), 43.5, 14.22 (2C), 14.17.
Preparation of Triethyl 1-Hydroxypropane-1,2,3-tricarboxylate (Triethyl Isocitrate, Mixture of Isomers) (39)
Iso-citric acid trisodium salt hydrate (1 g, 3.87 mmol), dried NaI (1.21 g, 8.07 mmol, 2.2 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 10 g), and abs. EtOH (35 mL) were refluxed for 48 h before Dowex was collected after filtration and washed with EtOH prior to the reaction mixture being evaporated to dryness in vacuo. The residue was dissolved in diethyl ether (20 mL) and washed three times with 0.5 M NaOH (8 mL), dried over MgSO4, and evaporated to dryness in vacuo. Triethyl isocitrate (311 mg, 29%, calculated based on anhydrous starting material) was obtained as a colorless viscous oil. 1H NMR (CDCl3): 4.37–4.34 (m, 1H), 4.33–4.24 (m, 2H), 4.20–4.10 (m, 4H), 3.53–4.49 (m, 1H), 3.16 (d, 1H, −OH, 3JHH = 5.4), 2.94–2.89 (m, 1H), 2.66–2.61 (m, 1H), 1.32 (t, 3H, 3JHH = 7.2), 1.27 (t, 3H, 3JHH = 7.2), 1.23 (t, 3H, 3JHH = 7.2). 13C NMR (CDCl3) δ 173.2, 171.9, 170.9, 70.8, 62.4, 61.4, 61.0, 45.0, 32.6, 14.3, 14.2, 14.1. MS (ESI+) calcd for C12H20O7 [M + H]+ 277.1282, found: 277.1287. NMR spectra in Supporting Information p. S3.
Preparation of Benzyl Acetate (40)
Benzyl alcohol (209 mg, 200 μL, 1.93 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 200 mg), and glacial acetic acid (1.5 mL) were stirred for 24 h at 80 °C before Dowex was collected after filtration and washed with DCM, and then the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in DCM (8 mL), washed with saturated NaHCO3 (2–3 mL), dried over MgSO4, and evaporated to dryness in vacuo. Benzyl acetate (246 mg, 85%) was obtained as a colorless liquid. 1H NMR (CDCl3): δ 7.39–7.31 (m, 5H), 5.11 (s, 2H), 2.11 (s, 3H). 13C NMR (CDCl3): δ 171.1, 136.1, 128.7 (2C), 128.41 (2C), 128.40, 66.5, 21.2.
Preparation of 3-(Acetyloxy)-2-phenylpropanoic Acid (41)
Tropic acid (100 mg, 0.60 mmol), dried NaI (18 mg, 0.12 mmol, 0.2 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 200 mg), and glacial acetic acid (1 mL) were stirred overnight (around 18 h) at 50 °C before Dowex was collected after filtration and washed with DCM, and the reaction mixture was evaporated to dryness in vacuo. 3-(Acetyloxy)-2-phenylpropanoic acid (124 mg, 99%) was obtained as a slightly brownish syrup. 1H NMR (CDCl3): δ 7.38–7.30 (m, 5H), 4.61–4.56 (m, 1H), 4.38–4.33 (m, 1H), 3.99–3.95 (m, 1 H), 2.04 (s, 3H). 13C NMR (CDCl3): δ 177.5, 171.0, 134.3, 129.1 (2C), 128.5, 128.3 (2C), 64.9, 50.7, 21.0.
Preparation of 3-(Isobutyryloxy)-2-phenylpropanoic Acid (42)
Tropic acid (200 mg, 1.20 mmol), dried NaI (18 mg, 0.12 mmol, 0.1 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 400 mg), and isobutyric acid (2 mL) were stirred for 24 h at 60 °C before Dowex was collected after filtration and washed with DCM, and then the reaction mixture was evaporated to dryness in vacuo. The residue was purified by silica column chromatography using hexane/ethyl acetate (1:1) as the eluent. 3-(Isobutyryloxy)-2-phenylpropanoic acid (124 mg, 44%) was obtained as an amorphous solid. 1H NMR (CDCl3): δ 7.38–7.29 (m, 5H), 4.59–4.53 (m, 1H), 4.40–4.35 (m, 1H), 2.51 (h, 1H, 3JHH = 7.0), 1.103 (d, 3H, 3JHH = 7.0), 1.100 (d, 3H, 3JHH = 7.0). 13C NMR (CDCl3): δ 176.9, 176.7, 134.4, 129.1 (2C), 128.39, 128.38 (2C), 64.6, 50.6, 34.0 19.0 (2C). MS (ESI+) calcd for C13H16O4 [M + H]+ 237.1121, found: 237.1124. NMR spectra in Supporting Information p. S4.
Preparation of 2-(Acetoxy)-2-phenylacetic Acid (43)
dl-Mandelic acid (100 mg, 0.66 mmol), dried NaI (10 mg, 0.067 mmol, 0.1 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 200 mg), and glacial acetic acid (1 mL) were stirred for 24 h at 90 °C before Dowex was collected after filtration and washed with DCM, and the reaction mixture was evaporated to dryness in vacuo. The residue was purified by silica column chromatography using ethyl acetate/MeOH (8:2) as the eluent (Rf. 0.8). 2-(Acetoxy)-2-phenylacetic acid (70 mg, 55%) was obtained as a white solid. 1H NMR (CD3OD): δ 7.51–7.46 (m, 2H), 7.40–7.32 (m, 3H), 5.86 (s, 1H), 2.14 (s, 3H). 13C NMR (CDCl3): δ 173.1 (very broad signal, hardly visible), 172.1, 136.3, 129.9, 129.6 (2C), 128.7 (2C), 76.6 (broad signal), 20.7.
Preparation of Octyl Acetate (44)
1-Octanol (2.0 mL, 1.65 g, 12.65 mmol), glacial acetic acid (1.44 mL, 1.51 g, 25.15 mmol, 2.0 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 1.0 g), and dried NaI (190 mg, 1.27 mmol, 0.1 equiv) were stirred for 2 h at 55 °C before Dowex was collected after filtration and washed with hexane (15 mL). Hexane was washed with 10% Na2S2O3 (sodium thiosulfate, 5 mL) and saturated NaHCO3 (20 mL), dried over MgSO4, and the hexane was removed in vacuo. Octyl acetate (1.82 g, 83%) was obtained as a colorless oil. 1H NMR (CDCl3): δ 4.05 (t, 2H, 3JHH = 6.8), 2.04 (s, 3H), 1.61 (qv, 2H), 1.38–1.22 (m, 10H), 0.88 (t, 3H). 13C NMR (CDCl3): δ 171.7, 64.8, 31.9, 29.35, 29.31, 28.7, 26.0, 22.8, 21.2, 14.2.
Preparation of But-3-en-1-yl Acetate (45)
3-Buten-1-ol (1.0 mL, 843 mg, 11.69 mmol), glacial acetic acid (1.35 mL, 1.42 g, 23.58 mmol, 2.0 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 350 mg), and dried NaI (75 mg, 0.50 mmol, 0.05 equiv) were stirred overnight (ca. 18 h) at 55 °C before Dowex was collected after filtration, and the reaction mixture was moved to a separation funnel. The reaction mixture was washed with saturated NaHCO3 (2–3 mL), the water layer (lower layer) was removed, and the ester phase was washed with a very small portion of 10% Na2S2O3 (sodium thiosulfate) and dried over MgSO4. But-3-en-1-yl acetate (698 mg, 52%) was obtained as a slightly yellow liquid. 1H NMR (CDCl3): δ 5.82–5.74 (m, 1H), 5.14–5.05 (m, 2H), 4.12 (t, 2H, 3JHH = 6.8), 2.40–2.35 (m, 2H), 2.04 (s, 3H). 13C NMR (CDCl3): δ 171.3, 134.1, 117.3, 63.7, 33.2, 21.1.
Preparation of 2-Hydroxy-4-methoxy-2-(2-methoxy-2-oxoethyl)-4-oxobutanoic Acid (Dimethyl Citrate) (46)
Citric acid (200 mg, 1.04 mmol), dried NaI (48 mg, 0.32 mmol, 0.3 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 300 mg), and MeOH (3 mL) were stirred for 24 h at room temperature before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. The solid residue was stirred in DCM (4 mL) for 2 h before centrifugation. Liquids were removed and the treatment was repeated once. Solids were dried in vacuo. Dimethyl citrate (138 mg, 60%) was obtained as a white powder. Mp 115–116 °C. 1H NMR (CDCl3): 3.67 (s, 6H), 2.97–2.93 (m, 2H), 2.84–2.80 (m, 2H). 13C NMR (CDCl3) δ 176.6, 172.1 (2C), 74.3, 52.5 (2C), 44.1 (2C).
Example of the Selective Acetylation of Cyclohexanol over Phenol
Cyclohexanol (110 μL, 106 mg, 1.06 mmol, 1 equiv), phenol (100 mg, 1.06 mmol, 1 equiv), glacial acetic acid (610 μL, 640 mg, 10.66 mmol, 10 equiv), dried Dowex 50W-X8 ion-exchange resin (H+-form, 200 mg), and dried NaI (16 mg, 0.11 mmol, 0.1 equiv) were stirred for 24 h at 50 °C. An NMR sample was taken from the reaction mixture, and 100% conversion of cyclohexanol to acetylated cyclohexanol was observed according to the 1H NMR spectrum (phenol conversion was 0%). 1H NMR (CDCl3): δ 4.77–4.71 (m, 1H), 2.04 (s, 3H), 1.88–1.81 (m, 2H), 1.75–1.68 (m, 2H), 1.57–1.51 (m, 1H), 1.44–1.31 (m, 4H), 1.28–1.20 (m, 1H).
Preparation of 2-Hydroxy-4-methoxy-4-oxobutanoic Acid (Malic Acid Monomethyl Ester, 47)
dl-Malic acid (500 mg, 3.73 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 500 mg), and MeOH (5 mL) were stirred for 2 h at room temperature before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. Residue was dissolved in ethyl acetate (3 mL) and added to a glass column filled with silica gel (2 cm diameter, 15 cm high) and eluted with 75 mL of ethyl acetate (this was done to remove unreacted starting material dl-malic acid from reaction mixture). Ethyl acetate was removed in vacuo and the residue was dissolved in chloroform (5 mL) and placed in the freezer overnight. The formed crystals were separated and dried in vacuo. 2-Hydroxy-4-methoxy-4-oxobutanoic acid (249 mg, 40%) was obtained as a white solid. Mp 76–77 °C. 1H NMR (CD3OD): δ 4.52–4.48 (m, 1H), 3.74 (s, 3H), 2.80–2.75 (m, 1H), 2.69–2.64 (m, 1H). 13C NMR (CD3OD): δ 175.2, 173.9, 68.6, 52.6, 39.9. MS (ESI+) calcd for C5H8O5 [M + H]+ 149.0445, found: 149.0449. NMR spectra in Supporting Information p. S5.
Example of the Selective Esterification of Phenylacetic Acid over Benzoic Acid and Their Isolation from the Reaction Mixture
Phenylacetic acid (100 mg, 0.73 mmol), benzoic acid (100 mg, 0.82 mmol), dried NaI (25 mg, 0.17 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 300 mg), and dried MeOH (3 mL) were stirred for 1.5 h at r.t. before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. The residue was dissolved in diethyl ether (5 mL), washed twice with 10% NaHCO3 (2 × 5 mL) and 10% Na2S2O3 (sodium thiosulfate, 0.5 mL), dried over MgSO4, and evaporated to dryness in vacuo. Methyl 2-phenylacetate (59 mg, 54%) was obtained as a colorless liquid. The combined NaHCO3 phases were carefully acidified with 2 M HCl (strong release of gases!) and extracted twice with diethyl ether (2 × 5 mL). The combined diethyl ether phases were dried over MgSO4 and evaporated to dryness in vacuo. Benzoic acid was obtained as a white solid. Methyl 2-phenylacetate: 1H NMR (CDCl3): δ 7.36–7.31 (m, 2H), 7.30–7.25 (m, 3H), 3.69 (s, 3H), 3.63 (s, 2H). Benzoic acid: 8.15–8.10 (m, 2H), 7.65–7.60 (m, 1H), 7.52–7.45 (m, 2H).
Example of the Selective Esterification of Fatty Acids (49) over Resin Acids (50) and Their Isolation from the Reaction Mixture
Prepurified, methanol-treated pine wood extract (40 g), dried NaI (2 g, 13 mmol), dried Dowex 50W-X8 ion-exchange resin (H+-form, 2.2 g), and dry MeOH (80 mL) were stirred at room temperature for 18 h before Dowex was collected after filtration and washed with MeOH, and the reaction mixture was evaporated to dryness in vacuo. The oily product was made basic with 1 M NaOH and washed three times with EtOAc (70 mL) to remove unsaturated fatty acid methyl esters. The remaining oily phase was acidified with 3 M HCl and extracted three times with EtOAc (50 mL). The organic fraction was evaporated in vacuo yielding a 31 g mixture of resin acids for further use (see the NMR spectra in the Supporting Information on pages S6–S7).
Dowex H+ Resin Handling in the Reusable Experiments
Dowex was collected by a Bühner funnel, washed with a large volume of MeOH, and dried in an oven for 1 h at 120 °C before reuse in the synthesis of 27 and 28.
Acknowledgments
This research was supported by the Jane and Aatos Erkko Foundation and by Business Finland, project ValueWoodChem 3280/31/2015. P.A.T. is grateful to Jane and Aatos Erkko Foundation’s support. The authors thank Maritta Salminkoski for her expert technical assistance in the syntheses, and Dr Marko Lehtonen and Miia Reponen for MS measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00790.
1H and 13C NMR spectra of all new compounds 23, 39, 42, and 47; 1H NMR spectra of resin acids (50) separation are presented just like the 1H NMR spectra of all other isolated esters (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gmelin L.Handbuch der Chemie, vol. 4: Handbuch der organischen Chemie (vol. 1); Karl Winter: Heidelberg, Baden (Germany), 1848; p 182. [Google Scholar]
- https://en.wikipedia.org/wiki/Ester (accessed February 11, 2019).
- Nelson D. L.; Cox M. M.. Lehninger Principles of Biochemistry, 3rd ed.; Worth Publishing: New York, 2000. [Google Scholar]
- Fernández V.; Guzman-Delgado P.; Graca J.; Santos S.; Gil L. Cuticle Structure in Relation to Chemical Composition: Re-assessing the Prevailing Model. Front. Plant Sci. 2016, 7, 427. 10.3389/fpls.2016.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.plasticscolor.com/en-US/Media/News/article/from-resin-to-product:-plastics-production,-uses-and-properties (accessed February 11, 2019).
- Espino-Díaz M.; Sepúlveda D. R.; González-Aguilar G.; Olivas G. I. Biochemistry of Apple Aroma: A Review. Food Technol. Biotechnol. 2016, 54, 375. 10.17113/ftb.54.04.16.4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.newworldencyclopedia.org/entry/Ester (accessed February 11, 2019).
- https://www.prweb.com/releases/2014/02/prweb11619424.htm (accessed February 11, 2019).
- Krieger M.; Scott M. P.; Matsudaira P. T.; Lodish H. F.; Darnell J. E.; Lawrence Z.; Kaiser C.; Berk A.. Section 4.1: Structure of Nucleic Acids, Molecular Cell Biology; W.H. Freeman and CO: New York, 2004. [Google Scholar]
- Otera J.; Nishikido J.. Esterification: Methods, Reactions, and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Nigiz F. U.; Hilmioglu N. D. Ethyl Lactate Synthesis by Catalytic Membranes in a Pervaporation-Assisted Membrane Reactor. Chem. Eng. Technol. 2018, 41, 836. 10.1002/ceat.201600620. [DOI] [Google Scholar]
- Zhou X.-Y.; Chen X. Na2CO3-Catalyzed O-Acylation of Phenols for the Synthesis of Aryl Carboxylates with Use of Alkenyl Carboxylates. Synlett 2018, 29, 2321. 10.1055/s-0037-1610265. [DOI] [Google Scholar]
- Finnveden M.; Brännström S.; Johansson M.; Malmström E.; Martinelle M. Novel sustainable synthesis of vinyl ether ester building blocks, directly from carboxylic acids and the corresponding hydroxyl vinyl ether, and their photopolymerization. RSC Adv. 2018, 8, 24716. 10.1039/C8RA04636K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebrián-García S.; Balu A. M.; Luque R. Ultrasound-Assisted Esterification of Valeric Acid to Alkyl Valerates Promoted by Biosilicified Lipases. Front. Chem. 2018, 6, 197. 10.3389/fchem.2018.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M.; Jiang L.; Niu F.; Zhang Y.; Sun X. A novel and highly efficient esterification process using triphenylphosphine oxide with oxalylchloride. R. Soc. Open Sci. 2018, 5, 171988 10.1098/rsos.171988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.cbc.ca/news/technology/marketplace-canadian-bottled-water-microplastics-1.4606182 (accessed February 11, 2019).
- Revel M.; Châtel A.; Mouneyrac C. Micro(nano)plastics: A threat to human health?. Curr. Opin. Environ. Sci. Health 2018, 1, 17. 10.1016/j.coesh.2017.10.003. [DOI] [Google Scholar]
- Turhanen P. A.; Vepsäläinen J. J. A Powerful tool for acid catalyzed organic addition and substitution reactions. RSC Adv. 2015, 5, 26218. 10.1039/C4RA17321J. [DOI] [Google Scholar]
- Turhanen P. A.; Vepsäläinen J. J. Preparation of useful building blocks, α-iodo- and bromoalkanols from cyclic ethers using the Dowex H+/NaX (X=I, Br) approach. RSC Adv. 2016, 6, 15937. 10.1039/C5RA20813K. [DOI] [Google Scholar]
- Park J.-Y.; Kim D.-K.; Lee J.-S. Esterification of free fatty acids using water-tolerable Amberlyst as heterogeneous catalyst. Bioresour. Technol. 2010, 101, S62. 10.1016/j.biortech.2009.03.035. [DOI] [PubMed] [Google Scholar]
- Diana; Sutijan; Rohmadi; Budiman A. Esterification of Indonesian turpentine using ion-exchange resin as solid acid catalyst. J. Eng. Sci. Technol. 2015, 4, 41. 10.7763/IPCBEE.2014.V74.14. [DOI] [Google Scholar]
- Ali S. H.; Tarakmah A.; Merchant S. Q.; Al-Sahhaf T. Synthesis of esters: Development of the rate expression for the Dowex 50 Wx8-400 catalyzed esterification of propionic acid with 1-propanol. Chem. Eng. Sci. 2007, 62, 3197. 10.1016/j.ces.2007.03.017. [DOI] [Google Scholar]
- Rojas A.; Ganesh T.; Walker A.; Dingledine R. Ethylatropine Bromide as a Peripherally Restricted Muscarinic Antagonist. ACS Chem. Neurosci. 2017, 8, 712. 10.1021/acschemneuro.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.-Y.; Tsai S.-W. Enzymatic hydrolytic resolution of (R,S)-tropic acid esters and (R,S)-ethyl α-methoxyphenyl acetate in biphasic media. J. Mol. Catal. B: Enzym. 2009, 57, 158. 10.1016/j.molcatb.2008.08.008. [DOI] [Google Scholar]
- Shao B.-H.; Xu X.-Z.; Wu Q.-Z.; Lu J.-D.; Fu X.-Y. Comparative Enantioseparation of 2-Arylpropionic Acid Esters on Cellulose Derivative and (S,S)-Whelk-O 1 Columns. J. Liq. Chromatogr. Relat. Technol. 2005, 28, 63. 10.1081/JLC-200038578. [DOI] [Google Scholar]
- Jo H.; Hassan A. H. E.; Jung S. Y.; Lee J. K.; Cho Y. S.; Min S.-J. Construction of 8-Azabicyclo[3.2.1]octanes via Sequential DDQ-Mediated Oxidative Mannich Reactions of N-Aryl Pyrrolidines. Org. Lett. 2018, 20, 1175. 10.1021/acs.orglett.8b00098. [DOI] [PubMed] [Google Scholar]
- Martínez-Rodríguez S.; Martínez-Gómez A. I.; Rodríguez-Vico F.; Clemente-Jiménez J. M.; Heras-Vázquez F. J. L. Natural Occurrence and Industrial Applications of d-Amino Acids: An Overview. Chem. Biodiversity 2010, 7, 1531. 10.1002/cbdv.200900245. [DOI] [PubMed] [Google Scholar]
- Akram M. Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biochem. Biophys. 2014, 68, 475. 10.1007/s12013-013-9750-1. [DOI] [PubMed] [Google Scholar]
- Turhanen P. A.; Demadis K. D.; Peräniemi S.; Vepsäläinen J. J. A Novel Strategy for the Preparation of Naturally Occurring Phosphocitrate and Its Partially Esterified Derivatives. J. Org. Chem. 2007, 72, 1468. 10.1021/jo061709c. [DOI] [PubMed] [Google Scholar]
- Reifenrath M.; Boles E. Engineering of hydroxymandelate synthases and the aromatic amino acid pathway enables de novo biosynthesis of mandelic and 4-hydroxymandelic acid with Saccharomyces cerevisiae. Metab. Eng. 2018, 45, 246. 10.1016/j.ymben.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Marchi C.; Fotiadu F.; Buono G. Diastereoselectivity of Borane Addition to Chiral Triquinphosphoranes. NMR, X-ray, and Molecular Modeling Studies. Organometallics 1999, 18, 915. 10.1021/om980774c. [DOI] [Google Scholar]
- Moore D. L.; Denton A. E.; Kohinke R. M.; Craig B. R.; Brenzovich W. E. Jr. Silica sulfuric acid as a highly efficient catalyst for the synthesis of diarylacetic acids. Synth. Commun. 2016, 46, 604. 10.1080/00397911.2016.1158269. [DOI] [Google Scholar]
- Queval P.; Caijo F.; Rouen M.; Tripoteau F.. Method for the synthesis of pheromones. PCT International Application, WO2018069146A1, Apr 19 2018.
- Altenbach H.-S.; Lange K.; Nandi S.; Ihizane R.; Jakob B.; Schneider M.. Process for preparation of citric acid mono- and diesters. PCT International Application, WO2013057110A1, Apr 25 2013.
- Md-Saleh S. R.; Chilvers E. C.; Kerr K. G.; Milner S. J.; Snelling A. M.; Weber J. P.; Thomas G. H.; Duhme-Klair A. K.; Routledge A. Synthesis of citrate-ciprofloxacin conjugates. Bioorg. Med. Chem. Lett. 2009, 19, 1496. 10.1016/j.bmcl.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Guo H.; Naser S. A.; Ghobrial G.; Phanstiel IV O. Synthesis and Biological Evaluation of New Citrate-Based Siderophores as Potential Probes for the Mechanism of Iron Uptake in Mycobacteria. J. Med. Chem. 2002, 45, 2056. 10.1021/jm0104522. [DOI] [PubMed] [Google Scholar]
- Anand R. C.; Milhotra V.; Milhotra A. Selective esterification of nonconjugated carboxylic acids in the presence of conjugated or aromatic carboxylic acids under mild conditions. J. Chem. Res. 1999, 6, 378. 10.1039/a901285k. [DOI] [Google Scholar]
- https://en.wikipedia.org/wiki/Malic_acid (accessed February 11, 2019).
- Papageorgiou C.; Benezra C. Use of enzymic hydrolysis of dimethyl malates for a short synthesis of tulipalin B and of its enantiomer. J. Org. Chem. 1985, 50, 1144. 10.1021/jo00207a054. [DOI] [Google Scholar]
- Keeling C. I.; Bohlmann J. Diterpene resin acids in conifers. Phytochemistry 2006, 67, 2415. 10.1016/j.phytochem.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Cermak S. C.; Evangelista R. L.; Kenar J. A.. Distillation of Natural Fatty Acids and Their Chemical Derivatives. In Distillation-Advances from Modeling to Applications; InTech: Rijeka, 2012; pp 109–142. [Google Scholar]
- Coumbarides G. S.; Eames J.; Weerasooriya N. A practical laboratory route to the synthesis of trideuteriomethyl-[13C] iodide. J. Labelled Compd. Radiopharm. 2003, 46, 291. 10.1002/jlcr.666. [DOI] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.