

Abstract
We report the synthesis of a structurally diverse amphiphilic dendrimer with oxidation and ultraviolet light-sensitive groups incorporated in the dendrimer interior. Convergent synthesis is utilized by reacting branched repeating units with a nonbranched functional molecule by two synthetic strategies, Passerini multicomponent reaction and azide–alkyne cycloaddition reaction. The periphery of dendrimer was functionalized by methoxy poly(ethylene glycol) to obtain a dendrimer with a hydrophobic core and hydrophilic peripheral chains. The G2-PEG dendrimer characterized by NMR, GPC, and MALDI-TOF MS for structural integrity and oxidation- and photo-triggered degradations of the G2-PEG dendrimer was investigated. The self-assembled morphology of the dendrimer in the presence of organic dye was also investigated by TEM and DLS analyses, together with dissipative particle dynamics simulation. The encapsulation of dye molecules in self-assembled nanospheres of the dendrimer and their responsive releases, triggered by the efficient disassembly of a dendrimer, have been demonstrated.
Introduction
Dendrimers are the class of synthetic polymers with a globular, well-defined branched structure with properties such as monodispersion, nanosize with a high density of surface functionality, internal cavities for guest encapsulation, and low intrinsic viscosity in solution.1,2 These properties of dendrimers make them an excellent choice for biomedical applications with suitable type and generation.3−5 Vögtle and co-workers6 first synthesized a dendrimer termed as cascade molecules in 1978; later on, Denkewalter, Tomalia, Newkome, Frechet, and co-workers independently carried out pioneering works for further development with a high order of complexity of these branched molecules and termed as dendrimers.7−10 Despite the progress of a dendrimer with improved features, the currently used dendrimers are nondegradable under physiological conditions, causing accumulation in cells and tissues, which induces cytotoxicity.11−13 The development of new synthetic methods such as orthogonal click chemistry, Michael reaction, multicomponent reactions, etc. in combination with convergent and divergent approaches provides excellent flexibility for construction of heterofunctional dendrimers14−21 with structural sophistication to meet current and future needs for therapeutic applications as well as drug and gene delivery.
Nanocarriers with more than one stimuli-responsive group are of great importance toward targeted, programmed, and controlled drug delivery by imparting into them the ability to respond to different environmental conditions to enhance their efficacy toward drug delivery.22−27 Among others, the nanocarriers responsive to reactive oxygen species (ROS)28−30 and ultraviolet (UV) light31,32 can act as promising theranostic agents for targeted delivery. The ROS-cleavable groups incorporated in the polymer backbone in response to stimuli degrade the bulk polymer into small molecules or change the amphiphilicity of the nanocarrier, resulting in disassembly with the release of encapsulate. In the literature, the polymer nanocarriers with oxidation-sensitive groups such as aryl boronic ester, thioether groups, polysulfides, and polyselenides reported disassembly either by degradation or solubility change.33 Most recently, aryl boronic ester sensitive to H2O2 oxidation was demonstrated with cleavage of the ester linkage.34 Along with this, light has gained attention as a stimulus since it can be applied with spatiotemporal precision without the use of reagents.35 The degradation of assembly is directly proportional to irradiation time and light intensity; hence, incorporated a wide range of light-responsive moieties in the polymer architectures.31,32 For drug delivery applications, the stimuli-responsive groups in suitable polymer architecture capable of forming self-assembled nanoparticles with low polydispersity and high payload capacity are crucial.36−40 The nanocarriers formed by dendritic amphiphiles are superior in terms of morphology and properties over micelles of linear surfactants.41,42
Passerini multicomponent reaction (P-MCR) is getting the attention of scientists working in the field of polymer chemistry because of catalyst-free ambient reaction conditions with high yields and simple purification methods. This P-MCR allows the easy way to incorporate environment-sensitive functional groups in the polymer backbone. A range of functional polymers are synthesized by P-MCR with structural diversity by a combination of monomers. Polymer architectures such as linear copolymers,34,43,44 hyperbranched polymers,45 and dendrimers20 are successfully synthesized with a controlled molecular weight (MW). However, there are very few reports published for dendrimers with functional groups placed in the interior46,47 due to synthetic challenges to work on two fronts: (1) growing dendrimer generations by incorporation of branching units and, at the same time, (2) introducing stimuli-responsive groups. P-MCR and click chemistry inspired us to design a synthetic methodology for a stimuli-responsive degradable dendrimer. We incorporated a chemically distinct ROS-responsive aryl boronic ester and UV-responsive o-nitrobenzyl groups in each generation of dendrimer so that, upon application of one stimulus, the other should remain intact to achieve stepwise degradation of the dendrimer. The dendrimer was confirmed by NMR and MALDI-TOF characterization for structural integrity. The degradation of the dendrimer at the aryl boronic ester position was achieved upon application of H2O2 as stimuli and, at the o-nitrobenzyl ester position, by UV irradiation. The degradation products were characterized by GPC, 1H NMR, and UV–Vis spectroscopy.
Results and Discussion
Dendrimer synthesis consists of hydrophobic repeating units with peripheral hydrophilic methoxy poly(ethylene glycol) (MPEG; MW = 600) chains to impart amphiphilicity in the macromolecule (Scheme 1). The stimuli-responsive groups such as o-nitrobenzyl (ONB), which is sensitive to UV light, and phenylboronic acid, which is responsive to oxidation, were incorporated, considering the significant difference in their cleavage rate to get control over degradation kinetics. The synthesis was achieved by stepwise manner, starting from P-MCR between a pinacol ester of 4-formylphenylboronic acid, 2-methyl-3-(prop-2-yn-1-yloxy)-2-((prop-2-yn-1-yloxy)methyl)propanoic acid (bis-MPA-dialkyne), and 1,6-diisocyanohexane in chloroform at 30 °C following the reported reaction conditions.34 This results in the formation of a G0.5 dendrimer by ester–amide linkage with four terminal alkyne groups with an 80% isolated yield. The G-1 ONB–aldehyde dendrimer was formed by copper-catalyzed azide–alkyne cycloaddition (CuAAC) between the G0.5 alkyne functional dendrimer and 5-(2-azidoethoxy)-2-nitrobenzaldehyde using tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) and Cu(I)I as a catalytic system. The pure G1 aldehyde dendrimer with four terminal formyl groups was obtained by silica gel column chromatography (78% yield). The second P-MCR between G1 aldehyde, 2,2-bis-MPA-dialkyne, and t-butyl isocyanide was performed in CH2Cl2 at 25 °C to get UV-responsive ONB moiety incorporated in the G-1.5 alkyne dendrimer with eight terminal alkyne groups (68% yield). Finally, a CuAAC click reaction between the G-1.5 dendrimer and MPEG–N3 was conducted using TBTA and Cu(I)I as a catalytic system in DMF at 25 °C for 18 h to get the amphiphilic G2-PEG dendrimer (62% yield). The dendrimer composed of 60% by weight of hydrophilic peripheral ethylene glycol chains and 40% hydrophobic part by weight as the core of the dendrimer. 1H NMR and 13C NMR characterized all dendrimer intermediate generations, and matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI-TOF MS) (Figures S1–S18 in the Supporting Information) and FT-IR (Figure S19 in the Supporting Information).
Scheme 1. Synthesis of G2-PEG Dendrimer by a Series of Passerini Multicomponent Reactions and Click Reactions.

The dendrimer is composed of interior heterofunctional stimuli-responsive groups and hydrophilic peripheral MPEG chains. 1H NMR spectrum (Figure S10 in the Supporting Information) and MALDI-TOF MS analysis (Figure 1) shows the structural integrity and purity of the G2-PEG dendrimer. In the 1H NMR spectrum, the integration of PEG chains and all other peaks such as newly formed methylene proton peaks are precisely matching with expected integrations. The MALDI-TOF MS analyses of all dendrimer generations show lines for a fragmentation product along with molecular ion peaks. The dendrimer generations show fragmentation at phenylboronic ester–amide linkage with breakage of the bond between benzylic carbon and oxygen (PhCH–O) and cleavage of benzylic (PhCH–O) bond from the ONB group. Here, we explicitly mention that, in the presence of H2O2 and upon UV irradiation, the ester bonds, not PhCH–O bonds, were cleaved (Figure 1). For an explanation, the G1.5 alkyne dendrimer MALDI-TOF MS spectrum is shown in Figure 1 with fragmentation peaks. The first fragmentation peak appears at 1869.96 g/mol by a cleavage bond between benzylic carbon atoms corresponding to the phenylboronic acid group, resulting in a cationic fragment that was detected by the MALDI-TOF MS spectrum. Another major fragmentation peak appears at 1098.60 g/mol by cleavage at the ONB group. The MALDI-TOF MS spectra for other dendrimer generations are included in Figures S20–S22 (Supporting Information).
Figure 1.

MALDI-TOF MS spectrum for G 1.5 alkyne.
For the final G2 dendrimer, we also observed fragmentation peaks in linear-mode MALDI TOF MS analysis due to cleavage of the dendrimer with the difference between cleaved populations around 1700 g/mol. In the case of inefficient click reaction, the difference would be 625 g/mol, indicating that the disintegration of the dendrimer extensively occurs. Lopp, So, and co-workers reported similar observations for the PAMAM dendrimer.48−50 The G2-PEG structural purity is also confirmed by gel permeation chromatography (GPC) (Mn = 8000 g mol–1; polydispersity index (PDI) = 1.01). The G2-PEG dendrimer is directly soluble in water and many common organic solvents such as methylene chloride, chloroform, tetrahydrofuran (THF), dimethylformamide, and dimethyl sulfoxide (DMSO).
Initially, we studied the H2O2-triggered degradation of the dendrimer by 1H NMR spectral analysis. The dendrimer was dissolved in a mixture of deuterated phosphate buffer solution (pH 7.4) and DMSO-d6 (10:1). DMSO-d6 was added for improved solubility of the dendrimer to detect hydrophobic protons. The excess of H2O2 solution (100 mM) was added and investigated with time by 1H NMR analysis (Figure 2). In the presence of H2O2, the phenylboronic pinacol ester was hydrolyzed by oxidation into a phenolic intermediate. As shown in Figure 2B, the 1H NMR spectrum shows an apparent shift in aromatic protons a and b to upfield regions 1a and 1b. The similar shift is also noted for a benzylic proton at the para position from phenylboronic acid c at δ 5.8 ppm to 1c at δ 5.65 ppm. The oxidative degradation of the boronic group initiates the self-immolation process from the phenolic intermediate, resulting in cleavage of the ester bond from ester–amide linkage into carboxylic acid and quinone methide. The quinone methide is highly reactive and is hydrolyzed into a benzyl alcohol derivative.
Figure 2.

(A) Proposed cleavage mechanism by H2O2 (100 mM) oxidation recorded in D2O and DMSO-d6 (10/2, v/v) at 25 °C. (B) Time-dependent H2O2-responsive degradation of dendrimer followed by 400 MHz 1H NMR spectroscopy.
Further, to confirm dendrimer degradation, G2-PEG in pH 7.4 phosphate buffer solution was treated with 100 mM H2O2 for 120 min, and the solvents were removed under vacuum. The residual solid was dissolved in THF and analyzed by GPC (Figure 3A). The GPC curves show degradation of the dendrimer into half-MW fractions with a shift in a peak at a higher retention time than the pure dendrimer (Mn = 4000; PDI = 1.05) and are precisely matching with expected MW by cleavage of two ester linkages in the dendrimer. In this process, the other ester linkages corresponding to the o-nitrobenzyl group remain intact with no shift in a peak in the 1H NMR spectrum corresponding to a benzylic proton at δ 6.5 ppm. The GPC curve also shows a peak shift to half the MW of the pure dendrimer instead of many fragmented peaks. These observations strongly suggest the degradation of the dendrimer by H2O2-triggered oxidation of phenylboronic moiety, which initiates the self-immolative process, not direct ester hydrolysis.
Figure 3.
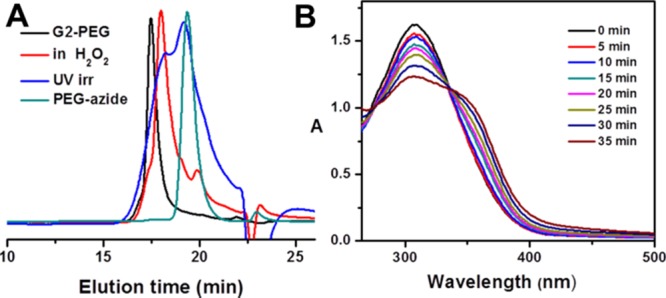
(A) GPC overlay for G2-PEG, after incubation for 3 h at 100 mM H2O2 in pH 7.4 PBS buffer, after UV irradiation in pH 7.4 buffer for 1 h and PEG azide. (B) UV–Vis spectra for 0.05 wt % G2-PEG after irradiation at different time intervals by 365 nm light.
Similarly, we studied the UV response of the o-nitrobenzyl group by UV–Vis spectroscopy, 1H NMR spectral analysis, and GPC. To study UV-triggered degradation, an aqueous solution of G2-PEG dendrimer (0.05 wt %) was irradiated at 365 nm light (100 W lamp) with monitoring over time by UV–Vis spectroscopy. The decrease in absorption intensity at 277 nm and the increase at 370 nm is due to the conversion of the nitrobenzyl group to the corresponding carbonyl group (Figure 3B). To further confirm the degradation of the dendrimer by GPC, 5 mg of G2-PEG dendrimer in 2 mL of pH 7.4 phosphate buffer solution was irradiated at 365 nm by UV light for 60 min. In the GPC curve of the irradiated sample, the peak corresponding to the pure dendrimer disappears entirely, and new peaks appear at low MW, indicating degradation of the dendrimer by cleavage of ester linkages corresponding to ONB groups (Figure 3A).
The photo-triggered degradation of the ester bond was also followed by 1H NMR spectroscopy with time (Figure S24). The decrease in the peak area of the benzylic proton corresponding to the o-nitrobenzyl group was observed due to the transfer of the proton by a radical mechanism with cleavage of ester linkage, as shown in the proposed mechanism (Figure S24). In this process, the peak corresponding to the benzylic proton para to phenylboronic acid remain intact, suggesting cleavage of ester linkages only corresponding to ONB groups by photoinitiated radical generation.
After structural characterization and responsive degradation of the dendrimer, we studied solution assemblies in aqueous solution, encapsulation, and responsive release of the model dye Nile red. The amphiphilic G2-PEG dendrimer with hydrophilic peripheral PEG chains and internal hydrophobic groups is expected to be self-assembled in aqueous solution. The 0.5 wt % G2 PEG dendrimer initially was prepared in water/DMF (9/1, v/v) by stirring at room temperature for 2 h; DLS analysis showed a particle size in the range of 100 nm and 1–2 μm, suggesting that annealing at higher temperature is needed to get a uniform morphology (Figure S23, sample a). A 0.5 wt % solution of the dendrimer was prepared by dissolving the dendrimer in water/DMF (9/1, v/v) solution with constant stirring at 80 °C for 2 h. The dendrimer solution was slowly cooled to room temperature and then subjected to dynamic light scattering (DLS) measurements; it shows solution morphologies with a hydrodynamic size (Dh) of 100 nm (Figure 5a). Transmission electron microscopy (TEM) revealed G2-PEG self-assembly into spherical micelles in a size range higher than as seen in DLS analysis, probably due to aggregation of nanoparticles in the drying process of TEM sample preparation. The effect of solvent proportion of water/DMF on solution morphology was studied by following the above sample preparation procedure. The self-assembled morphologies showed an increase in size with water/DMF (7/3, v/v) and water/DMF (5/5, v/v), whereas no self-assembly occurred in pure DMF solution (Figure S23, samples b–d).
Figure 5.

(a) TEM image with DLS size distribution curve (inset) and (b) Nile red release in the presence of 0.0, 0.1, 1.0, and 10.0 mM H2O2 in pH 7.4 phosphate buffer solution.
To understand the formation of micelles in the absence and presence of a sample hydrophobic drug, Nile red, dissipative particle dynamics (DPD) simulations were conducted for the system consisting of the dendrimer, Nile red, and mixed solvents of water and DMF. The mass and volume of each bead is 108 amu and 180 Å3, respectively, and the cutoff radius rc between two beads is 8.14 Å. In the DPD method, a set of soft interacting beads that is large on the atomistic scale but is still macroscopically small is used to simulate a fluid system. The underlying principle of DPD theory is well-described in previous reports.51−53 The DPD bead-spring model is parameterized by setting up the interactions between all different species. The DPD interaction parameters are obtained in two steps: a determination of the Flory–Huggins interaction parameters, followed by a conversion to DPD parameters. Here, the Flory–Huggins interaction parameter (χij) was calculated from the solubility parameters
| 1 |
where R is the gas constant, T is the absolute temperature, and v is the volume per mole beads. The DPD repulsion parameters aij can be related to the Flory–Huggins solubility parameter for a density value, in reduced units, of 3 as follows49,50
| 2 |
where the aij values are in reduced units. As illustrated in Figure 4a, the eight-arm polymer consists of nine types of beads (A to I), and Nile red consists of three types of beads (K, L, and M); also, the solubility parameter of each bead is derived by using Synthia or cohesive energy density calculations using Forcite Plus, COMPASS, and Amorphous Cell in Materials Studio 5.0 (Accelrys). The interaction parameters aij are calculated according to eq 2 and illustrated in Table 1. The diagonal elements used by Groot and Rabone are chosen such that a bead fluid has the compressibility of water.49 A cubic simulation box with periodic boundary condition was applied in all directions. A box of 30 × 30 × 30 rc3 is enough to avoid the finite size effects, and the integration time step of 0.05 was small enough for our system to get thermodynamic equilibrium. The simulation steps of 200,000 were used. All simulations were carried out using the DPD method in Mesocite module of the commercial software package Materials Studio.
Figure 4.

(a) Molecular structures and coarse-grained models of polymer, Nile red, water, and DMF and (b) configurations of polymer micelles in the absence (i–viii) and presence (ix–xii) of Nile red at different polymer (P)/water (W)/DMF (D)/Nile red (N) volume ratios, where Nile reds are presented in the CPK model, and water and DMF beads are not shown for clarity.
Table 1. Interaction Parameters aij between Different Beads in Figure 4a.
| aij | A | B | C | D | E | F | G | H | I | DMF | K | L | M | H2O |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 25.00 | |||||||||||||
| B | 28.06 | 25.00 | ||||||||||||
| C | 25.10 | 29.15 | 25.00 | |||||||||||
| D | 26.49 | 25.28 | 30.27 | 25.00 | ||||||||||
| E | 25.23 | 29.97 | 25.05 | 28.71 | 25.00 | |||||||||
| F | 27.18 | 25.07 | 29.00 | 25.11 | 29.92 | 25.00 | ||||||||
| G | 26.09 | 32.80 | 25.74 | 31.59 | 25.41 | 33.17 | 25.00 | |||||||
| H | 28.67 | 25.03 | 31.24 | 25.62 | 32.37 | 25.25 | 36.26 | 25.00 | ||||||
| I | 29.63 | 25.16 | 32.65 | 26.12 | 33.90 | 25.59 | 38.13 | 25.07 | 25.00 | |||||
| DMF | 33.64 | 26.42 | 38.38 | 28.80 | 40.02 | 27.75 | 45.39 | 26.34 | 25.80 | 25.00 | ||||
| K | 25.02 | 28.56 | 25.02 | 26.84 | 25.12 | 27.60 | 25.82 | 29.21 | 30.24 | 34.46 | 25.00 | |||
| L | 25.38 | 26.28 | 25.82 | 25.36 | 26.21 | 25.73 | 27.77 | 26.68 | 27.35 | 30.38 | 25.57 | 25.00 | ||
| M | 26.81 | 34.59 | 26.12 | 31.59 | 25.75 | 32.97 | 25.09 | 35.64 | 37.25 | 43.37 | 26.46 | 28.87 | 25.00 | |
| H2O | 234.77 | 187.16 | 305.52 | 251.19 | 312.84 | 242.50 | 334.98 | 228.08 | 220.50 | 196.35 | 238.85 | 217.30 | 275.70 | 25.00 |
It is known that amphiphilic polymers self-assemble to form micelles in aqueous solution when the concentration is higher than its critical micelle concentration. Several morphologies of micelles consist of a polymer, water, and DMF with different volume ratios. All beads distributed randomly at the beginning of the simulation aggregate to form small clusters first and then turn into larger aggregates, depending on the water-to-DMF ratio. At 5 and 10% volumes of the G2-PEG dendrimer in the absence of Nile red, the dendrimer beads tend to aggregate to form spherical micelles at the low volume ratios of DMF (Figure 4b). The hydrophobic centers of the dendrimer are distributed inside the micelle-forming hydrophobic core, and hydrophilic PEG spreads around the surface, forming a protective shell. As the DMF-to-water ratio increases, the spherical aggregates change to ring- and sheet-like structures. No specific morphologies were formed at the volume ratio of DMF > water. From the DPD simulation results in the absence of Nile red, we found that the dendrimer aggregates to stable spherical micelles at the volume ratio of water/DMF > 9.
The similar morphological developments of the dendrimer were observed in the DPD simulations in the presence of 1% Nile red (Figure 4b(ix)–(xii)). When the volume ratio of dendrimer/water/DMF/Nile red is 5.0:86.0:8.0:1.0, spherical micelles with a stable structure with PEG shell are formed at the early period of simulation with less than 20,000 steps. Extra simulation steps to 200,000 gave no significant changes in the aggregate morphology. This is in good agreement with the experimental results (see TEM image in Figure 5a). The Nile red beads gradually diffuse into the micelles and finally are distributed in the core of micelle formed by the dendrimer beads (see the cross-sectional view in Figure 4b(ix)), indicating that the micelle formed by the dendrimer is a reasonable carrier for a hydrophobic drug such as Nile red. Due to the increase in dendrimer content from 5 to 10% at the similar DMF-to-water ratio, the stable spherical micelles collapse to smaller micelles. The increase of the relative amount of DMF yields a fibril shape. It is interesting to note that no specific configurations of aggregates are observed and that the Nile red beads are not stably distributed inside of aggregates when DMF is a primary solvent (see the morphology obtained at P/W/D/N = 5.0:8.0:88.0:1.0).
For dye encapsulation and release studies, 1 mg of Nile red solution in DMF was added to 0.5 wt % G2-PEG in pH 7.4 phosphate buffer solution and DMF mixture in 9:1 (v/v) ratio and stirred at 80 °C for 2 h. After slowly cooling the solution temperature to room temperature, the Nile red encapsulation in dendrimer was studied by UV–visible spectroscopy. The drug loading capacity of the micelles was determined as 12%, and the drug loading efficiency was 41% by UV–visible spectroscopy. The size and shape of the self-assembled morphology were similar to those self-assembled in the absence of Nile red (Figure 5a). The Nile red molecules are surrounded by the hydrophobic block located in the core of the micelles, which might lead to improved hydrophobic Nile red encapsulation capacity and efficiency via inter- and intramolecular hydrophobic interactions.
A H2O2-triggered dye release by the disintegration of micelles is studied by fluorescence spectroscopy, and the Nile red encapsulated micellar solution is treated with different concentrations (0–10 mM) of H2O2 solution. The controlled experiment performed without adding H2O2 showed a constant Nile red intensity, whereas a decrease in Nile red fluorescence intensity was observed in the presence of H2O2 as an indication for dye release with the disintegration of micelles (Figure 5b). At 10 mM H2O2, a faster decrease in Nile red intensity was observed compared to 1 mM and 0.1 mM H2O2 due to faster degradation of micelles. About 58% decrease in Nile red intensity was observed in 2 h at 10 mM H2O2, while 28 and 10% decreases were recorded at 1 and 0.1 mM, respectively, at same time scale. In 24 h, the Nile red release reached 80% for both 10 and 1 mM H2O2, whereas for 0.1 mM, the release reached only 40%. The experimental data show a slow degradation of micelles in the presence of H2O2 (24 h) compared to the disintegration of micelles by UV irradiation (60 min).
Conclusions
In conclusion, we designed and prepared a stimuli-responsive heterofunctional degradable G2-PEG dendrimer in straightforward steps utilizing a series of P-MCR and click chemistry. The degradability of the resulting amphiphilic dendrimer can be tuned by H2O2 concentration and UV irradiation time. A fast degradation was observed under UV light irradiation, whereas slow degradation was observed in the presence of H2O2. Since the degradation kinetics can be controlled by UV irradiation and oxidation condition, the dendrimer can be a good encapsulant for the controlled release of encapsulate.
Experimental Section
Materials and Methods
See the Supporting Information.
Synthetic Procedures
Synthesis of G0.5 Alkyne
In a Schlenk tube, 1,6-diisocyanohexane (0.2 g, 1.47 mmol), 4-formylbenzeneboronic acid pinacol ester (0.75 g, 3.23 mmol), and bis-MPA-dialkyne (0.679 g, 3.23 mmol) were dispersed in 5 mL of chloroform and stirred at 30 °C for 96 h under inert atmosphere. The reaction mixture was diluted with 50 mL of chloroform and washed with water for several times. The organic phase was dried over anhydrous MgSO4 and purified by column chromatography using dichlomethane/methanol (95:5) as an eluent to obtain a colorless viscous liquid. Yield = 1.2 g (80%).
1H NMR (400 MHz, chloroform-d) δ = 7.80 (d, J = 8.2 Hz, 4H), 7.47 (d, J = 8.2 Hz, 4H), 6.97–6.90 (m, 2H), 6.10 (s, 2H), 4.24–4.09 (m, 8H), 3.82 (t, J = 8.6 Hz, 4H), 3.69–3.60 (m, 4H), 3.32–3.32 (m, 2H), 3.35–3.12 (m, 4H), 2.47 (t, J = 2.2, 6.7 Hz, 4H), 1.54–1.41 (m, 4H), 1.37–1.32 (m, 30 H). 13C NMR (101 MHz, chloroform-d) δ = 172.3, 168.1, 138.3, 134.9, 126.4, 83.8, 75.2, 72.0, 58.8, 48.2, 39.1, 29.4, 26.2, 24.8, 18.0.
Synthesis of G1 Aldehyde
In a Schlenk tube, G0.5 alkyne (1.0 g, 0.98 mmol) and 5-(2-azidoethoxy)-2-nitrobenzaldehyde (1.15 g, 4.90 mmol) were dissolved in DMF. The solution was purged with nitrogen for 30 min. In another vial, TBTA (0.052 g, 0.098 mmol) was dissolved in 1 mL of DMF and purged by nitrogen for 15 min, then, CuI (0.018 g, 0.098 mmol) was added and further purged for additional 10 min. This catalytic solution was added to the above reaction mixture and stirred for 18 h at room temperature. The reaction was quenched by precipitation in diethyl ether. The crude product was purified by column chromatography using dichloromethane/methanol (9:1) as an eluent. Yield = 1.5 g (78%).
1H NMR (400 MHz, chloroform-d) δ = 10.42 (s, 4 H), 8.11 (dd, J = 1.6, 9.0 Hz, 4 H), 7.79–7.64 (m, 6 H), 7.43–7.36 (m, 4 H), 7.32–7.24 (m, 6 H), 7.16–6.94 (m, 6 H), 6.00 (s, 2 H), 4.91–4.71 (m, 8 H), 4.67–4.44 (m, 14 H), 3.81–3.57 (m, 8 H), 3.25–2.84 (m, 4 H), 1.69 (br. s., 8 H), 1.32 (s, 24 H), 1.24 (s, 6 H). 13C NMR (101 MHz, chloroform-d) δ = 188.2, 172.8, 168.1, 162.0, 144.7, 142.8, 138.3, 134.5, 129.2, 127.4, 126.3, 123.8, 118.8, 114.0, 110.0, 83.9, 75.7, 75.0, 72.3, 67.2, 65.8, 64.8, 49.2, 48.4, 38.9, 29.1, 24.8, 18.1.
Synthesis of G1.5 Alkyne
In a 10 mL Schlenk tube, G1 aldehyde (1.2 g, 0.61 mmol), bis-MPA-dialkyne (0.641 g, 3.05 mmol), and t-butyl isocyanide (0.253 g, 3.05 mmol) were dispersed in dichloromethane and stirred at 25 °C for 96 h under inert atmosphere. The reaction mixture was precipitated in diethyl ether. Further purification was carried out by silica gel column chromatography using dichlomethane/methanol (9:1) as an eluent to obtain a faint yellow solid. Yield = 1.3 g (68%).
1H NMR (400 MHz, chloroform-d) δ = 8.03 (d, 4 H), 7.77 (d, 4 H), 7.74–7.66 (m, 4H), 7.45 (d, 4H), 7.11 (m, 6H), 6.90 (m, 4H), 6.62(d, 4H), 6.39 (s, 4H), 6.04 (s, 2H), 4.75 (m, 8H), 4.61 (m, 8H), 4.44 (m, 8H), 4.12 (m, 16H), 3.55–3.80 (m, 26H), 2.90–3.25 (m, 4H), 2.43 (m, 8H), 1.50–1.80 (m, 8H), 1.33 (d, 24 H), 1.25 (s, 12H), 1.22 (s, 6H). 13C NMR (101 MHz, chloroform-d) δ = 181.1, 172.8, 172.6, 165.7, 161.3, 141.6, 134.9, 133.6, 130.5, 127.8, 126.4, 116.2, 114.4, 86.6, 83.9, 79.2, 75.1, 72.5, 71.9, 58.8, 58.7, 51.7, 48.4, 39.1, 29.7, 29.2, 28.5, 24.8, 18.1, 17.9.
Synthesis of G2-PEG by Click Reaction
In a Schlenk tube, G2 alkyne (0.5 g, 0.16 mmol) and MPEG-azide (0.99 g, 1.59 mmol) were dissolved in DMF. The solution was purged with nitrogen for 30 min. In another vial TBTA (0.0085 g, 0.016 mmol) was dissolved in 1 mL of DMF and purged with nitrogen for 15 min; then, Cu(I)I (0.003 g, 0.016 mmol) was added and further purged for additional 10 min. This catalytic solution was added to the above reaction mixture and stirred for 18 h at room temperature. The reaction was quenched by precipitation in diethyl ether. The crude product was passed through a short plug of silica using 10% methanol in dichloromethane as an eluent to get pure G2-PEG as a colorless viscous liquid. Yield = 0.8 g (62%).
1H NMR (400 MHz, chloroform-d) δ = 8.0 (d, 4 H), 7.6–7.85 (m, 16 H), 7.3–7.45 (m, 8H), 7.05 (m, 6H), 6.90 (m, 4H), 6.53(m, 8H), 6.01 (s, 2H), 4.75 (m, 8H), 4.55 (m, 42H), 3.80 (m, 16H), 3.65 (m, 400H) 2.90–3.25 (m, 4H), 1.50–1.80 (m, 8H), 1.30 (d, 24 H), 1.25 (s, 12H), 1.22 (s, 6H).
The drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated by measuring the absorption of Nile red at 550 nm by using the following formulas
Acknowledgments
The Basic Science Research Program supported this work through the National Research Foundation of Korea (2018R1D1A1A09081809). The authors also thank the BK21 PLUS Program for the partial financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00384.
Chemicals and methods, experimental procedures, NMR spectra, MALDI-TOF spectra, UV spectra, FT-IR spectra, and a table of the interaction parameters for DPD simulations (PDF)
Author Contributions
All authors equally contributed to the manuscript writing. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Astruc D.; Boisselier E.; Ornelas C. Dendrimers Designed for Functions: From Physical, Photophysical, and Supramolecular Properties to Applications in Sensing, Catalysis, Molecular Electronics, Photonics, and Nanomedicine. Chem. Rev. 2010, 110, 1857–1959. 10.1021/cr900327d. [DOI] [PubMed] [Google Scholar]
- Bosman A. W.; Janssen H. M.; Meijer E. W. About Dendrimers: Structure, Physical Properties, and Applications. Chem. Rev. 1999, 99, 1665–1688. 10.1021/cr970069y. [DOI] [PubMed] [Google Scholar]
- Sikwal D. R.; Kalhapure R. S.; Govender T. An Emerging Class of Amphiphilic Dendrimers for Pharmaceutical and Biomedical Applications: Janus Amphiphilic Dendrimers. Eur. J. Pharm. Sci. 2017, 97, 113–134. 10.1016/j.ejps.2016.11.013. [DOI] [PubMed] [Google Scholar]
- Kono K. Dendrimer-Based Bionanomaterials Produced by Surface Modification, Assembly and Hybrid Formation. Polym. J. 2012, 44, 531–540. 10.1038/pj.2012.39. [DOI] [Google Scholar]
- Kaminskas L. M.; Porter C. J. H. Targeting the Lymphatics Using Dendritic Polymers (Dendrimers). Adv. Drug Delivery Rev. 2011, 63, 890–900. 10.1016/j.addr.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Buhleier E. W.; Wehner W.; Vögtle F. Cascade and Nonskid-Chain-Like Syntheses of Molecular Cavity Topologies. Synthesis 1978, 2, 155–158. [Google Scholar]
- Denkewalter R. G.; Kolc J.; Lukasavage W. J.. Surface Modifying Agents, Metal Chelating Agents, Substrates for Drugs. US Pat 4,289,872, 1981.
- Tomalia D. A.; Baker H.; Dewald J.; Hall M.; Kallos G.; Martin S.; Roeck J.; Ryder J.; Smith P. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polym. J. 1985, 17, 117–132. 10.1295/polymj.17.117. [DOI] [Google Scholar]
- Newkome G. R.; Yao Z.; Baker G. R.; Gupta V. K. Micelles. Part 1. Cascade Molecules: A New Approach to micelles. A [27]-arborol. J. Org. Chem. 1985, 50, 2003–2004. 10.1021/jo00211a052. [DOI] [Google Scholar]
- Hawker C. J.; Frechet J. M. J. Preparation of Polymers with Controlled Molecular Architecture. A New Convergent Approach to Dendritic Macromolecules. J. Am. Chem. Soc. 1990, 112, 7638–7647. 10.1021/ja00177a027. [DOI] [Google Scholar]
- Jain K.; Kesharwani P.; Gupta U.; Jain N. K. Dendrimer Toxicity: Let’s Meet the Challenge. Int. J. Pharm. 2010, 394, 122–142. 10.1016/j.ijpharm.2010.04.027. [DOI] [PubMed] [Google Scholar]
- Barth R. F.; Adams D. M.; Soloway A. H.; Alam F.; Darby M. V. Boronated Starburst Dendrimer-Monoclonal Antibody Immunoconjugates: Evaluation as a Potential Delivery System for Neutron Capture Therapy. Bioconjugate Chem. 1994, 5, 58–66. 10.1021/bc00025a008. [DOI] [PubMed] [Google Scholar]
- Malik N.; Wiwattanapatapee R.; Klopsch R.; Lorenz K.; Frey H.; Weener J. W.; Meijer E. W.; Paulus W.; Duncan R. Dendrimers: Relationship between Structure and Biocompatibility in Vitro, and Preliminary Studies on the Biodistribution of 125I-Labelled Polyamidoamine Dendrimers in Vivo. J. Controlled Release 2000, 65, 133–148. 10.1016/S0168-3659(99)00246-1. [DOI] [PubMed] [Google Scholar]
- Sowinska M.; Urbanczyk-Lipkowska Z. Advances in the Chemistry of Dendrimers. New J. Chem. 2014, 38, 2168–2203. 10.1039/c3nj01239e. [DOI] [Google Scholar]
- Walter M. V.; Malkoch M. Simplifying the Synthesis of Dendrimers: Accelerated Approaches. Chem. Soc. Rev. 2012, 41, 4593–4609. 10.1039/c2cs35062a. [DOI] [PubMed] [Google Scholar]
- Arseneault M.; Wafer C.; Morin J. F. Recent Advances in Click Chemistry Applied to Dendrimer Synthesis. Molecules 2015, 20, 9263–9294. 10.3390/molecules20059263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Taton D.; Ibarboure E.; Chaikof E. L.; Gnanou Y. Janus-Type Dendrimer-like Poly ( Ethylene Oxide ) S. J. Am. Chem. Soc. 2008, 130, 11662–11676. 10.1021/ja7103119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Xiao Q.; Sherman S. E.; Muncan A.; Ramos Vicente A. D. M.; Wang Z.; Hammer D. A.; Williams D.; Chen Y.; Pochan D. J.; Vértesy S.; André S.; Klein M. L.; Gabius H.-J.; Percec V. Glycodendrimersomes from Sequence-Defined Janus Glycodendrimers Reveal High Activity and Sensor Capacity for the Agglutination by Natural Variants of Human Lectins. J. Am. Chem. Soc. 2015, 137, 13334–13344. 10.1021/jacs.5b08844. [DOI] [PubMed] [Google Scholar]
- Ma X.; Tang J.; Shen Y.; Fan M.; Tang H.; Radosz M. Facile Synthesis of Polyester Dendrimers from Sequential Click Coupling of Asymmetrical Monomers. J. Am. Chem. Soc. 2009, 131, 14795–14803. 10.1021/ja9037406. [DOI] [PubMed] [Google Scholar]
- Deng X.-X.; Du F.-S.; Li Z.-C. Combination of Orthogonal ABB and ABC Multicomponent Reactions toward Efficient Divergent Synthesis of Dendrimers with Structural Diversity. ACS Macro Lett. 2014, 3, 667–670. 10.1021/mz500207z. [DOI] [PubMed] [Google Scholar]
- Jee J. A.; Spagnuolo L. A.; Rudick J. G. Convergent Synthesis of Dendrimers via the Passerini Three-Component Reaction. Org. Lett. 2012, 14, 3292–3295. 10.1021/ol301263v. [DOI] [PubMed] [Google Scholar]
- Schattling P.; Jochum F. D.; Theato P. Multi-Stimuli Responsive Polymers – the All-in-One Talents. Polym. Chem. 2014, 5, 25–36. 10.1039/C3PY00880K. [DOI] [Google Scholar]
- Zhuang J.; Gordon M. R.; Ventura J.; Li L.; Thayumanavan S. Multi-Stimuli Responsive Macromolecules and Their Assemblies. Chem. Soc. Rev. 2013, 42, 7421–7435. 10.1039/c3cs60094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guragain S.; Bastakoti B. P.; Malgras V.; Nakashima K.; Yamauchi Y. Multi-Stimuli-Responsive Polymeric Materials. Chem. – Eur. J. 2015, 21, 13164–13174. 10.1002/chem.201501101. [DOI] [PubMed] [Google Scholar]
- Stuart M. A. C.; Huck W. T. S.; Genzer J.; Müller M.; Ober C.; Stamm M.; Sukhorukov G. B.; Szleifer I.; Tsukruk V. V; Urban M.; Winnik F.; Zauscher S.; Luzinov I.; Minko S. Emerging Applications of Stimuli-Responsive Polymer Materials. Nat. Mater. 2010, 9, 101–113. 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- Cheng R.; Meng F.; Deng C.; Klok H.-A.; Zhong Z. Dual and Multi-Stimuli Responsive Polymeric Nanoparticles for Programmed Site-Specific Drug Delivery. Biomaterials 2013, 34, 3647–3657. 10.1016/j.biomaterials.2013.01.084. [DOI] [PubMed] [Google Scholar]
- Wang H.; Huang Q.; Chang H.; Xiao J.; Cheng Y. Stimuli-Responsive Dendrimers in Drug Delivery. Biomater. Sci. 2016, 4, 375–390. 10.1039/C5BM00532A. [DOI] [PubMed] [Google Scholar]
- Lippert A. R.; Van De Bittner G. C.; Chang C. J. Boronate Oxidation as a Bioorthogonal Reaction Approach for Studying the Chemistry of Hydrogen Peroxide in Living Systems. Acc. Chem. Res. 2011, 44, 793–804. 10.1021/ar200126t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapeinos C.; Pandit A. Physical, Chemical, and Biological Structures Based on ROS-Sensitive Moieties That Are Able to Respond to Oxidative Microenvironments. Adv. Mater. 2016, 28, 5553–5585. 10.1002/adma.201505376. [DOI] [PubMed] [Google Scholar]
- Kim K. S.; Lee D.; Song C. G.; Kang P. M. Reactive Oxygen Species-Activated Nanomaterials as Theranostic Agents. Nanomedicine 2015, 10, 2709–2723. 10.2217/nnm.15.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohy J.-F.; Zhao Y. Photo-Responsive Block Copolymer Micelles: Design and Behavior. Chem. Soc. Rev. 2013, 42, 7117–7129. 10.1039/c3cs35469e. [DOI] [PubMed] [Google Scholar]
- Zhao Y. Light-Responsive Block Copolymer Micelles. Macromolecules 2012, 45, 3647–3657. 10.1021/ma300094t. [DOI] [Google Scholar]
- Lallana E.; Tirelli N. Oxidation-Responsive Polymers: Which Groups to Use, How to Make Them, What to Expect from Them (Biomedical Applications). Macromol. Chem. Phys. 2013, 214, 143–158. 10.1002/macp.201200502. [DOI] [Google Scholar]
- Cui Y.; Zhang M.; Du F.-S.; Li Z.-C. Facile Synthesis of H2O2-Cleavable Poly(ester-Amide)s by Passerini Multicomponent Polymerization. ACS Macro Lett. 2016, 6, 11–15. 10.1021/acsmacrolett.6b00833. [DOI] [PubMed] [Google Scholar]
- Sugiura S.; Sumaru K.; Ohi K.; Hiroki K.; Takagi T.; Kanamori T. Photoresponsive Polymer Gel Microvalves Controlled by Local Light Irradiation. Sens. Actuators, A 2007, 140, 176–184. 10.1016/j.sna.2007.06.024. [DOI] [Google Scholar]
- Hsu C.-H.; Dong X.-H.; Lin Z.; Ni B.; Lu P.; Jiang Z.; Tian D.; Shi A.-C.; Thomas E. L.; Cheng S. Z. D. Tunable Affinity and Molecular Architecture Lead to Diverse Self-Assembled Supramolecular Structures in Thin Films. ACS Nano 2015, 10, 919–929. 10.1021/acsnano.5b06038. [DOI] [PubMed] [Google Scholar]
- Ren J. M.; McKenzie T. G.; Fu Q.; Wong E. H. H.; Xu J.; An Z.; Shanmugam S.; Davis T. P.; Boyer C.; Qiao G. G. Star Polymers. Chem. Rev. 2016, 116, 6743–6836. 10.1021/acs.chemrev.6b00008. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Grayson S. M. Approaches for the Preparation of Non-Linear Amphiphilic Polymers and Their Applications to Drug Delivery. Adv. Drug Delivery Rev. 2012, 64, 852–865. 10.1016/j.addr.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Stals P. J. M.; Li Y.; Burdyńska J.; Nicolaÿ R.; Nese A.; Palmans A. R. A.; Meijer E. W.; Matyjaszewski K.; Sheiko S. S. How Far Can We Push Polymer Architectures?. J. Am. Chem. Soc. 2013, 135, 11421–11424. 10.1021/ja400890v. [DOI] [PubMed] [Google Scholar]
- Tan W. S.; Zhu Z.; Sukhishvili S. A.; Rubner M. F.; Cohen R. E. Effect of Block Copolymer Architecture on the Thermally Induced Swelling of Micelle-Containing Multilayer Thin Films. Macromolecules 2011, 44, 7767–7774. 10.1021/ma201397d. [DOI] [Google Scholar]
- Thota B. N. S; Urner L. H; Haag R. Supramolecular Architectures of Dendritic Amphiphiles in Water. Chem. Rev. 2016, 116, 2079–2102. 10.1021/acs.chemrev.5b00417. [DOI] [PubMed] [Google Scholar]
- Rosen B. M; Wilson C. J; Wilson D. A; Peterca M; Imam M. R; Percec V. Dendron-Mediated Self-Assembly, Disassembly, and Self-Organization of Complex Systems. Chem. Rev. 2009, 109, 6275–6540. 10.1021/cr900157q. [DOI] [PubMed] [Google Scholar]
- Kreye O.; Tóth T.; Meier M. A. R. Introducing Multicomponent Reactions to Polymer Science: Passerini Reactions of Renewable Monomers. J. Am. Chem. Soc. 2011, 133, 1790–1792. 10.1021/ja1113003. [DOI] [PubMed] [Google Scholar]
- Li L.; Deng X. X.; Li Z. L.; Du F. S.; Li Z. C. Multifunctional Photodegradable Polymers for Reactive Micropatterns. Macromolecules 2014, 47, 4660–4667. 10.1021/ma501019c. [DOI] [Google Scholar]
- Deng X.-X.; Cui Y.; Du F.-S.; Li Z.-C. Functional Highly Branched Polymers from Multicomponent Polymerization (MCP) Based on the ABC Type Passerini Reaction. Polym. Chem. 2014, 5, 3316–3320. 10.1039/c3py01705b. [DOI] [Google Scholar]
- Kang T.; Amir R. J.; Khan A.; Ohshimizu K.; Hunt J. N.; Sivanandan K.; Montañez M. I.; Malkoch M.; Ueda M.; Hawker C. J. Facile Access to Internally Functionalized Dendrimers through Efficient and Orthogonal Click Reactions. Chem. Commun. 2010, 46, 1556. 10.1039/b921598k. [DOI] [PubMed] [Google Scholar]
- Antoni P.; Hed Y.; Nordberg A.; Nyström D.; Von Holst H.; Hult A.; Malkoch M. Bifunctional Dendrimers: From Robust Synthesis and Accelerated One-Pot Postfunctionalization Strategy to Potential Applications. Angew. Chem., Int. Ed. 2009, 48, 2126–2130. 10.1002/anie.200804987. [DOI] [PubMed] [Google Scholar]
- Subbi J.; Aguraiuja R.; Tanner R.; Allikmaa V.; Lopp M. Fragmentation of Poly(amidoamine) Dendrimers in Matrix-Assisted Laser Desorption. Eur. Polym. J. 2005, 41, 2552–2558. 10.1016/j.eurpolymj.2005.05.031. [DOI] [Google Scholar]
- Peterson J.; Allikmaa V.; Subbi J.; Pehk T.; Lopp M. Structural Deviations in Poly(amidoamine) Dendrimers: A MALDI-TOF MS Analysis. Eur. Polym. J. 2003, 39, 33–42. 10.1016/S0014-3057(02)00188-X. [DOI] [Google Scholar]
- So H.; Lee J.; Han S. Y.; Oh H. B. MALDI in-Source Decay Mass Spectrometry of Polyamidoamine Dendrimers. J. Am. Soc. Mass Spectrom. 2012, 23, 1821–1825. 10.1007/s13361-012-0445-4. [DOI] [PubMed] [Google Scholar]
- Groot R.D.; Rabone K. L. Mesoscopic Simulation of Cell Membrane Damage, Morphology Change, and Rupture by Nonionic Surfactants. Biophys. J. 2001, 81, 725–736. 10.1016/S0006-3495(01)75737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot R. D.; Warren P. B. Dissipative Particle Dynamics: Bridging the Gap between Atomistic and Mesoscopic Simulation. J. Chem. Phys. 1997, 107, 4423–4435. 10.1063/1.474784. [DOI] [Google Scholar]
- Hoogerbrugge P. J.; Koelman J. M. V. A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. Europhys. Lett. 1992, 19, 155–160. 10.1209/0295-5075/19/3/001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.