

Abstract
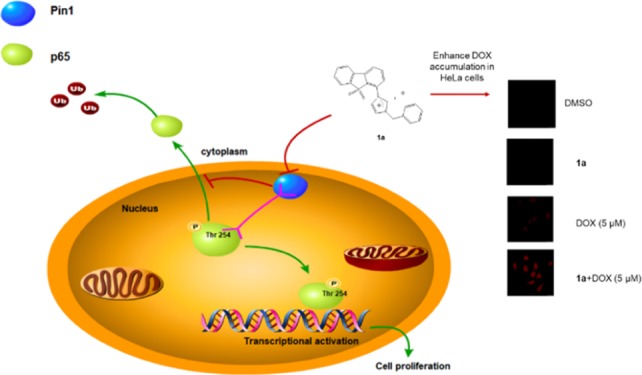
The peptidyl-prolyl isomerase Pin1 is correlated with the progression of cervical cancer via regulating numerous oncogenic and tumor suppressor pathways. p65 is a crucial regulator of tumorigenesis that is regulated by Pin1, and p65 signaling suppression can enhance the antitumor efficacy of doxorubicin (DOX). Here, we utilized a structural mimicry approach to synthesize a series of dibenzothiophene analogues and evaluated their ability to inhibit Pin1 activity. Compound 1a was identified as a potent Pin1 inhibitor that inhibited p65 signaling in vitro and in cervical cancer cells. Moreover, compound 1a enhanced the cytotoxicity of DOX in cervical cancer cells via reducing p65 nuclear accumulation and enhancing DOX uptake. These compounds are promising scaffolds for developing more potent Pin1 inhibitors against cervical cancer, either alone or in combination with anticancer drugs such as DOX.
Introduction
Cervical cancer is the third most common malignancy among women and the second most frequent cause of cancer death worldwide.1 Surgical resection and radiotherapy are the standard of care for the treatment of cervical cancer, however, patients with advanced tumors frequently fail to respond to treatments.2,3 Targeted therapy is an attractive approach to treat advanced tumors. However, blocking only a single pathway is often insufficient to eradicate advanced tumors, especially aggressive or drug-resistant tumors because of the activation of redundant and/or alternative oncogenic pathways.4,5 In cervical cancer, the dysregulation of numerous oncogenes and tumor suppressor genes are often mediated through phosphorylation events. Pin1 is a peptidyl-prolyl isomerase that is correlated with the progression of cervical cancer by regulating numerous signal pathways via phosphorylation.6,7 In cancer cells, overexpression of Pin1 promotes numerous tumor oncogenes, including the p65 subunit of nuclear factor-kappa B (NF-κB), by catalyzing the cis–trans isomerization of pSer/Thr-Pro motifs.8 This has stimulated the development of Pin1 inhibitors for the potential treatment of cancer, including cervical cancer.9 Examples of Pin1 small-molecule inhibitors reported in the literature include a shikimic acid derivative discovered by virtual screening,10 a naphthoquinone juglone derivative found in walnut trees,11 and a covalent Pin1 inhibitor KPT-6566.12PiB, a fused tetracyclic tetraone, inhibited Pin1 and suppressed the growth of cancer cells.10,13,14 However, to date, existing Pin1 inhibitors are unable to efficiently enter cells to inhibit Pin1 function in vivo. All-trans retinoic acid is the only Pin1 inhibitor that has had some successes in clinical use, being approved for the treatment of acute promyelocytic leukemia.5 Thus, new Pin1 inhibitors to treat cervical cancers are urgently desired. In addition, Pin1 might be a potential target for potentiating the potency of existing anticancer drugs. Knockdown of Pin1 enhanced the sensitivity of HeLa cells to cisplatin, while Pin1 overexpression led to an adverse effect.15 Other reports have shown that inhibition of p65 translocation can enhance doxorubicin (DOX)-induced apoptosis in carcinoma cells by the increase of intracellular DOX.16−19 DOX is a broad-spectrum antitumor drug that is used for the treatment of advanced cervical cancer. However, the cardiotoxicity of DOX limits its clinical use.20−23 Therefore, the discovery of new Pin1 inhibitors that can target Pin1 to inhibit p65 might be a potential strategy to potentiate the activity of DOX in cervical cancer.
Results and Discussion
A 3-fluorophenylalanine derivative containing a benzothiophene group inhibited Pin1 in vitro with nanomolar potency but was inactive in cells.24 The benzothiophene motif was also present in a non-natural peptide inhibitor of Pin1, which bound to the Pin1 active site with high specificity and potency but yet also showed no effect in cells.25 Although benzothiophene motifs are frequently present in prodrugs, and the benzothiophene core appears sufficiently hydrophobic that one might expect that it would pass through cell membranes,26,27 and the phosphate group present in these two benzothiophene-containing compounds might have impaired cellular permeability, leading to reduced cellular activity. To overcome this issue, we considered the addition of the imidazole motif, which is known to increase the cell permeability of many existing drugs.28,29 The ionizable property of the imidazole group confers favorable electronic characteristics that mediates cell penetration as well as supramolecular interactions with specific biological targets.30−39 Therefore, in our rational design strategy, three groups (1–3) of compounds containing the dibenzothiophene and imidazole motifs (Figure 1) were synthesized through the carbon–nitrogen Ullmann coupling reaction, followed by the oxidation of the benzothiophene motif and subsequent quaternarization of the imidazole group. All compounds 1a–e possess imidazole substituents at the C4 position of the dibenzothiophene scaffold. Compounds 2a–c increase the aromatic bulk of the C4 group by changing from imidazole to benzimidazole. Finally, compounds 3a–c have the imidazole substituent at the C2 position instead of at C4.
Figure 1.

Chemical structures of compounds 1a–3c.
These compounds were subsequently tested at 100 μM for their ability to decrease Pin1 activity using a PPIase activity assay. Based on the PPIase activity assay results (Figure 2), a preliminary structure–activity relationship (SAR) analysis could also be performed. Quaternarization of compounds generally led to improved Pin1 inhibitory activity, except in the case of the benzimidazole-substituent series where the neutral compound 2a was most active. This suggests that electronic properties may play an important role in guiding the compounds to the active site of the protein. Compound 1a, bearing a benzyl substituent on the imidazole group, was superior to compound 1b (methyl substituent) and 1d (ethyl substituent), suggesting that the substitution pattern on the imidazole group was important for activity. The C2-substituted compounds 3a–c (average inhibition: 49.5%) were generally inferior to the C4-substituted compounds 1a–e (average inhibition: 70.4%), suggesting that the steric effect of the imidazole motif was important for determining bioactivity. Moreover, sulfoxides were generally superior to (e.g., 1b vs 1e, 2c vs 2b, or 3a vs 3c), indicating that the oxidation state of the dibenzothiophene scaffold is also important. Finally, the methylimidazolium iodide salts were more active than the neutral imidazole compounds (e.g., 1b. vs 1c, 3c vs 3b), with the exception of salt 2c which was slightly less potent than the neutral benzimidazole 2a.
Figure 2.

Inhibition of Pin1 activity by compounds 1a–3c and PiB. Pin1 activity was determined using a PPIase activity assay. Error bars represent standard deviation of the means of the results from three independent experiments.
Among the tested compounds, compound 1a was considered to be the most promising Pin1 antagonist as it inhibited Pin1 activity by 80% at 100 μM (Figure 2). A dose–response experiment indicated an EC50 value of ca. 5 μM for compound 1a, which was comparable with PiB which showed an EC50 value of ca. 10 μM under parallel conditions (Figure S2). The synthesis of compound 1a is shown in Scheme 1. The compound displayed high stability in DMSO-d6 for up to 7 days as shown by 1H NMR spectroscopy (Figure S1).
Scheme 1. Synthesis of Compound 1a: (i) BuLi, THF, n-Hexane, −40 to 0 °C, 6 h; BrCH2CH2Br, −78 to 20 °C, 14 h; (ii) 1H-Imidazole, CuI, K2CO3, l-Proline, DMF; (iii) Hydrogen Peroxide, Glacial Acetic Acid; and (iv) (Bromomethyl)benzene, CH3CN; Methanol, NaI.

Pin1 is overexpressed in several kinds of cancer, and ablation of Pin1 activity can reduce tumor growth.12 To evaluate the effects of compound 1a in cellulose, we first explored whether Pin1 is engaged by 1a in HeLa cell lysates. The cellular thermal shift assay (CETSA) was performed to quantify the extent of drug engagement to Pin1 within HeLa cell lysates. After lysates of HeLa cells were incubated with compound 1a for 1 h, the aliquots were heated at different temperatures ranging from 45 to 70 °C for 8 min. The level of Pin1 remaining in the soluble fraction was determined by Western blotting. In the results, compound 1a increased Pin1 stability compared to the dimethyl sulfoxide (DMSO)-treated control (ΔTm: 1a: 8.8 °C), suggesting that compound 1a can engage Pin1 even in the complex biochemical environment of the cellular lysates (Figure 3a,b). A dose experiment with compound 1a (0–100 μM) revealed a Kd value of 5.7 μM for Pin1 protein at 61 °C (Figure S3).
Figure 3.
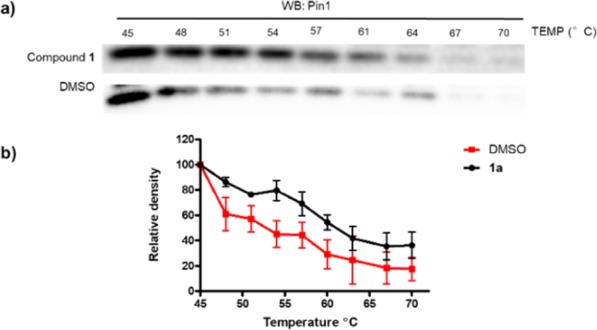
Compound 1a (100 μM) stabilizes Pin1 in HeLa cell lysates as determined by CETSA. (a) Pin1 content in the soluble fraction as revealed by Western blotting. (b) Plot of band intensity of Pin1 in the soluble fraction against temperature. The data were normalized to the Pin1 level of the control group at 45 °C and are expressed as the means ± SEM of three individual experiments.
Next, the effect of compound 1a against the proliferation of cancer cells and normal cells was assessed. Cervical cancer (HeLa) cells and normal liver (LO2) cells were treated with different doses of compound 1a for 72 h. The results showed that compound 1a decreased the cell viability of HeLa cells in a dose-dependent manner, with an IC50 value of 9.4 μM (Figure S4a), while it showed lower cytotoxicity in normal cells with an IC50 value of 39.8 μM (Figure S5). To further explore whether the cytotoxicity of compound 1a in HeLa cells was due to Pin1 inhibition, Pin1 knockdown was performed using siRNA. Notably, compound 1a showed reduced cytotoxicity (IC50 > 100 μM) in HeLa cells after Pin1 knockdown compared with normal HeLa cells (Figure S4a). In addition, we transfected Pin1 plasmid10 into embryonic kidney (293T) cells to generate a Pin1-overexpressing cell line. Intriguingly, compound 1a showed higher cytotoxicity in transfected 293T cells compared with nontransfected 293T cells (Figure S4b). Finally, compound 1a had no effect on the Pin1 content in HeLa cells after 12 h (Figure S7), suggesting that the inhibition of cell viability was not dependent on alteration of Pin1 expression levels. Taken together, these results suggest that compound 1a induces cell death, at least partly, due to the inhibition of Pin1 activity.
In cancer cells, Pin1 regulates numerous tumor suppressors and oncogenes by catalyzing the cis–trans isomerization of pSer/Thr-Pro motifs.8 p65, as a subunit of the transcription factor NF-κB, is a crucial regulator of many physiological and pathophysiological processes, including control of the adaptive and innate immune responses, inflammation, proliferation, tumorigenesis, and apoptosis.40 Although small-molecule NF-κB inhibitors have been tested as anticancer drugs either alone or in combination with classical chemotherapeutics to counteract-activated NF-κB-dependent anti-apoptotic behaviors, most of these target IKK, the negative regulator of NF-κB.40 It is not desirable to block NF-κB signaling for prolonged periods of time because the transcription factor also plays an important role in the maintenance of host defense responses. Therefore, intervening selectively with p65 transcriptional activity without completely blocking NF-κB activation might be a superior strategy for therapeutic intervention.
The unique substrate specificity of Pin1 toward specific pSer/Thr-Pro motifs is controlled by its C-terminal PPIase domain and N-terminal WW domain. Pin1 binds and isomerizes the pThr254-Pro motif of p65, resulting in enhanced p65 stabilization and nuclear accumulation, which is associated with human malignancies.41 Thus, we hypothesized that the inhibition of Pin1 activity to interrupt the Pin1–p65 interaction may represent a potential anticancer strategy. Encouragingly, 1a disrupted the binding of Pin1 to p65 in HeLa cells as observed by an immunoprecipitation assay (Figure 4a) and also decreased the accumulation of phosphor-p65(Thr254) levels in the nucleus (Figure 4b). Moreover, a half-life assay was performed to evaluate p65 stability. After incubation of HeLa cells with compound 1a (10 μM) for 12 h, cells were treated with 50 mg/mL cycloheximide, an antibiotic that inhibits new protein biosynthesis, for different time periods and p65 protein levels were analyzed. The results showed that with compound 1a, the half-life of p65 was about 180 min, whereas only about 20% of p65 was degraded after 180 min in the absence of compound 1a (Figure S6). This result suggests that compound 1a could reduce the stability of p65 and promote its degradation in HeLa cells, which we presume is related to its ability to target Pin1 activity.41 To expand our investigation beyond p65, we also measured the protein levels of other Pin1 substrates including Erk1/2, c-Jun, and PKM2. Compound 1a reduced Erk1/2, c-Jun, and PKM2 protein levels in HeLa cells (Figure S7a), phenocopying Pin1 ablation with Pin siRNA10 (Figure S7b). This result suggests that compound 1a could impact multiple oncogenic pathways controlled by Pin1 in order to exert its anticancer effects.
Figure 4.

Inhibition of Pin1–p65 interaction in cells: (a) 1a suppressed the Pin1–p65 interaction in HeLa cells as revealed by co-immunoprecipitation. HeLa cells were treated with the indicated concentration of 1a for 12 h. Protein lysates were incubated with antip65 and magnetic beads. The precipitated proteins were revealed by Western blotting with anti-Pin1 antibodies. 20% protein lysates were labeled as input were revealed by Western blotting with anti-p65 and anti-GAPDH antibodies. (b) 1a decreased the nuclear phospho-p65 level and had no effect on the cytoplasmic p65 level in HeLa cells.
The therapeutic efficacy of many anticancer drugs is limited by their adverse effects on healthy cells.15,42,43 Combination therapies may improve efficacy and lower toxicity allowing anticancer drugs to be utilized as lower dosages.44,45 DOX, an anthracycline derivative, is used for the treatment of a variety of malignancies including cervical cancer because of its broad spectrum antitumor activity.20−22,46,47 Inhibition of NF-κB activation attenuates resistance to DOX.22 Thus, we hypothesized that compound 1a could be combined with DOX to improve its efficacy and decrease adverse effects on healthy cells. Using the MTT assay,48−50 compound 1a was found to improve DOX efficacy in HeLa cells with combination index (CI) <1, suggesting synergism between compound 1a and DOX (Figure 5). Considering Pin1 contributes to cisplatin resistance in cervical cancer,15 we also measured the combination cytotoxicity of cisplatin with compound 1a. However, no synergistic effect was found (Figure S8). To further investigate the mechanism of cytotoxicity efficacy enhancement of DOX by compound 1a, we monitored p65 accumulation in treated HeLa cells. DOX increased p65 accumulation in the nucleus, while cotreatment with 1a reversed this effect (Figure S9). Moreover, 1a had no effect on the drug resistance-related protein P-glycoprotein (Figure S9). This suggests that the improvement of DOX efficacy by 1a may be related to its ability to suppress nuclear accumulation of p65. Additionally, 1a could increase the intracellular accumulation of DOX in HeLa cells, as revealed by an increase of DOX autofluorescence in cells cotreated with compound 1a and DOX (Figure S10). Flow cytometric analysis also showed that compound 1a augmented the intracellular concentration of Dox (Figure S11), suggesting that the improvement of DOX efficacy in HeLa cells may also be attributed to enhanced DOX accumulation by 1a. Overall, 1a acts as an enhancer of DOX efficacy in HeLa cells, which we presume is related to its ability to enhance intracellular DOX accumulation while lowering p65 accumulation in the nucleus. These findings suggest that 1a, when combined with low doses of DOX, has the potential to provide more efficient therapeutic effects against cervical cancer with lower general toxicity.
Figure 5.

1a enhances DOX efficacy. HeLa cells were treated with DOX in the presence or absence of 1a for 72 h, and cell viability was measured using the MTT. CI values are calculated. D1 and D2 are the concentrations of 1a and DOX used in the combination, and DLX,1 and DLX,2 are the concentrations of a single drug to produce the same effect.
Conclusions
In summary, this study has identified compound 1a as a Pin1 inhibitor for potential cervical cancer therapy. Compound 1a was identified using a preliminary PPIase activity screening of an in-house library of 11 compounds (1a–3c) containing benzothiophene and imidazole motifs. Preliminary SAR analysis indicated that quaternarization of the imidazole group, the nature of the imidazole substituent, and oxidation level of the dibenzothiophene scaffold were all determinants of Pin1 activity. In terms of mechanism, we demonstrated that compound 1a can interrupt the interaction between Pin1 and p65, thus reducing p65 nuclear accumulation and inhibiting the growth of HeLa cells. Moreover, compound 1a showed synergistic activity with DOX via reducing p65 accumulation and enhancing DOX uptake, which could allow the anticancer drug to be used at lower and safer dosages. To our knowledge, 1a represents the first Pin1 inhibitor for cervical cancer therapy. We anticipate that compound 1a may function as a novel scaffold for further development of more potent Pin1 inhibitors against Pin1-overexpressing cancers such as cervical cancer.
Experimental Section
General Experimental Section
Chemicals
Dibenzo[b,d]thiophene and 2-bromodibenzo[b,d]thiophene are purchased from J&K Chemical Ltd. 1H-imidazole and 1H-benzo[d]imidazole are purchased from Aladdin Chemical Ltd. (Shanghai). Unless stated otherwise, all the other general reagents and chemicals used in this work are obtained commercially and without further purification.
Cells and Reagents
HeLa, 293T, and LO2 cells were preserved by our laboratory. Fetal bovine serum and Dulbecco’s modified Eagle’s medium were obtained from Gibco BRL (Gaithersburg, MD, USA). All the compounds were dissolved in DMSO at a stock concentration of 10 mM.
Pin1 Activity Assay
The inhibition of Pin1 activity was assayed by a SensoLyte Green fluorimetric Pin1 activity assay kit. Briefly, a mixture containing the test compound (10 μL), enzyme solution (10 μL, 5 μg/mL), and Pin1 developer solution (30 μL) was mixed and added to wells of a 96-well plate. The solutions were incubated for 30 min at room temperature in the dark. Pin1 activity was determined by measuring the fluorescence intensity at Ex/Em = 490 nm/520 nm.
Data Analysis
All data were reported as the means of at least three separate experiments. Group comparisons between the control group and various drug treatment groups were done by one-way ANOVA using GraphPad Prism software (Prism).
Acknowledgments
This work is supported by Hong Kong Baptist University (FRG2/17-18/003), the Health and Medical Research Fund (HMRF/14150561), the National Natural Science Foundation of China (21575121 and 21775131), the Hong Kong Baptist University Century Club Sponsorship Scheme 2018, the Interdisciplinary Research Matching Scheme (RC-IRMS/16-17/03), Interdisciplinary Research Clusters Matching Scheme (RC-IRCs/17-18/03), Collaborative Research Fund (C5026-16G), SKLEBA and HKBU Strategic Development Fund (SKLP_1718_P04), the Science and Technology Development Fund, Macao SAR (0072/2018/A2), the University of Macau (MYRG2016-00151-ICMS-QRCM and MYRG2018-00187-ICMS), Natural Science Foundation of Jiangsu Province (BK20150283), and National Natural Science Foundation of China (NSFC no. 21505097).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00281.
General methods, experimental protocols, synthesis of molecules, and characterization data (PDF)
Author Contributions
∥ K.-J.W. and X. L. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer statistics, 2017. Ca-Cancer J. Clin. 2017, 67, 7–30. 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Agarwal R.; Kaye S. B. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502. 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- Goff B. A.; Muntz H. G.; Paley P. J.; Tamimi H. K.; Koh W.-J.; Greer B. E. Impact of surgical staging in women with locally advanced cervical cancer. Gynecol. Oncol. 1999, 74, 436–442. 10.1006/gyno.1999.5472. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Wei S.; Kozono S.; Kats L.; Nechama M.; Li W.; Guarnerio J.; Luo M.; You M.-H.; Yao Y.; Kondo A.; Hu H.; Bozkurt G.; Moerke N. J.; Cao S.; Reschke M.; Chen C.-H.; Rego E. M.; Lo-Coco F.; Cantley L. C.; Lee T. H.; Wu H.; Zhang Y.; Pandolfi P. P.; Zhou X. Z.; Lu K. P. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457. 10.1038/nm.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. P.; Zhou X. Z. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904. 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- Liou Y.-C.; Zhou X. Z.; Lu K. P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011, 36, 501–514. 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.; Hunter T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014, 24, 1033. 10.1038/cr.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theuerkorn M.; Fischer G.; Schiene-Fischer C. Prolyl cis/trans isomerase signalling pathways in cancer. Curr. Opin. Pharmacol. 2011, 11, 281–287. 10.1016/j.coph.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Wu K.-J.; Zhong H.-J.; Yang G.; Wu C.; Huang J.-M.; Li G.; Ma D.-L.; Leung C.-H. Small Molecule Pin1 Inhibitor Blocking NF-κB Signaling in Prostate Cancer Cells. Chem.—Asian J. 2018, 13, 275–279. 10.1002/asia.201701216. [DOI] [PubMed] [Google Scholar]
- Hennig L.; Christner C.; Kipping M.; Schelbert B.; Rücknagel K. P.; Grabley S.; Küllertz G.; Fischer G. Selective Inactivation of Parvulin-Like Peptidyl-Prolylcis/transIsomerases by Juglone. Biochemistry 1998, 37, 5953–5960. 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- Campaner E.; Rustighi A.; Zannini A.; Cristiani A.; Piazza S.; Ciani Y.; Kalid O.; Golan G.; Baloglu E.; Shacham S.; Valsasina B.; Cucchi U.; Pippione A. C.; Lolli M. L.; Giabbai B.; Storici P.; Carloni P.; Rossetti G.; Benvenuti F.; Bello E.; D’Incalci M.; Cappuzzello E.; Rosato A.; Del Sal G. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun. 2017, 8, 15772. 10.1038/ncomms15772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida T.; Takamiya M.; Takahashi M.; Miyashita H.; Ikeda H.; Terada T.; Matsuo Y.; Shirouzu M.; Yokoyama S.; Fujimori F.; Hunter T. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem. Biol. 2003, 10, 15–24. 10.1016/s1074-5521(02)00310-1. [DOI] [PubMed] [Google Scholar]
- Zhou X. Z.; Lu K. P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463. 10.1038/nrc.2016.49. [DOI] [PubMed] [Google Scholar]
- Wang T.; Liu Z.; Shi F.; Wang J. Pin1 modulates chemo-resistance by up-regulating FoxM1 and the involvements of Wnt/β-catenin signaling pathway in cervical cancer. Mol. Cell. Biochem. 2016, 413, 179–187. 10.1007/s11010-015-2651-4. [DOI] [PubMed] [Google Scholar]
- Kim M. R.; Choi H. S.; Yang J. W.; Park B. C.; Kim J.-A.; Kang K. W. Enhancement of vascular endothelial growth factor–mediated angiogenesis in tamoxifen-resistant breast cancer cells: role of Pin1 overexpression. Mol. Cancer Ther. 2009, 8, 1535–7163. 10.1158/1535-7163.mct-08-1061. [DOI] [PubMed] [Google Scholar]
- Yeh P. Y.; Yeh K.-H.; Chuang S.-E.; Song Y. C.; Cheng A.-L. Suppression of MEK/ERK Signaling Pathway Enhances Cisplatin-induced NF-κB Activation by Protein Phosphatase 4-mediated NF-κB p65 Thr Dephosphorylation. J. Biol. Chem. 2004, 279, 26143–26148. 10.1074/jbc.m402362200. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj M.; Barbu V.; Fillet M.; Chariot A.; Relic B.; Jacobs N.; Gielen J.; Merville M.-P.; Bours V. NF-κB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 2003, 22, 90. 10.1038/sj.onc.1206056. [DOI] [PubMed] [Google Scholar]
- Lee J. S.; Hong E. K. Agaricus blazei Murill enhances doxorubicin-induced apoptosis in human hepatocellular carcinoma cells by NFκB-mediated increase of intracellular doxorubicin accumulation. Int. J. Oncol. 2011, 38, 401–408. 10.3892/ijo.2010.852. [DOI] [PubMed] [Google Scholar]
- Li J.-M.; Wang Y.-Y.; Zhao M.-X.; Tan C.-P.; Li Y.-Q.; Le X.-Y.; Ji L.-N.; Mao Z.-W. Multifunctional QD-based co-delivery of siRNA and doxorubicin to HeLa cells for reversal of multidrug resistance and real-time tracking. Biomaterials 2012, 33, 2780–2790. 10.1016/j.biomaterials.2011.12.035. [DOI] [PubMed] [Google Scholar]
- Li J.; Wang Y.; Zhu Y.; Oupický D. Recent advances in delivery of drug-nucleic acid combinations for cancer treatment. J. Controlled Release 2013, 172, 589–600. 10.1016/j.jconrel.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashikawa K.; Shishodia S.; Fokt I.; Priebe W.; Aggarwal B. B. Evidence that activation of nuclear factor-κB is essential for the cytotoxic effects of doxorubicin and its analogues. Biochem. Pharmacol. 2004, 67, 353–364. 10.1016/j.bcp.2003.08.039. [DOI] [PubMed] [Google Scholar]
- Welander C. E.; Homesley H. D.; Barrett R. J. Combined interferon alf a and doxorubicin in the treatment of advanced cervical cancer. Am. J. Obstet. Gynecol. 1991, 165, 284–291. 10.1016/0002-9378(91)90080-b. [DOI] [PubMed] [Google Scholar]
- Guo C.; Hou X.; Dong L.; Dagostino E.; Greasley S.; Ferre R.; Marakovits J.; Johnson M. C.; Matthews D.; Mroczkowski B.; Parge H.; VanArsdale T.; Popoff I.; Piraino J.; Margosiak S.; Thomson J.; Los G.; Murray B. W. Structure-based design of novel human Pin1 inhibitors (I). Bioorg. Med. Chem. Lett. 2009, 19, 5613–5616. 10.1016/j.bmcl.2009.08.034. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Daum S.; Wildemann D.; Zhou X. Z.; Verdecia M. A.; Bowman M. E.; Lücke C.; Hunter T.; Lu K.-P.; Fischer G.; Noel J. P. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem. Biol. 2007, 2, 320–328. 10.1021/cb7000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malamas M. S.; Sredy J.; Moxham C.; Katz A.; Xu W.; McDevitt R.; Adebayo F. O.; Sawicki D. R.; Seestaller L.; Sullivan D.; Taylor J. R. Novel benzofuran and benzothiophene biphenyls as inhibitors of protein tyrosine phosphatase 1B with antihyperglycemic properties. J. Med. Chem. 2000, 43, 1293–1310. 10.1021/jm990560c. [DOI] [PubMed] [Google Scholar]
- Mourey R. J.; Burnette B. L.; Brustkern S. J.; Daniels J. S.; Hirsch J. L.; Hood W. F.; Meyers M. J.; Mnich S. J.; Pierce B. S.; Saabye M. J.; Schindler J. F.; South S. A.; Webb E. G.; Zhang J.; Anderson D. R. A Benzothiophene Inhibitor of Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 Inhibits Tumor Necrosis Factor Production and Has Oral Anti-Inflammatory Efficacy in Acute and Chronic Models of Inflammation. J. Pharmacol. Exp. Ther. 2010, 333, 797–807. 10.1124/jpet.110.166173. [DOI] [PubMed] [Google Scholar]
- Sharma D.; Narasimhan B.; Kumar P.; Judge V.; Narang R.; De Clercq E.; Balzarini J. Synthesis, antimicrobial and antiviral evaluation of substituted imidazole derivatives. Eur. J. Med. Chem. 2009, 44, 2347–2353. 10.1016/j.ejmech.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Iguchi K.; Usui S.; Ishida R.; Hirano K. Imidazole-induced cell death, associated with intracellular acidification, caspase-3 activation, DFF-45 cleavage, but not oligonucleosomal DNA fragmentation. Apoptosis 2002, 7, 519–525. 10.1023/a:1020691026578. [DOI] [PubMed] [Google Scholar]
- Kilpin K. J.; Dyson P. J. Enzyme inhibition by metal complexes: concepts, strategies and applications. Chem. Sci. 2013, 4, 1410–1419. 10.1039/c3sc22349c. [DOI] [Google Scholar]
- Wolber G.; Seidel T.; Bendix F.; Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discovery Today 2008, 13, 23–29. 10.1016/j.drudis.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Ma D.-L.; Chan D. S.-H.; Wei G.; Zhong H.-J.; Yang H.; Leung L. T.; Gullen E. A.; Chiu P.; Cheng Y.-C.; Leung C.-H. Virtual screening and optimization of Type II inhibitors of JAK2 from a natural product library. Chem. Commun. 2014, 50, 13885–13888. 10.1039/c4cc04498c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Fu L.; Yeo H.; Zhu J.-L.; Chou C.-K.; Kou Y.-H.; Yeh S.-F.; Gullen E.; Austin D.; Cheng Y.-C. Inhibition of hepatitis B virus gene expression and replication by helioxanthin and its derivative. Antiviral Chem. Chemother. 2005, 16, 193–201. 10.1177/095632020501600305. [DOI] [PubMed] [Google Scholar]
- Glas A.; Bier D.; Hahne G.; Rademacher C.; Ottmann C.; Grossmann T. N. Constrained Peptides with Target-Adapted Cross-Links as Inhibitors of a Pathogenic Protein-Protein Interaction. Angew. Chem., Int. Ed. 2014, 53, 2489–2493. 10.1002/anie.201310082. [DOI] [PubMed] [Google Scholar]
- Bier D.; Bartel M.; Sies K.; Halbach S.; Higuchi Y.; Haranosono Y.; Brummer T.; Kato N.; Ottmann C. Small-Molecule Stabilization of the 14-3-3/Gab2 Protein-Protein Interaction (PPI) Interface. ChemMedChem 2016, 11, 911–918. 10.1002/cmdc.201500484. [DOI] [PubMed] [Google Scholar]
- Bartel M.; Schäfer A.; Stevers L. M.; Ottmann C. Small molecules, peptides and natural products: getting a grip on 14-3-3 protein-protein modulation. Future Med. Chem. 2014, 6, 903–921. 10.4155/fmc.14.47. [DOI] [PubMed] [Google Scholar]
- Wang W.; Dong Z.-Z.; Yang G.; Leung C.-H.; Lin S.; Ma D.-L. A long-lived iridium(iii) chemosensor for the real-time detection of GHB. J. Mater. Chem. B 2017, 5, 2739–2742. 10.1039/c6tb03396b. [DOI] [PubMed] [Google Scholar]
- Castaneda M.; Chen L.; Pradhan L.; Li S.; Zein R.; Lee Y.; Lim H.-S.; Nam H.-J.; Lee J. A Forkhead Box Protein C2 Inhibitor: Targeting Epithelial-Mesenchymal Transition and Cancer Metastasis. ChemBioChem 2018, 19, 1359–1364. 10.1002/cbic.201800022. [DOI] [PubMed] [Google Scholar]
- Shin M.-K.; Hyun Y.-J.; Lee J. H.; Lim H.-S. Comparison of Cell Permeability of Cyclic Peptoids and Linear Peptoids. ACS Comb. Sci. 2018, 20, 237–242. 10.1021/acscombsci.7b00194. [DOI] [PubMed] [Google Scholar]
- Karin M.; Yamamoto Y.; Wang Q. M. The IKK NF-κB system: a treasure trove for drug development. Nat. Rev. Drug Discovery 2004, 3, 17. 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- Ryo A.; Suizu F.; Yoshida Y.; Perrem K.; Liou Y.-C.; Wulf G.; Rottapel R.; Yamaoka S.; Lu K. P. Regulation of NF-κB Signaling by Pin1-Dependent Prolyl Isomerization and Ubiquitin-Mediated Proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- Chen Z.-S.; Tiwari A. K. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011, 278, 3226–3245. 10.1111/j.1742-4658.2011.08235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z.; Tiwari A. K.; Patel A. S.; Fu L.-W.; Chen Z.-S. Roles of sildenafil in enhancing drug sensitivity in cancer. Cancer Res. 2011, 71, 3735–3738. 10.1158/0008-5472.can-11-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foucquier J.; Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anreddy N.; Gupta P.; Kathawala R.; Patel A.; Wurpel J.; Chen Z.-S. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules 2014, 19, 13848–13877. 10.3390/molecules190913848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregeau N. L.; Wang Y.; Pon R. T.; Wylie W. A.; Lown J. W. Characterization of a CPI-Lexitropsin conjugate-oligonucleotide covalent complex by 1H NMR and restrained molecular dynamics simulation. J. Am. Chem. Soc. 1995, 117, 8917–8925. 10.1021/ja00140a004. [DOI] [Google Scholar]
- Yuqiang W.; Liren H.; Wright S. C.; Larrick J. W. Doxorubicin and DNA minor groove-binding oligopeptide conjugates as anticancer agents(1). European journal of medicinal chemistry 143 (2018): 1021-1027. (2). Chemical science 8.7 (2017): 4756-4763. (3) Cancer letters 396 (2017): 76-84.. Gene 1994, 149, 63–67. 10.1016/0378-1119(94)90413-8. [DOI] [PubMed] [Google Scholar]
- Wu K.-J.; Zhong H.-J.; Li G.; Liu C.; Wang H.-M. D.; Ma D.-L.; Leung C.-H. Structure-based identification of a NEDD8-activating enzyme inhibitor via drug repurposing. Eur. J. Med. Chem. 2018, 143, 1021–1027. 10.1016/j.ejmech.2017.11.101. [DOI] [PubMed] [Google Scholar]
- Liu L.-J.; Wang W.; Huang S.-Y.; Hong Y.; Li G.; Lin S.; Tian J.; Cai Z.; Wang H.-M. D.; Ma D.-L.; Leung C.-H. Inhibition of the Ras/Raf interaction and repression of renal cancer xenografts in vivo by an enantiomeric iridium(III) metal-based compound. Chemical Science 2017, 8, 4756–4763. 10.1039/C7SC00311K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T.-S.; Wang W.; Zhong H.-J.; Dong Z.-Z.; Huang Q.; Mok S. W. F.; Leung C.-H.; Wong V. K. W.; Ma D.-L. An anti-prostate cancer benzofuran-conjugated iridium(III) complex as a dual inhibitor of STAT3 and NF-κB. Cancer Lett. 2017, 396, 76–84. 10.1016/j.canlet.2017.03.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.