

Abstract

We have synthesized and theoretically calculated 5-methylisoindolo[2,1-a]quinoline derivatives as novel near-infrared absorption dyes via a ruthenium-catalyzed one-pot metathesis/oxidation/1,3-dipolar cycloaddition protocol. The reactivity in 1,3-dipolar cycloaddition was governed by the electronic effect of aromatic ring substituents. Substrates with an electron-withdrawing group on the aromatic ring afforded higher yields. The maximal absorption wavelength of 3,5-dimethyl-11-phenylisoindolo[2,1-a]quinoline-7,10-dione and 11-(4-methoxyphenyl)-5-methylisoindolo[2,1-a]quinoline-7,10-dione in MeOH increased to 736 and 737 nm, although that of 3a was 727 nm.
Introduction
New applications of near-infrared (NIR) dyes have sparked recent interest in these substances with strong NIR absorption, proving an important and useful phenomenon. While fluorescence is particularly applicable to labeling in microscopy,1 organic molecules with eminent NIR absorption have attracted interest owing to the wide range of applications for their optical and electronic properties.2,3 Nowadays, nonmetallic organic dyes have many potential advantages, such as being lightweight, low-cost, and easily processed with colorful and transparent features. These classical metal-free NIR organic dyes4 include squaraines,5 cyanines,6 and the particularly well-known rhodamines,7 boron-dipyrromethenes8 and diketopyrrolopyrroles9 (Figure 1). These structures have received increasing attention from both industry and academic research. However, the range of the reported NIR dye molecular skeletons is limited, making the development of novel NIR dye core structures highly desirable. Although NIR chromophores constructed from novel nitrogen-containing polyheterocycles10,11e have been discovered recently, methods for obtaining these desired NIR dyes have yet to be established. One such approach to obtaining the desired NIR dyes is through the discovery of novel chromophores.
Figure 1.

Metal-free NIR absorption chromophores.
In our search for a novel and efficient ruthenium-catalyzed one-pot reaction involving both metathesis and nonmetathesis reactions,11,12 we previously developed a one-pot ring-closing metathesis (RCM)/oxidation/1,3-dipolar cycloaddition protocol to prepare various isoindolo[2,1-a]quinolines 3 from N-allyl-2-alkenylaniline derivatives 1 (Scheme 1).13 The key intermediate in this reaction is likely to be azomethine ylide I derived from 1,2-dihydroquinoline 2.
Scheme 1. Our Previous Work: One-Pot Ring-Closing Metathesis (RCM)/Oxidation/1,3-Dipolar Cycloaddition Protocol.

The isoindolo[2,1-a]quinoline motif (Figure 2) is attractive because of its biological properties.14 For instance, 5,11-dioxoisoindolo[2,1-a]quinolines are analogs of berberine alkaloids that have been shown to have an inhibitory effect against N2-induced hypoxia14a and inhibit human topoisomerase II.14b Meanwhile, 7,10-dioxoisoindolo[2,1-a]quinolines, first synthesized by our group,13 are functional dyes. In particular, we found that compound 3 showed a variety of colors when exchanging the R1 substituent (R1 = Ph, Me), including blue, yellow, and red (Scheme 1 and Figure 3), despite the core structure of 3 being the same as that of the 7,10-dioxoisoindolo[2,1-a]quinoline system.
Figure 2.

Structures of isoindolo[2,1-a]quinolines.
Figure 3.

Colors of compounds 3a–c (500 μM in toluene).
Blue compound 3a (Figure 3), which bears a methyl substituent at the 5-position of isoindolo[2,1-a]quinoline, clearly showed absorption peaks in the NIR region with a maximum at 673–720 nm (Figure 4). In contrast, the maximal absorption wavelengths of yellow compound 3b and red compound 3c containing phenyl substituents at the 5-position absorbed at 447–452 nm and 477–494 nm, respectively.
Figure 4.

Absorption spectra of compounds 3a–c (200–900 nm; 50 μM in CHCl3).
Considering that the chemical yield of 5-methylisoindolo[2,1-a]quinoline 3a from corresponding precursor 1a was low, and that 3a was a novel metal-free NIR absorption dye, we decided to optimize the synthetic conditions to afford 3a and synthesize 5-methylisoindolo[2,1-a]quinoline derivatives 3 with different substituents on the aromatic ring to better understand the relationship between the structure and NIR absorption. We also performed theoretical calculation of the newly synthesized 5-methylisoindolo[2,1-a]quinoline derivatives 3 (Scheme 2).
Scheme 2. This Work: Synthesis, Absorbance Properties, and Theoretical Calculation of 5-Methylisoindolo[2,1-a]quinolines 3.

Results and Discussion
N-Allyl-N-benzylaniline derivatives 1 were prepared systematically and efficiently. Our tandem catalysis strategy was first investigated using N-allyl-N-benzylaniline derivatives 1a, 1,4-benzoquinone, and ruthenium carbene catalysts. We first optimized the reaction conditions of the one-pot reaction, as shown in Table 1. Entries 2–4 show that a higher loading of the Grubbs II catalyst (20 mol %) was not effective, with a 1 mol % loading proving sufficient for this one-pot reaction. Furthermore, benzene was successfully substituted with less-toxic toluene as the solvent (entry 5), affording 3a in 18% yield in a shorter reaction time of 10 min (entry 6). We next optimized the reaction concentration. When the one-pot reaction was performed in a 0.1 M solution, 3a was obtained in 5% yield with a cross metathesis product obtained in 28% yield (entry 7). The chemical yield of 3a was not improved under glovebox conditions (entry 8). We next applied these optimal conditions (entry 6) to the synthesis of substituted derivatives of 5-methylisoindolo[2,1-a]quinoline 3.
Table 1. Optimization of the One-Pot RCM/Oxidation/1,3-Dipolar Cycloaddition Protocol.

| RCM |
1,3-dipolar cycloaddition |
isolated yield | |||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | Grubbs II (mol %) | solvent | temp. (°C) | conc. (M) | time (min) | temp. (°C) | conc. (M) | time (min) | (%, two steps) |
| 1 | 10 | benzene | reflux | 0.01 | 30 | reflux | 0.01 | 60 | 15 |
| 2 | 20 | benzene | reflux | 0.01 | 30 | reflux | 0.01 | 60 | 11 |
| 3 | 5 | benzene | reflux | 0.01 | 30 | reflux | 0.01 | 60 | 16 |
| 4 | 1 | benzene | reflux | 0.01 | 30 | reflux | 0.01 | 60 | 18 |
| 5 | 1 | toluene | 80 | 0.01 | 30 | 80 | 0.01 | 60 | 18 |
| 6 | 1 | toluene | 80 | 0.01 | 10 | 80 | 0.01 | 30 | 18 |
| 7a | 1 | toluene | 80 | 0.1 | 10 | 80 | 0.1 | 30 | 5 |
| 8b | 1 | toluene | 80 | 0.01 | 10 | 80 | 0.01 | 30 | 15 |
Cross metathesis product was also isolated in 28% yield.
Reaction was performed in a glovebox.
Experiments performed to establish the substrate scope are summarized in Scheme 3. The RCM of 1 proceeded quantitatively, as determined by thin layer chromatography (TLC) analysis, regardless of the substituent attached to the aromatic ring. However, 1,3-dipolar cycloaddition and oxidation were affected by the substituent on the aromatic ring. Therefore, the electronic effects of substituents on the aromatic ring were responsible for the substrate reactivity toward 1,3-dipolar cycloaddition. Higher yields of one-pot reaction products 3d, 3e, 3f, 3g, and 3h were obtained using substrates with an electron-withdrawing group on the aromatic ring. 3,5-Dimethylisoindolo[2,1-a]quinoline (3j) was synthesized in 17% yield. Although generation of 3i and 3k was observed by TLC, these products were unstable and decomposed during purification. Naphthylamine derivative 1l was also converted to pentacyclic compound 3l in 44% yield. 1,4-naphthoquinone was also used as a 1,3-dipolarophile in the reaction, affording pentacyclic compound 3m. We also synthesized 3n and 3o from the corresponding precursor in 6 and 12% yields, respectively.
Scheme 3. Scope of One-Pot RCM/Oxidation/1,3-Dipolar Cycloaddition Protocol.

The all-obtained isoindolo[2,1-a]quinolines 3 were blue-colored solids. The color of the solution of compound 3 in CHCl3 is shown in the Supporting Information S1 (Figure S1). Compounds 3d–3h containing electron-withdrawing groups formed blue-colored solutions in CHCl3. Meanwhile, solutions of methyl-substituted tetracyclic compound (3j) and benzo[g]isoindolo[2,1-a]quinoline (3l) were light-green to green in color. The solution of 3m in CHCl3, synthesized from 1f and 1,4-naphtoquinone, had a deep blue color. The color of a solution of 3n and 3o in CHCl3 was blue.
We then investigated the absorption profiles of compounds 3 in CHCl3 (Figure 5) and in MeOH (Figure 6). The maximal absorption wavelengths (λex) and molar absorption coefficients (ε) of compounds 3 are shown in Table 2. Except for 3l, all compounds 3 showed similar absorption spectra in the visible region in CHCl3. The maximal absorption wavelength of 3d, with a chloride substituent at the 4-position of the isoindolo[2,1-a]quinoline, was extended in the same way as that of 3a in both solvents. However, other chloro-substituted compounds 3e–3g had lower maximal absorption wavelengths and molar absorption coefficients than 3a in all solvents. A bromo-substituent (3h) affected the absorption characteristics in a similar fashion to the chloro-substituent (3e). In contrast, the maximal absorption wavelength of 3j, with a methyl substituent at position 3 of the isoindolo[2,1-a]quinoline, was red-shifted compared to that of 3e. Particularly in MeOH, the maximal absorption wavelength of 3j was extended up to 736 nm. The weaker absorption of benzo[g]quinoline derivative 3l in CHCl3 was observed at a shorter wavelength. The maximal absorption wavelength of 3n, with a p-methoxyphenyl substituent at the 11-position of the isoindolo[2,1-a]quinoline, was red-shifted compared to that of 3a. Particularly in MeOH, the maximal absorption wavelength of 3n was extended up to 737 nm. In contrast, the maximal absorption wavelength of 3o, with a p-chlorophenyl substituent at the 11-position of the isoindolo[2,1-a]quinoline, was shorter than that of 3a. An electron-donating group on the aromatic ring or heterocycle affected the maximum absorbance wavelength longer. Molar absorption coefficients were very low, that is, <10 000.
Figure 5.

Absorption spectra of compounds 3 (250–1100 nm; 50 μM in CHCl3).
Figure 6.
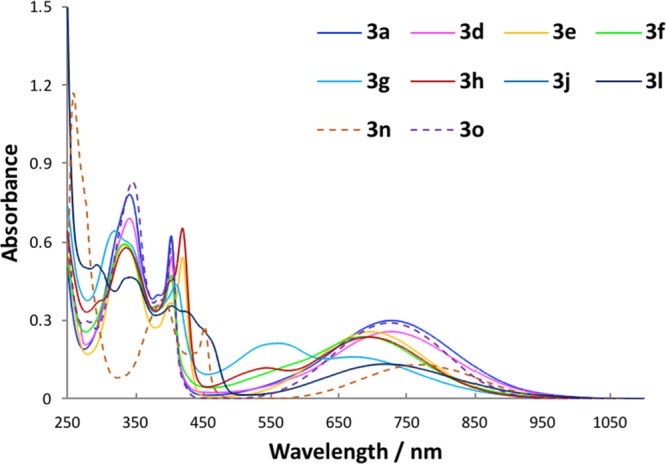
Absorption spectra of compounds 3 (250–1100 nm; 50 μM in MeOH).
Table 2. Maximal Absorption Wavelengths and Molar Absorption Coefficients of Compounds 3 (250–1100 nm; 50 μM in CHCl3 or MeOH).
| CHCl3 |
MeOH |
|||||||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | λex (nm) | abs. | ε | λex (nm) | abs. | ε | |
| 3a | H | H | 722 | 0.376 | 7530 | 726 | 0.301 | 6020 |
| 3d | 4-Cl | H | 722 | 0.389 | 7770 | 727 | 0.258 | 5160 |
| 3e | 3-Cl | H | 688 | 0.250 | 5010 | 698 | 0.258 | 5160 |
| 3f | 2-Cl | H | 691 | 0.288 | 5760 | 688 | 0.239 | 3780 |
| 3g | 1-Cl | H | 690 | 0.287 | 5740 | 673 | 0.161 | 3230 |
| 3h | 3-Br | H | 685 | 0.301 | 6010 | 697 | 0.237 | 4750 |
| 3j | 3-Me | H | 726 | 0.298 | 5960 | 736 | 0.255 | 5100 |
| 3l | 715 | 0.134 | 2690 | 715 | 0.134 | 2690 | ||
| 3n | H | OMe | 724 | 0.292 | 5833 | 737 | 0.119 | 2379 |
| 3o | H | Cl | 714 | 0.099 | 1971 | 717 | 0.132 | 2638 |
In addition, we calculated all the above compounds 3a, 3d–h, 3j, 3l, 3m, and 3p (Figure 7)13 in order to understand a structure and an absorption–wavelength relationships using time-dependent density functional theory (TD-DFT). All calculations were performed using Gaussian 09 (Rev. C) software. The ground-state geometries were optimized using the DFT method with B3LYP/6-31G(d). Harmonic frequency calculations were carried out to confirm that these geometries are stable with no imaginary frequencies. To obtain excited state energies, we calculated vertical excitation energies by the TD-DFT method with TD-B3LYP/6-31G(d). We compared experimental and calculated excitation energies in the electron volt unit. Although the calculated wavelength underestimated the experimental peak absorption wavelength, we found the linearity between the calculated and the experimental maximum absorption wavelength.
Figure 7.

Structure of 3p. Absorption wavelength of 3p is 663 nm.
Figure 8 shows the correlation between the calculated and the experimental absorption wavelength in CHCl3. In Figure 8, the line is determined by the least squares method using the regression equation. y = 1.0x + b. The differences in calculated and experimental energies (1.7 vs 2.6 eV) might come from the shape and dyes’ structures and charge-transfer band. According to the regression analysis (Figure 8), the compounds outside the 95% confidence interval were 3a, 3j, 3g, and 3p. Especially, 3g seems to be an outlier. The difference of 3g from other compounds is that R1 is Cl-substituent. This big difference of experimental wavelength of 3g and calculation might be caused by the repulsion between the Cl and phenyl group which can produce the distortion of the chromophore structure. These calculated results show that the TD-DFT approach is an effective method to understand the relationships between the structure and the absorption property.
Figure 8.

Correlation between experimental and calculated wavelength excitation energies.
We investigated the absorption profiles of 3j in various solvents in Figure 9. A higher maximal absorption wavelength was observed for polar solvents, as shown in Table 3, such as alcohol or DMSO. In dioxane, the maximal absorption wavelength was about 50 nm lower than that in MeOH. The molar absorption coefficient was stronger in CHCl3 and toluene than in other solvents.
Figure 9.

Absorption spectra of 3j in various solvents (250–1100 nm; 50 μM).
Table 3. Maximal Absorption Wavelengths and Molar Absorption Coefficients of 3j in Various Solvents (250–1100 nm; 50 μM).
| λex (nm) | abs. | ε | |
|---|---|---|---|
| MeOH | 736 | 0.255 | 5100 |
| EtOH | 733 | 0.250 | 5010 |
| DMSO | 732 | 0.247 | 4940 |
| CHCl3 | 726 | 0.298 | 5960 |
| DMF | 722 | 0.239 | 4770 |
| CH3CN | 709 | 0.231 | 4620 |
| acetone | 707 | 0.247 | 4940 |
| AcOEt | 697 | 0.239 | 4770 |
| toluene | 693 | 0.262 | 5250 |
| dioxane | 689 | 0.243 | 4850 |
Finally, we also synthesized 6-methylisoindolo[2,1-a]quinoline 5, a novel isomer of the methyl substituent, from N-allyl-N-(2-methylallyl)-2-vinylaniline 4 via our one-pot strategy in 53% yield (Scheme 4). The structure of 5 was determined by single-crystal X-ray diffraction (Figure 10). According to X-ray diffraction, there is no steric repulsion between the methyl substituent at position 6 and the carbonyl group at position 6 of isoindolo[2,1-a]quinoline. Thus, isoindolo[2,1-a]quinoline chromophore of 5 has a planar structure.
Scheme 4. Synthesis of 6-methylisoindolo[2,1-a]quinoline 5.

Figure 10.

X-ray structure of 6-methylisoindolo[2,1-a]quinoline 5.
A solution color of the 6-methylisoindolo[2,1-a]quinoline 5 in MeOH is red, although that of 5-methylisoindolo[2,1-a]quinoline 3a is blue (Figure 11). As both compounds 3a and 5 had the same core isoindolo[2,1-a] system, we considered that the methyl substituent’s electronic effects to the pi-conjugate system were dramatically changed dependent on the methyl substituent’s position on the pi-conjugate system. We then investigated the absorption profiles of compounds 3a and 5 in MeOH (Figure 12). The maximal absorption wavelength of compound 5 in MeOH was 474 nm. Compound 5 has no absorption in the NIR region. A molar absorption coefficient of 5 was 6730 M–1 cm–1, which is higher than that of 3a. From these results, we surmised that the most important thing for absorbing light in the NIR region was the methyl substituent at position 5 of the isoindolo[2,1-a]quinoline chromophore.15
Figure 11.

Colors of compound 3a and 5 (50 μM in MeOH).
Figure 12.

Absorption spectra of 3a and 5 in MeOH (250–1100 nm; 50 μM).
In summary, we synthesized isoindolo[2,1-a]quinoline derivatives 3d–3h, 3j, 3l, 3n, and 3o as novel NIR absorption dyes and found that, among them, 3j and 3n were the red-shifted NIR dye in MeOH. We also found that the maximal absorption wavelength of isoindolo[2,1-a]quinolines 3 was affected by solvent polarity and that TD-DFT was effective to understand the structure and the absorption–wavelength relationships of 3.
Experimental Section
General Information
Chemicals and solvents were either purchased from commercial suppliers or purified by standard techniques. All reactions were performed under a N2 atmosphere unless otherwise noted. For TLC, silica gel plates Merck 60 F254 were used. 1H NMR were recorded at 300, 400, and 500 MHz. 13C NMR spectra were recorded at 101 and 126 MHz. Chemical shifts are given in ppm relative to tetramethylsilane (TMS) and the coupling constants J are given in Hz. The spectra were recorded in CDCl3 as the solvent at room temperature unless otherwise noted. TMS served as the internal standard (δ = 0 ppm) for 1H NMR and CDCl3 was used as the internal standard (δ = 77.0 ppm) for 13C NMR. Column chromatography was performed with silica gel 60N (spherical, neutral, 63–210 μm, Kanto Chemical Co., Inc.), flash silica gel 60 (spherical, acid, 40–50 μm, Kanto Chemical Co., Inc.), and flash silica gel 60N (spherical, neutral, 40–50 μm, Kanto Chemical Co., Inc.) unless otherwise noted. Melting points were determined on the heated plate and are uncorrected. HRMS (m/z) was measured using a MALDI (matrix-assisted laser desorption/ionization)-TOF(time-of-flight) spectrometer unless otherwise noted. 3a, 3b, and 3c are known compounds.13
General Procedure for Preparation of N-Allyl-N-benzyl-o-isopropenylaniline 1
Procedure A for Synthesizing N-Benzyl-o-isopropenylaniline (Reductive Amination)16
To a solution of o-isopropenylaniline derivatives (1.0 equiv), PhCHO (1.2 equiv) and AcOH (1.2 equiv) in toluene (0.1 M) were added NaBH(OAc)3 (2.0 equiv) and the reaction mixture was stirred at rt for a while and then at refluxing temperature for 12 h. After the reaction mixture was cooled to 0 °C, saturated aqueous Na2CO3 was added to the reaction mixture. The organic compounds were extracted with AcOEt. The organic phases were washed with brine, dried over Na2SO4, and concentrated in vacuo. The obtained residue was prepurified by filtration through a short-pad silica gel with n-hexane/AcOEt = 24:1 as an eluent to give a crude compound containing N-benzyl-o-isopropenylaniline.
Procedure B for Synthesis of N-Benzyl-o-isopropenylaniline (Alkylation)
A solution of o-isopropenylaniline derivatives (1.0 equiv) in THF (0.1 M) was cooled to −78 °C and was stirred. To a stirred mixture, n-BuLi (2.6 M in n-hexane, 1.0 equiv) was added dropwise and the whole mixture was stirred for 15 min. To a stirred mixture, BnBr (1.0 equiv) was added dropwise and the stirred mixture was warmed to rt. After 1 h, the reaction was quenched by the addition of water. The organic compounds were extracted with AcOEt. The organic phases were washed with brine, dried over Na2SO4, and concentrated in vacuo. The obtained residue was prepurified by filtration through a short-pad silica gel with n-hexane/AcOEt = 24:1 as an eluent to give a crude compound containing N-benzyl-o-isopropenylaniline.
Synthesis of N-Allyl-N-benzyl-o-isopropenylaniline (Allylation)
A solution of
crude N-benzyl-o-isopropenylaniline
in 1,4-dioxane (0.5 M) was cooled to 0 °C and was stirred. To
the stirred mixture, NaH (60% in mineral oil, 2.0 equiv) was added
and the whole mixture was stirred for 15 min. To a stirred mixture,
allyl bromide (2.0 equiv) and tetrabutyl ammonium fluoride (50 mol
%) were added and the reaction mixture was stirred at 100 °C
for 12 h. After the reaction mixture was cooled to 0 °C, saturated
K2CO3 was added to the reaction mixture in MeOH.
The organic compounds were extracted with AcOEt. The organic phases
were washed with water, brine, dried over Na2SO4, and concentrated in vacuo. The obtained residue was subjected to
column chromatography (neutral acid flash silica gel, n-hexane → n-hexane/AcOEt = 49:1) to give
a colorless or brown oil.
N-Allyl-N-benzyl-3-chloro-2-isopropenylaniline (1d)
Following the general procedure A, 1d (706 mg, 2.37 mmol, 68%) was obtained as a yellow oil from S1d (587 mg, 3.50 mmol).
1H NMR (CDCl3, 400 MHz): δ 7.35–7.25 (5H, m), 7.16 (1H, dd, J = 8.2, 1.4 Hz), 7.12 (1H, dd, J = 7.8,
7.8 Hz), 6.96 (1H, dd, J = 7.8, 1.4 Hz), 5.79 (1H,
ddt, J = 17.4, 9.6, 6.0 Hz), 5.45 (1H, s), 5.12 (1H,
dt, J = 17.4, 1.4 Hz), 5.12 (1H, dt, J = 9.6, 1.4 Hz), 5.00 (1H, s), 4.18 (2H, br s), 3.60 (2H, d, J = 6.0 Hz), 2.18 (3H, s); 13C{1H}
NMR (CDCl3, 101 MHz): δ 150.3, 142.1, 138.7, 138.3,
134.8, 133.2, 128.8, 128.1, 127.5, 127.0, 124.4, 121.1, 117.7, 117.5,
58.2, 55.3, 23.1; HRMS (MALDI): calcd for C19H21NCl, 298.1357 [(M + H)+]; found, 298.1354.
N-Allyl-N-benzyl-4-chloro-2-isopropenylaniline (1e)
Following the general procedure A, 1e (167 mg, 0.561 mmol, 56%) was obtained as a yellow oil from S2e (168 mg, 1.00 mmol).
1H NMR
(CDCl3, 400 MHz): δ 7.29–7.20 (3H, m), 7.18–7.13
(3H, m), 7.09 (1H, ddd, J = 8.7, 2.7, 1.4 Hz), 6.80
(1H, d, J = 8.7 Hz), 5.74 (1H, ddt, J = 17.5, 10.8, 6.0 Hz), 5.15–5.05 (4H, m), 4.17 (2H, s), 3.57
(2H, d, J = 6.0 Hz), 2.21 (3H, s); 13C{1H} NMR (CDCl3, 101 MHz): δ 146.6, 146.5,
140.0, 137.8, 134.4, 130.1, 128.9, 128.2, 127.18, 127.15, 127.0, 122.4,
117.9, 115.5, 56.1, 54.4, 22.0; HRMS (MALDI): calcd for C19H21NCl, 298.1357 [(M + H)+]; found, 298.1354.
N-Allyl-N-benzyl-5-chloro-2-isopropenylaniline (1f)
Following the general procedure A, 1f (1.28 g, 4.30 mmol, 64%) was obtained as a yellow oil from S2f (1.13 g, 6.74 mmol).
1H NMR (CDCl3, 500 MHz): δ 7.31–7.28 (2H, m), 7.26–7.23
(1H, m), 7.22–7.19 (2H, m), 7.09 (1H, d, J = 8.1 Hz), 6.94 (1H, dd, J = 8.1, 1.8 Hz), 6.88
(1H, d, J = 1.8 Hz), 5.75 (1H, ddt, J = 16.6, 10.3, 6.6 Hz), 5.16–5.07 (4H, m), 4.22 (2H, s), 3.61
(2H, d, J = 6.6 Hz), 2.21 (3H, s); 13C{1H} NMR (CDCl3, 126 MHz): δ 149.2, 146.7,
137.8, 136.5, 134.0, 133.0, 131.3, 128.9, 128.2, 127.1, 122.1, 121.2,
118.1, 115.2, 55.7, 54.0, 22.0; HRMS (MALDI): calcd for C19H21NCl, 298.1357 [(M + H)+]; found, 298.1358.
N-Allyl-N-benzyl-2-chloro-6-isopropenylaniline (1g)
Following the general procedure B, 1g (122 mg, 0.409 mmol, 41%) was obtained as a yellow oil from S2g (168 mg, 1.00 mmol).
1H NMR
(CDCl3, 400 MHz): δ 7.31–7.20 (6H, m), 7.00
(1H, s), 6.99 (1H, s), 5.87 (1H, ddt, J = 16.9, 10.1,
6.9 Hz), 5.13 (1H, d, J = 0.9 Hz), 5.12 (1H, d, J = 16.9 Hz), 5.00 (1H, d, J = 10.1 Hz),
4.79 (1H, d, J = 0.9 Hz), 4.25 (2H, br s), 3.75 (2H,
d, J = 6.9 Hz), 2.06 (3H, s); 13C{1H} NMR (CDCl3, 101 MHz): δ 146.0, 145.7,
144.6, 139.3, 136.1, 134.3, 129.7, 128.9, 128.5, 128.0, 126.7, 125.5,
117.0, 115.3, 56.6, 55.8, 24.3; HRMS (MALDI): calcd for C19H21NCl, 298.1357 [(M + H)+]; found, 298.1353.
N-Allyl-N-benzyl-4-bromo-2-isopropenylaniline (1h)
Following the general procedure A, 1h (240 mg, 0.701 mmol, 70%) was obtained as a pale yellow oil from S2h (212 mg, 1.00 mmol).
1H
NMR (CDCl3, 400 MHz): δ 7.31–7.20 (5H, m),
7.19–7.13 (2H, m), 6.74 (1H, d, J = 8.7 Hz),
5.73 (1H, ddt, J = 17.4, 10.2, 6.4 Hz), 5.14–5.06
(4H, m), 4.18 (2H, s), 3.57 (2H, d, J = 6.4 Hz),
2.21 (3H, s); 13C{1H} NMR (CDCl3,
101 MHz): δ 147.1, 146.5, 140.3, 137.8, 134.3, 132.9, 130.1,
128.8, 128.2, 127.0, 122.7, 117.9, 115.6, 114.9, 56.0, 54.3, 21.9;
HRMS (MALDI): calcd for C19H21NBr, 342.0852
[(M + H)+]; found, 342.0851.
N-Allyl-N-benzyl-2-isopropenyl-3-methylaniline (1i)
Following the general procedure A, 1i (259 mg, 0.934 mmol, 52%) was obtained as a pale yellow oil from S2i (265 mg, 1.80 mmol).
1H
NMR (CDCl3, 500 MHz, 60 °C): δ 7.29–7.23
(3H, m), 7.22–7.17 (1H, m), 7.09–7.03 (1H, m), 6.94–6.90
(2H, m), 5.77 (1H, ddt, J = 17.2, 10.9, 6.9 Hz),
5.31 (1H, d, J = 1.7 Hz), 5.05 (1H, d, J = 12.0 Hz), 5.02 (1H, dq, J = 1.7, 1.2 Hz), 4.79
(1H, d, J = 8.0 Hz), 4.79 (1H, s), 4.12 (2H, br s),
3.56 (2H, br s), 2.26 (3H, s, minor rotamer 2.27), 2.09 (3H, s, minor
rotamer 2.11); 13C{1H} NMR (CDCl3, 101 MHz): δ 148.7, 144.5, 140.3, 139.0, 136.1, 135.5, 128.9,
128.0, 126.8, 126.5, 125.5, 120.2, 117.1, 115.8, 58.9, 55.3, 23.7,
20.4; HRMS (MALDI): calcd for C20H24N, 278.1903
[(M + H)+]; found, 278.1896.
N-Allyl-N-benzyl-2-isopropenyl-4-methylaniline (1j)
Following the general procedure A, 1j (88.8 mg, 0.320 mmol, 31%) was obtained as a pale yellow oil from S2j (151 mg, 1.03 mmol).
1H
NMR (CDCl3, 400 MHz): δ 7.28–7.21 (5H, m),
7.00–6.94 (2H, m), 6.83 (1H, d, J = 7.8 Hz),
5.76 (1H, ddt, J = 16.9, 10.5, 6.4 Hz), 5.11–5.03
(4H, m), 4.15 (2H, s), 3.56 (2H, d, J = 6.4 Hz),
2.28 (3H, s), 2.24 (3H, s); 13C{1H} NMR (CDCl3, 101 MHz): δ 147.8, 145.7, 138.7, 138.6, 134.9, 131.8,
131.1, 129.0, 128.1, 128.0, 126.8, 121.1, 117.4, 114.5, 56.6, 54.5,
22.6, 20.6; HRMS (MALDI): calcd for C20H24N,
278.1903 [(M + H)+]; found, 278.1908.
N-Allyl-N-benzyl-2-isopropenyl-4-methoxylaniline (1k)
Following the general procedure A, 1k (613 mg, 2.09 mmol, 60%) was obtained as a pale yellow oil prepared from S2h (571 mg, 3.50 mmol).
1H NMR (CDCl3, 400 MHz): δ 7.29–7.25
(2H, m), 7.23–7.20 (3H, m), 6.87 (1H, d, J = 8.6 Hz), 6.75 (1H, d, J = 3.1 Hz), 6.71 (1H,
dd, J = 8.6, 3.1 Hz), 5.77 (1H, ddt, J = 16.0, 11.0, 6.4 Hz), 5.12 (1H, s), 5.08 (1H, d, J = 11.0 Hz), 5.07 (1H, d, J = 16.0 Hz), 5.03 (1H,
s), 4.09 (2H, s), 3.78 (3H, s), 3.51 (2H, d, J =
6.4 Hz), 2.25 (3H, s); 13C{1H} NMR (CDCl3, 126 Hz): δ 155.2, 147.5, 141.6, 140.8, 138.5, 135.1,
129.1, 128.0, 126.8, 122.6, 117.4, 115.7, 114.7, 112.1, 57.5, 55.4,
55.0, 22.8; HRMS (MALDI): calcd for C20H24NO,
294.1852 [(M + H)+]; found, 294.1847.
N-Allyl-N-benzyl-3-isopropenylnaphthalen-2-amine (1l)
Following the general procedure A, 1l (161 mg, 0.514 mmol, 41%) was obtained as a colorless solid from S2i (229 mg, 1.25 mmol).
1H NMR (CDCl3, 400 MHz): δ 7.73 (1H, d, J = 8.2 Hz), 7.64 (1H, d, J = 7.8 Hz), 7.64 (1H, s), 7.37 (1H, ddd, J = 7.8, 7.4, 1.4 Hz), 7.32 (1H, ddd, J = 8.2, 7.4, 1.4 Hz), 7.29–7.18 (5H, m), 7.21 (1H, s), 5.79 (1H, ddt, J = 17.0, 10.1, 6.4 Hz), 5.22 (1H, s), 5.18 (1H, q, J = 1.2 Hz), 5.11 (1H, d, J = 10.1 Hz), 5.11 (1H, dd, J = 17.0, 1.8 Hz), 4.33 (2H, s), 3.70 (2H, d, J = 6.4 Hz), 2.31 (3H, s); 13C{1H} NMR (CDCl3, 126 Hz): δ 147.8, 146.7, 139.2, 138.1, 134.5, 133.3, 129.7, 129.04, 128.97, 128.2, 127.4, 126.9, 126.5, 125.8, 124.2, 117.8, 117.4, 114.8, 56.1, 54.2, 22.7; HRMS (MALDI): calcd for C23H24N, 314.1903 [(M + H)+]; found, 314.1905; mp 90.5–91.0 °C (recrystallized from n-hexane/AcOEt, colorless column).
N-Allyl-N-(4-methoxybenzyl)-2-isopropenylaniline (1m)
Following the general procedure A and allylation, 1m (720 mg, 2.45 mmol, 49% in 2 steps) was obtained as a yellow oil from commercial isopropenylaniline (0.63 mL, 5.00 mmol).
1H NMR (CDCl3, 500 MHz): δ 7.18–7.13 (2H, m), 7.10 (2H, d, J = 8.6 Hz), 6.96 (1H, ddd, J = 5.7, 5.7, 1.0 Hz), 6.89 (1H, d, J = 8.0 Hz), 6.80 (2H, d, J = 8.6 Hz), 5.79–5.71 (1H, m) 5.10–5.02 (4H, m), 4.14(2H, s), 3.78 (3H, s), 3.58 (2H, d, J = 6.3 Hz), 2.23 (3H, s); 13C{1H} NMR (CDCl3, 126 Hz): δ 158.5, 148.2, 147.8, 138.6, 134.9, 130.4, 130.3, 130.1, 127.4, 122.3, 121.2, 117.4, 114.6, 113.4, 55.4, 55.2, 54.0, 22.3; HRMS (MALDI): calcd for C20H24NO, 294.1852 [(M + H)+]; found, 294.1851.
N-Allyl-N-(4-chlorobenzyl)-2-isopropenylaniline (1n)
Following the general procedure A and allylation, 1n (1.15 g, 3.85 mmol, 64% in 2 steps) was obtained as a yellow oil from commercial isopropenylaniline (0.85 mL, 6.00 mmol).
1H NMR (CDCl3, 400 MHz): δ 7.23 (2H, d, J = 8.3 Hz), 7.16 (2H, m), 7.12 (2H, d, J = 8.7 Hz), 6.97 (1H, dd, 8.0 Hz, 6.9 Hz), 6.87 Hz (1H, d, J = 6.9 Hz), 5.81–5.71 (1H, m), 5.12–5.06 (4H, m), 4.16 (2H, s), 3.58 (2H, d, J = 6.4 Hz), 2.22 (3H, s); 13C{1H} NMR (CDCl3, 126 Hz): δ 147.8, 147.6, 138.6, 136.8, 134.6, 132.5, 130.4, 130.2, 128.2, 127.5, 122.5, 121.1, 117.7, 114.8, 55.3, 54.5, 22.4; HRMS (MALDI): calcd for C19H21NCl, 298.1357 [(M + H)+]; found, 298.1356.
General Procedure for Preparation of Isoindolo[2,1-a]quinoline 3
To a solution of N-allyl-2-alkenylaniline derivatives 1 (1.0 equiv) in
toluene (0.01 M) was added Grubbs II (1 mol %) and the reaction mixture
was stirred at 80 °C for 10 min. After completion of the RCM
(monitored by TLC), quinone (10 equiv) was added to the reaction mixture.
The reaction mixture was stirred at 80 °C for another 30 min
and then cooled to room temperature. To the reaction mixture saturated
aqueous NH4Cl was added. The organic compounds were extracted
with AcOEt. The organic phases were washed with brine, dried over
Na2SO4, and concentrated in vacuo. The obtained
residue was subjected to column chromatography (neutral flash silica
gel, n-hexane/AcOEt = 9:1 → 3:1; 2nd time, n-hexane/toluene/AcOEt = 6:18:1) to give 3.
4-Chloro-5-methyl-11-phenylisoindolo[2,1-a]quinoline-7,10-dione (3d)
Following the general procedure, 3d (13.0 mg, 0.0350 mmol, 35%) was prepared obtained as a blue solid from 1d (29.8 mg, 0.100 mmol).
1H NMR (CDCl3, 400 Hz): δ 9.06 (1H, d, J = 8.2 Hz), 7.45 (1H, dd, J = 8.2, 8.2
Hz), 7.40–7.30 (3H, m), 7.28–7.23 (1H, m), 6.99 (1H,
d, J = 7.3 Hz), 6.88 (1H, d, J =
10.1 Hz), 6.76 (1H, d, J = 10.1 Hz), 6.53 (1H, d, J = 7.8 Hz), 6.27 (1H, d, J = 6.9 Hz),
4.96 (3H, s); 13C{1H} NMR (CDCl3,
101 MHz, 50 °C): δ 187.0, 184.7, 142.7, 142.2, 139.9, 138.2,
135.6, 134.5, 134.2, 134.1, 131.7, 130.4, 129.3, 128.2, 126.7, 118.3,
117.3, 111.0, 107.0, 105.6, 56.5; HRMS (MALDI): calcd for C23H15NO2Cl, 372.0776 [(M + H)+]; found,
372.0786; mp 172.0–173.0 °C (recrystallized from CHCl3, blue needle).
3-Chloro-5-methyl-11-phenylisoindolo[2,1-a]quinoline-7,10-dione (3e)
Following the general procedure, 3e (15.7 mg, 0.0422 mmol, 42%) was obtained as a blue solid from 1e (29.8 mg, 0.100 mmol).
1H NMR
(CDCl3, 500 MHz): δ 7.48 (1H, d, J = 8.6 Hz), 7.37–7.32 (3H, m), 7.20 (2H, d, J = 7.4 Hz), 7.03 (1H, d, J = 7.4 Hz), 7.02 (1H,
d, J = 10.3 Hz), 6.74 (1H, d, J =
10.3 Hz), 6.53 (1H, d, J = 9.2 Hz), 6.36 (1H, d, J = 7.4 Hz), 4.99 (3H, s); 13C{1H}
NMR (CDCl3, 126 Hz): δ 186.3, 182.1, 142.1, 140.6,
138.5, 137.5, 136.8, 133.8, 133.52, 133.50, 131.1, 130.2, 129.3, 128.3,
126.5, 122.3, 122.2, 110.7, 107.2, 106.5, 56.8; HRMS (MALDI): calcd
for C23H14NO2Cl, 371.0708 [M+]; found, 371.0705; mp 175.0–176.0 °C (recrystallized
from CHCl3, blue needle).
2-Chloro-5-methyl-11-phenylisoindolo[2,1-a]quinoline-7,10-dione (3f)
Following the general procedure, 3f (14.0 mg, 0.0376 mmol, 38%) was obtained as a blue solid from 1f (29.8 mg, 0.100 mmol).
1H NMR
(CDCl3, 400 MHz): δ 9.11 (1H, d, J = 1.8 Hz), 7.42–7.34 (3H, m), 7.30–7.23 (2H, m), 6.98
(1H, d, J = 7.3 Hz), 6.89 (1H, d, J = 10.3 Hz), 6.78 (1H, d, J = 10.3 Hz), 6.50 (1H,
d, J = 1.8 Hz), 6.30 (1H, d, J =
7.3 Hz), 4.93 (3H, s); 13C{1H} NMR (CDCl3, 126 Hz, 60 °C): δ 186.5, 184.5, 142.6, 141.6,
140.9, 138.8, 137.7, 135.8, 134.7, 134.4, 133.8, 129.4, 128.7, 128.5,
126.9, 117.6, 116.6, 111.6, 107.6, 105.9, 56.6; HRMS (MALDI): calcd
for C23H14NO2Cl, 371.0708 [M+]; found, 371.0694; mp 175.5–177.0 °C (recrystallized
from CHCl3, blue needle).
1-Chloro-5-methyl-11-phenylisoindolo[2,1-a]quinoline-7,10-dione (3g)
Following the general procedure, 3g (8.2 mg, 0.0221 mmol, 22%) was obtained as a blue solid from 1g (29.8 mg, 0.100 mmol).
1H NMR
(CDCl3, 500 MHz): δ 9.06 (1H, d, J = 9.7 Hz), 7.48 (1H, d, J = 9.7 Hz), 7.34–7.22
(5H, m), 6.89 (1H, d, J = 10.3 Hz), 6.79 (1H, d, J = 10.3 Hz), 6.78 (1H, s), 6.27 (1H, d, J = 7.4 Hz), 5.43 (3H, s); 13C{1H} NMR (CDCl3, 126 Hz): δ 186.5, 185.2, 142.4, 140.7, 140.4, 137.7,
136.8, 135.0, 134.7, 134.4, 132.5, 132.4, 128.9, 127.9, 127.0, 119.3,
119.2, 113.3, 112.5, 109.0, 58.5; HRMS (MALDI): calcd for C23H14NO2Cl, 371.0708 [M+]; found,
371.0699; mp 171.0–172.5 °C (recrystallized from CHCl3, blue needle).
3-Bromo-5-methyl-11-phenylisoindolo[2,1-a]quinoline-7,10-dione (3h)
Following the general procedure, 3h (17.5 mg, 0.0420 mmol, 42%) was obtained as a blue solid from 1h (34.2 mg, 0.100 mmol).
1H NMR
(CDCl3, 400 MHz): δ 7.68 (1H, dd, J = 8.7, 1.4 Hz), 7.40–7.30 (3H, m), 7.21 (2H, d, J = 7.3 Hz), 7.05 (1H, dd, J = 6.6, 1.4 Hz), 7.03
(1H, d, J = 10.1 Hz), 6.75 (1H, d, J = 10.1 Hz), 6.47 (1H, dd, J = 8.7, 1.4 Hz), 6.41
(1H, dd, J = 7.3, 1.8 Hz), 5.01 (3H, s); 13C{1H} NMR (CDCl3, 126 Hz): δ 186.4, 181.7,
142.1, 140.3, 139.0, 137.43, 137.39, 136.7, 133.8, 133.6, 132.0, 131.3,
129.4, 129.3, 128.4, 126.50, 126.46, 126.40, 123.2, 110.8, 107.5,
106.8, 56.8; HRMS (MALDI): calcd for C23H14NO2Br, 415.0202 [M+]; found, 415.0202; mp 173.0–175.0
°C (recrystallized from AcOEt, blue needle).
3-Bromo-5-methyl-11-phenylisoindolo[2,1-a]quinoline-7,10-dione (3j)
Following the general procedure, 3j (5.9 mg, 0.0168 mmol, 17%) was obtained as a green solid from 1j (27.7 mg, 0.100 mmol).
1H NMR
(CDCl3, 500 MHz): δ 7.39–7.31 (5H, m), 7.22
(2H, d, J = 7.4 Hz), 7.06 (1H, d, J = 7.4 Hz), 6.96 (1H, d, J = 10.3 Hz), 6.72 (1H,
d, J = 10.3 Hz), 6.62 (1H, d, J =
8.0 Hz), 6.37 (1H, d, J = 7.4 Hz), 5.03 (3H, s),
2.11 (3H, s); 13C{1H} NMR (CDCl3,
126 MHz): δ 187.2, 182.7, 142.3, 141.5, 137.6, 137.5, 137.0,
134.4, 134.3, 133.4, 131.9, 130.2, 129.2, 128.1, 126.9, 126.5, 122.6,
110.3, 106.8, 106.1, 56.6, 24.1; HRMS (MALDI): calcd for C24H17NO2, 351.1254 [M+]; found, 351.1239;
mp 181.0–182.0 °C (recrystallized from AcOEt, blue needle).
6-Methyl-14-phenylbenzo[g]isoindolo[2,1-a]quinoline-1,4-dione (3l)
Following the general procedure, 3l (17.0 mg, 0.0439 mmol, 44%) was obtained as a green solid from 1l (31.3 mg, 0.100 mmol).
1H NMR (CDCl3, 500 MHz): δ 7.91 (1H, d, J = 8.6 Hz), 7.52 (1H, d, J = 7.4 Hz), 7.51 (1H, s), 7.42 (1H, dd, J = 7.4, 7.4 Hz), 7.38–7.35 (2H, m), 7.33–7.28 (2H, m), 7.21 (1H, ddd, J = 6.9, 6.9, 1.2 Hz), 7.02 (1H, d, J = 10.0 Hz), 6.95 (1H, d, J = 7.4 Hz), 6.83 (1H, d, J = 10.0 Hz), 6.73 (1H, s), 6.18 (1H, d, J = 7.4 Hz), 4.99 (3H, s); 13C{1H} NMR (CDCl3, 126 MHz): δ 186.2, 184.9, 141.9, 140.2, 137.9, 136.4, 135.8, 135.0, 134.7, 134.6, 132.0, 129.9, 129.3, 129.2, 128.3, 128.1, 126.8, 126.6, 125.0, 123.7, 122.1, 114.3, 104.2, 103.9, 56.6; HRMS (MALDI): calcd for C27H17NO2, 387.1254 [M+]; found, 387.1269, mp 199.0–200.0 °C (recrystallized from CHCl3, green needle).
2-Chloro-5-methyl-13-phenylbenzo[5,6]isoindolo[2,1-a]quinoline-7,12-dione (3m)
Following the general procedure, 3m (7.6 mg, 0.0180 mmol, 18%) was obtained as a blue solid from 1f (29.8 mg, 0.100 mmol).
1H NMR (CDCl3, 500 MHz): δ 9.38 (1H, d, J = 1.8 Hz), 8.30 (1H, d, J = 6.4 Hz), 8.19 (1H, d, J = 7.3 Hz), 7.77 (1H, dd, J = 7.8, 1.4 Hz), 7.69 (1H, dd, J = 7.8, 1.4 Hz), 7.52 (1H, d, J = 5.0 Hz), 7.42–7.34 (3H, m), 7.27 (1H, s), 6.98 (1H, d, J = 7.3 Hz), 6.54 (1H, d, J = 1.8 Hz), 6.34 (1H, d, J = 7.3 Hz), 4.94 (3H, s); 13C{1H} NMR (CDCl3, 126 MHz): δ 183.2, 180.8, 141.6, 140.5, 139.5, 136.8, 136.6, 135.1, 134.5, 134.4, 134.0, 132.1, 131.3, 128.4, 127.8, 127.2, 126.4, 126.1, 125.2, 117.1, 115.2, 110.3, 106.8, 105.4, 55.0; HRMS (MALDI): calcd for C27H16NO2Cl, 421.0864 [M+]; found, 421.0861, mp 291.0–292.0 °C (recrystallized from DMSO, blue needle).
11-(4-Methoxyphenyl)-5-methylisoindolo[2,1-a]quinoline-7,10-dione (3n)
Following general procedure, 3n (10.8 mg, 0.0294 mmol, 6%) was prepared as a blue solid from 1m (149 mg, 0.500 mmol).
1H NMR (CDCl3, 400 MHz): δ 9.07 (1H, d, J = 8.7 Hz), 7.47 (1H, dd, J = 8.4, 7.8 Hz), 7.19 (2H, d, J = 8.7 Hz), 6.98 (1H, d, J = 7.4 Hz), 6.90 (2H, d, J = 8.7 Hz), 6.88 (1H, d, J = 8.6 Hz), 6.75 (1H, d, J = 10.1 Hz), 6.57 (1H, d, J = 8.2 Hz), 6.26 (1H, d, J = 7.3 Hz), 4,89 (3H, s), 3.79 (3H, s); 13C{1H} NMR (CDCl3, 101 MHz): δ 187.2, 184.6, 159.5, 142.9, 142.4, 139.8, 138.2, 134.1, 134.0, 131.8, 130.4, 128.1, 125.9, 117.8, 117.1, 114.6, 111.0, 107.0, 105.5, 56.1, 55.3, 29.7; HRMS (MALDI): calcd for C24H17NO3, 367.1208 [M+]; found, 367.1198; mp 254.0–255.0 °C (recrystallized from AcOEt, blue needle).
11-(4-Chlorophenyl)-5-methylisoindolo[2,1-a]quinoline-7,10-dione (3o)
Following general procedure, 3o (8.3 mg, 0.022 mmol, 4%) was prepared as a blue solid from 1n (149 mg, 0.500 mmol).
1H NMR (CDCl3, 400 MHz): δ 9.06 (1H, d, J = 8.7 Hz), 7.45 (1H, dd, J = 8.2, 8.7
Hz), 7.34 (2H, d, J = 8.7 Hz), 7.28 (1H, s), 7.20
(2H, d, 8.7 Hz), 6,96 (1H, d, 6.9 Hz), 6.89 (1H, d, 10.1 Hz), 6.77
(1H, d, 10.1 Hz), 6.45 (1H, d, 7.8 Hz), 6.27 (1H, d, 7.3 Hz), 4.92
(2H, s); 13C{1H} NMR (CDCl3, 100
MHz): δ 187.0, 184.8, 142.7, 142.1, 139.5, 138.1, 135.4, 134.10,
134.06, 132.9, 131.7, 130.3, 129.5, 127.9, 118.2, 117.3, 111.1, 107.2,
105.5, 56.0, 29.7; HRMS(MALDI): calcd for C23H14NO2Cl, 371.0713 [M+]; found, 371.0706; mp 275.0–276.0
°C (recrystallized from AcOEt, blue needle).![]()
N-Benzyl-N-(2-methylallyl)-2-vinylaniline (4)
To a solution of N-benzyl-2-vinylaniline13 (88.0 mg, 0.598 mmol) in DMF (1.2 mL) was added 3-chloro-2-methyl-1-propene (117 μL, 1.20 mmol), K2CO3 (207 mg, 1.50 mmol) and TBAI (221 mg, 0.598 mmol) and the whole was stirred at 120 °C overnight. After the reaction mixture was cooled to rt, to the reaction mixture was added sat. K2CO3 in MeOH. The organic compounds were extracted with AcOEt. The organic phases were washed with water, brine, dried over Na2SO4 and concentrated in vacuo. The obtained residue was subjected to preparative gel permeation liquid chromatography (CHCl3) to give 4 (42.1 mg, 0.160 mmol, 27%) as a colorless oil.
1H NMR (CDCl3, 500
MHz): δ 7.50 (1H, dd, J = 7.8, 1.4 Hz), 7.31–7.15
(6H, m), 7.12 (1H, ddd, J = 7.8, 7.8, 1.4 Hz), 7.00
(1H, dd, J = 7.8, 7.8 Hz), 6.89 (1H, dd, J = 7.8, 1.4 Hz), 5.69 (1H, dd, J = 17.9,
1.4 Hz), 5.26 (1H, dd, J = 11.0, 1.4 Hz), 4.93 (1H,
s), 4.88 (1H, s), 4.14 (2H, s), 3.51 (2H, s), 1.72 (3H, s); 13C{1H} NMR (CDCl3, 126 MHz, 60 °C): δ
149.2, 142.8, 138.4, 134.9, 133.3, 128.7, 128.1, 128.0, 127.9, 126.9,
126.8, 121.9, 113.4, 113.2, 59.2, 57.5, 20.8; HRMS (MALDI): calcd
for C19H22N, 264.1747 [(M + H)+];
found, 264.1745.
6-Methyl-isoindolo[2,1-a]quinoline-7,10-dione (5)
To a solution of N-benzyl-N-(2-methylallyl)aniline derivatives 4 (27.7 mg, 0.100 mmol) in toluene (10 mL), Grubbs II (8.5 mg, 0.010 mmol) was added and the reaction mixture was stirred at 80 °C for 10 min. After completion of the RCM (monitored by TLC), 1,4-benzoquinone (108 mg, 1.00 mmol) was added to the reaction mixture. The reaction mixture was stirred at 80 °C for another 30 min and then cooled to room temperature. Saturated aqueous NH4Cl was added to the reaction mixture. The organic compounds were extracted with AcOEt. The organic phases were washed with brine, dried over Na2SO4, and concentrated in vacuo. The obtained residue was subjected to column chromatography (neutral flash silica gel, n-hexane/AcOEt = 9:1 → 3:1) to give 5 (17.8 mg, 0.0527 mmol, 53%) as a red solid.
1H NMR (CDCl3, 500 MHz): δ 7.39–7.31 (5H, m), 7.22 (2H, d, J = 7.4 Hz), 7.06 (1H, d, J = 7.4 Hz), 6.96 (1H, d, J = 10.3 Hz), 6.72 (1H, d, J = 10.3 Hz), 6.62 (1H, d, J = 8.0 Hz), 6.37 (1H, d, J = 7.4 Hz), 5.03 (2H, s), 2.11 (3H, s); 13C{1H} NMR (CDCl3, 126 MHz): δ 183.3, 181.2, 142.5, 138.2, 135.4, 134.1, 132.9, 132.8, 130.2, 129.6, 129.4, 128.9, 128.2, 127.5, 126.8, 126.5, 125.8, 121.8, 118.8, 113.0, 24.1; HRMS (MALDI): calcd for C23H15NO2, 337.1097 [M+]; found, 337.1098; mp 212.0–213.0 °C (recrystallized from n-hexane/AcOEt, red column).
Experiments for Photochemical Properties Measurements17
A 10 mM DMSO stock solution of each compound was prepared. Each spectrum was measured by using diluted stock solution of the desired solvent. A quartz cuvette containing the prepared solution was held on a cell holder placed in the light path of a monochromator. UV/vis/NIR spectra were recorded in the range from 250 to 1100 nm. The set-up parameters were as follows: UV/vis bandwidth: 2 nm, NIR bandwidth: 8 nm, UV/vis response: 0.06 s, NIR response: 0.06 s, path length: 1 cm, scan speed: 1000 nm/min, and grating and detector switch-over: 850 nm.
Acknowledgments
This work was partially supported by a Grant-in-Aid from JSPS Research Fellow (grant no. JP 16J05524), from JSPS KAKENHI (grant no. JP A16H010260) for Precisely Designed Catalysts with Customized Scaffolding, A262880510, T15K149760, and T15KT00630; the Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (MEXT), JST ACT-C grant number JPMJCR12YM, Japan, AMED under grant number 17am0101084j0001 and Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00315.
The authors declare no competing financial interest.
Supplementary Material
References
- a Weissleder R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19, 316–317. 10.1038/86684. [DOI] [PubMed] [Google Scholar]; b Hilderbrand S. A.; Weissleder R. Near-infrared fluorescence: application to in vivo molecular imaging. Curr. Opin. Chem. Biol. 2010, 14, 71–79. 10.1016/j.cbpa.2009.09.029. [DOI] [PubMed] [Google Scholar]; c Escobedo J. O.; Rusin O.; Lim S.; Strongin R. M. NIR dyes for bioimaging applications. Curr. Opin. Chem. Biol. 2010, 14, 64–70. 10.1016/j.cbpa.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fabian J.; Nakazumi H.; Matsuoka M. Near-infrared absorbing dyes. Chem. Rev. 1992, 92, 1197–1226. 10.1021/cr00014a003. [DOI] [Google Scholar]; b Weil T.; Vosch T.; Hofkens J.; Peneva K.; Müllen K. The Rylene Colorant Family-Tailored Nanoemitters for Photonics Research and Applications. Angew. Chem., Int. Ed. 2010, 49, 9068–9093. 10.1002/anie.200902532. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9252 - 9278.10.1002/ange.200902532; c Qian G.; Wang Z. Y. Near-Infrared Organic Compounds and Emerging Applications. Chem.—Asian J. 2010, 5, 1006–1029. 10.1002/asia.200900596. [DOI] [PubMed] [Google Scholar]
- a Mayerhöffer U.; Deing K.; Gruß K.; Braunschweig H.; Meerholz K.; Würthner F. Outstanding Short-Circuit Currents in BHJ Solar Cells Based on NIR-Absorbing Acceptor-Substituted Squaraines. Angew. Chem., Int. Ed. 2009, 48, 8776–8779. 10.1002/anie.200903125. [DOI] [PubMed] [Google Scholar]; b Imahori H.; Umeyama T.; Ito S. Large π-Aromatic Molecules as Potential Sensitizers for Highly Efficient Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1809–1818. 10.1021/ar900034t. [DOI] [PubMed] [Google Scholar]; c Bürckstümmer H.; Kronenberg N. M.; Meerholz K.; Würthner F. Near-Infrared Absorbing Merocyanine Dyes for Bulk Heterojunction Solar Cells. Org. Lett. 2010, 12, 3666–3669. 10.1021/ol101492q. [DOI] [PubMed] [Google Scholar]; d Qian G.; Qi J.; Davey J. A.; Wright J. S.; Wang Z. Y. Family of Diazapentalene Chromophores and Narrow-Band-Gap Polymers: Synthesis, Halochromism, Halofluorism, and Visible-Near Infrared Photodetectivity. Chem. Mater. 2012, 24, 2364–2372. 10.1021/cm300938s. [DOI] [Google Scholar]
- For recent review, see:Brogdon P.; Cheema H.; Delcamp J. H. Near-Infrared-Absorbing Metal-Free Organic, Porphyrin, and Phthalocyanine Sensitizers for Panchromatic Dye-Sensitized Solar Cells. ChemSusChem 2018, 11, 86–103. 10.1002/cssc.201701441. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Hu L.; Yan Z.; Xu H. Advances in synthesis and application of near-infrared absorbing squaraine dyes. RSC Adv. 2013, 3, 7667–7676. 10.1039/c3ra23048a. [DOI] [Google Scholar]; b Qin C.; Wong W.-Y.; Han L. Squaraine Dyes for Dye-Sensitized Solar Cells: Recent Advances and Future Challenges. Chem.—Asian J. 2013, 8, 1706–1719. 10.1002/asia.201300185. [DOI] [PubMed] [Google Scholar]
- Sun W.; Guo S.; Hu C.; Fan J.; Peng X. Recent Development of Chemosensors Based on Cyanine Platforms. Chem. Rev. 2016, 116, 7768–7817. 10.1021/acs.chemrev.6b00001. [DOI] [PubMed] [Google Scholar]
- Sibrian-Vazquez M.; Escobedo J. O.; Lowry M.; Fronczek F. R.; Strongin R. M. Field Effects Induce Bathochromic Shifts in Xanthene Dyes. J. Am. Chem. Soc. 2012, 134, 10502–10508. 10.1021/ja302445w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Mack J.; Yang Y.; Shen Z. Structural modification strategies for the rational design of red/NIR region BODIPYs. Chem. Soc. Rev. 2014, 43, 4778–4823. 10.1039/c4cs00030g. [DOI] [PubMed] [Google Scholar]
- Grzybowski M.; Gryko D. T. Diketopyrrolopyrroles: Synthesis, Reactivity, and Optical Properties. Adv. Opt. Mater. 2015, 3, 280–320. 10.1002/adom.201400559. [DOI] [Google Scholar]
- a Xie J.; Shi K.; Cai K.; Zhang D.; Wang J.-Y.; Pei J.; Zhao D. A NIR dye with high-performance n-type semiconducting properties. Chem. Sci. 2016, 7, 499–504. 10.1039/c5sc03045e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chai Z.; Wu M.; Fang M.; Wan S.; Xu T.; Tang R.; Xie Y.; Mei A.; Han H.; Li Q.; Li Z. Similar or Totally Different: the Adjustment of the Twist Conformation Through Minor Structural Modification, and Dramatically Improved Performance for Dye-Sensitized Solar Cell. Adv. Energy Mater. 2015, 5, 1500846. 10.1002/aenm.201500846. [DOI] [Google Scholar]; c Pei K.; Wu Y.; Islam A.; Zhang Q.; Han L.; Tian H.; Zhu W. Constructing High-Efficiency D-A−π-A-Featured Solar Cell Sensitizers: a Promising Building Block of 2,3-Diphenylquinoxaline for Antiaggregation and Photostability. ACS Appl. Mater. Interfaces 2013, 5, 4986–4995. 10.1021/am400754d. [DOI] [PubMed] [Google Scholar]; d Yao Z.; Zhang M.; Li R.; Yang L.; Qiao Y.; Wang P. A Metal-Free N-Annulated Thienocyclopentaperylene Dye: Power Conversion Efficiency of 12% for Dye-Sensitized Solar Cells. Angew. Chem., Int. Ed. 2015, 54, 5994–5998. 10.1002/anie.201501195. [DOI] [PubMed] [Google Scholar]; e Ni J.-S.; Yen Y.-C.; Lin J. T. Organic dyes with a fused segment comprising benzotriazole and thieno[3,2-b]pyrrole entities as the conjugated spacer for high performance dye-sensitized solar cells. Chem. Commun. 2015, 51, 17080–17083. 10.1039/c5cc07105d. [DOI] [PubMed] [Google Scholar]
- a Arisawa M.; Terada Y.; Takahashi K.; Nakagawa M.; Nishida A. Development of Isomerization and Cycloisomerization with Use of a Ruthenium Hydride withN-Heterocyclic Carbene and Its Application to the Synthesis of Heterocycles. J. Org. Chem. 2006, 71, 4255–4261. 10.1021/jo060308u. [DOI] [PubMed] [Google Scholar]; b Arisawa M.; Terada Y.; Takahashi K.; Nakagawa M.; Nishida A. Non-metathesis Reactions of Ruthenium Carbene Catalysts and Their Application to the Synthesis of Nitrogen-Containing Heterocycles. Chem. Rec. 2007, 7, 238–253. 10.1002/tcr.20121. [DOI] [PubMed] [Google Scholar]; c Kato H.; Ishigame T.; Oshima N.; Hoshiya N.; Shimawaki K.; Arisawa M.; Shuto S. One-Pot Ring-Closing Metathesis (RCM)/Oxidation by an Assisted Tandem Ruthenium Catalysis for the Synthesis of 2-Quinolones. Adv. Synth. Catal. 2011, 353, 2676–2680. 10.1002/adsc.201100303. [DOI] [Google Scholar]; d Fujii Y.; Takehara T.; Suzuki T.; Fujioka H.; Shuto S.; Arisawa M. One-Pot Olefin Isomerization/Aliphatic Enamine Ring-Closing Metathesis/Oxidation/1,3-Dipolar Cycloaddition for the Synthesis of Isoindolo[1,2-a]isoquinolines. Adv. Synth. Catal. 2015, 357, 4055–4062. 10.1002/adsc.201500680. [DOI] [Google Scholar]; e Yamashita K.; Fujii Y.; Yoshioka S.; Aoyama H.; Tsujino H.; Uno T.; Fujioka H.; Arisawa M. Development of an Enyne Metathesis/Isomerization/Diels-Alder One-Pot Reaction for the Synthesis of a Novel Near-Infrared (NIR) Dye Core. Chem.—Eur. J. 2015, 21, 17491–17494. 10.1002/chem.201502313. [DOI] [PubMed] [Google Scholar]; f Yoshioka S.; Fujii Y.; Tsujino H.; Uno T.; Fujioka H.; Arisawa M. One-pot enyne metathesis/Diels-Alder/oxidation to six-membered silacycles with a multi-ring core: discovery of novel fluorophores. Chem. Commun. 2017, 53, 5970–5973. 10.1039/c7cc02788e. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Zieliński G. K.; Grela K. Tandem Catalysis Utilizing Olefin Metathesis Reactions. Chem.—Eur. J. 2016, 22, 9440–9454. 10.1002/chem.201505136. [DOI] [PubMed] [Google Scholar]; b Ríos-Lombardía N.; G-Álvarez J.; G-Sabín J. One-Pot Combination of Metal- and Bio-Catalysis in Water for the Synthesis of Chiral Molecules. Catalysts 2018, 8, 75. 10.3390/catal8020075. [DOI] [Google Scholar]; c Fuwa H.; Sasaki M. Exploiting Ruthenium Carbene-Catalyzed Reactions in Total Synthesis of Marine Oxacyclic Natural Products. Bull. Chem. Soc. Jpn. 2016, 89, 1403–1415. 10.1246/bcsj.20160224. [DOI] [Google Scholar]
- Arisawa M.; Fujii Y.; Kato H.; Fukuda H.; Matsumoto T.; Ito M.; Abe H.; Ito Y.; Shuto S. One-Pot Ring-Closing Metathesis/1,3-Dipolar Cycloaddition through Assisted Tandem Ruthenium Catalysis: Synthesis of a Dye with Isoindolo[2,1-a]quinoline Structure. Angew. Chem., Int. Ed. 2013, 52, 1003–1007. 10.1002/anie.201206765. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1037 - 1041.10.1002/ange.201206765
- a Ishihara Y.; Kiyota Y.; Goto G. Synthesis of isoindolo(2,1-a)quinoline derivatives and their effects on N2-induced hypoxia. Chem. Pharm. Bull. 1990, 38, 3024–3030. 10.1248/cpb.38.3024. [DOI] [PubMed] [Google Scholar]; b Sui Z.; Altom J.; Nguyen V. N.; Fernandez J.; Bernstein J. I.; Hiliard J. J.; Barrett J. F.; Podlogar B. L.; Ohemeng K. A. Synthesis and Inhibitory Activity of Novel Tri- and Tetracyclic Quinolines against Topoisomerases. Bioorg. Med. Chem. 1998, 6, 735–742. 10.1016/s0968-0896(98)00030-3. [DOI] [PubMed] [Google Scholar]
- The difference between the absorption spectra of compounds 3b, 3c, and 5 the rest of the 3× series might come from dihedral angle between the substituent on position 5 of isoindolo[2,1-a]quinoline and the heterocycle.
- When we tried to synthesize 1f and 1h by procedure A, an electrocyclic reaction was proceeded to obtained trisubstituted quinoline. Considering this result, we synthesized 1f and 1h by procedure.Zhang X.; Xu X.; Yu L.; Zhao Q. Bronsted acid-mediated reactions of aldehydes with 2-vinylaniline and biphenyl-2-amine. Tetrahedron Lett. 2014, 55, 2280–2282. 10.1016/j.tetlet.2014.02.090. [DOI] [Google Scholar]
- Kobayashi T.; Urano Y.; Kamiya M.; Ueno T.; Kojima H.; Nagano T. Highly Activatable and Rapidly Releasable Caged Fluorescein Derivatives. J. Am. Chem. Soc. 2007, 129, 6696–6697. 10.1021/ja070376d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.