


Abstract
Calvarial bone healing remains difficult but may be improved by stimulating chondrogenesis of implanted stem cells. To simultaneously promote chondrogenesis and repress adipogenesis of stem cells, we built a CRISPRai system that comprised inactive Cas9 (dCas9), two fusion proteins as activation/repression complexes and two single guide RNA (sgRNA) as scaffolds for recruiting activator (sgRNAa) or inhibitor (sgRNAi). By plasmid transfection and co-expression in CHO cells, we validated that dCas9 coordinated with sgRNAa to recruit the activator for mCherry activation and also orchestrated with sgRNAi to recruit the repressor for d2EGFP inhibition, without cross interference. After changing the sgRNA sequence to target endogenous Sox9/PPAR-γ, we packaged the entire CRISPRai system into an all-in-one baculovirus for efficient delivery into rat bone marrow-derived mesenchymal stem cells (rBMSC) and verified simultaneous Sox9 activation and PPAR-γ repression. The activation/inhibition effects were further enhanced/prolonged by using the Cre/loxP-based hybrid baculovirus. The CRISPRai system delivered by the hybrid baculovirus stimulated chondrogenesis and repressed adipogenesis of rBMSC in 2D culture and promoted the formation of engineered cartilage in 3D culture. Importantly, implantation of the rBMSC engineered by the CRISPRai improved calvarial bone healing. This study paves a new avenue to translate the CRISPRai technology to regenerative medicine.
INTRODUCTION
Calvarial bone healing proceeds through intramembranous ossification whereby bone develops directly from mesenchymal progenitors (1), but successful healing of large calvarial bone defects is difficult (2). Although gene therapy in combination with cell therapy utilizing bone marrow-derived mesenchymal stem cells (BMSC) or adipose-derived stem cells (ASC) hold promise (1), satisfactory calvarial bone healing remains challenging. In contrast, complete healing of long bone (e.g. femora) appears to be easier (3), which proceeds through a distinct endochondral ossification pathway that involves chondrogenic differentiation of mesenchymal progenitors and formation of a cartilage template. We previously demonstrated that stimulating chondrogenic differentiation of ASC can switch the differentiation pathway from intramembranous to endochondral ossification and improve calvarial bone healing in vivo (4). However, BMSC and ASC may differentiate towards adipogenic, chondrogenic or osteogenic lineages. Intricate control of differentiation favorably towards chondrogenic, instead of adipogenic, pathway may be desired for calvarial bone healing. Since Sox9 and PPAR-γ are master transcription factors governing chondrogenesis and adipogenesis, respectively, and PPAR-γ inhibits Sox9 (5), simultaneous Sox9 activation and PPAR-γ inhibition in BMSC or ASC may promote calvarial bone healing.
CRISPR is a powerful RNA-guided genome editing tool that involves ectopic expression of Cas9 nuclease and a chimeric single guide RNA (sgRNA) comprising the spacer sequence to recognize the DNA target and the scaffold motif for Cas9 binding (6,7). This system was repurposed for CRISPR interference (CRISPRi) by using a catalytically inactive Cas9 (dCas9), which orchestrates with sgRNA to sterically block the transcription of target genes (8). The repression efficiency was enhanced by fusing dCas9 with transcription repressors such as KRAB (9). In addition, CRISPR activation (CRISPRa) was developed to stimulate target gene expression, by fusing dCas9 with transcription activators such as VP64 (9). The magnitude of stimulation was further enhanced by fusing dCas9 with a tandem array of peptides (10), epigenome modifier (11) or with a tripartite activator VPR (12). Alternatively, Zhang et al. (13) developed a synergistic activation mediator (SAM) system that comprises (i) dCas9-VP64, (ii) engineered sgRNA containing two copies of MS2 RNA hairpin that interacts with MS2 coat protein (MCP), and (iii) MPH fusion protein comprising MCP, p65 and heat shock factor 1 (HSF1) as the activation complex. After co-expression in the same cell, dCas9-VP64, sgRNA and MPH associate together to activate endogenous genes more potently than dCas9-VP64 alone.
Meanwhile, Qi and colleagues turned sgRNA into a scaffold by extending the sgRNA sequence with RNA aptamers to recruit orthogonal RNA binding proteins such as MCP and Com (14). MCP was fused with VP64 (MCP-VP64) to serve as the activation complex while Com was fused with KRAB (Com-KRAB) to serve as the repression complex. By expressing dCas9, MCP-VP64 and Com-KRAB as well as scaffold RNA to recruit MCP and Com, this approach enabled simultaneous gene activation and inhibition in yeast and HEK293 cells (14).
CRISPRi and CRISPRa have been exploited for diverse applications including genome-scale genetic screen (10,15–17), disease modeling (18), genetic interaction mapping (19), cell signaling engineering (20) and cell fate regulation (21–23). However, both CRISPRi and CRISPRa have yet to be harnessed for tissue regeneration in animal studies. Neither has the system simultaneously activating/repressing genes (14) been used for tissue engineering.
Since calvarial bone healing can be improved by stimulating stem cell chondrogenesis (4), we aimed to simultaneously activate Sox9 and inhibit PPAR-γ in BMSC, in attempts to activate chondrogenesis and repress adipogenesis, and hence favorably direct the differentiation pathway towards chondrogenesis. To this end, we developed a CRISPRai system that consists of dCas9, MPH, Com-KRAB, activating sgRNA (sgRNAa) that binds dCas9 and recruits MPH for activation, as well as inhibiting sgRNA (sgRNAi) that binds dCas9 and recruits Com-KRAB for inhibition. We showed that the CRISPRai system was able to simultaneously activate and repress reporter and endogenous genes. The entire CRISPRai system was packaged into an all-in-one baculovirus and efficiently delivered into BMSC for concurrent Sox9 activation and PPAR-γ inhibition. The CRISPRai system stimulated chondrogenesis while repressing adipogenesis in 2D culture, and promoted formation of engineered cartilage in 3D culture. Importantly, implantation of the CRISPRai-engineered BMSC augmented the healing of calvarial bone defects. The CRISPRai technology holds promise for translation to regenerative medicine and may be adapted to diverse applications.
MATERIALS AND METHODS
Stem cells isolation and cell culture
Six-weeks-old SD rats (≈151–175 g, Lesco Biotech, Taiwan) were euthanized by CO2 inhalation. rBMSC (24) and rASC (25) were isolated from bone marrow and subcutaneous fat pad, respectively, as described and cultured in α-MEM (Gibco) supplemented with 10% fetal bovine serum (FBS, Hyclone), 100 IU/ml penicillin, 100 IU/streptomycin and 4 ng/ml fibroblast growth factor basic (FGF, Peprotech). Cells of passage 3–5 were used for subsequent experiments. CHO DUXB11 cells were cultured in DMEM/F12 medium (Gibco) supplemented with 10% FBS, 100 IU/ml penicillin, 100 IU/ml streptomycin and 100× HT supplement (Gibco).
For chondroinduction, the culture medium was changed to chondroinductive medium (DMEM high glucose containing 1% FBS, 100 IU/ml penicillin, 100 IU/ml streptomycin, 350 × 10−3 M l-proline, 10−6 M l-ascorbate-2-phosphate, 10−7 M dexamethasone, ITS+1, 10 ng/ml transforming growth factor-β1 (TGF-β1, PeproTech). To augment adipogenesis, the cells were cultured in adipoinductive medium (StemPro Adipogenesis Differentiation Kit, Gibco).
Construction of plasmids
Construction of the reporter plasmid pmC-d2E is described in Supporting Info. To construct the plasmid pCRISPRai, we first replaced the KRAB-P2A-Zeocin fragment in pCMV-dCas9-KRAB-P2A-Zeocin (26) with two chemically synthesized porcine teschovirus-1 2A (P2A) sequences to yield pCMV-dCas9-P2A-P2A. Com-KRAB (CK) fragment and WPRE-SV40 terminator-loxP (WAL) fragment were PCR-amplified from pJZC78 (Addgene #62339 (14)) and pBacLEBW (25), respectively, and inserted into pCMV-dCas9-P2A-P2A downstream of the second P2A to generate pCMV-dCas9-P2A-P2A-CK-WAL. The gene fragment encoding MCP-p65-HSF1 (MPH) was PCR-amplified from pMS2-P65-HSF1_GFP (Addgene #61423 (13)) and inserted into pCMV-dCas9-P2A-P2A-CK-WAL downstream of the first P2A, yielding pCMV-dCas9-MPH-CK-WAL. The sequence encoding loxP-CMV enhancer-rat EF-1α (rEF-1α) promoter was PCR-amplified from pBacLEBW and inserted into pCMV-dCas9-MPH-CK-WAL to replace CMV promoter. As a result, the CRISPRai module (dCas9, MPH and CK) was driven by the rEF-1α promoter and flanked by loxP sites. The resultant plasmid was designated pCRISPRai.
To construct psgRNAa, we PCR-amplified from psgRNA(MS2) (Addgene #61424 (13)) a complete gene cassette comprising human U6 (hU6) promoter, spacer insertion linker and the sgRNA scaffold with two copies of MS2 binding aptamers inserted at the tetraloop and stem loop 2. The entire cassette was cloned into TA vector to contain an XbaI site upstream and NheI-XhoI sites downstream of the cassette for subsequent assembly of multiple sgRNAs. To construct psgRNAi, the hU6 promoter and spacer insertion linker were PCR-amplified from psgRNA(MS2), and the sgRNA scaffold containing Com hairpin at the 3′ end was PCR-amplified from pJZC78. These two amplicons were fused together by overlap PCR and cloned into TA vector, yielding psgRNAi.
Spacer sequences on sgRNAa/sgRNAi were designed using online tool (www.benchling.com), with windows from −400 to −50 (for activation) and from −50 to +300 (for repression) relative to the transcription start site of target genes (10). The 20-nt spacer sequences with the highest targeting efficiency and specificity scores were chosen (Supplementary Table S1), chemically synthesized and inserted into the spacer insertion linker in psgRNAa or psgRNAi.
Preparation of baculoviruses
To prepare the donor plasmids for baculovirus construction, the gene fragment encoding the entire loxP-flanking CRISPRai module was subcloned from pCRISPRai to the baculovirus donor plasmid pFastBac™ Dual (Thermo) to yield pBac-CRISPRai. Next we designed four different spacer sequences targeting Sox9 and four different spacer sequences targeting PPAR-γ (PPAR-γ has two isoforms: PPAR-γ1 and -γ2; thus we designed two spacers to target PPAR-γ1 and another two spacers to target PPAR-γ2). The four Sox9-targeting spacers were separately cloned into psgRNAa, and the four Sox9-targeting sgRNAa were assembled as a sgRNAa array in a single plasmid by BioBrick assembly. The resultant sgRNAa array was cloned into pBac-CRISPRai to yield pBac-aS. Similarly, the four PPAR-γ-targeting spacers were separately cloned into psgRNAi and the resultant sgRNAi were assembled as a sgRNAi array in a single plasmid by BioBrick assembly. The resultant sgRNAi array was cloned into pBac-CRISPRai to yield pBac-iP. Likewise, the sgRNAa and sgRNAi arrays were assembled into a sgRNAai array (consisting of four Sox9-targeting sgRNAa and four PPAR-γ-targeting sgRNAi) in another plasmid and subcloned to pBac-CRISPRai to yield pBac-aS-iP.
Donor plasmids pBac-aS, pBac-iP and pBac-aS-iP were used to generate the corresponding baculoviruses Bac-aS, Bac-iP and Bac-aS-iP using the Bac-To-Bac system (Thermo). All these three baculoviruses contained two loxP sites to flank the CRISPRai module and sgRNA arrays. The baculovirus Bac-Cre that expressed Cre recombinase was constructed previously (24). All baculovirus vectors were amplified by infecting insect cell Sf-9. Virus titers were determined by end-point dilution assay and expressed as plaque forming units (pfu)/ml (27).
Baculovirus transduction
Baculovirus transduction of rASC and rBMSC was conducted as described earlier (27) with minor modifications. Briefly, cells were seeded to six-well plate (1 × 105 cells/well), 10-cm dish (1 × 106 cells/dish) or 15-cm dish (2.5 × 106 cells/dish) for qRT-PCR, western blot and scaffold seeding, respectively. After overnight culture, the cells were incubated with the virus solution at an optimized multiplicity of infection (MOI) and shaken gently on a rocking plate at room temperature. After 6 h of transduction, the virus solution was replaced with α-MEM medium containing 3 mM sodium butyrate and cells continued to be cultured. After 16 h, the cells were trypsinized for experiments, or the spent medium was replaced with fresh α-MEM medium with medium exchange every 3 days. Alternatively, the medium was replaced with adipoinductive or chondroinductive medium, with half of the medium being exchanged every 3 days.
Fluorescence microscopy and flow cytometry
CHO DUXB11 cells were seeded to six-well plates (5 × 105 cells/well) overnight and co-transfected with 1000 ng pmc-d2E, 500 ng pCRISPRai and 1000 ng psgRNAa/psgRNAi mixture using Lipofectamine 3000 (Invitrogen). At 1 day post-transfection, the fluorescence of mCherry and d2EGFP was photographed using a fluorescence microscope (ECLIPSE TS100-F, Nikon). Alternatively, the cells were trypsinized at 2 days post-transfection and analyzed by flow cytometry (FACSCalibur™, BD Biosciences). Data were collected from 10 000 counts using FL1 channel (530/30 band pass filter) for d2EGFP and FL2 channel (585/42 band pass filter) for mCherry and normalized to those of control (ϕ) group.
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA was extracted from the cells using Quick-RNA™ Miniprep kit (Zymo Research) and reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA was subsequently subjected to quantitative real-time PCR (StepOnePlus, Applied Biosystems) using primers specific to Sox9, PPAR-γ, Acan, Col2a1, Col10a1, C/ebpα and Fabp4 (Supplementary Table S2). All the data were analyzed using gapdh as the internal control and normalized to those of mock-transduced cells to yield the relative expression levels.
Western blot, Alcian blue staining and Oil Red O staining
The details of western blot, Alcian blue staining and Oil Red O staining are described in Supporting Info.
Preparation of cell/scaffold constructs
Gelatin sponge scaffolds were prepared by cutting the Spongostan™ gelatin sponge (porosity≈97%, cat#MS0003, Ethicon) into disks (diameter≈6 mm) and subsequent immersion in saline solution for 30 min. rBMSC cultured in 15-cm dishes were mock-transduced (Mock group) or co-transduced with Bac-aS-iP/Bac-Cre (CRISPRai group) as described above. The cells were trypsinized at 1 day post-transduction (dpt), seeded onto the gelatin scaffold (5 × 106 cells/scaffold) and allowed to stand for 4 h. The rBMSC/scaffold constructs were added with chondroinductive medium, cultured overnight, and implanted into animals next day (see the following). Alternatively, the rBMSC/scaffold constructs continued to be cultured in chondroinductive medium with half of the medium exchanged every 3 days.
Qualitative and quantitative analysis of engineered cartilage
After culturing the rBMSC/scaffold constructs in chondroinductive medium for 1 or 7 days (i.e. at 2 or 8 dpt), the constructs were photographed. At 8 dpt, the constructs were harvested for fixation in 4% aqueous phosphate formaldehyde (Macron) for 1 day at room temperature. Fixed constructs were dehydrated, embedded in paraffin, and sliced into 10-μm-thick sections. The sections were stained with Gill's Hematoxylin V (Muto Pure Chemicals) and Eosin Alcohol Stain Solution (Muto Pure Chemicals) or with Safranin O (Sigma).
Alternatively, the constructs harvested at 8 dpt were frozen overnight at −80°C and crushed with a 1.5-cc disposable pestle. To determine GAG content, each crushed construct was incubated with 1 ml papain solution (Sigma-Aldrich) at 65°C for 3 h. Half of the papain-digested sample was collected for total DNA content analysis using Quant-iTTM Picogreen® dsRNA Reagent and Kits (Invitrogen). The other half was centrifuged (10 000 × g, 5 min) and the GAG in the supernatant was measured using the Blyscan™ Sulfated Glycosaminoglycan Assay Kit (Biocolor). The OD of treated samples was read on Multiskan EX (Thermo Scientific) at 620 nm.
To quantify collagen type II (Col II), the constructs were similarly crushed, but were homogenized in the buffer (0.05 M acetic acid and 0.5 M sodium chloride) for 1 h, digested with pepsin (Sigma) at 4°C overnight and digested again with elastase (Sigma) at 4°C overnight. Half of the digested sample was collected for total DNA content determination. The other half was centrifuged (10 000 × g, 5 min) and the supernatant was assayed using ELISA-based Type II Collagen Detection Kit (Chondrex). The OD of treated samples was read on Multiskan EX at 450 nm. Total GAG and Col II contents were normalized to total DNA content of the corresponding construct.
Surgical procedures
All animal experiments were performed in compliance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology, Taiwan). The experimental protocols were approved by the Institutional Animal Care and Use Committee of National Tsing Hua University. The rBMSC/scaffold constructs were prepared and cultured overnight prior to implantation. The SD rats were anesthetized by intramuscular injection of Zoletil 50 (25 mg/kg body weight, Virbac Animal Health) and 2% Rompun® (0.15 ml/kg body weight, Bayer Health Care), followed by intramuscular injection of the antibiotic cefazolin (160 mg/kg body weight). A midline sagittal incision (2 cm) in the scalp was made to expose the parietal bone, and the pericranium was removed by blunt scraping. A critical-size (6 mm in diameter) defect in the middle of parietal bone was created using a disposable biopsy punch (Integra Miltex) without disturbing the underlying dura mater. To minimize damage to the scull and adjacent blood vessels, sterile saline solution was intermittently sprayed to lower the temperature when we created the defects. The constructs were implanted onto the defect and gently pressed, followed by suturing with 4–0 absorbable stitch (Polysorb™, Coviden). The animals received second intramuscular injection of cefazolin and a topical administration of neomycin and bacitracin zinc at the surgery site.
μCT imaging analysis
Regeneration of the calvarial bone defects was scanned at 4 and 8 weeks post-surgery using Nano SPECT/CT (Cold Spring Harbor). The scanned data were processed using PMOD software (PMOD Technologies) to extract bone area (mm2), bone volume (mm3) and bone density (average Hounsfield Unit, HU) within a chosen disk-shaped volume of interest (VOI, 6 mm in diameter and 1 mm in height) representing the original defect. The data were normalized to the original defect area (28.3 mm2), volume (28.3 mm3) and density (≈4600 HU) to yield the percentage of bone regeneration. Amira software (Visualization Science Group) was utilized to reconstruct 3D projection of the defects.
Histological and immunohistochemical staining
After μCT scanning at week 8, we sacrificed the rats and removed the calvarial bones. The bone specimens were fixed in 4% aqueous phosphate formaldehyde for 3 days at room temperature and subsequently decalcified by immersing in Osteosoft® (Merck) for 14 days. Decalcified specimens were washed in PBS before undergoing dehydration, paraffin embedding, and sagittal slicing into 10-μm-thick sections. Sections were then rehydrated and stained with H&E.
Alternatively, rehydrated sections were subjected to trypsin treatment for 1 h at 37°C for antigen retrieval, followed by blocking in 5% skimmed milk and immunohistochemical staining. The primary antibodies were rabbit anti-BSP (1:200, Abcam) and mouse anti-OCN (1:200, Abcam). The secondary antibodies were goat anti-rabbit IgG-HRP (1:5000, GeneTex) and goat anti-mouse IgG-HRP (1:5000, Invitrogen). The sections were developed with hydrogen peroxide and 3,3′‐diaminobenzidine (Sigma) and counterstained with Eosin for visualization.
Statistical analysis
All in vitro data were representative of at least 3 independent culture experiments. All quantitative data are expressed as means±standard deviations (SD) and were analyzed using student's t-test or One-way ANOVA. P < 0.05 was considered significant.
RESULTS
Design and validation of CRISPRai system
To explore the feasibility of concurrent activation and inhibition of selected genes, we first constructed a reporter plasmid pmC-d2E that co-expressed mCherry and d2EGFP (Figure 1A). We next constructed pCRISPRai that expressed the CRISPRai module comprising the dCas9 regulator, MCP-p65-HSF1 (MPH) as the activation complex (13) and Com-KRAB as the repression complex (14). We further constructed a set of psgRNAa and psgRNAi plasmids for mCherry activation and d2EGFP inhibition, respectively (Figure 1B). Following the sgRNA 2.0 design developed by Zhang and coworkers (13), psgRNAa expressed the activating sgRNA (sgRNAa) that had two MS2 binding aptamers appended to the sgRNAa scaffold for MPH recruitment and the spacer targeting the template (T1) or non-template (NT1) strands of mCherry cassette (Figure 1A). psgRNAi expressed the inhibiting sgRNA (sgRNAi) following the design described by Qi and colleagues (14), with the Com binding aptamer appended at the 3′ end of the scaffold for Com-KRAB recruitment and the spacer targeting the non-template (NT2 or NT3) strands of d2EGFP cassette (Figure 1A). We envision that dCas9 can associate with sgRNAa to recruit MPH for mCherry activation while dCas9 also associates with sgRNAi to recruit Com-KRAB for d2EGFP suppression (Figure 1B). As controls, we also constructed psgRNAa and psgRNAi expressing sgRNAa/sgRNAi with only the scaffold but no spacer.
Figure 1.

Design and validation of CRISPRai system. (A) Illustration of reporter plasmid. mCherry was driven by PGK promoter; d2EGFP was driven by SV40 promoter. NT1, T1, NT2 and NT3 indicate the sgRNA targeting positions. (B) Plasmids used for the CRISPRai system. pCRISPRai co-expressed the CRISPRai module (dCas9, MPH and Com-KRAB) under the rat EF-1α promoter with a WPRE sequence at the 3′ end. dCas9, MPH and Com-KRAB were separated by P2A sequences and contained the SV40 nuclear localization signal (NLS). psgRNAa and psgRNAi expressed the sgRNAa and sgRNAi under the hU6 promoter. sgRNAa comprised the spacer targeting NT1 or T1 on the PGK promoter and the scaffold sequences containing two MS2 binding aptamers at the tetraloop and stem loop 2. sgRNAi comprised the spacer targeting NT2 or NT3 on the d2EGFP cassette and the scaffold sequence with Com binding aptamer at the 3′ end. dCas9 can associate with sgRNAa to recruit MPH for mCherry activation while dCas9 also associates with sgRNAi to recruit Com-KRAB for d2EGFP suppression. (C) Fluorescence microscopic images. The CHO DUXB11 cells were co-transfected with pmC-d2E, pCRISPRai and different combinations of psgRNAa and psgRNAi, and observed at 1 day post-transfection. (D, E) Flow cytometry analysis of mCherry and d2EGFP expression at day 2. The mean fluorescence intensity (MFI) of each group was normalized to that control group (ϕ). ****P < 0.0001. ***P < 0.001. *P < 0.05. Bar, 500 μm. The fold change of mCherry activation and percentages of d2EGFP suppression are shown above the bars. The data represent means ± SD of three independent culture experiments.
We co-transfected CHO cells with pmC-d2E, pCRISPRai and different combinations of psgRNAa/psgRNAi, followed by fluorescence microscopy (Figure 1C) and flow cytometry (Figure 1D and E) analyses. Our data confirmed robust mCherry and d2EGFP expression in the control group (ϕ) co-transfected with pmc-d2E, pCRISPRai and psgRNAa/psRNAi devoid of spacer sequences. Compared with the control group (ϕ), psgRNAa targeting the mCherry cassette with T1+NT1 conferred mCherry activation for 3.5-fold (Figure 1D), without significant compromise on d2EGFP expression (Figure 1E). Conversely, targeting the d2EGFP cassette (NT2, NT3 or NT2+NT3) with psgRNAi significantly knocked down d2EGFP expression to ≈8–9% (Figure 1E) without markedly affecting mCherry expression (Figure 1D). As such, the CRISPRai module indeed orchestrated with the sgRNAa/sgRNAi to execute gene activation/inhibition, without mutual interference. Critically, targeting both mCherry and d2EGFP cassettes (T1+NT1+NT2+NT3) enabled concomitant mCherry activation (2.9-fold) and d2EGFP suppression (9.9%) without mutual interference, proving that the CRISPRai system conferred orthogonal, simultaneous gene activation and suppression in mammalian cells.
CRISPRai delivered by baculovirus activated Sox9 and repressed PPAR-γ in stem cells
Since non-viral transfection of stem cells is inefficient, we chose to deliver the CRISPRai system using baculovirus, which enables delivery of large genetic cargo into primary and stem cells at high efficiency (27–30). We previously constructed a Cre/loxP-based baculovirus system in which one baculovirus harbors the transgene cassette flanked by loxP sites while the other expresses the Cre recombinase (31). Co-transduction of cells with the two baculoviruses leads to Cre recognition of the loxP sequences, excision of the transgene cassette off the baculovirus genome, formation of episomal DNA minicircle encompassing the transgene cassette (Supplementary Figure S1) and prolongs/enhances transgene expression (24,25,32).
We constructed three baculoviruses (Figure 2A) which all expressed identical CRISPRai module (dCas9, MPH and Com-KRAB under rat EF-1α promoter), but expressed different sgRNAa/sgRNAi. Bac-aS expressed four sgRNAa targeting four positions of rat Sox9 promoter for activation; Bac-iP expressed four sgRNAi targeting four positions of rat PPAR-γ gene for inhibition; and Bac-aS-iP expressed four Sox9-targeting sgRNAa and four PPAR-γ-targeting sgRNAi. All three baculoviruses contained two loxP sites to flank the CRISPRai module and sgRNA array. The entire transgene cassette in Bac-aS-iP was up to 12.6 kb.
Figure 2.

CRISPRai delivered by baculovirus activated Sox9 and repressed PPAR-γ in stem cells. (A) Baculovirus vectors and targeting sites. Bac-aS expressed four sgRNAa targeting four positions of rat Sox9 promoter. Bac-iP expressed four sgRNAi targeting four positions of rat PPAR-γ gene. Bac-aS-iP expressed four Sox9-targeting sgRNAa and four PPAR-γ-targeting sgRNAi. All these three baculoviruses contained two loxP sites to flank the CRISPRai module and sgRNA. Bac-Cre expressed Cre recombinases. (B, C) Sox9 and PPAR-γ expression. rBMSC and rASC were mock-transduced (Mock group) or transduced with Bac-aS, Bac-iP or Bac-aS-iP at MOI 300 and cultured in α-MEM. The expression levels were measured by qRT-PCR at 3 dpt and normalized to those of Mock group. (D, E) Kinetics of Sox9 and PPAR-γ expression. rBMSC were mock-transduced (Mock group), singly transduced with Bac-aS-iP (MOI 300) or co-transduced with Bac-aS-iP and Bac-Cre at MOI 300/100 and cultured in α-MEM. The expression levels were measured by qRT-PCR at 3 dpt and normalized to those of Mock group. The data represent means± SD of three independent culture experiments. ****P < 0.0001. *** P < 0.001. ** P < 0.01. *P < 0.05. ns, not significant (P > 0.05).
We first transduced rat ASC (rASC) or BMSC (rBMSC) with one of the three baculoviruses and analyzed Sox9 (Figure 2B) and PPAR-γ (Figure 2C) expression by qRT-PCR, using mock-transduced cells as the reference. In rBMSC, Bac-aS significantly upregulated Sox9 expression (7.8-fold) without suppressing PPAR-γ expression, while Bac-iP knocked down PPAR-γ expression to 25.8% without disturbing Sox9 expression. Importantly, Bac-aS-iP concurrently upregulated Sox9 expression (17.1-fold) and inhibited PPAR-γ expression (to 30.3%) in rBMSC. In rASC, Bac-aS and Bac-aS-iP potently activated Sox9 expression (Figure 2B), but none of the three vectors knocked down PPAR-γ expression (Figure 2C).
Given the success of Bac-aS-iP to activate Sox9 and suppress PPAR-γ in rBMSC, we explored whether the magnitude and duration of gene activation/suppression could be prolonged and enhanced, by co-transducing rBMSC with Bac-aS-iP and Bac-Cre (Figure 2A) which expressed Cre to recognize loxP sites for episomal minicircle formation (Supplementary Figure S1). As controls, rBMSC were mock-transduced or singly transduced with Bac-aS-iP. qRT-PCR analysis showed that Bac-aS-iP transduction alone effectively enhanced Sox9 expression (Figure 2D) and suppressed PPAR-γ expression (Figure 2E) in the first 7 days, but the regulation diminished after 14 days post-transduction (dpt). Bac-aS-iP/Bac-Cre co-transduction further enhanced and prolonged Sox9 expression, which increased to ≈107.9-fold and 16.8-fold at 3 and 7 dpt, and remained at 4.6-fold at 21 dpt (Figure 2D). Bac-aS-iP/Bac-Cre co-transduction also knocked down PPAR-γ expression, though the magnitude and duration of suppression were not significantly (ns, Figure 2D) different when compared with Bac-aS-iP transduction only.
Sox9/PPAR-γ regulation by CRISPRai enhanced chondrogenesis and inhibited adipogenesis
In vitro chondrogenic differentiation of BMSC is characterized by upregulation of aggrecan (Acan) and collagen IIa1 (Col2a1) as well as accumulation of glycosaminoglycan (GAG), but at later stage cells tend to undergo undesired hypertrophy as characterized by collagen Xa1 (Col10a1) upregulation (33). Conversely, adipogenesis governed by PPAR-γ activates downstream Fabp4 and C/ebpα and subsequently stimulates oil droplet formation (34). To evaluate whether simultaneous Sox9 activation and PPAR-γ inhibition promoted chondrogenesis and repressed adipogenesis in vitro, we co-transduced rBMSC with Bac-aS-iP/Bac-Cre (designated as CRISPRai group). We also mock-transduced rBMSC as the control (Mock group).
qRT-PCR analysis confirmed that CRISPRai upregulated both Acan (1.9-fold, Figure 3A) and Col2a1 (122-fold, Figure 3B) and significantly downregulated Col10a1 (Figure 3C) after 14 days. The Alcian blue staining performed after 21 days illustrated more evident sulfated GAG accumulation in the CRISPRai group than in the Mock group (Figure 3D). After 7 days culture in adipoinductive medium, conversely, CRISPRai significantly inhibited the expression of FABP4 and C/EBPα in the mRNA and protein levels, as unveiled by qRT-PCR (Figure 3E and F), Western blot (Figure 3G) and ensuing quantitative analysis (Figure 3H and I). In accord, Oil Red O staining (Figure 3J) and quantitative analysis (Figure 3K) attested that CRISPRai abolished the accumulation of oil droplet. Figure 3 confirms that CRISPRai-mediated Sox9 activation/PPAR-γ inhibition enhanced rBMSC chondrogenesis and inhibited adipogenesis.
Figure 3.

Sox9/PPAR-γ regulation by CRISPRai enhanced chondrogenesis and inhibited adipogenesis. (A–C) qRT-PCR analysis of Acan, Col2a1 and Co10a1. (D) Alcian blue staining. (E, F) qRT-PCR analysis of Fabp4 and C/ebpα. (G–I) Western blot and ensuing semi-quantitative analysis of FABP4 and C/EBPα. (J, K) Oil Red O staining and subsequent quantitative analysis. rBMSC in six-well plates were mock-transduced (Mock group) or co-transduced with Bac-aS-iP and Bac-Cre at MOI 300/100 (CRISPRai group). The cells were cultured in chondroinductive medium for 14 days for qRT-PCR (A–C) or for 21 days for Alcian blue staining (D). For adipogenesis, the cells were cultured in adipoinductive medium for 7 days for qRT-PCR (E, F) and Western blot (G–I) or for 14 days for Oil Red O staining (J, K). The data represent means ± SD of three independent culture experiments. ****P < 0.0001. ** P < 0.01. *P < 0.05.
Sox9/PPAR-γ regulation by CRISPRai promoted the formation of engineered cartilage
Chondroinduction of BMSC cultured in 3D facilitates accumulation of cartilage-specific extracellular matrix (ECM) and formation of engineered articular cartilage (35). To assess the formation of engineered cartilage, we seeded the mock-transduced (Mock group) or transduced cells (CRISPRai group) into chondroconductive gelatin scaffold, and cultured the cell/scaffold constructs in chondroinductive medium. At 8 dpt, the CRISPRai constructs deposited apparently more glassy ECM-like materials on the surface than the Mock constructs (Figure 4A). In accord, the CRISPRai group accumulated more ECM and cartilage-specific GAG in the CRISPRai group (Figure 4B and C). Quantitative GAG analysis (Figure 4D) and Col II-specific ELISA (Figure 4E) further attested that the CRISPRai group deposited more GAG and Col II than the Mock group. These data demonstrated that CRISPRai-mediated Sox9 activation/PPAR-γ inhibition promoted the formation of engineered articular cartilage in 3D culture.
Figure 4.

Sox9/PPAR-γ regulation by CRISPRai promoted the formation of engineered cartilage. (A) Appearance of rBMSC/scaffold construct. (B) H&E staining. (C) Safranin O staining. (D) GAG content. (E) Col II content. rBMSC cultured in 15-cm dish were mock-transduced (Mock group) or co-transduced (CRISPRai group) as in Figure 3, seeded into porous gelatin scaffold (diameter = 6 mm) at 1 dpt and cultured in 12-well plates using chondroinductive medium. The constructs were photographed at 2 and 8 dpt. The constructs were harvested at 8 dpt and sectioned for H&E staining (n = 3) or Safranin O staining (n = 3). Triangles indicate scaffold material. Arrows indicate ECM. Alternatively, the constructs were harvested at 8 dpt for analysis of GAG (n = 3) and Coll II (n = 3) contents. ****P < 0.0001. ***P < 0.001.
CRISPRai-mediated gene regulation promoted calvarial bone regeneration in vivo
To evaluate whether CRISPRai-stimulated chondrogenesis augmented calvarial bone formation, we seeded the Bac-aS-iP/Bac-Cre-transduced (CRISPRai group, n = 6) or mock-transduced (Mock group, n = 6) rBMSC into gelatin scaffolds as in Figure 4, and implanted the cell/scaffold constructs into the critical-size (6 mm in diameter) calvarial bone defects in rats. The frontal (Figure 5A), sagittal (Figure 5B) and top (Figure 5C) views of μCT imaging demonstrated negligible bone growth and bridging in the Mock group at weeks 4 (W4) and 8 (W8), highlighting the difficulty to repair critical-size calvarial defects simply using rBMSC. Nonetheless, the CRISPRai group evidently ameliorated bone growth, extension and bridging (Figure 5A and B), which enabled the formation of a large bone island from the periphery at W4 and bone ingrowth at W8 (Figure 5C). Quantitative analyses using the μCT images showed that at W8 the Mock group only filled ≈1.9% of the original defect area (Figure 5D), ≈1.0% of the volume (Figure 5E), and the density (Figure 5F) was ≈0.3% that of the original defect. In contrast, at W8 the CRISPRai constructs significantly improved the calvarial bone repair, filling ≈28.6% of defect area and ≈12.3% of defect volume, with the nascent bone density reaching ≈14.3% that of the original defect.
Figure 5.

In vivo Bone healing evaluated by μCT. The frontal (A), sagittal (B) and top (C) views of μCT imaging. (D–F) Quantitative analysis of bone area, volume and density. We seeded the Bac-aS-iP/Bac-Cre-transduced (CRISPRai group, n = 6) or mock-transduced (Mock group, n = 6) rBMSC into gelatin scaffolds as in Figure 4, and implanted the cell/scaffold constructs into the critical-size (6 mm in diameter) calvarial bone defects in rats. At weeks 4 and 8, the rats were subjected to μCT scanning to obtain the nascent bone area, volume and density. The percentages of healing, as calculated by dividing the nascent bone area (or volume, density) within the defect by those in the original defect, are shown above the bars (D–F). ****P < 0.0001. *** P < 0.001. ** P < 0.01. *P < 0.05.
H&E staining of bone specimens harvested at W8 illustrated that the defect in the Mock group was primarily filled with soft fibrous tissue with abundant fibroblasts (FB, Figure 6A). Besides fibrous tissue, the CRISPRai group also contained nascent bone (NB), abundant osteoblasts (OB) lining the bone matrix surface and blood vessel (BV)-like structures occupied by nucleus-lacking eosin-stained cells, suggesting vascularization within the new bone tissue. Furthermore, osteocalcin (OCN) is a bone formation marker while bone sialoprotein (BSP) is an indicator of intermediate bone formation process wherein osteoblast differentiation ceases and matrix mineralization begins (1). The immunostaining (Figure 6B and C) revealed accumulation of more BSP and OCN in the CRISPRai group than in the Mock group, confirming the improved formation of mineralized bone.
Figure 6.
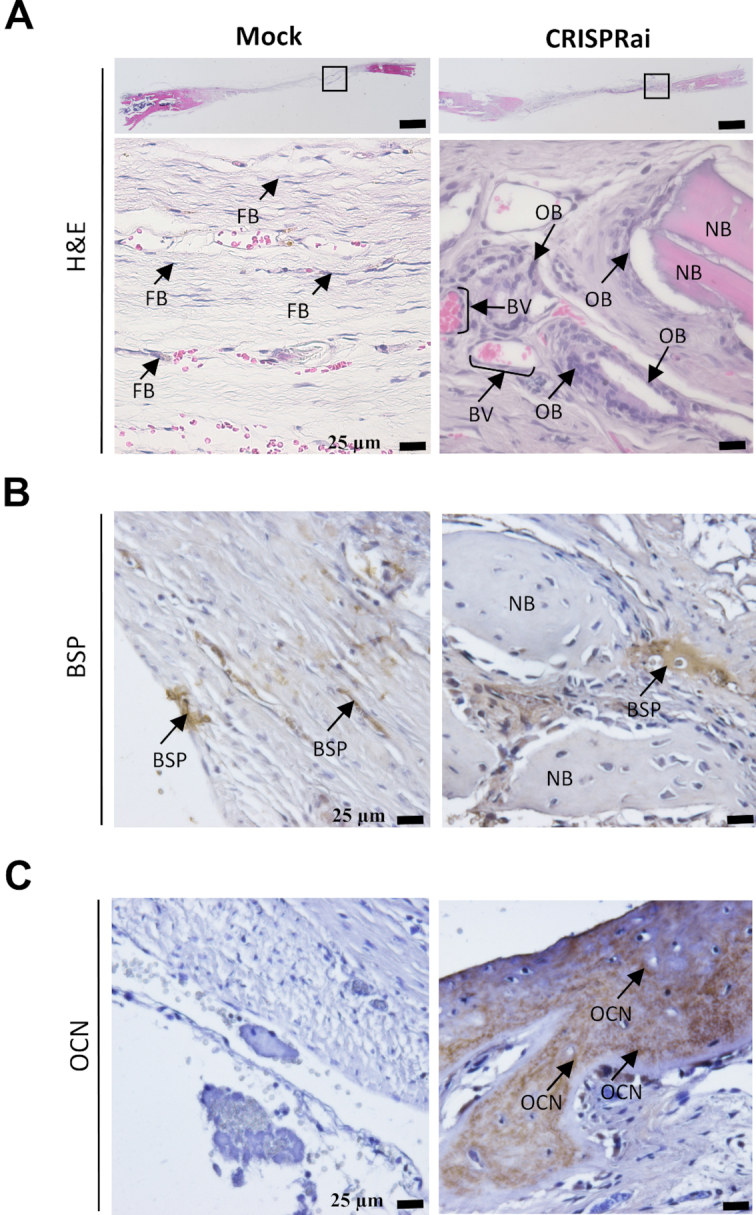
Bone healing evaluated by histology. After μCT imaging, the defect specimens were harvested at week 8, fixed, and decalcified for paraffin-embedded sectioning. The sections (10 μm) were subjected to (A) H&E staining (n = 5) or immunohistochemical staining for (B) BSP (n = 5) and (C) OCN (n = 5). FB, fibroblast. OB, osteoblast. NB, nascent bone. BV, blood vessel.
DISCUSSION
To date, CRISPRa has been harnessed to induce neuronal differentiation of embryonic stem cells (15), mouse embryonic fibroblasts (MEF) (21) or induced pluripotent stem cells (iPSC) (12,36), reprogram MEF (23) or fibroblast (37) to iPSC, as well as induce MSC differentiation into adipogenic lineage (38). CRISPRi has also been exploited for gene knockdown in pluripotent stem cells to regulate cell fate (22) or induce spatiotemporal mosaic self-patterning (22). These studies focus on in vitro cell fate reprogramming, but do not demonstrate in vivo applications. Only recently was CRISPRa system used to treat mouse models of muscular dystrophy (39) and transfected into the rat corneal endothelium to promote wound healing (40). Meanwhile, CRISPR-mediated simultaneous gene activation/repression is only implemented to redirect metabolic network in yeast (14) or dissect genetic interactions in mammalian cells (41), but has yet to be translated to in vivo tissue regeneration in animal studies.
In this study, we developed the CRISPRai system (Figure 1) by adopting the scaffold RNA concept proposed by Qi and co-workers for simultaneous gene activation and repression (14). Using this design, the sgRNA can encode information for target gene recognition and for recruiting a specific repressor or activator protein. As such, we designed the sgRNAi and Com-KRAB for gene repression as described (14). Notably, instead of using MCP-VP64 fusion developed by Qi et al. for gene activation (14), we exploited the SAM system (the sgRNAa and MPH complex) developed by Zhang and colleagues (13) due to its proven robustness in gene activation (42) and applications in genetic screens (17) and induced cell differentiation (36). However, the SAM system involves the expression of dCas9-VP64 as the regulator, which may hinder gene repression in the CRISPRai system because VP64 is a transcription activator. To maintain the modularity of CRISPRai, we chose to express dCas9 as the master regulator. The CRISPRai design allowed us to concurrently activate one gene with dCas9/sgRNAa/MPH while repress another gene with dCas9/sgRNAi/Com-KRAB without mutual crosstalk (Figure 1).
Besides, CRISPRa/CRISPRi systems are commonly delivered using plasmids (13,43), lentivirus (10,38,44) or adeno-associated virus (AAV) (39). For instance, CRISPRa is delivered using lentivirus for activation of endogenous adipogenic genes and induction of BMSC differentiation into adipocyte-like cells (38). CRISPRa is also carried using AAV to activate utrophin to ameliorate muscular dystrophy symptoms in mouse models (39). Conversely, CRISPRi is delivered using AAV for in vivo gene repression and treatment of retinitis pigmentosa (45). However, transfection of plasmids into stem cells is notoriously inefficient, while both AAV and lentivirus have a limited packaging capacity (≈4.7–5 kb for AAV and ≈8 kb for lentivirus), rendering it difficult to carry the entire CRISPRa/CRISPRi system (e.g. dCas9, MPH and Com-KRAB) in a single vector. Consequently, the dCas9 regulator, effector and sgRNA are often split into separate viral vectors (21,38,39) and concurrent delivery of all CRISPRa/CRISPRi components to the same cells is necessary, and challenging (46). Furthermore, lentivirus integrates the gene cassettes into chromosome, which is undesired from the perspective of regenerative medicine.
Here, we employed the baculovirus vector for CRISPRai delivery and ex vivo genetic modification of stem cells. In contrast to aforementioned vectors, baculovirus has a packaging capacity of at least 38 kb (47) and can accommodate all essential elements of the CRISPRai system, including the CRISPRai module and sgRNA array (≈12.6 kb), into a single vector (e.g. Bac-aS-iP, Figure 2A). Baculovirus infects insects in nature and is non-pathogenic to humans, but can transduce various stem cells at exceedingly high transduction efficiencies (e.g. >95% for BMSC (28) and ASC (48)). Bac-aS-iP alone is able to concurrently and robustly activate Sox9 and suppress PPAR-γ expression in rBMSC (Figure 2B and C). Within the transduced cells, baculovirus genome exists as an episome (49) and degrades with time (28), hence the transgene expression is transient (30). By using a Cre/loxP-based hybrid baculovirus system (Bac-aS-iP/Bac-Cre), we were able to potentiate and prolong the gene activation/repression effect (Figure 2D) in rBMSC, stimulate chondrogenesis and repress adipogenesis of transduced rBMSC in 2D culture (Figure 3) as well as promote the formation of engineered cartilage in 3D culture (Figure 4). Implantation of rBMSC engineered with the CRISPRai system significantly improved the calvarial bone healing (Figures 5 and 6) by improving the in vivo chondrogenesis (Supplementary Figure S2). These data not only echo our previous finding that augmenting the chondrogenesis of ASC improves calvarial bone healing after implantation (4), but also demonstrate for the first time that CRISPRa and CRISPRi can be combined to stimulate tissue regeneration. In comparison with tissue engineering approaches that typically promote regeneration by overexpressing a growth factor (or cocktail) such as BMP-6 (3), BMP-2 (50) or TGF-β (35) and only allows one direction of regulation, the CRISPRai system delivered by baculovirus enables bidirectional regulation and potentially allows for multiplexing activation/repression at the same time in the same cell, hence providing a more flexible and effective tool than recombinant DNA transfer for tissue regeneration.
Note, however, that we previously unveiled that baculovirus-engineered stem cells elicit anti-transgene immune responses in vivo after implantation into bone defects, although the responses were mild and did not compromise the bone healing (51). Conversely, Cas9 protein can elicit immune responses (52) and pre-existing adaptive immune responses against Cas9 was recently uncovered in humans (53). Furthermore, the sgRNA itself may trigger innate immune responses in human and murine cells (54). As such, it is not surprising that implantation of rBMSC expressing the CRISPRai system elicits immune responses against the CRISPRai system or baculovirus. Whether the immune responses are potent enough to compromise the bone healing or provoke other side effects remains to be investigated.
Aside from the application in tissue regeneration, our CRISPRai system may be readily adapted to other applications. First, CRISPRai can be used to govern the cell fate of other stem cells by simultaneously promoting one lineage while attenuating the other. For instance, iPSC can differentiate into cells belonging to 3 germ layers: endoderm (e.g. hepatocytes), mesoderm (e.g. osteoblast) and ectoderm (e.g. neural cells). To yield neuronal cells from iPSC, one may use the CRISPRai system to stimulate neural differentiation by activating genes encoding neural transcription factors (e.g. Ngn2) while suppressing genes promoting mesodermal (e.g. bmp4) or endodermal (e.g. activin A) differentiation. Second, CRISPR-mediated genome editing efficiency can be improved by enhancing homology direct repair (HDR) while inhibiting non-homologous end joining (NHEJ) (55). Recent evidence also unveils that Cas9-induced DNA double strand break triggers P53-dependent cytotoxicity, hence impairing genome editing efficiency of iPSC (56). It is thus tempting to exploit the CRISPRai system to upregulate HDR pathway genes (e.g. Rad51) and concurrently downregulate p53 and NHEJ pathway genes (e.g. Ku80) to improve the genome editing efficiency. Third, CRISPR-mediated knockout and CRISPRa-mediated gene activation were recently coupled for interrogation of pairwise gene interactions (57). However, CRISPR-mediated knockout may trigger P53-dependent cell death, leading to biased data interpretation. Using a pool of sgRNAa and sgRNAi libraries, CRISPRai may be repurposed to decipher the crosstalk between gene pairs at the genome scale while avoiding the problem caused by CRISPR-mediated knockout.
Fourth, CRISPRai may be harnessed for cell engineering to enhance product yield/productivity. For instance, CHO cell is commonly used for biopharmaceutics production and cell line optimization requires an orchestrated increase in the expression of enzymes that convert precursors into the desired product, and simultaneous repression of enzymes that divert these precursors toward byproducts. Our CRISPRai system may be used to develop superior producer CHO cell line by activating target pathway genes while concurrently repressing genes that divert precursors/intermediates to competing pathways. Fifth, synthetic circuits based on CRISPRa and CRISPRi have been devised, in which orthogonal dCas9 derived from different species are fused with VPR or KRAB for activation/suppression (58). Such system allows for the construction of AND, OR, NAND, and NOR dCas9 logic operators (58). With the ability to simultaneously turn ON/OFF genes, our CRISPRai system may be adapted for conditional gene activation/repression and be used to build logic gates to control cell fate or classify cell type (59,60).
Note, however, that the current system did not repress PPAR-γ in rASC as efficiently as in rBMSC (Figure 2C), probably because PPAR-γ is a master transcription factor driving adipogenesis and its expression in rASC was too high to be knocked down using Com-KRAB and sgRNAi. The inefficient repression may be attributed to sgRNAi which was designed by fusing the Com binding aptamer at the 3′ end of the sgRNA scaffold. Despite being functional, this design appears to be inferior to the sgRNAa design (13) whereby the protein binding aptamer is appended to the sgRNA tetraloop and stem loop 2. As such, the repression may be improved by moving the Com-binding aptamer to the sgRNA tetraloop/stem loop 2. Moreover, fusion of DNA methyltransferase such as DNMT3a to dCas9 induces target DNA methylation and gene repression (61). As such, DNMT3a may be fused to Com to replace KRAB or fused to Com-KRAB to form a tripartite repressor complex (Com-KRAB-DNMT3a) to synergize the inhibitory effect. Very recently, it was also shown that fusion of transcription repression domain of MeCP2 to dCas9-KRAB significantly enhances the repression effect (62). The repression efficiency of the CRISPRai system may be improved by fusing KRAB-MeCP2 to Com to form a tripartite repressor complex (Com-KRAB-MeCP2).
In conclusion, we developed a CRISPRai system for programmable, simultaneous activation and repression of reporter and endogenous genes. Using the all-in-one baculovirus vector for efficient delivery into rBMSC, the CRISPRai system was able to concurrently activate Sox9 and repress PPAR-γ. In conjunction with the hybrid Cre/loxP-based baculovirus, the CRISPRai system augmented chondrogenesis and repressed adipogenesis in 2D culture, promoted the formation of engineered cartilage in 3D culture and improved the healing of calvarial bone healing in vivo. This study paves a new avenue to translate the CRISPRai technology to bone tissue engineering and regenerative medicine.
Supplementary Material
ACKNOWLEDGEMENTS
Authors contributions: V.A.T. conducted the experiments and wrote the paper. M.N.H. conducted the experiments. N.T.K.N. conducted experiments. M.W.L. conducted the experiments. C.C.S. conducted the experiments. Y.C.H. designed the project and wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Science and Technology [MOST 105-2923-E-007-002-MY3, 106-2622-E-007-014-CC1, 107-3017-F-007-002, 107-2622-E-007-003-CC1, 107-2221-E-007-046-MY3, 108-3017-F-007-003]; Frontier Research Center on Fundamental and Applied Sciences of Matters; Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education [MOE 108QR001I5, 107QR001I5, 107Q2529E1], Taiwan. Funding for open access charge: Ministry of Science and Technology [MOST 105-2923-E-007-002-MY3, 106-2622-E-007-014-CC1, 107-3017-F-007-002, 107-2622-E-007-003-CC1, 107-2221-E-007-046-MY3, 108-3017-F-007-003]; Frontier Research Center on Fundamental and Applied Sciences of Matters; Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education [MOE108QR001I5, 107QR001I5, 107Q2529E1], Taiwan.
Conflict of interest statement. None declared.
REFERENCES
- 1. Santo V.E., Gomes M.E., Mano J.F., Reis R.L.. Controlled release strategies for bone, cartilage, and osteochondral engineering-part I: recapitulation of native tissue healing and variables for the design of delivery systems. Tissue Eng. Part B Rev. 2013; 19:308–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Szpalski C., Barr J., Wetterau M., Saadeh P.B., Warren S.M.. Cranial bone defects: current and future strategies. Neurosurg. Focus. 2010; 29:E8. [DOI] [PubMed] [Google Scholar]
- 3. Bez M., Sheyn D., Tawackoli W., Avalos P., Shapiro G., Giaconi J.C., Da X., David S.B., Gavrity J., Awad H.A. et al.. In situ bone tissue engineering via ultrasound-mediated gene delivery to endogenous progenitor cells in mini-pigs. Sci. Transl. Med. 2017; 9:eaal3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin C.-Y., Chang Y.-H., Li K.-C., Lu C.-H., Sung L.-Y., Yeh C.-L., Lin K.-J., Huang S.-F., Yen T.-C., Hu Y.-C.. The use of ASCs engineered to express BMP2 or TGF-β3 within scaffold constructs to promote calvarial bone repair. Biomaterials. 2013; 34:9401–9412. [DOI] [PubMed] [Google Scholar]
- 5. Ren X., Zheng D., Guo F., Liu J., Zhang B., Li H., Tian W.. PPARγ suppressed Wnt/β-catenin signaling pathway and its downstream effector SOX9 expression in gastric cancer cells. Med. Oncol. 2015; 32:91. [DOI] [PubMed] [Google Scholar]
- 6. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013; 152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. et al.. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C. et al.. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014; 159:647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hilton I.B., D’Ippolito A.M., Vockley C.M., Thakore P.I., Crawford G.E., Reddy T.E., Gersbach C.A.. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015; 33:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., P R Iyer E., Lin S., Kiani S., Guzman C.D., Wiegand D.J. et al.. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015; 12:326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H. et al.. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015; 517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zalatan J.G., Lee M.E., Almeida R., Gilbert L.A., Whitehead E.H., La Russa M., Tsai J.C., Weissman J.S., Dueber J.E., Qi L.S. et al.. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015; 160:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y., Yu C., Daley T.P., Wang F., Cao W.S., Bhate S., Lin X., Still C. 2nd, Liu H., Zhao D. et al.. CRISPR activation screens systematically identify factors that drive neuronal fate and reprogramming. Cell Stem Cell. 2018; 23:758–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bester A.C., Lee J.D., Chavez A., Lee Y.R., Nachmani D., Vora S., Victor J., Sauvageau M., Monteleone E., Rinn J.L. et al.. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell. 2018; 173:649–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joung J., Engreitz J.M., Konermann S., Abudayyeh O.O., Verdine V.K., Aguet F., Gootenberg J.S., Sanjana N.E., Wright J.B., Fulco C.P. et al.. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017; 548:343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mandegar M.A., Huebsch N., Frolov E.B., Shin E., Truong A., Olvera M.P., Chan A.H., Miyaoka Y., Holmes K., Spencer C.I. et al.. CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell. 2016; 18:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Du D., Roguev A., Gordon D.E., Chen M., Chen S.-H., Shales M., Shen J.P., Ideker T., Mali P., Qi L.S. et al.. Genetic interaction mapping in mammalian cells using CRISPR interference. Nat. Methods. 2017; 14:577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Y.C., Han J.H., Chen Z.C., Wu H.W., Dong H.S., Nie G.H.. Engineering cell signaling using tunable CRISPR-Cpf1-based transcription factors. Nat. Commun. 2017; 8:2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Black J.B., Adler A.F., Wang H.-G., D’Ippolito A.M., Hutchinson H.A., Reddy T.E., Pitt G.S., Leong K.W., Gersbach C.A.. Targeted epigenetic remodeling of endogenous loci by CRISPR/Cas9-based transcriptional activators directly converts fibroblasts to neuronal cells. Cell Stem Cell. 2016; 19:406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Libby A.R., Joy D.A., So P.L., Mandegar M.A., Muncie J.M., Mendoza-Camacho F.N., Weaver V.M., Conklin B.R., McDevitt T.C.. Spatiotemporal mosaic self-patterning of pluripotent stem cells using CRISPR interference. Elife. 2018; 7:e36045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu P., Chen M., Liu Y., Qi L.S., Ding S.. CRISPR-based chromatin remodeling of the endogenous Oct4 or Sox2 locus enables reprogramming to pluripotency. Cell Stem Cell. 2018; 22:252–261. [DOI] [PubMed] [Google Scholar]
- 24. Li K.-C., Chang Y.-H., Yeh C.-L., Hu Y.-C.. Healing of osteoporotic bone defects by baculovirus-engineered bone marrow-derived MSCs expressing microRNA sponges. Biomaterials. 2016; 74:155–166. [DOI] [PubMed] [Google Scholar]
- 25. Lo S.-C., Li K.-C., Chang Y.-H., Hsu M.-N., Sung L.-Y., Vu T.A., Hu Y.-C.. Enhanced critical-size calvarial bone healing by ASCs engineered with Cre/loxP-based hybrid baculovirus. Biomaterials. 2017; 124:1–11. [DOI] [PubMed] [Google Scholar]
- 26. Shen C.-C., Sung L.-Y., Lin S.-Y., Lin M.-W., Hu Y.-C.. Enhancing protein production yield from CHO Cells by CRISPR interference (CRISPRi). ACS Synth. Biol. 2017; 6:1509–1519. [DOI] [PubMed] [Google Scholar]
- 27. Sung L.-Y., Chen C.-L., Lin S.-Y., Li K.-C., Yeh C.-L., Chen G.-Y., Lin C.-Y., Hu Y.-C.. Efficient gene delivery into cell lines and stem cells using baculovirus. Nat. Protoc. 2014; 9:1882–1899. [DOI] [PubMed] [Google Scholar]
- 28. Chen C.-Y., Wu H.-H., Chen C.-P., Chern S.-R., Hwang S.-M., Huang S.-F., Lo W.-H., Chen G.-Y., Hu Y.-C.. Biosafety assessment of human mesenchymal stem cells engineered by hybrid baculovirus vectors. Mol. Pharmaceut. 2011; 8:1505–1514. [DOI] [PubMed] [Google Scholar]
- 29. Airenne K.J., Hu Y.-C., Kost T.A., Smith R.H., Kotin R.M., Ono C., Matsuura Y., Wang S., Yla-Herttuala S.. Baculovirus: an insect-derived vector for diverse gene transfer applications. Mol. Ther. 2013; 21:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen H.-C., Sung L.-Y., Lo W.-H., Chuang C.-K., Wang Y.-H., Lin J.-L., Hu Y.-C.. Combination of baculovirus-mediated BMP-2 expression and rotating-shaft bioreactor culture synergistically enhances cartilage formation. Gene Ther. 2008; 15:309–317. [DOI] [PubMed] [Google Scholar]
- 31. Sung L.-Y., Chen C.-L., Lin S.-Y., Hwang S.-M., Lu C.-H., Li K.-C., Lan A.-S., Hu Y.-C.. Enhanced and prolonged baculovirus-mediated expression by incorporating recombinase system and in cis elements: a comparative study. Nucleic Acids Res. 2013; 41:e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsu M.-N., Liao H.-T., Li K.-C., Chen H.-H., Yen T.-C., Makarevich P., Parfyonova Y., Hu Y.-C.. Adipose-derived stem cell sheets functionalized by hybrid baculovirus for prolonged GDNF expression and improved nerve regeneration. Biomaterials. 2017; 140:189–200. [DOI] [PubMed] [Google Scholar]
- 33. Huey D.J., Hu J.C., Athanasiou K.A.. Unlike bone, cartilage regeneration remains elusive. Science. 2012; 338:917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ahfeldt T., Schinzel R.T., Lee Y.-K., Hendrickson D., Kaplan A., Lum D.H., Camahort R., Xia F., Shay J., Rhee E.P. et al.. Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 2012; 14:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu C.-H., Yeh T.-S., Yeh C.-L., Fang Y.-H.D., Sung L.-Y., Lin S.-Y., Yen T.-C., Chang Y.-H., Hu Y.-C.. Regenerating cartilages by engineered ASCs: Prolonged TGF-β3/BMP-6 expression improved articular cartilage formation and restored zonal structure. Mol. Ther. 2014; 22:186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li S.L., Zhang A.Q., Xue H.P., Li D.L., Liu Y.. One-step piggyBac transposon-based CRISPR/Cas9 activation of multiple genes. Mol. Ther.-Nucleic Acids. 2017; 8:64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weltner J., Balboa D., Katayama S., Bespalov M., Krjutškov K., Jouhilahti E.-M., Trokovic R., Kere J., Otonkoski T.. Human pluripotent reprogramming with CRISPR activators. Nat. Commun. 2018; 9:2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Furuhata Y., Nihongaki Y., Sato M., Yoshimoto K.. Control of adipogenic differentiation in mesenchymal stem cells via endogenous gene activation using CRISPR-Cas9. ACS Synth. Biol. 2017; 6:2191–2197. [DOI] [PubMed] [Google Scholar]
- 39. Liao H.-K., Hatanaka F., Araoka T., Reddy P., Wu M.-Z., Sui Y., Yamauchi T., Sakurai M., O’Keefe D.D., Núñez-Delicado E. et al.. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell. 2017; 171:1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang Y.K., Hwang J.S., Chung T.Y., Shin Y.J.. SOX2 activation using CRISPR/dCas9 promotes wound healing in corneal endothelial cells. Stem Cells. 2018; 36:1851–1862. [DOI] [PubMed] [Google Scholar]
- 41. Jost M., Chen Y.W., Gilbert L.A., Horlbeck M.A., Krenning L., Menchon G., Rai A., Cho M.Y., Stern J.J., Prota A.E. et al.. Combined CRISPRi/a-based chemical genetic screens reveal that Rigosertib is a microtubule-destabilizing agent. Mol. Cell. 2017; 68:210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chavez A., Tuttle M., Pruitt B.W., Ewen-Campen B., Chari R., Ter-Ovanesyan D., Haque S.J., Cecchi R.J., Kowal E.J.K., Buchthal J. et al.. Comparison of Cas9 activators in multiple species. Nat. Methods. 2016; 13:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bao Z.H., Jain S., Jaroenpuntaruk V., Zhao H.M.. Orthogonal genetic regulation in human cells using chemically induced CRISPR/Cas9 activators. ACS Synth. Biol. 2017; 6:686–693. [DOI] [PubMed] [Google Scholar]
- 44. Chakraborty S., Ji H., Kabadi A.M., Gersbach C.A., Christoforou N., Leong K.W.. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Rep. 2014; 3:940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moreno A.M., Fu X., Zhu J., Katrekar D., Shih Y.-R.V., Marlett J., Cabotaje J., Tat J., Naughton J., Lisowski L. et al.. In situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation. Mol. Ther. 2018; 26:1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Komor A.C., Badran A.H., Liu D.R.. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017; 168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheshenko N., Krougliak N., Eisensmith R.C., Krougliak V.A.. A novel system for the production of fully deleted adenovirus vectors that does not require helper adenovirus. Gene Ther. 2001; 8:846–854. [DOI] [PubMed] [Google Scholar]
- 48. Lin C.-Y., Chang Y.-H., Kao C.-Y., Lu C.-H., Sung L.-Y., Yen T.-C., Lin K.-J., Hu Y.-C.. Augmented healing of critical-size calvarial defects by baculovirus-engineered MSCs that persistently express growth factors. Biomaterials. 2012; 33:3682–3692. [DOI] [PubMed] [Google Scholar]
- 49. Luo W.-Y., Lin S.-Y., Lo K.-W., Hung C.-L., Chen C.-Y., Chang C.-C., Hu Y.-C.. Adaptive immune responses elicited by baculovirus and impacts on subsequent transgene expression in vivo. J. Virol. 2013; 87:4965–4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen H.-C., Lee H.-P., Sung M.-L., Liao C.-J., Hu Y.-C.. A novel rotating-shaft bioreactor for two-phase cultivation of tissue-engineered cartilage. Biotechnol. Prog. 2004; 20:1802–1809. [DOI] [PubMed] [Google Scholar]
- 51. Lin C.-Y., Lin K.-J., Li K.-C., Sung L.-Y., Hsueh S., Lu C.-H., Chen G.-Y., Chen C.-L., Huang S.-F., Yen T.-C. et al.. Immune responses during healing of massive segmental femoral bone defects mediated by hybrid baculovirus-engineered ASCs. Biomaterials. 2012; 33:7422–7434. [DOI] [PubMed] [Google Scholar]
- 52. Chew W.L., Tabebordbar M., Cheng J.K.W., Mali P., Wu E.Y., Ng A.H.M., Zhu K., Wagers A.J., Church G.M.. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods. 2016; 13:868–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. et al.. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019; 25:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim S., Koo T., Jee H.-G., Cho H.-Y., Lee G., Lim D.-G., Shin H.S., Kim J.-S.. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 2018; 28:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ye L., Wang C., Hong L., Sun N., Chen D., Chen S., Han F.. Programmable DNA repair with CRISPRa/i enhanced homology-directed repair efficiency with a single Cas9. Cell Discov. 2018; 4:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C. et al.. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018; 24:939–946. [DOI] [PubMed] [Google Scholar]
- 57. Najm F.J., Strand C., Donovan K.F., Hegde M., Sanson K.R., Vaimberg E.W., Sullender M.E., Hartenian E., Kalani Z., Fusi N. et al.. Orthologous CRISPR–Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 2018; 36:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao Y., Xiong X., Wong S., Charles E.J., Lim W.A., Qi L.S.. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods. 2016; 13:1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lin M.-W., Tseng Y.-W., Shen C.-C., Hsu M.-N., Hwu J.-R., Chang C.-W., Yeh C.-J., Chou M.-Y., Wu J.-C., Hu Y.-C.. Synthetic switch-based baculovirus for transgene expression control and selective killing of hepatocellular carcinoma cells. Nucleic Acids Res. 2018; 46:e93–e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wroblewska L., Kitada T., Endo K., Siciliano V., Stillo B., Saito H., Weiss R.. Mammalian synthetic circuits with RNA binding proteins for RNA-only delivery. Nat. Biotechnol. 2015; 33:839–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vojta A., Dobrinic P., Tadic V., Bockor L., Korac P., Julg B., Klasic M., Zoldos V.. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016; 44:5615–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yeo N.C., Chavez A., Lance-Byrne A., Chan Y., Menn D., Milanova D., Kuo C.C., Guo X., Sharma S., Tung A. et al.. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods. 2018; 15:611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.