


Abstract
Proliferating cell nuclear antigen (PCNA) is a sliding clamp that acts as a central co-ordinator for mismatch repair (MMR) as well as DNA replication. Loss of Elg1, the major subunit of the PCNA unloader complex, causes over-accumulation of PCNA on DNA and also increases mutation rate, but it has been unclear if the two effects are linked. Here we show that timely removal of PCNA from DNA by the Elg1 complex is important to prevent mutations. Although premature unloading of PCNA generally increases mutation rate, the mutator phenotype of elg1Δ is attenuated by PCNA mutants PCNA-R14E and PCNA-D150E that spontaneously fall off DNA. In contrast, the elg1Δ mutator phenotype is exacerbated by PCNA mutants that accumulate on DNA due to enhanced electrostatic PCNA–DNA interactions. Epistasis analysis suggests that PCNA over-accumulation on DNA interferes with both MMR and MMR-independent process(es). In elg1Δ, over-retained PCNA hyper-recruits the Msh2–Msh6 mismatch recognition complex through its PCNA-interacting peptide motif, causing accumulation of MMR intermediates. Our results suggest that PCNA retention controlled by the Elg1 complex is critical for efficient MMR: PCNA needs to be on DNA long enough to enable MMR, but if it is retained too long it interferes with downstream repair steps.
INTRODUCTION
Maintenance of genome stability requires accurate replication of DNA coupled with constant surveillance by the repair machinery. Proliferating cell nuclear antigen (PCNA) acts as a central co-ordinator for DNA repair as well as DNA replication (1). PCNA is important for mismatch repair (MMR) and required at multiple steps during the MMR process (2–5). However, how PCNA regulation affects MMR is not fully understood. In particular, it is unknown if PCNA residence time on DNA is important for MMR.
PCNA is a ring-shaped homotrimeric complex that encircles DNA to act as a sliding clamp, ensuring processivity of DNA polymerases. It also operates as a platform for recruitment of numerous other proteins involved in DNA replication, DNA damage repair, mismatch repair, and chromatin structure and assembly (1). PCNA levels on DNA are regulated by loading and unloading. During DNA replication, the hetero-pentameric Replication Factor C (RFC) complex, composed of largest subunit Rfc1 and smaller subunits Rfc2, 3, 4 and 5, loads PCNA at primer-template junctions as synthesis of each Okazaki fragment initiates (6–8). After completion of each Okazaki fragment, the Elg1 RFC-like complex (Elg1-RLC), which comprises the Elg1 subunit associated with the Rfc2–5 subunits, functions to unload PCNA from the lagging strand (9–12). In the absence of Elg1, PCNA over-accumulates on recently replicated chromatin in the wake of replication forks (11). The role of the Elg1-RLC in PCNA unloading appears to be conserved in humans, since ATAD5 (the mammalian ortholog of Elg1) is required for proper removal of PCNA from chromatin in human cell lines (13,14).
Elg1 is critical for genome maintenance. In budding yeast, loss of the ELG1 gene causes gross chromosomal rearrangements, increased sister chromatid recombination, defective sister chromatid cohesion, derailed telomere length maintenance and sensitivity to the DNA alkylating drug methyl methanesulfonate (MMS) (15–21). The requirement for Elg1 in genome maintenance is conserved in higher eukaryotes, since mice with reduced expression of ATAD5 exhibit genome instability and have a high tumor incidence (22). In humans, somatic mutations in ATAD5 have been found in primary endometrial tumors, and ATAD5 was identified as a susceptibility locus for invasive epithelial ovarian cancer (22,23). PCNA over-accumulation on DNA is the main cause of genome instability observed in yeast cells lacking Elg1 (24,25). In addition to the defects described above, yeast cells lacking Elg1 exhibit increased mutation rates (26), suggesting that Elg1 is required for suppressing spontaneous mutations. However, it is unknown if the increase in mutation rate in elg1Δ is the consequence of over-accumulation of PCNA on DNA, and whether it arises from defective MMR.
The MMR pathway corrects mispaired bases resulting from replication errors (2–5). Defects in MMR cause increased mutation rates and result in the development of different cancers including Lynch syndrome (27). In eukaryotic MMR, mispaired bases are recognized by two partially redundant heterodimers of MutS-related proteins, Msh2–Msh6 and Msh2–Msh3 (2–5). The Msh2–Msh6 complex primarily recognizes base-base mispairs and small insertion/deletion mispairs, whereas the Msh2–Msh3 complex tends to recognize larger insertions/deletions as well as some single base mispairs (28). After binding to a mismatch and adenosine triphosphate, these MutS homologs are converted into closed clamp forms that trap DNA and can slide along it (29,30). The MutS homolog then recruits a MutL endonuclease homolog complex (Mlh1–Pms1 or Mlh1–Mlh3 in Saccharomyces cerevisiae), targeting repair to the newly synthesized DNA strand along with accessory factors including PCNA, RFC, DNA polymerase delta, RPA and exonuclease 1 (Exo1) (2–4,31–33). Two different pathways are proposed to act at the MMR excision step—an ‘Exo1-dependent’ pathway where Exo1 removes mispairs, or an ‘Exo1-independent’ pathway where Mlh1-Pms1 endonuclease cleaves multiple times to remove mispairs (34,35).
PCNA is involved in multiple steps of MMR. Msh3 and Msh6 interact with PCNA through PCNA-interacting peptide (PIP) motifs (31,36–37). Yeast cells expressing Msh3 and Msh6 mutants that lack PIP motifs exhibit substantial reduction of the MMR activity, indicating that the PIP motif plays an important, although not completely essential, role in MMR (31,37). It has been proposed that the PIP motif of Msh6 tethers Msh2–Msh6 to PCNA on replication forks and is important for replication machinery-coupled mispair recognition (38,39). Msh2–Msh6 also inhibits PCNA unloading through its PIP motif, maintaining the post-replicative temporal window for MMR (40). PCNA retained or loaded on DNA induces activation of the human MutL endonuclease homolog in a strand specific, mismatch- and MutS homolog-dependent manner (41). The orientation of PCNA on DNA determines strand specificity of MMR, directing removal of the mispaired bases specifically from the newly synthesized DNA strand (40,41). Many PCNA mutants in yeast exhibit increased mutation rates in the absence of Exo1, suggesting a central role for PCNA in the Exo1-independent MMR pathway (35). These findings suggest that PCNA coordinates multiple reactions in MMR, but it is not known how PCNA over-retention affects MMR and whether timely PCNA removal is important.
Here, we demonstrate that yeast cells lacking Elg1 exhibit increased mutation rate caused by PCNA over-retention on DNA after DNA replication. In general, premature unloading of PCNA causes increased mutation rates, but dissociation-prone PCNA mutants PCNA-R14E and PCNA-D150E can attenuate the elg1Δ mutator phenotype. In contrast, the elg1Δ mutator phenotype is exacerbated by retention-prone PCNA mutants that we identify in this study. Epistasis analysis suggests that PCNA over-accumulation interferes with both MMR and MMR-independent process(es). We focused on the effect of PCNA accumulation on MMR, and found that PCNA over-retained behind replication forks over-recruits Msh2–Msh6 to chromatin through the Msh6 PIP-box motif. PCNA over-retained on DNA prevents Msh2–Msh6- and, to a lesser extent, Msh2–Msh3-dependent MMR, and leads to accumulation of MMR intermediates. Our results suggest that control of PCNA retention time by the Elg1 complex is critical for efficient mismatch repair.
MATERIALS AND METHODS
Overexpression dominant negative screening
A total of 61 missense mutations were introduced into the POL30 gene, which was present on a high-copy plasmid and under the control of the constitutive ADH promoter (Supplementary Table S1). Individual residues were selected for mutagenesis based on sequence conservation, and focused mainly on charged amino acids since those amino acids were more likely to mediate protein–protein interactions (42). The plasmids for overexpression of PCNA mutants were transformed into wild-type strain or an elg1Δ mutant strain, which retain the wild-type copy of POL30 in the genome. Whole cell extracts (WCE) were prepared from exponentially growing cultures, and PCNA and its SUMOylated forms were detected by Western blot using anti-PCNA antibody.
Yeast strains
Saccharomyces cerevisiae strains used are listed in Supplementary Table S2. Epitope tagging and gene disruption were carried out using standard polymerase chain reaction (PCR)-based gene-insertion methods (43). PCNA point mutants were constructed either by replacing wild-type PCNA with mutant PCNA (24) or by using a CRISPR-Cas9 genome editing system (44). The PIP mutants of MSH3 and MSH6, and a chimera msh6(3MBD) were constructed by the CRISPR-Cas9 system. Plasmids and oligonucleotides used for CRISPR-Cas9 genome editing are listed in Supplementary Tables S1 and S3, respectively. For over-expression of Msh2, Msh3 and Msh6, the yeast strains RDKY5964 and CJY1 were transformed with multicopy plasmids carrying Msh2, Msh3 or Msh6 (45). For microscopy studies, the C-terminus of Pms1 was tagged with four tandem copies of green fluorescent protein (GFP) amplified from the pSM1023 plasmid.
Preparation of whole-cell extracts and chromatin-enriched fractions and western blotting
WCE and chromatin-enriched fractions were prepared as described previously (11). Western blotting and quantification were performed as described previously (46). Antibodies used were mouse monoclonal anti-PCNA (ab70472, Abcam), rabbit polyclonal anti-histone H3 (ab46765, Abcam), mouse monoclonal anti-Flag M2 (F1804, Sigma), mouse monoclonal anti-HA (HA.11 clone 16B12, Covance) and Rat monoclonal anti-GFP (3H9, #029762, Chromotek).
Fluctuation analysis of mutation rates
Mutation rates were determined using the lys2–10A and hom3–10 frameshift reversion and the CAN1 inactivation assays, by fluctuation analysis (28,47) and the Ma-Sandri-Sarkar (MSS) maximum-likelihood method (48,49). Each mutation rate was determined using at least 11 independent cultures. Similarly sized colonies grown 2 days at 30°C on YPD plates were transferred to 2–5 ml of liquid YPD and further incubated overnight. The cells were plated on YPD to count viable cells after appropriate dilution, and SD-Lys or SD-Thr to measure the lys2–10A reversion and the hom3–10 reversion, respectively. For the CAN1 inactivation assay, the cells were plated on SD-Arg to count the viable colonies after appropriate dilution and SD-Arg + 60 μg/ml canavanine to measure the inactivation of the CAN1 gene. Colonies were counted after 3 days. In the lys2–10A reversion assay for cells over-expressing Msh2, Msh3, and Msh6, SD-Ura-Trp-Leu plates and liquid medium and SD-Ura-Trp-Leu-Lys plates were used. Since cells over-expressing Msh2, Msh3 and Msh6 grow slowly, those cells were incubated for 3 days on the plates and for 2 days in the liquid medium. The 95% confidence intervals were calculated as described previously (48). Comparisons of mutation rates were evaluated using 95% confidence intervals.
Purification, crystallization and structure determination of PCNA-D17K and PCNA-D21K
PCNA point mutations D17K and D21K were introduced by site-directed mutagenesis (50) in a modified pET-28 vector that contains an N-terminal 6-His tag and a PreScission Protease™ cleavage site. PCNA mutants D17K and D21K were expressed in Escherichia coli and purified as previously described (51). For crystallization, 0.9 μl of protein (between 15 and 20 mg/ml) was mixed with an equal volume of well solution in 24-well hanging drop trays at room temperature. The well solutions used were 50 mM sodium citrate pH 5.3 and 1.7 M (NH4)2SO4 for PCNA-D17K, and 50 mM sodium citrate pH 5.6, 2 M (NH4)2SO4 for PCNA-D21K. Crystals appeared within 1 day and continued to grow for 5 days. Crystals were briefly swiped through Paratone N cryo-protectant and frozen at 100K in the cryostream. Diffraction data were collected on a Rigaku system with a Saturn 944 CCD detector. Indexing, integration and scaling was performed with HKL3000 (52). The structure was solved by molecular replacement using PHASER (53) with wild-type PCNA as the search model (PDB ID: 4YHR) (54). Refinement and model building were carried out using phenix.refine (55) and Coot (56). The atomic coordinates of PCNA-D17K and PCNA-D21K were deposited to the protein databank (PDB ID: 6D0R and 6D0Q, respectively). The recent cocrystal of human PCNA bound to a duplex of DNA (PDB ID: 5L7C) (57) was used to model the presence of DNA in the PCNA ring. The wild-type yeast PCNA structure (PDB ID: 4YHR) was superimposed onto human PCNA. The structural model of yeast PCNA bound to duplex DNA (51) was not used for two reasons: (i) the DNA duplex is poorly resolved in the crystal structure, and (ii) the DNA is tilted at too sharp of an angle for proper PCNA sliding.
Chromatin immunoprecipitation (ChIP)-qPCR
To synchronize replication fork movements between cells, cells were released from a cdc7–1 block into S phase at 16°C and collected 15 min after release, as performed previously (11). Chromatin immunoprecipitation (ChIP) of Msh6–6HA or PCNA-3myc was performed as described (58) using mouse monoclonal anti-HA (HA.11 clone 16B12, Covance) or mouse monoclonal anti-myc (M047–3, MBL) antibody, respectively. ChIP and corresponding input samples were analyzed by LightCycler 480 II (Roche) using Light cycler SYBR Green master reagent (Roche). ChIP efficiency at each locus was calculated as the mean of three technical replicates.
Cell imaging
Exponentially growing cells were washed and immobilized on Concanavalin A (C-2631, Sigma) coated μ-Slide (chambered coverslip) with 8 wells (80826, ibidi). The cells are covered with Synthetic complete media and imaged on a Deltavision (Applied Precision) microscope with 100 × 1.35NA objective. Sixteen 0.3 μm z-sections were acquired and image processing and foci count were performed using imageJ.
RESULTS
Increased mutation rate in elg1Δ is dependent on PCNA accumulation on DNA
PCNA accumulation on DNA in elg1Δ causes genome instability, including increased recombination of sister chromatids and direct repeats, and elongation of telomeres (24,25). It has been suggested that loss of ELG1 also results in elevated mutation rate (26). We first tested if PCNA accumulation on DNA causes an elevated mutation rate in elg1Δ. To analyze mutation rate, we measured the reversion of +1 frameshift mutations in the LYS2 (lys2–10A) and the HOM3 genes (hom3–10) (28,47). Consistent with a previous report (26), loss of ELG1 resulted in elevated reversion rates in the Lys+ and Hom+ assays (Figure 1A; Supplementary Figure S1A and Table S4). The lys2–10A allele is characterized by a 67-bp ‘InsE’ insertion containing a homonucleotide run of 10 adenine nucleotides at the position 3015 nt (Supplementary Figure S1B). Sequencing the LYS2 gene of the revertants arising in wild-type and elg1Δ confirmed that all reversion events tested involved the deletion of one nucleotide, mainly a single A in the run of 10 As, to restore the correct open reading frame (Supplementary Figure S1B). Reversion was not due to pop-out recombination between 6-bp short repeats that flank the InsE element. To test if the increase in reversion rate in elg1Δ is dependent on PCNA accumulation on DNA, we utilized the disassembly-prone PCNA mutant pol30-D150E (Figure 1A and B). This mutation disrupts the trimer interface, decreasing PCNA trimer stability in vitro (35), and preventing PCNA accumulation on chromatin in vivo even in an elg1Δ mutant (as demonstrated by the failure of PCNA-D150E to accumulate on chromatin in elg1Δ; (24,35). The pol30-D150E mutation itself caused an ∼9-fold increase in reversion rate (Figure 1A, column 3). In the disassembly prone mutant pol30-D150E background, loss of ELG1 did not increase the reversion rate compared to ELG1+ (Figure 1A, columns 3 and 4), but rather, the disassembly-prone mutant pol30-D150E moderately rescued the increased mutation rate observed in elg1Δ (Figure 1A, columns 2 and 4). These results support the idea that the increased mutation rate in elg1Δ is dependent on PCNA accumulation on DNA.
Figure 1.

Increased mutation rate in elg1Δ is dependent on over-accumulation of PCNA, not SUMOylated PCNA, on DNA after DNA replication. (A) Mutation rate analysis was performed to measure the reversion rate of lys2–10A to LYS+ phenotype. Fold increases over wild-type are shown above the histogram. Error bars, 95% confidence intervals. (B) Structure of the PCNA trimer (PDB ID: 3K4X) (51). The positions mutated are highlighted. (C) The inner ring surface PCNA mutants (K13E, R14E, K20E, C22Y and K217E) and the trimer instability PCNA mutants (L68S and C81R) do not accumulate on chromatin in elg1Δ. WCE and chromatin-enriched fractions (Chromatin) were prepared from cells expressing PCNA mutants in log phase. The PCNA mutants are the only copy of PCNA in these cells. PCNA and histone H3 (loading control) were detected by Western blotting. (D) Structure of the PCNA trimer (PDB ID: 3K4X) (51). The positions mutated are highlighted. (E) Mutation rates of ELG1+ and elg1Δ in the wild-type and dissociation-prone PCNA mutants backgrounds at the lys2–10A locus. Fold increases over wild-type are shown above. Error bars, 95% confidence intervals. *, no overlapping of error bars; ns, overlapping of error bars. Mutation rates for ELG1+ and elg1Δ in wild-type PCNA background are as shown in panel A, repeated for ease of reference here and in subsequent Figures 1F, 2D and 3B. (F) Structure of the PCNA trimer (PDB ID: 3K4X) (51). The positions mutated are highlighted. Mutation rate of ELG1+ and elg1Δ in the wild-type and unSUMOylatable PCNA mutants backgrounds at the lys2–10A locus. Fold increase over wild-type is shown above the histogram. Error bars, 95% confidence intervals. (G) Mutation rates of the cell-cycle-regulated alleles of ELG1 (24) at the lys2–10A locus. Fold increases over ELG1+ are shown above. Error bars, 95% confidence intervals.
To see a clearer rescue in reversion rate in elg1Δ by disassembly-prone mutants, we sought to identify PCNA mutants that fall off DNA spontaneously but do not show a severe mutator phenotype. To this end, we performed a biochemical version of a previously developed protocol that employed overexpression dominant negative (ODN) screening to identify separation-of-function mutations in telomerase subunits (42,59). Briefly, a set of 61 PCNA point mutants (see ‘Materials and Methods’ section) was over-expressed from a multi-copy plasmid in the presence of endogenous PCNA, and chromatin-bound PCNA was assessed by examining the amount of SUMOylated PCNA present in WCE by western blotting. SUMO-PCNA provides a useful proxy for chromatin-bound PCNA since SUMOylation is DNA dependent (9,60). In the elg1Δ background, most over-expressed wild-type PCNA and PCNA mutants were SUMOylated efficiently, but 16 PCNA mutants (K13E, R14E, K20E, C22Y/E, L68S/E, K77E, C81R, R110E, E113K, K117E, E143K, K146E, D150K and K217E) exhibited reduced amounts of their SUMOylated forms in WCE (Supplementary Figure S1C), consistent with the possibility that these mutants fall off DNA spontaneously. It is likely that PCNA with mutations near the trimer interface (K77E, C81R, R110E, E113K, K117E, E143K, K146E and D150K) are disassembly-prone. To examine further the phenotypes caused by the mutations in PCNA, we constructed strains expressing each PCNA mutant as the only copy of the POL30 gene, expressed from its endogenous locus. To test if the inner ring mutants are dissociation-prone, we constructed pol30-K13E, pol30-R14E, pol30-K20E, pol30-C22Y and pol30-K217E as well as two known trimer instability mutants pol30-L68S and pol30-C81R as controls. We found that all seven mutants we tested did exhibit a ‘dissociation-prone’ phenotype, as assessed by amounts of PCNA in chromatin fractions (Figure 1C lower panel and D). Screening among these mutants, most exhibited substantially elevated mutation rates (see the next section and Figure 1E), making them unsuitable for testing whether they could rescue the elevated mutation rate of elg1Δ. However the mutant pol30-R14E, where R14 locates on the inner ring surface (Figure 1B), by itself only slightly elevated mutation rate (Figure 1A, column 5). Combining pol30-R14E with elg1Δ largely rescued the elevated mutation rate of the single elg1Δ mutant (Figure 1A, column 2 and 6). These results strongly suggest that the increased mutation rate in elg1Δ is dependent on PCNA accumulation on DNA.
Premature unloading of PCNA from DNA causes an increase in mutation rate
In addition to the trimer instability mutants pol30-L68S and pol30-C81R, several PCNA mutants with changes on the inner ring (K13E, R14E, K20E, C22Y and K217E) showed a dissociation-prone phenotype (Figure 1C and D). However, at least three of them (K13E, C22Y and K217E) were previously shown to form stable trimers in vitro (35,61). The phenotypes are consistent with the idea that these inner ring mutants can form stable trimers when not DNA-associated, but cannot be retained on chromatin due to inner ring mutations that potentially disrupt interaction with DNA. As previously reported, the inner ring mutants pol30-K13E, pol30-C22Y and pol30-K217E as well as the trimer instability mutants pol30-L68S and pol30-C81R display a mutator phenotype (Figure 1E) (35). All dissociation-prone mutants tested here, including new mutants pol30-R14E and pol30-K20E, exhibit an elevated mutation rate, suggesting that premature unloading of PCNA from DNA contributes to an increase in mutation rate. In these dissociation-prone mutant backgrounds, deletion of ELG1 did not cause any further increase in mutation rate (Figure 1A and E), consistent with the idea that the elevated mutation rate observed in elg1Δ is dependent on PCNA accumulation on DNA.
SUMOylation of PCNA is not required for increased mutation rates in elg1Δ
PCNA accumulated on DNA in elg1Δ becomes hyper-SUMOylated (Figure 1C, lane 2) (9). We tested if SUMOylation of PCNA contributes to the increased mutation rate in elg1Δ. In the unSUMOylatable pol30-KK127,164RR mutant background, deletion of ELG1 still increased the mutation rate (Figure 1F). Because this unSUMOylatable PCNA mutant accumulates on DNA in elg1Δ (9), these results suggest that accumulation of unmodified PCNA on DNA is sufficient to increase the mutation rate in elg1Δ. These results also suggest that the increased mutation rate in elg1Δ at the lys2–10A locus is not due to error-prone DNA polymerases, which are recruited to DNA via PCNA that is ubiquitinated at K164.
Timely unloading of PCNA by Elg1-RLC during S phase prevents increase in mutation rate
In the absence of Elg1, PCNA accumulates on DNA in the wake of replication forks during S phase and is retained on DNA even in G2/M phase (11,24). Using cell-cycle-regulated alleles of ELG1 (24), we tested in which cell-cycle phase PCNA accumulation on DNA causes increased mutation rate. We observe that Elg1 expressed in S phase (S-ELG1) almost completely rescues the mutator phenotype of elg1Δ (Figure 1G). Elg1 expressed in G2/M phase (G2/M-ELG1) exhibits a partial rescue phenotype, but Elg1 expressed in G1 phase (G1-ELG1) does not. These results suggest that PCNA retention on DNA after DNA replication increases the mutation rate, and timely unloading of PCNA by Elg1-RLC during S phase is important for limiting mutations.
Identification of new PCNA mutants that are retained on DNA and increase the mutation rate
To show that PCNA accumulation on DNA, and not a different effect of elg1Δ, is the cause of the increase in mutation rate, we used ODN screening to identify PCNA mutants that are retained on DNA even in the presence of Elg1. Briefly, we selected as candidates mutants that exhibit increased amounts of their SUMOylated forms in WCE when over-expressed, even in the presence of Elg1 (Supplementary Figure S2A and B). We then constructed strains expressing each PCNA mutant as the only copy of the POL30 gene, expressed from its endogenous locus, and found two PCNA mutants PCNA-D17K and PCNA-D21K that accumulate on DNA even with Elg1 present (Figure 2A–C). PCNA-D21K is unloaded from DNA slowly compared to WT PCNA, and is retained on DNA during S phase and even in G2/M phase (Supplementary Figure S2C). Elg1 can physically interact with PCNA-D17K and actually shows enhanced interaction with PCNA-D21K (Supplementary Figure S2D), suggesting that accumulation of those PCNA mutants on DNA is not due to a loss of interaction with the Elg1-RLC unloader. These retention-prone mutants increase the mutation rate even in the presence of Elg1 (Figure 2D, columns 3 and 5), suggesting that PCNA accumulation on DNA alone and/or the D17K and D21K mutations themselves can increase the mutation rate. Deletion of ELG1 causes further accumulation of those mutants on DNA (Figure 2B and C). In contrast to the dissociation-prone mutant backgrounds, deletion of ELG1 further increases the mutation rate in those retention-prone mutant backgrounds (Figure 2D, columns 4 and 6). These results, together with those in Figure 1, suggest that PCNA over-accumulation on DNA, not loss of other functions of Elg1, increases the rate of mutation.
Figure 2.

Identification of new PCNA mutants that are retained on DNA due to enhanced electrostatic interaction and cause increase in mutation rate. (A) Structure of the PCNA trimer (PDB ID: 3K4X) (51). The positions mutated are highlighted. (B) The inner ring surface PCNA mutants (D17K and D21K) accumulate on chromatin. WCE and chromatin-enriched fractions (Chromatin) were prepared from cells expressing PCNA mutants in log phase. The PCNA mutants are the only copy of PCNA in these cells. PCNA and histone H3 (loading control) were detected by Western blotting. (C) Quantification of chromatin-bound PCNA and PCNA mutants. Average of two experiments of total PCNA on chromatin (normalized to histone H3), relative to WT, is shown. Error bars, SDs. (D) Mutation rates of ELG1+ and elg1Δ in the wild-type and retention-prone PCNA mutants backgrounds at the lys2–10A locus. Fold increases over wild-type are shown above. Error bars, 95% confidence intervals. (E) 2.8 Å resolution crystal structure of PCNA-D21K (green, PDB ID: 6D0Q) compared with a hybrid model of WT yeast PCNA (purple, PDB ID: 4YHR) with DNA modeled based on a human PCNA:DNA cocrystal structure (PDB ID: 5L7C). Inset shows that lysine 21 could simultaneously interact with two DNA phosphate groups. (F) 2.85 Å crystal structure of PCNA-D17K (tan, PDB ID: 6D0R) compared with a hybrid model of WT yeast PCNA (purple, PDB ID: 4YHR) bound to DNA (PDB ID: 5L7C). Inset shows that lysine 17 is positioned within the major groove of DNA, but also induces a conformational change helix 1 and an adjacent loop.
Structure of the retention-prone PCNA mutants
To determine the molecular mechanism by which these PCNA variants retain their interaction with DNA and Elg1, we determined their structures using x-ray crystallography (Supplemental Table S5). The structures (at 2.80- and 2.85-Å resolution for PCNA-D21K and PCNA-D17K, respectively) reveal that the mutations alter the inner pore of the PCNA ring (Figure 2E and F). In both cases, the PCNA pore becomes more electropositive due to the swap of a negative charge to a positive charge (Supplemental Figure S3). To examine whether the electrostatic changes might strengthen interaction with DNA, we modeled the presence of DNA within the yeast PCNA ring using the recent cocrystal structure of human PCNA bound to a short DNA duplex (57). The D21K mutation positions lysine 21 to interact favorably in a bidentate manner with two backbone phosphates of DNA (Figure 2E). For D17K, lysine 17 is positioned within the major groove of DNA (Figure 2F), where the electrostatic interaction with DNA is also enhanced. Thus, both mutants are expected to have enhanced interactions with DNA, although through slightly differing contacts. For the D21K variant, we observed no significant conformational changes relative to wild-type PCNA (Cα RMSD ∼0.48 Å), indicating that the binding of PCNA partner proteins should not be perturbed. The D17K mutation on the other hand disrupts a salt bridge network within the PCNA pore, resulting in partial unraveling of helix 1, and a conformational change of an adjacent loop (residues 20 through 25; Figure 2F). Because this conformational change occurs on the primary interaction face of PCNA, the D17K variant could potentially perturb binding of PCNA partner proteins. To avoid this complication, we focused our further analyses primarily on the D21K variant.
PCNA over-accumulation on DNA prevents Msh2–Msh6-dependent mismatch repair at the lys2-10A locus
We next investigated how PCNA over-accumulation on DNA increases the mutation rate. In particular, we tested whether PCNA over-accumulation on DNA prevents mismatch repair. To this end, we first measured mutation rates in the absence of the mismatch repair proteins Msh3 and Msh6. If PCNA accumulation on DNA increases mutation rate only by preventing mismatch repair, then deletion of ELG1 will not further increase mutation rate of an msh3Δ msh6Δ double mutant (which lacks both Msh2–Msh3 and Msh2–Msh6 mismatch recognition complexes). If PCNA accumulation on DNA leads to mutation through other mechanism(s) (e.g. by allowing polymerase slippage and/or preventing DNA polymerase proofreading), then deletion of ELG1 will synergistically increase the mutation rate with msh3Δ msh6Δ. An msh3Δ msh6Δ mutant exhibits an ∼5800-fold increase in mutation rate in the lys2–10A assay (Figure 3A). Deletion of ELG1 did not cause further increase in mutation rate in the msh3Δ msh6Δ mutant background, indicating that an elg1Δ mutant is epistatic with an msh3Δ msh6Δ mutant (Figure 3A). These results suggest that loss of Elg1 prevents MMR.
Figure 3.

Over-retained PCNA prevents MMR and also affects MMR-independent process(es). (A) Mutation rates of ELG1+ and elg1Δ in wild-type, msh3Δ and msh6Δ backgrounds at the lys2–10A locus. Fold increases over wild-type are shown above. The number in the brackets, fold changes over ELG1+ in each mutant background. Error bars, 95% confidence intervals. *, no overlapping of error bars; ns, overlapping of error bars. (B) Mutation rates of elg1Δ, pol30-D21K and pol30-M644G in MSH2+ and msh2Δ backgrounds at the lys2–10A locus. Fold increases over wild-type are shown above. The number in the brackets, fold changes over a single msh2Δ. Error bars, 95% confidence intervals. *, no overlapping of error bars; ns, overlapping of error bars. (C) Mutation rates of ELG1+ and elg1Δ in wild-type, msh2Δ, pol30-R14E and msh2Δ pol30-R14E backgrounds at the CAN1 locus. Fold increases over wild-type are shown above. The number in the brackets, fold changes over ELG1+ in each mutant background. Error bars, 95% confidence intervals. *, no overlapping of error bars; ns, overlapping of error bars.
We next tested whether deletion of ELG1 affects Msh2–Msh3 or Msh2–Msh6-mediated repair, or both. In the lys2–10A assay, deletion of ELG1 causes an ∼5.5-fold increase in mutation rate in the msh3Δ background where Msh6 is present, but only an ∼1.2-fold increase in the msh6Δ background where Msh3 is present (Figure 3A). In the hom3–10 assay, we observed similar effects (Supplementary Figure S4A). Our results indicate that PCNA accumulation on DNA prevents mainly the Msh2–Msh6-dependent mismatch repair pathway.
Similar to msh3Δ msh6Δ, combining elg1Δ with msh2Δ did not cause a synergistic increase in the mutation rate in the lys2–10A assay (Figure 3B). In contrast, the pol2-M644G mutation exhibits a synergistic effect when combined with msh2Δ (Figure 3B and Supplementary Figure S4B) (38). This observation confirms that high mutation rates are measurable using the lys2–10A assay, and moreover implies that loss of Elg1 increases mutation rates not by facilitating DNA replication errors (at least, not at the level of errors caused by pol2-M644G) but rather by preventing MMR. Consistent with this idea, no synergistic increase in mutation rate was observed on combining the retention-prone mutant pol30-D21K with msh2Δ (Figure 3B).
When testing mutation rates using the hom3–10 assay, loss of Elg1 did cause a slight but significant increase in mutation rate in the msh2Δ mutant background (Figure 3B; Supplementary Figure S4B and Table S4). This observation leaves open the possibility that loss of Elg1 prevents Msh2-dependent MMR, and simultaneously impacts an additional pathway.
PCNA over-accumulation on DNA increases mutation rate also through MMR-independent process(es)
We next tested if over-accumulated PCNA also affects MMR at the CAN1 locus. The CAN1 inactivation assay measures a broader spectrum of mutation types (including small and large insertion/deletion and translocation that cause loss of Can1 function) compared to the lys2–10A assay which detects single nucleotide deletion mainly within a run of A or T. Combining elg1Δ with msh2Δ did cause a synergistic increase in mutation rate in the CAN1 inactivation assay (Figure 3C), in contrast to our finding at the lys2–10A assay. Sequencing can1 from canavanine resistant mutants confirmed the majority of mutations to be single nucleotide deletions or substitutions, as opposed to large deletions or translocations (Supplementary Figure S5), despite the fact that a single elg1Δ mutant exhibits elevated recombination levels (15–17). At this locus deletion of ELG1 may therefore affect MMR-independent repair or stimulate nucleotide misincorporation to increase the mutation rate. In the dissociation-prone mutant pol30-R14E and the double msh2Δ pol30-R14E backgrounds, deletion of ELG1 did not increase the mutation rate in the CAN1 assay (Figure 3C), suggesting that PCNA accumulation on DNA in elg1Δ remains the cause of increased mutation rate, affecting MMR-independent process(es). Note that we do not know if MMR is also affected by PCNA accumulation on DNA in the CAN1 locus in our experimental condition.
Davidson et al. previously reported that the mutator phenotype of the elg1Δ single mutant and the elg1Δ pol30-flag strain observed at the CAN1 locus was attenuated by deletion of the transcriptional factor SWI4, which mediates expansion of dNTP pools (62). They proposed that increased dNTP levels are correlate with increased mutagenesis (62). We test if deletion of SWI4 also attenuates the mutator phenotype of the elg1Δ mutant observed in lys2–10A locus. In the swi4Δ background, deletion of ELG1 still increased the mutation rate at the lys2–10A locus, although deletion of SWI4 reduced the mutation rates both in ELG1+ and elg1Δ (Supplementary Figure S4C). This result suggests that while dNTP pools might affect the mutation rate at the lys2–10A, the increased mutation rate caused by deletion of ELG1 at the lys2–10A is not due to expansion of dNTP pools. In contrast to the lys2–10A locus, the increased mutation rate in elg1Δ at the CAN1 locus is suppressed by deleting SWI4 (62), consistent with the idea that the downstream effect of PCNA accumulation on DNA differs at the lys2–10A and the CAN1 loci. In this study we focus our further analyses on the effect of PCNA accumulation on MMR-dependent pathway at the lys2–10A locus.
Over-retained PCNA hyper-recruits the MutS homolog to chromatin through its PIP motif, affecting MMR
We next tested how PCNA over-accumulation on DNA perturbs Msh2-dependent mismatch repair. We envisaged three potential scenarios: (i) over-retained PCNA acts as obstacle and prevents Msh2–Msh6 from sliding along DNA as it scans for mismatches, (ii) over-retained PCNA causes inappropriate recruitment of the MutS homolog (via its PIP-box motif) to DNA where there is no mismatch or (iii) over-retained PCNA prevents mismatch repair indirectly through inappropriate nucleosome deposition by the PIP-box-containing histone chaperone CAF-1.
Single-molecule analysis using DNA curtains has shown that the Msh2–Msh3 complex can ‘hop over’ obstacles on DNA, while Msh2–Msh6 complex cannot (63). A Msh6 chimera with its mispair-binding domain (MBD) replaced by that of Msh3 hops over obstacles on DNA (63,64). However, in this chimera msh6(3MBD) background, we observe that deletion of ELG1 still caused an ∼8.5-fold increase in mutation rate (Figure 4A). This result makes it unlikely that prevention of Msh2–Msh6 sliding along DNA by accumulated PCNA is the primary cause of mutation rate increase in the elg1Δ mutant.
Figure 4.

Over-retained PCNA prevents MMR dependently on PIP motifs in the MutS homologs at the lys2–10A locus. (A) Schematic structure of a chimeric protein Msh6(3MBD) and mutation rate in the msh6(3MBD) background at the lys2–10A locus. Error bars, 95% confidence intervals. Mutation rates for ELG1+ and elg1Δ are as shown in Figure 3A, repeated for ease of reference here and in subsequent Figures 4B and 5I, (B) Mutation rates in the msh3PIP and msh6PIP mutants backgrounds at the lys2–10A locus. The mutation sites in the PIP motifs are shown. Error bars, 95% confidence intervals. *, no overlapping of error bars; ns, overlapping of error bars.
We then tested the second scenario that PCNA over-accumulated on DNA over-recruits the MutS homolog to DNA, affecting mismatch repair. To prevent over-recruitment of the MutS homologs to DNA via PCNA, we mutated the PIP-box motifs of Msh3 and Msh6. In the Msh3 Msh6 double PIP mutant (msh3PIP msh6PIP) background, deletion of ELG1 did not further increase the mutation rate (Figure 4B), suggesting that over-recruitment of the MutS homologs to DNA through PCNA contributes to the increased mutation rate in elg1Δ. Note that the mutation rate of the msh3PIP msh6PIP mutant is low enough to test an additive effect (not just a synergistic effect) with elg1Δ. In the single msh3PIP background where Msh6 can be over-recruited to DNA via PCNA, deletion of ELG1 caused a 9.8-fold increase in mutation rate, while the single msh6PIP background where Msh3 can be over-recruited to DNA caused a 5.7-fold increase (Figure 4B), further evidence that PCNA accumulation on DNA affects the Msh6-dependent pathway to a greater extent than Msh3-dependent pathway. The reason why elg1Δ is epistatic with msh6Δ (Figure 3A) but not with msh6PIP (Figure 4B) might be that Msh3 over-recruited by over-retained PCNA may interfere with the Msh6-dependent pathway where Msh6PIP can be still functional in MMR.
We then tested if Msh2–Msh6 is hyper-recruited to over-retained PCNA in elg1Δ cells. Western blot analysis of chromatin fractions suggested that Msh6 is over-recruited to chromatin in the elg1Δ mutant (Figure 5A and B) and the over-recruitment is dependent on the Msh6 PIP motif (Figure 5C and D). SILAC-based quantitative proteomic analysis has also shown over-recruitment of Msh2 and Msh6 to chromatin in elg1Δ in the presence of HU (Figure 5B) (46).
Figure 5.

Over-retained PCNA over-recruits Msh2–Msh6 to chromatin, affecting mismatch repair. (A) Msh6 accumulates on chromatin in elg1Δ, compared to ELG1+. WCE and chromatin-enriched fractions (Chromatin) were prepared from cells expressing Msh6–6HA in log phase. Msh6–6HA, PCNA and histone H3 (loading control) were detected by western blotting. (B) Quantification of chromatin-bound Msh6, normalized to histone H3 (average of two independent clones shown in panel A) and chromatin-bound Msh2 and Msh6 in cells in hydroxyurea by SILAC quantitative proteomics performed previously (46). Error bars, SDs. (C) Msh6PIP does not accumulate on chromatin in elg1Δ, compared to ELG1+. WCE and chromatin-enriched fractions (Chromatin) were prepared from msh6PIP msh3PIP mutants expressing Msh6PIP-6HA in log phase. Msh6PIP-6HA, PCNA and histone H3 (loading control) were detected by Western blotting. (D) Quantification of chromatin-bound Msh6PIP (normalized to histone H3) shown in panel C. Average of two independent clones is shown. Error bars, SDs. (E) ChIP-seq analysis of PCNA performed previously (11). PCNA distribution around ARS607 on chromosome VI is shown. PCNA is unloaded behind replication forks in ELG1+ but retained in elg1Δ in S phase (15 min after release from a cdc7–1 block at 16°C). Black square, region quantified by ChIP-qPCR in panels F and G. (F) ChIP-qPCR analysis of Msh6 and PCNA for early origin ARS607 in the presence and absence of Elg1. ChIP was performed using cells arrested in alpha-factor (G1) or collected 15 min after release from a cdc7–1 block into S phase at 16°C (S). Error bars, SDs of three technical replicates. (G) ChIP-qPCR analysis of Msh6 for ARS607 in the msh6PIP mutant or the disassembly-prone PCNA mutant pol30-D150E backgrounds. ChIP was performed using cells collected 15 min after release from a cdc7–1 block into S phase at 16°C. Three independent isolates of msh6PIP elg1Δ were shown. Error bars, SDs of three technical replicates. (H) Effect of over-expression of Msh2, Msh3 and Msh6 or their PIP mutants on mutation rate at the lys2–10A locus. Msh2, Msh3 and Msh6 or their PIP mutants are over-expressed under their own promoters from three multicopy plasmids. Error bars, 95% confidence intervals. (I) Mutation rates of ELG1+ and elg1Δ in wild-type and exo1Δ backgrounds at the lys2–10A locus. Error bars, 95% confidence intervals.
To confirm and further examine over-recruitment of Msh6 to over-retained PCNA in elg1Δ at a specific locus, we performed ChIP-qPCR. ChIP-seq analysis previously performed showed that PCNA is generally retained on chromatin in the wake of replication forks in elg1Δ (11), including over-retention near ARS607 15 min after release into S phase (Figure 5E), at which point PCNA has already been unloaded in WT cells. To synchronize replication fork movement between cells, cells were released from a cdc7–1 block into S phase (as performed previously; (11). Consistent with Figure 5E, ChIP-qPCR analysis showed that, in the absence of Elg1, PCNA is over-retained at ARS607 15 min after release into S phase (Figure 5F). We found that Msh6, like PCNA, accumulates at ARS607 15 min after release into S phase in elg1Δ (Figure 5F). We observed similar results at ARS305 (Supplementary Figure S6). Over-recruitment of Msh6 to ARS607 in elg1Δ largely depends on the Msh6 PIP motif, since signal is greatly reduced in the msh6PIP mutant background (Figure 5G). A residual level of Msh6PIP accumulation on DNA in elg1Δ suggests some degree of PIP-independence of Msh6 retention when PCNA is over-retained. The over-recruitment of Msh6 to ARS607 in elg1Δ is however completely dependent on PCNA over-retention, since in the disassembly-prone pol30-D150E background the Msh6 signal in elg1Δ is reduced to similar level as in ELG1+ (Figure 5G). Overall, these results indicate that in an elg1Δ mutant Msh6 is hyper-recruited to over-retained PCNA at replicated regions, via its PIP motif. Consistent with our results, knockdown of human Elg1 (ATAD5) also causes PCNA-dependent accumulation of Msh2 on chromatin (13).
Moreover, we observed that over-expression of Msh2, Msh3 and Msh6 from multicopy plasmids caused an increase in mutation rate (Figure 5H, columns 1 and 3) and deletion of ELG1 results in further increase in mutation rate (Figure 5H, columns 3 and 4), consistent with the idea that over-recruitment of the MutS homolog to chromatin is the cause of the increased mutation rate. Over-expression of Msh2 with Msh3PIP and Msh6PIP has less impact on mutation rates, compared to the corresponding wild-type proteins (Figure 5H, columns 3 and 5). The elg1Δ mutation did however increase mutation rate in the context of the PIP mutant protein over-expression (Figure 5H, columns 5 and 6). This increase presumably reflects our observation that the over-expressed PIP mutant proteins do show some accumulation on DNA in elg1Δ, as shown in ChIP-qPCR (Figure 5G).
We then tested the third possibility that histone deposition inappropriately competes with MMR through over-recruitment of the CAF-1 histone chaperone. CAF-1 also has a PIP-box motif and accumulates on chromatin in elg1Δ (46). However, we did not observe any rescue of the mutator phenotype of elg1Δ by deleting a PIP-box-containing CAF-1 subunit (Supplementary Figure S4D and E). Although we cannot entirely exclude the possibility that PCNA accumulation on DNA affects mismatch repair indirectly through other means, our results support the second scenario, in which PCNA over-retained on DNA after replication over-recruits the MutS homolog to DNA through its PIP motif, affecting efficiency of mismatch repair.
PCNA over-retained on DNA prevents Exo1-independent mismatch repair
After an initial endonuclease cleavage event by the MutL homolog, mispairs are removed by either Exo1-dependent or -independent pathways. PCNA has a central role in the Exo1-independent pathway probably through driving progressive excision by the PCNA-activated MutL homolog endonuclease (35). We tested if PCNA over-retained on DNA in elg1Δ prevents Exo1-independent MMR. Deletion of ELG1 causes further increase in mutation rate when combined with a deletion of EXO1 (Figure 5I), suggesting that PCNA over-retained on DNA in elg1Δ prevents Exo1-independent mismatch repair. Note that we do not know if PCNA over-retained on DNA also prevents Exo1-dependent mismatch repair.
PCNA over-retained on DNA leads to accumulation of MMR intermediates
To further test whether deletion of ELG1 affects MMR, we monitored the recruitment and residence of Mlh1-Pms1 endonuclease, which acts downstream of mismatch recognition by either Msh2–Msh3 or Msh2–Msh6. Cells lacking Elg1 had elevated, rather than reduced, levels of Pms1 foci (Figure 6A and B), indicating that PCNA over-retained on DNA does not prevent recruitment of Mlh1–Pms1. Moreover, we observed accumulation of Pms1 on chromatin in elg1Δ by western blot (Supplementary Figure S7). These results suggest that deletion of ELG1 causes accumulation of Mlh1–Pms1 on DNA.
Figure 6.
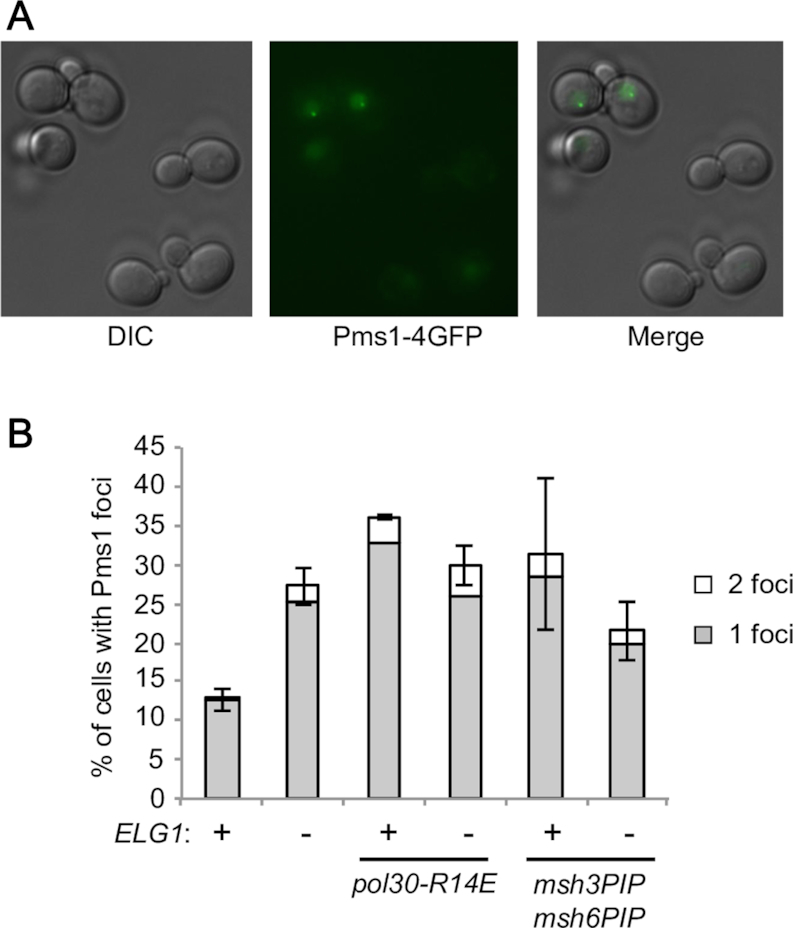
The elg1Δ mutant exhibits accumulation of Pms1 foci dependent on PCNA over-retention on DNA and PIP motifs of Msh3 and Msh6. (A) Representative image of Pms1-GFP foci. (B) Plot shows percentage of cells with Pms1 foci for ELG1+ and elg1Δ in the wild-type and indicated mutant backgrounds. The average value for two independent experiments is presented. Error bars, SDs.
We next tested if increase of Pms1 foci in elg1Δ is caused by PCNA over-retention on DNA. In the dissociation-prone mutant pol30-R14E background, deletion of ELG1 did not further elevate Pms1 foci (Figure 6B), suggesting that accumulation of Pms1 foci in elg1Δ is caused by PCNA over-retention. To test if the observed increase of Pms1 foci in elg1Δ is caused by over-recruitment of the MutS homologs to over-retained PCNA, we monitored Pms1 foci in msh3PIP msh6PIP. In the msh3PIP msh6PIP background, deletion of ELG1 does not change level of Pms1 foci significantly (Figure 6B). These results suggest that accumulation of Pms1 foci in elg1Δ is caused by over-recruitment of the MutS homologs via their PIP-box motifs to over-retained PCNA.
It has been previously suggested that Mlh1-Pms1 foci increase when Mlh1-Pms1 is retained longer on mispaired sites due to the defects in the MMR pathway downstream of Mlh1-Pms1 recruitment, and/or when there are more Mlh1-Pms1 recruitment events due to increase in the number of mispaired bases (38). We suspect that increased Pms1 foci in elg1Δ is caused by the defects in the MMR pathway downstream of Mlh1-Pms1 recruitment, rather than increase in the number of mispaired bases (if the latter is the case, we would see increase of Pms1 foci caused by deletion of ELG1 even in the PIP mutant background). We propose that PCNA accumulation on DNA in elg1Δ affects the MMR pathway downstream of Mlh1-Pms1 recruitment, causing increase of MMR intermediates.
Taken together, the results shown here suggest that PCNA over-retained behind replication forks in elg1Δ over-recruits the MutS homolog to DNA and affects steps downstream of Mlh1-Pms1 recruitment, leading to accumulation of MMR intermediates and a consequent increase in mutation rate (Figure 7).
Figure 7.

A model for the role of PCNA and Elg1-RLC in mismatch repair and the effect of derailed PCNA regulation on mismatch repair. (A) In wild-type, Elg1-RLC unloads free PCNA from DNA after completion of Okazaki fragment processing on the lagging strand, but does not unload PCNA bound to Msh2–Msh6. This will allow factors involved in mismatch repair to work around mispair sites. After removal of mispaired nucleotides by Mlh1-Pms1 and Exo1, Mlh1-Pms1 will dissociate from DNA. (B) In elg1Δ, PCNA over-accumulates on DNA, leading to over-recruitment of Msh2–Msh6 to chromatin, which in turn, causes accumulation of repair intermediate. PCNA over-accumulation on DNA also affects MMR-independent process(es) to increase mutation rates. (C) In disassembly/dissociation-prone PCNA mutants, PCNA will dissociate from DNA prematurely, causing slow or no activation of Mlh1-Pms1 by PCNA.
DISCUSSION
Cells lacking the PCNA unloader Elg1-RLC exhibit genome instability caused by PCNA over-retained on DNA (24). Cells lacking Elg1 also exhibit a mutator phenotype (26), but it was unknown how deletion of ELG1 leads to an increased mutation rate or whether PCNA retention time on DNA matters for MMR. In this study, by isolating and analysing PCNA mutants with particular properties, we found that PCNA over-retained on DNA after replication is the cause of the increased mutation rate in elg1Δ. Epistasis analysis suggest that PCNA accumulation on DNA interferes with both MMR and MMR-independent processes. We further analyzed the effect of PCNA accumulation on MMR, and found that PCNA over-retained on DNA in elg1Δ over-recruits the Msh2–Msh6 complex to DNA and leads to hyper-accumulation of MMR intermediate Mlh1-Pms1 foci. We propose that PCNA over-retained behind replication forks over-recruits the MutS homolog to DNA unnecessarily interfering with downstream steps of mismatch repair (Figure 7). Increased mutation rates in disassembly/dissociation-prone PCNA mutants observed in this study suggest that premature unloading of PCNA also results in increased mutation rates. Taken together, we conclude that proper retention of PCNA on DNA controlled by Elg1-RLC is critical for efficient MMR. PCNA has to be retained on DNA long enough to enable MMR, but if it is retained too long it interferes with downstream repair steps (Figure 7).
How does PCNA accumulation on DNA prevent mismatch repair? In wild-type, Elg1-RLC may unload free PCNA that is not associated with other proteins, so that Msh6-bound PCNA is not unloaded. Interestingly, exemption of Msh6-bound PCNA from unloading has also been proposed based on experiments in the Xenopus egg extract system (40) (Figure 7A). This temporary retention of PCNA that is interacting with Msh6 would allow MMR proteins to work properly around mispairs and remove them quickly. In elg1Δ, PCNA is over-retained on DNA due to lack of unloading by Elg1-RLC, where we propose it over-recruits PCNA binding proteins including Msh6 (Figure 7B). The over-retained PCNA with the MutS homolog stimulates hyper-accumulation of repair intermediate Mlh1-Pms1 foci, potentially deregulating downstream MMR steps. One possibility is that PCNA bound to the MutS homolog physically occupies the DNA around mispair sites, interfering with the normal rounds of nicking by Mlh1-Pms1 endonuclease (Figure 7B). In addition to the Exo1-independent pathway, PCNA over-retention on DNA might affect the Exo1-dependent pathway by inhibiting the action of Exo1 at steps downstream of Mlh1-Pms1 recruitment. Consistent with our model that hyper-recruitment of Msh2–Msh6 to PCNA is the cause of the increased mutation rate, a very recent study shows co-overexpression of Msh2 and Msh6 increases the mutation rate at lys2–14A in a PCNA interaction-dependent manner (as co-overexpression of Msh2 and the Msh6 PIP mutant does not confer a mutator phenotype) (65).
Over-retained PCNA may interfere with other PCNA-related DNA processes such as nucleosome deposition by histone chaperone CAF-1. CAF-1 has a PIP-box motif and accumulates on chromatin in elg1Δ (46). Recent work shows that PCNA accumulation causes over-recruitment of CAF-1 to chromatin in elg1Δ, leading to defective gene silencing (66). However, the mechanism that disrupts gene silencing differs from that involved in MMR. Janke et al. proposed that over-retained PCNA causes the CAF-1 pool to become sequestered from active replication forks in elg1Δ, because over-expression of CAF-1 rescues the defect in gene silencing in elg1Δ (66). Our study however shows that over-expression of the MutS homologs does not rescue the elg1Δ mutator phenotype, but rather increases mutation rates further (Figure 5H). This result suggests that sequestering the MutS homolog is not the cause of increased mutation rate in elg1Δ. Hence, over-retained PCNA disrupts both MMR and gene silencing, but through different mechanisms. It remains unknown how over-retained PCNA causes other DNA abnormalities in elg1Δ, e.g. increased recombination and telomere elongation.
Does PCNA over-retention affect Msh2-independent process(es) as well as Msh2-dependent MMR? In the msh2Δ background, deletion of ELG1 causes a slight, but significant, increase in mutation rate in the hom3–10 assays (although not a significant increase in the lys2–10A assays) (Figure 3B; Supplementary Figure S4B Table S4). This observation leaves open the possibility that PCNA over-retention prevents Msh2-independent mismatch removal, possibly by base excision repair and/or nucleotide excision repair. In human cells, knockdown of human Elg1 (ATAD5) causes chromatin accumulation of Msh2, together with other replication/repair proteins including FEN1, LIG1 and MRE11 (13), whose over-recruitment might interfere with removal of mispairs. Results of the CAN1 inactivation assay suggest that PCNA over-accumulation on DNA can increase mutation rate also through MMR-independent process(es) (Figure 3C and B). The observed differences between the lys2–10A and the CAN1 loci in the effects of PCNA over-accumulation probably reflect more frequent slippage events in a long homonucleotide run at the lys2–10A locus, where repair largely relies on MMR (47). The CAN1 inactivation assay in contrast detects many mutation types that can be caused by the defects in MMR-independent repair, such as single nucleotide deletions or base substitutions. These could reflect for example defects in base excision repair or nucleotide excision repair (67), which could potentially also be affected by PCNA over-accumulation. Alternatively, over-accumulation of ubiquitinated PCNA might over-recruit error-prone DNA polymerases to induce mispaired bases at the CAN1 locus. The mutation pattern (indel vs substitution) in the double elg1Δ msh2Δ mutant is not clearly different from that in the single msh2Δ mutant, but we observed a slight increase of substitution in the double elg1Δ msh2Δ mutant, compared to the single msh2Δ mutant (Supplementary Figure S5). Those substitution occurs mainly at G or C, suggesting that oxidative damage (8-oxo-dG) might be related to those substitutions. How PCNA accumulation on DNA in elg1Δ increases spontaneous mutation at the CAN1 locus remains to be investigated.
All the disassembly/dissociation-prone PCNA mutants tested in this study exhibited increased mutation rate, suggesting that proper retention of PCNA on DNA after DNA replication is important for mismatch repair. When PCNA dissociates from DNA prematurely, it will fail to stimulate MutL homolog endonuclease activity (Figure 7C). We, however, observed substantial variation in the rate of mutation among the dissociation-prone mutants. This may be because the mutants have different abilities to bind the MutS homolog or to stimulate the MutL homolog (35). In addition, the variation may reflect different duration times of dissociation-prone PCNA mutants on DNA, with the mutants that are retained slightly longer on DNA promoting mismatch repair to a greater extent than the mutants that quickly fall off DNA.
In this study, we identified various new dissociation-prone and retention-prone PCNA mutants, providing useful tools to investigate PCNA-related DNA transactions. Previously identified disassembly prone PCNA mutants have mutations at the trimer interface, causing trimer instability as shown by gel filtration analysis of PCNA-C81R and PCNA-E143K (35) and by native gel electrophoresis of PCNA-C81R, PCNA-E113G and PCNA-G178S (61,68). We performed ODN screening and isolated PCNA mutants that fall off DNA spontaneously in elg1Δ. As expected, we observed a disassembly-prone phenotype in PCNA mutants that have mutations on the trimer interface (K77E, C81R, R110E, E113K, K117E, K143K, K146E and D150K) (Figure 1 and Supplementary Figure S1C). Interestingly, we observed that mutations on the PCNA inner ring surface also cause a dissociation-prone phenotype (Figure 1 and Supplementary Figure S1C) although at least three of them have previously shown to form stable trimers in vitro (35,61). The new inner surface mutants have arginine/lysine to aspartic acid/glutamic acid substitutions: K13E, R14E, K20E and K217E. Crystal structures of yeast and human PCNA revealed that positively charged amino acids on the PCNA inner ring surface are in contact with DNA (51,57). Taken together, these results suggest that while inner surface mutants are able to form stable trimers, they are not well retained on DNA due to the mutations on the inner surface that disrupt electrostatic interactions with DNA. As C22Y does not convert a charged residue it is not in this category, but the crystal structure of PCNA-C22Y revealed that the C22Y substitution alters the alpha-helices that comprise the inner ring, altering the position of DNA-contacting residue K217 (61).
It has been reported that acetylation of lysine residues at the inner surface of PCNA is induced by DNA lesions, and the cohesion acetyltransferase Eco1 targets lysine 20 on the inner surface of the PCNA ring (69). Interestingly, the acetylation-mimicking mutant PCNA-K20Q shows a reduction of PCNA SUMOylated forms (69). This implies that PCNA-K20Q is a dissociation-prone mutant like PCNA-K20E shown in our study. There is the possibility that cells actively control PCNA retention on DNA by acetylating lysine residues at the inner surface of PCNA by Eco1 in response to DNA damage, which would be important for homologous recombination (69).
As well as dissociation-prone PCNA mutants, we identified new PCNA mutants that accumulate on DNA (Figure 2). Retention-prone mutants PCNA-D17K and PCNA-D21K have point mutations on the DNA-interacting region of inner ring. The aspartic acid to lysine substitutions in those PCNA mutants result in enhanced electrostatic interactions between PCNA and DNA, as illustrated by their crystal structures (Figure 2). Our structural analysis and modeling indicates that the D21K variant strengthens direct interactions with the DNA backbone, without a substantial perturbation of the PCNA structure. The PCNA-D21K mutant provides an excellent tool for testing how PCNA retention on DNA affects cellular function.
PCNA association with DNA needs to be spatially and temporally regulated to coordinate the action of many replisome-associated proteins and repair proteins. Our study shows that PCNA retention time controlled by Elg1-RLC is crucial for suppressing spontaneous mutations. Timely regulation of PCNA is important not only for mismatch repair but also other DNA transactions, including recombination, telomere maintenance and gene silencing. Elg1-RLC is the key factor for clearing up the workshop occupied by the functional ‘tool belt’ PCNA protein, and prolonged PCNA residence causes multiple problems in numerous DNA transactions. Both the unloading function of Elg1 and the MMR pathway appear to be conserved in mammals. Knockdown of human Elg1 (ATAD5) causes accumulation not just of PCNA but also the accumulation of other replication proteins on chromatin in a PCNA-dependent manner (13). Over-recruitment of PCNA-interacting proteins may disturb DNA replication and repair, potentially causing the defects observed in ATAD5 knockdown cells, which include chromosome instability and increased spontaneous DNA damage, as well as high tumor incidence in mice with reduced ATAD5 expression (14,22,70). Overexpression of PCNA, Msh2 and Msh6 has been observed in many cancers and is associated with deleterious outcomes and phenotypes (65). Since knockdown of ATAD5 also causes PCNA-dependent accumulation of Msh2 on chromatin (13), its function in MMR may well be conserved in mammals and important for preventing tumor development, a possibility for further investigation.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession number: 6D0R (PCNA-D17K) and 6D0Q (PCNA-D21K).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Richard Kolodner and Eric Alani for strains and plasmids. We thank Anne Donaldson, Alexander Lorenz and Catherine Johnson from University of Aberdeen for careful reading of the manuscript. We thank Annabelle Duff and Veronika Petrova for assisting with the mutation rate assays, and Duru Cosar for assisting with crystal structure analysis. We appreciate assistance from staff of the Microscopy and Histology Core Facility and the qPCR facility at the University of Aberdeen.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Medical Research Council (MRC) Career Development Fellowship [L019698/1 to T.K.]; American Cancer Society Research Scholar Award [Grant #440685 to B.A.K.]; National Institute of General Medical Sciences [R01 GM127776 to B.A.K.]; National Institutes of Health grant [R01 GM106060 to V.L.]. Swiss National Science Foundation Postdoc Mobility Fellowship (to C.G.). Funding for open access charge: Medical Research Council via University of Aberdeen Open Access Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Moldovan G.L., Pfander B., Jentsch S.. PCNA, the maestro of the replication fork. Cell. 2007; 129:665–679. [DOI] [PubMed] [Google Scholar]
- 2. Kunkel T.A., Erie D.A.. Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet. 2015; 49:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiricny J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iyer R.R., Pluciennik A., Burdett V., Modrich P.L.. DNA mismatch repair: functions and mechanisms. Chem. Rev. 2006; 106:302–323. [DOI] [PubMed] [Google Scholar]
- 5. Kolodner R.D., Marsischky G.T.. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999; 9:89–96. [DOI] [PubMed] [Google Scholar]
- 6. Bowman G.D., O’Donnell M., Kuriyan J.. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. 2004; 429:724–730. [DOI] [PubMed] [Google Scholar]
- 7. Gomes X.V., Burgers P.M.. ATP utilization by yeast replication factor C. I. ATP-mediated interaction with DNA and with proliferating cell nuclear antigen. J. Biol. Chem. 2001; 276:34768–34775. [DOI] [PubMed] [Google Scholar]
- 8. Kelch B.A., Makino D.L., O’Donnell M., Kuriyan J.. How a DNA polymerase clamp loader opens a sliding clamp. Science. 2011; 334:1675–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kubota T., Nishimura K., Kanemaki M.T., Donaldson A.D.. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell. 2013; 50:273–280. [DOI] [PubMed] [Google Scholar]
- 10. Kubota T., Myung K., Donaldson A.D.. Is PCNA unloading the central function of the Elg1/ATAD5 replication factor C-like complex. Cell Cycle. 2013; 12:2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kubota T., Katou Y., Nakato R., Shirahige K., Donaldson A.D.. Replication-Coupled PCNA unloading by the Elg1 complex occurs Genome-wide and requires okazaki fragment ligation. Cell Rep. 2015; 12:774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu C., Gan H., Han J., Zhou Z.-X., Jia S., Chabes A., Farrugia G., Ordog T., Zhang Z.. Strand-Specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol. Cell. 2014; 56:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee K.Y., Fu H., Aladjem M.I., Myung K.. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013; 200:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shiomi Y., Nishitani H.. Alternative replication factor C protein, Elg1, maintains chromosome stability by regulating PCNA levels on chromatin. Genes Cells. 2013; 18:946–959. [DOI] [PubMed] [Google Scholar]
- 15. Kanellis P., Agyei R., Durocher D.. Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr. Biol. 2003; 13:1583–1595. [DOI] [PubMed] [Google Scholar]
- 16. Bellaoui M., Chang M., Ou J., Xu H., Boone C., Brown G.W.. Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. EMBO J. 2003; 22:4304–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ben-Aroya S., Koren A., Liefshitz B., Steinlauf R., Kupiec M.. ELG1, a yeast gene required for genome stability, forms a complex related to replication factor C. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9906–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smolikov S., Mazor Y., Krauskopf A.. ELG1, a regulator of genome stability, has a role in telomere length regulation and in silencing. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:1656–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Banerjee S., Myung K.. Increased genome instability and telomere length in the elg1-deficient Saccharomyces cerevisiae mutant are regulated by S-phase checkpoints. Eukaryot. Cell. 2004; 3:1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parnas O., Zipin-Roitman A., Mazor Y., Liefshitz B., Ben-Aroya S., Kupiec M.. The ELG1 clamp loader plays a role in sister chromatid cohesion. PLoS One. 2009; 4:e5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maradeo M.E., Skibbens R.V.. The Elg1-RFC clamp-loading complex performs a role in sister chromatid cohesion. PLoS One. 2009; 4:e4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bell D.W., Sikdar N., Lee K.Y., Price J.C., Chatterjee R., Park H.D., Fox J., Ishiai M., Rudd M.L., Pollock L.M. et al.. Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLos Genet. 2011; 7:e1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuchenbaecker K.B., Ramus S.J., Tyrer J., Lee A., Shen H.C., Beesley J., Lawrenson K., McGuffog L., Healey S., Lee J.M. et al.. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 2015; 47:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson C., Gali V.K., Takahashi T.S., Kubota T.. PCNA retention on DNA into G2/M phase causes genome instability in cells lacking Elg1. Cell Rep. 2016; 16:684–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shemesh K., Sebesta M., Pacesa M., Sau S., Bronstein A., Parnas O., Liefshitz B., Venclovas C., Krejci L., Kupiec M.. A structure-function analysis of the yeast Elg1 protein reveals the importance of PCNA unloading in genome stability maintenance. Nucleic Acids Res. 2017; 45:3189–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang M.E., Rio A.G., Nicolas A., Kolodner R.D.. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:11529–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kastrinos F., Stoffel E.M.. History, genetics, and strategies for cancer prevention in Lynch syndrome. Clin. Gastroenterol. Hepatol. 2014; 12:715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marsischky G.T., Filosi N., Kane M.F., Kolodner R.. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996; 10:407–420. [DOI] [PubMed] [Google Scholar]
- 29. Gradia S., Subramanian D., Wilson T., Acharya S., Makhov A., Griffith J., Fishel R.. hMSH2–hMSH6 forms a Hydrolysis-Independent sliding clamp on mismatched DNA. Mol. Cell. 1999; 3:255–261. [DOI] [PubMed] [Google Scholar]
- 30. Mendillo M.L., Mazur D.J., Kolodner R.D.. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J. Biol. Chem. 2005; 280:22245–22257. [DOI] [PubMed] [Google Scholar]
- 31. Flores-Rozas H., Clark D., Kolodner R.D.. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat. Genet. 2000; 26:375–378. [DOI] [PubMed] [Google Scholar]
- 32. Flores-Rozas H., Kolodner R.D.. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prolla T.A., Pang Q., Alani E., Kolodner R.D., Liskay R.M.. MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science. 1994; 265:1091–1093. [DOI] [PubMed] [Google Scholar]
- 34. Amin N.S., Nguyen M.N., Oh S., Kolodner R.D.. exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol. Cell. Biol. 2001; 21:5142–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goellner E.M., Smith C.E., Campbell C.S., Hombauer H., Desai A., Putnam C.D., Kolodner R.D.. PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol. Cell. 2014; 55:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shell S.S., Putnam C.D., Kolodner R.D.. The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Mol. Cell. 2007; 26:565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clark A.B., Valle F., Drotschmann K., Gary R.K., Kunkel T.A.. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J. Biol. Chem. 2000; 275:36498–36501. [DOI] [PubMed] [Google Scholar]
- 38. Hombauer H., Campbell C.S., Smith C.E., Desai A., Kolodner R.D.. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell. 2011; 147:1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haye J.E., Gammie A.E.. The eukaryotic mismatch recognition complexes track with the replisome during DNA synthesis. PLos Genet. 2015; 11:e1005719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kawasoe Y., Tsurimoto T., Nakagawa T., Masukata H., Takahashi T.S.. MutSalpha maintains the mismatch repair capability by inhibiting PCNA unloading. Elife. 2016; 5:e15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pluciennik A., Dzantiev L., Iyer R.R., Constantin N., Kadyrov F.A., Modrich P.. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:16066–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lubin J.W., Rao T., Mandell E.K., Wuttke D.S., Lundblad V.. Dissecting protein function: an efficient protocol for identifying separation-of-function mutations that encode structurally stable proteins. Genetics. 2013; 193:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Longtine M.S., McKenzie A. 3rd, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R.. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998; 14:953–961. [DOI] [PubMed] [Google Scholar]
- 44. Laughery M.F., Hunter T., Brown A., Hoopes J., Ostbye T., Shumaker T., Wyrick J.J.. New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast. 2015; 32:711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chakraborty U., George C.M., Lyndaker A.M., Alani E.. A delicate balance between repair and replication factors regulates recombination between divergent DNA sequences in saccharomyces cerevisiae. Genetics. 2016; 202:525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kubota T., Hiraga S., Yamada K., Lamond A.I., Donaldson A.D.. Quantitative proteomic analysis of chromatin reveals that Ctf18 acts in the DNA replication checkpoint. Mol. Cell Proteomics. 2011; 10:doi:10.1074/mcp.M110.005561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tran H.T., Keen J.D., Kricker M., Resnick M.A., Gordenin D.A.. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 1997; 17:2859–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosche W.A., Foster P.L.. Determining mutation rates in bacterial populations. Methods. 2000; 20:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sarkar S., Ma W.T., Sandri G.H.. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica. 1992; 85:173–179. [DOI] [PubMed] [Google Scholar]
- 50. Liu H., Naismith J.H.. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008; 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McNally R., Bowman G.D., Goedken E.R., O’Donnell M., Kuriyan J.. Analysis of the role of PCNA-DNA contacts during clamp loading. BMC Struct. Biol. 2010; 10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997; 276:Academic Press; 307–326. [DOI] [PubMed] [Google Scholar]
- 53. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. LuCore S.D., Litman J.M., Powers K.T., Gao S., Lynn A.M., Tollefson W.T., Fenn T.D., Washington M.T., Schnieders M.J.. Dead-End elimination with a polarizable force field repacks PCNA structures. Biophys. J. 2015; 109:816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D.. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 57. De March M., Merino N., Barrera-Vilarmau S., Crehuet R., Onesti S., Blanco F.J., De Biasio A.. Structural basis of human PCNA sliding on DNA. Nat. Commun. 2017; 8:13935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Katou Y., Kanoh Y., Bando M., Noguchi H., Tanaka H., Ashikari T., Sugimoto K., Shirahige K.. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003; 424:1078–1083. [DOI] [PubMed] [Google Scholar]
- 59. Lubin J.W., Tucey T.M., Lundblad V.. Using separation-of-function mutagenesis to define the full spectrum of activities performed by the Est1 telomerase subunit in Vivo. Genetics. 2018; 208:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parker J.L., Bucceri A., Davies A.A., Heidrich K., Windecker H., Ulrich H.D.. SUMO modification of PCNA is controlled by DNA. EMBO J. 2008; 27:2422–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dieckman L.M., Boehm E.M., Hingorani M.M., Washington M.T.. Distinct structural alterations in proliferating cell nuclear antigen block DNA mismatch repair. Biochemistry. 2013; 52:5611–5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davidson M.B., Katou Y., Keszthelyi A., Sing T.L., Xia T., Ou J., Vaisica J.A., Thevakumaran N., Marjavaara L., Myers C.L. et al.. Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J. 2012; 31:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brown M.W., Kim Y., Williams G.M., Huck J.D., Surtees J.A., Finkelstein I.J.. Dynamic DNA binding licenses a repair factor to bypass roadblocks in search of DNA lesions. Nat. Commun. 2016; 7:10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shell S.S., Putnam C.D., Kolodner R.D.. Chimeric Saccharomyces cerevisiae Msh6 protein with an Msh3 mispair-binding domain combines properties of both proteins. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:10956–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chakraborty U., Dinh T.A., Alani E.. Genomic instability promoted by overexpression of mismatch repair factors in yeast: A model for understanding cancer progression. Genetics. 2018; 209:439–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Janke R., King G.A., Kupiec M., Rine J.. Pivotal roles of PCNA loading and unloading in heterochromatin function. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E2030–E2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Swanson R.L., Morey N.J., Doetsch P.W., Jinks-Robertson S.. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999; 19:2929–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dieckman L.M., Washington M.T.. PCNA trimer instability inhibits translesion synthesis by DNA polymerase η and by DNA polymerase δ. DNA Repair. 2013; 12:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Billon P., Li J., Lambert J.P., Chen Y., Tremblay V., Brunzelle J.S., Gingras A.C., Verreault A., Sugiyama T., Couture J.F. et al.. Acetylation of PCNA sliding surface by eco1 promotes genome stability through homologous recombination. Mol. Cell. 2017; 65:78–90. [DOI] [PubMed] [Google Scholar]
- 70. Sikdar N., Banerjee S., Lee K-y., Wincovitch S., Pak E., Nakanishi K., Jasin M., Dutra A., Myung K.. DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity. Cell Cycle. 2009; 8:3199–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession number: 6D0R (PCNA-D17K) and 6D0Q (PCNA-D21K).