


Abstract
SPT6 is a conserved elongation factor that is associated with phosphorylated RNA polymerase II (RNAPII) during transcription. Recent transcriptome analysis in yeast mutants revealed its potential role in the control of transcription initiation at genic promoters. However, the mechanism by which this is achieved and how this is linked to elongation remains to be elucidated. Here, we present the genome-wide occupancy of Arabidopsis SPT6-like (SPT6L) and demonstrate its conserved role in facilitating RNAPII occupancy across transcribed genes. We also further demonstrate that SPT6L enrichment is unexpectedly shifted, from gene body to transcription start site (TSS), when its association with RNAPII is disrupted. Protein domains, required for proper function and enrichment of SPT6L on chromatin, are subsequently identified. Finally, our results suggest that recruitment of SPT6L at TSS is indispensable for its spreading along the gene body during transcription. These findings provide new insights into the mechanisms underlying SPT6L recruitment in transcription and shed light on the coordination between transcription initiation and elongation.
INTRODUCTION
It is well known that SPT6 is a transcription elongation factor, as evidenced by its physical association with elongating RNAPII (1–3) and ability to enhance elongation in vitro (4) and in vivo (5). The Src homology 2 (SH2) domain of SPT6 recognizes and binds to phosphorylated serine 2 and tyrosine 1 repeats within the C-terminal domain (CTD) of RNA polymerase II (RNAPII), as well as to the phosphorylated linker region preceding the CTD (3,5–7). Deletion or mutation of SH2 disrupts the interaction between SPT6 and RNAPII (3,8,9) and significantly reduces the occupancy levels of SPT6 and RNAPII at transcribed genic regions (3,9). Genetic and genomic studies in yeasts have indicated the role of SPT6 and other elongation factors in controlling intragenic initiation (10–12). Recently, it was found that SPT6 is also involved in regulation of genic initiation and that mutation of SPT6 caused the reduced occupancy of TFIIB at genic promoters (13).
In Arabidopsis, there are two versions of SPT6: SPT6 (At1g63210) and SPT6-like (SPT6L) (At1g65440) (14). The transcript of SPT6 was barely detectable in most of the tissues (15) and no visible phenotype was observed in spt6 mutants (14), suggesting that SPT6 may not play an essential role in transcription. On the other hand, SPT6L appears to be commonly expressed (15) and mutations of SPT6L led to the formation of aberrant apical-basal axis and embryonic lethality (14). Furthermore, SPT6L can be co-purified with RNAPII and other elongation factors (15). These findings indicate the potential roles of SPT6L in regulating transcription.
In this study, we profiled the genome-wide occupancy of SPT6L and demonstrated its functional conservation in transcription elongation. When analyzing the global association between SPT6L and RNAPII, intriguingly, we found that the enrichment of SPT6L was shifted from the transcribed region to transcription start site (TSS) in the absence of its association with RNAPII. We further generated a series of domain deletions and showed that the HtH and YqgF domains of SPT6L are not only required for its TSS enrichment but also for the distribution along gene bodies. Overall, our results suggest a possible scenario where SPT6L may be initially recruited at TSS and then spread to the gene body during transcription. In summary, our findings reveal novel mechanisms underlying the recruitment of SPT6L into the transcription machinery.
MATERIALS AND METHODS
Plant materials and growth conditions
The spt6l heterozygous seeds (SALK_016621), previously described (14), were obtained from the Arabidopsis Biological Resource Center (ABRC) at Ohio State University. Three formerly generated transgenic lines: ProREF6:REF6-GFP (16), ProACC1:ACC1-GFP (17) and 35S:GFP (16) were used. All Arabidopsis seeds used were in the Columbia (Col-0) background. Plants were grown either on half-strength Murashige and Skoog ( MS) medium (0.5× MS salts, 1.5% [w/v] sucrose, and 0.8% agar [pH 5.8]) or in soil under 16 h/8 h light/dark cycle at 23 °C. For the inhibitor treatment, 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole (DRB) (10010302, Cayman Chemical), flavopiridol (10009197, Cayman Chemical), or triptolide (11973, Cayman Chemical) was added to the media at a final concentration of 100, 10 and 10 μM, respectively. For the short-time treatment, 10-day-old seedlings were sprayed with 10 μM flavopiridol and grown on
MS) medium (0.5× MS salts, 1.5% [w/v] sucrose, and 0.8% agar [pH 5.8]) or in soil under 16 h/8 h light/dark cycle at 23 °C. For the inhibitor treatment, 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole (DRB) (10010302, Cayman Chemical), flavopiridol (10009197, Cayman Chemical), or triptolide (11973, Cayman Chemical) was added to the media at a final concentration of 100, 10 and 10 μM, respectively. For the short-time treatment, 10-day-old seedlings were sprayed with 10 μM flavopiridol and grown on  MS plates for 1 h. For the heat shock treatment, seeds were germinated and grown on
MS plates for 1 h. For the heat shock treatment, seeds were germinated and grown on  MS plates for 7 days at 23 °C. The plates were moved to 17 °C for 3 days, after which time, the seedlings were subjected to a heat shock treatment for 1 h at 27 °C. The primers used for genotyping are listed in Supplementary Table S1.
MS plates for 7 days at 23 °C. The plates were moved to 17 °C for 3 days, after which time, the seedlings were subjected to a heat shock treatment for 1 h at 27 °C. The primers used for genotyping are listed in Supplementary Table S1.
Plasmid construction for plant transformation
Due to the repetitive nature of the 3′ end sequence of SPT6L, we combined PCR amplification and direct DNA synthesis approaches to clone the full-length SPT6L genomic region and its 2 kb upstream regulatory sequence. Specifically, part1 (from −2009 bp to +6594 bp; − and + are relative to ATG) and part2 (from +5247 bp to +8443 bp) were PCR-amplified from genomic DNA and cloned into the Gateway entry vector pDONR221 according to the manufacturer's instructions (Invitrogen, Cat.: 12535-019). Part3 (from +6889 bp to +7380 bp, including the repetitive sequence) was synthesized by GenScript (www.genscript.com) and also inserted into pDONR221. Lastly, the entire sequence was assembled by sequential digestions and ligations as follows: first, part2-part3 (AvrII and PvuI) and then, part2-3-part1 (XhoI and PvuI), and cloned into pDONR221 (ProSPT6L:SPT6L-pDONR221). Finally, to generate the fusion construct with GFP (ProSPT6L:SPT6L-GFP), the assembled and sequence-confirmed SPT6L fragment was inserted into the destination vector pMDC107 (18) according to the manufacturer's instructions (Invitrogen, Cat.: 12535-019). All of the domain deletion mutants were generated based on the ProSPT6L:SPT6L-pDONR221 construct by using a site-directed mutagenesis protocol (Q5® Site-Directed Mutagenesis Kit Protocol (E0554)). For tagging biotin to RNAPII, we generated a new binary vector with pMDC123 (18) backbone by replacing the Gateway cassette with a newly assembled cassette containing the following components: genomic sequence of NRPB1 (including 2308 bp upstream of ATG), DNA sequence of biotin ligase recognition peptide (BLRP), Nos terminator, sequence of proACT2:BirA, and Nos terminator. The primers are listed in Supplementary Table S1.
Plant transformation
All of the sequence-confirmed constructs were transferred into Agrobacterium GV3101. Plant transformations were performed as previously described (19). After obtaining transgenic lines, the morphological phenotypes and GFP signals, for each construct, were examined by using at least three independent single insertion lines (determined by segregation ratio).
Confocal microscopy
To detect green fluorescence signals, root tips were cut from 7-day-old seedlings and transferred onto glass slides with 50 μl propidium iodide (PI) solution (1 μg/ml). The green fluorescence was detected by confocal microscopy (Leica) with excitation at 488 nm and emission at 505–525 nm.
Analysis of transcript levels
Total RNA extraction, cDNA synthesis, and real-time qPCR were performed as previously described (17). The primers are listed in Supplementary Table S1. The WT RNA-seq data are obtained from our previous work (17). Transcripts were grouped into eight subgroups (Supplementary Dataset 1), from high to low, based on their FPKM values (after conversion to logarithm value [log10]). Finally, the SPT6L ChIP signals were plotted separately for each of the gene groups.
Co-immunoprecipitation and immunoblot
Seventy-five milligrams of 10-day-old seedlings, grown on  MS medium, were homogenized to fine powder in a mixer mills and dissolved in 300 μl lysis buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 2.5 mM EDTA, 1 mM DTT, 0.1% TritonX-100, 0.1 mM PMSF, and Protease inhibitor) for 20 min at 4°C. Supernatants were collected after centrifuging at 14 000 × g, 4°C for 10 min. For the Western blot, the supernatants were mixed with 2× boiled SDS loading buffer and then loaded onto SDS-PAGE gels. For the co-IP, 20 μl anti-GFP μMACS micro-beads (lot: 5180928110) were added to the supernatants and gently shaken for 1 h at 4 °C. Following the protocol of μMACS GFP Isolation Kit (130-091-125, MACS), the interacting proteins were eluted and loaded onto SDS-PAGE gels. For the affinity purification, 20 μl streptavidin magnetic beads (S1420S, NEB) were added into the supernatants and gently shaken for 1 h at 4 °C. The associated proteins were washed five times with lysis buffer and eluted with boiled SDS loading buffer. All the blotting, immunoprecipitation, and affinity purification were performed three times with independent samples. The antibodies used in this study are listed as follows: anti-GFP (ab290, Abcam, lot: GR240324), anti-RNAPII [8WG16] (ab817, anti-RNAPII CTD repeat YSPTSPS, Abcam, lot: GR313984), anti-RNAPIISer2P (ab5095, Abcam, lot: GR309257), anti-biotin (ab53494, Abcam, lot: GR3197003-8) and anti-Actin (AS13 2640, Agrisera).
MS medium, were homogenized to fine powder in a mixer mills and dissolved in 300 μl lysis buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 2.5 mM EDTA, 1 mM DTT, 0.1% TritonX-100, 0.1 mM PMSF, and Protease inhibitor) for 20 min at 4°C. Supernatants were collected after centrifuging at 14 000 × g, 4°C for 10 min. For the Western blot, the supernatants were mixed with 2× boiled SDS loading buffer and then loaded onto SDS-PAGE gels. For the co-IP, 20 μl anti-GFP μMACS micro-beads (lot: 5180928110) were added to the supernatants and gently shaken for 1 h at 4 °C. Following the protocol of μMACS GFP Isolation Kit (130-091-125, MACS), the interacting proteins were eluted and loaded onto SDS-PAGE gels. For the affinity purification, 20 μl streptavidin magnetic beads (S1420S, NEB) were added into the supernatants and gently shaken for 1 h at 4 °C. The associated proteins were washed five times with lysis buffer and eluted with boiled SDS loading buffer. All the blotting, immunoprecipitation, and affinity purification were performed three times with independent samples. The antibodies used in this study are listed as follows: anti-GFP (ab290, Abcam, lot: GR240324), anti-RNAPII [8WG16] (ab817, anti-RNAPII CTD repeat YSPTSPS, Abcam, lot: GR313984), anti-RNAPIISer2P (ab5095, Abcam, lot: GR309257), anti-biotin (ab53494, Abcam, lot: GR3197003-8) and anti-Actin (AS13 2640, Agrisera).
Chromatin immunoprecipitation (ChIP)
ChIP was performed as previously described (17,20) with minor modifications. For each biological replicate, five grams of 10-day-old Arabidopsis seedlings (one gram for spt6l and spt6l SPT6LΔSH2-GFP seedlings), grown on  MS medium, were collected. Protein A Dynabeads (Invitrogen) were pre-incubated with the antibody (5 μl for 50 μl beads) for at least 6 h, at 4 °C in a rotor. After removing excess or unbound antibody, the pre-cleaned chromatin (cleaned by incubating with Dynabeads alone) was added to the antibody bound Dynabeads. Since variations generated from sonication may affect the comparison of SPT6L and RNAPII occupancy levels around TSS, the sheared chromatin was equally divided into either two or four tubes and then subjected to different antibodies (two tubes for anti-GFP and anti-RNAPII in ChIP-seq; or four tubes for anti-GFP, anti-RNAPII, anti-RNAPIIS2P and anti-IgG in ChIP-qPCR). ChIP libraries were prepared by using the NEBNext® Ultra™ DNA Library Prep Kit (E7370S) following the manufacturer's instructions, and subsequently used for Illumina single-end sequencing. Two and three biological replicates were performed for ChIP-seq and ChIP-qPCR, respectively. All of the domain deleted SPT6L samples (except single tSH2 deletion) were performed in the spt6l heterozygous background. The primers used for ChIP-qPCR are listed in Supplementary Table S1.
MS medium, were collected. Protein A Dynabeads (Invitrogen) were pre-incubated with the antibody (5 μl for 50 μl beads) for at least 6 h, at 4 °C in a rotor. After removing excess or unbound antibody, the pre-cleaned chromatin (cleaned by incubating with Dynabeads alone) was added to the antibody bound Dynabeads. Since variations generated from sonication may affect the comparison of SPT6L and RNAPII occupancy levels around TSS, the sheared chromatin was equally divided into either two or four tubes and then subjected to different antibodies (two tubes for anti-GFP and anti-RNAPII in ChIP-seq; or four tubes for anti-GFP, anti-RNAPII, anti-RNAPIIS2P and anti-IgG in ChIP-qPCR). ChIP libraries were prepared by using the NEBNext® Ultra™ DNA Library Prep Kit (E7370S) following the manufacturer's instructions, and subsequently used for Illumina single-end sequencing. Two and three biological replicates were performed for ChIP-seq and ChIP-qPCR, respectively. All of the domain deleted SPT6L samples (except single tSH2 deletion) were performed in the spt6l heterozygous background. The primers used for ChIP-qPCR are listed in Supplementary Table S1.
ChIP-seq data analysis
The sequenced reads were aligned to the TAIR10 assembly using the Bowtie2 program (21) with default settings. The unmapped reads and PCR duplicates were removed by converting bam to bed format and running the MACS2 ‘filterdup’ function, respectively. After removing unmapped reads and PCR duplicates, peaks were called by using the MACS2 program (22) with the following settings (-g 135000000, -broad, and -broad-cutoff 0.01). A summary of reads numbers is listed in Supplementary Table S2. Only highly reproducible peaks, across two biological replicates (Irreproducible Discovery Rate, IDR ≤ 0.01), were kept. Common genes were identified by using PeakAnalyzer (23). Coverage files (BigWig files), for all samples, were converted from bam files by using bamCoverage (from deeptools2) (24) with the following settings (-bs 10 –effectiveGenomeSize 135000000 –normalizeUsing RPGC –ignoreDuplicates -e 300 –samFlagExclude 1796). Pearson correlation across two biological replicates was calculated by using deeptools2 with the following settings: multiBigwigSummary bins -bs 100; and plotCorrelation –c pearson. All of the correlation values can be found in Supplementary Figure S1. The averaged coverage file from two biological replicates was generated by running a GitHub script (http://wresch.github.io/2014/01/31/merge-bigwig-files.html). Heatmaps and mean density plots were generated with deeptools2 (settings indicated in Figure legends). Visualization of coverage files was carried out with a web-based genome browser (ENPG, www.plantseq.org). The genome-wide profiles of histone methylation and acetylation marks were obtained from published ChIP-seq datasets (17,25). The RNAPII pausing index was calculated by modifying a previously described formula (26):
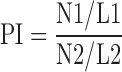 |
N1: the number of RNAPII reads in TSS ± 250 bp of genes; N2: the number of RNAPII reads in gene body (from +251 to TTS); L1: 500 bp; L2: length of gene body (from +251 to TTS). Genes with lengths shorter than 500 bp were excluded. We also removed any gene whose promoter (1 kb upstream to TSS) overlapped with neighboring genes. Four groups of genes can be found in Supplementary Dataset 2.
RESULTS
SPT6L co-occupies genome-wide with RNAPII over highly transcribed genes
To gain insights into the functions of SPT6L in plants, we sought to profile its genome-wide occupancy. To that end, we first obtained the previously characterized spt6l mutant (SALK_016621) (14). We observed that, when cultured on  MS agar plates, the spt6l (also referred to as spt6l−/− homozygous mutants, hereafter) seeds could germinate but with undeveloped roots, deformed cotyledons, and spike-like structures at shoot apex (Figure 1A and B; see Supplementary Figures S2A and B for transcript analysis). The spt6l seedlings were unable to develop true leaves and set seeds either on MS agar or in soil. The mutant phenotypes of spt6l could be rescued by the wild type version of the SPT6L gene (fused with GFP) driven by its native promoter (ProSPT6L:SPT6L-GFP) (Figure 1C).
MS agar plates, the spt6l (also referred to as spt6l−/− homozygous mutants, hereafter) seeds could germinate but with undeveloped roots, deformed cotyledons, and spike-like structures at shoot apex (Figure 1A and B; see Supplementary Figures S2A and B for transcript analysis). The spt6l seedlings were unable to develop true leaves and set seeds either on MS agar or in soil. The mutant phenotypes of spt6l could be rescued by the wild type version of the SPT6L gene (fused with GFP) driven by its native promoter (ProSPT6L:SPT6L-GFP) (Figure 1C).
Figure 1.

SPT6L associates with transcribed genes and promotes transcription (A) to (C) 10-day-old WT (A), spt6l (B) and spt6l ProSPT6L:SPT6L-GFP (C) seedlings. Bar = 0.5 mm. (D) Mean density of SPT6L occupancy at the SPT6L target genes. Plotting regions were scaled to the same length as follows: 5′ ends (–2.0 kb to transcription starting site [TSS]) and 3′ ends (transcription termination site [TTS] to downstream 2.0 kb) were not scaled, and the gene bodies were scaled to 3 kb. Y-axis represents the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapping bins, averaged over two replicates (SPT6L) or one replicate (Input). The number of genes were indicated (n). (E) Reads correlation between SPT6L and RNAPII ChIP-seq (Pearson correlation value was indicated). The entire genome was equally divided into 100 bp non-overlapping bins and the numbers of reads were averaged over two replicates. (F) Mean density of RNAPII occupancy in WT and spt6l mutant at SPT6L target genes. Plotting regions were the same as in Figure 1D. Y-axis values were the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapped bins, averaged over two replicates or one replicate (for inputs in WT and spt6l background). (G) Mean density of SPT6L occupancy at different transcription groups. By sorting the transcript level of each gene from high to low, all Arabidopsis genes were clustered into eight groups. Y-axis values are the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapped bins, averaged over two replicates.
We examined the subcellular localization of SPT6L-GFP and observed the GFP signals in the nuclei (Supplementary Figures S2C–G), which is consistent with its expected role in transcription elongation as demonstrated in other organisms (2,4,5,7,8). Next, we profiled the genome-wide occupancy of SPT6L by Chromatin Immunoprecipitation Sequencing (ChIP-seq) and found that SPT6L was mainly recruited to the transcribed regions of genes (Figure 1D). This occupancy pattern resembles the previously revealed profiles of RNAPII in plants (27). Thus, we examined the potential association of SPT6L with transcription in plants. Firstly, by plotting published profiles of histone modifications (17,25) on SPT6L-occupied genes (Supplementary Dataset 3), we found that SPT6L-occupied genes were mainly marked with active histone modifications such as histone acetylation and trimethylation of lysine 4 at histone 3 (H3K4me3), but not the repressive histone marks such as H3K27me3 (Supplementary Figure S2H). Secondly, the occupancies of SPT6L and RNAPII were highly correlated in genome-wide (Figure 1E) and the enrichment of RNAPII was decreased in spt6l (Figure 1F). Thirdly, the occupancy intensity of SPT6L was positively associated with transcript levels (Figure 1G). In summary, these data indicate that Arabidopsis SPT6L likely plays similar roles, in transcription, as its homologs in other species.
SPT6L enriched at transcription start sites in the absence of its association with RNAPII
In yeasts, SPT6 interacts with both the phosphorylated C-terminal domain (CTD) of RNAPII and the phosphorylated linker region preceding the CTD via its two tandem SH2 domains (tSH2) (5–7). While deletion of the tSH2 domain disrupts the interaction of SPT6 with RNAPII and significantly reduces the occupancy of SPT6 and RNAPII along genes (3,9), low levels of SPT6ΔtSH2 still can be detected at the transcribed regions (3,9), suggesting a RNAPII-independent recruitment of SPT6. To examine whether plant SPT6L can also be recruited to genes in a tSH2 independent manner, we made a transgenic line expressing a tSH2-deleted version of SPT6L tagged with GFP (ProSPT6L:SPT6LΔtSH2-GFP) in the spt6l+/− background (also referred to as heterozygous mutant background; Supplementary Figure S3A). We then performed co-immunoprecipitation (Co-IP) experiments with the transgenic line and found that deletion of the tSH2 domain indeed impaired the interaction between SPT6L and RNAPIIS2P (Figure 2A). Of note, due to the lack of antibodies that can recognize the phosphorylated linker region of RNAPII, we used an antibody raised against the phosphorylated serine 2 of CTD for the Co-IP experiments. To further confirm that SPT6ΔtSH2 no longer interacts with RNAPII, we generated a construct for expressing both the BirA ligase and NRPB1 (large subunit of RNAPII, At4g35800) tagged with a peptide (biotin ligase recognition peptide [BLRP]) which can be specifically recognized by BirA. The construct was then introduced into WT as well as the ProSPT6L:SPT6L-GFP and ProSPT6L:SPT6LΔtSH2-GFP transgenic backgrounds. Using the new transgenic plants, we performed an affinity purification followed by blotting experiment. As shown in Supplementary Figure S3B, there was no detectable interaction between SPT6LΔtSH2 and NRPB1.
Figure 2.

SPT6L enriched at TSS in a RNAPII independent manner (A) Co-immunoprecipitation performed on SPT6L-GFP and SPT6LΔtSH2-GFP. Immunoprecipitation (IP) and Western blot (WB) were performed using specified antibodies. (B–D) The morphological phenotypes of 5-day (B), 10-day (C) and 30-day (D) old spt6l seedlings. Bar = 0.5 mm. (E–G) The morphological phenotypes of 5-day (E), 10-day (F) and 30-day (G) old spt6l SPT6LΔtSH2 seedlings. Bar = 0.5 mm. (H) Heatmaps of RNAPII, SPT6L, and SPT6LΔtSH2 reads measured by ChIP-seq in wide-type (WT), spt6l, and spt6l SPT6LΔSH2 backgrounds or after treating with 10 μl flavopiridol for 1 h, over the same regions as shown in Figure 1D. From top to bottom, the plotted genomic regions were sorted by RNAPII signal strength in WT. The plotted values are the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapped bins, averaged over two replicates. (I) Screenshot of representative peaks at chromosome 1 in genome browser (ENPG, www.plantseq.org) to visualize peak changes. Y-axis represents the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapping bins. Black arrows indicate TSS enriched ChIP signals. (J) Mean density of RNAPII, SPT6L, and SPT6LΔtSH2 occupancy at the SPT6L target genes, which were grouped into four groups based on the pausing index (PI, for formula see Materials and Methods) of RNAPII at each gene. The number of genes in each group was indicated. Y-axis represents the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapped bins, averaged over two replicates. Reads were plotted on 2 kb upstream and downstream of TSS.
Noticeably, unlike the severe defects observed in the spt6l mutants (Figure 2B–D), the spt6l SPT6LΔtSH2-GFP seedlings were able to grow bigger and develop small true leaves (Figure 2E–G). In line with the morphological phenotype, the introduction of SPT6LΔtSH2 also partially rescued the genome-wide occupancy of RNAPII (Figure 2H–J). These findings indicate that SPT6LΔtSH2 still retains some of its capacity in facilitating RNAPII transcription.
We next asked whether the tSH2-deleted SPT6L can still be recruited to chromatin. To address this question, we profiled the genome-wide occupancy of SPT6LΔtSH2 in the spt6l background. In contrast to the occupancy pattern observed for the full-length SPT6L, that spreads over the entire transcribed regions of genes, the signals of SPT6LΔtSH2 were unexpectedly enriched only at the transcription start sites (TSS) (Figure 2H–J). This occupancy pattern is different from the observed occupancy of SPT6ΔtSH2 in yeasts, where tSH2-deleted SPT6 can still weakly spread all over the transcribed regions (3,6,9). Since SPT6 is involved in the regulation of chromatin structure in yeasts (28–31), the unpredicted TSS enrichment of SPT6LtΔSH2 might be caused by altered chromatin structure. To rule out this possibility, we performed a ChIP-seq analysis of SPT6LΔtSH2 in the spt6l+/− background and a similar pattern was detected (Supplementary Figure S3C). Another arguable point could be that the potential changes in protein folding might lead to the TSS enrichment. To assess this possibility, we tried to examine the profiles of SPT6L after inhibiting transcription initiation or elongation. We applied three widely used transcription inhibitors (32): DRB, flavopiridol, and triptolide. Among them, triptolide inhibits transcription initiation, while both DRB and flavopiridol prevent the entry of RNAPII into transcription elongation (32). Although all three inhibitors are effective in yeast and animal cells (32), only the treatment of flavopiridol affected plant growth with the recommended concentration (Supplementary Figure S3D). It has been shown that flavopiridol inhibits transcription elongation by blocking the kinase activity of positive transcription elongation factor b (P-TEFb) (33). Therefore, we further examined the temporal response of seedlings to flavopiridol. As shown in Supplementary Figure S3E, the phosphorylation level of RNAPII at serine 2 was reduced within 1 h of treatment. We then examined whether the application of flavopiridol would compromise the interaction of SPT6L with RNAPII. The transgenic plants expressing the BLRP-tagged NRPB1, in the ProSPT6L:SPT6L-GFP background, were treated with flavopiridol and then subsequently used for an affinity purification followed by blotting experiment. As shown in Supplementary Figure S3F, treatment with flavopiridol indeed attenuated the interaction between SPT6L and RNAPII. Finally, we profiled the genome-wide occupancy of SPT6L after 1 h flavopiridol treatment. As shown in Figure 2H, we detected enrichment of SPT6L at both TSS and TTS (transcription stop sites). Since flavopiridol mainly inhibits kinase activity rather than promotes dephosphorylation (33), the TTS enrichment of SPT6L was likely due to the residual phosphorylated RNAPII around TTS. Indeed, we found that the SPT6L–RNAPII interaction was not completely abolished after flavopiridol treatment (Supplementary Figure S3F), showing the existence of some phosphorylated RNAPII. This pharmacological study complements our genetic work and suggests that SPT6L can be targeted to TSS in the absence of its interaction with RNAPII.
The unexpected TSS enrichment of SPT6L drew our attention to its potential effects on RNAPII occupancy around TSS. Since the amount of RNAPII around TSS is determined by the equilibrium of new entry and quick release, thus, to quantify and compare the amount of RNAPII around TSS, we reasoned that it would be helpful to take RNAPII pausing at promoter-proximal regions into account. Promoter-proximal pausing has been well-documented, in Drosophila and mammalian cells, when RNAPII pauses right after the TSS following initiation and waits for regulatory signals to continue transcription (34,35). To distinguish the pausing strength at different genes, researchers introduced the concept of pausing index (PI), which is defined as the ratio of pausing peak density to gene body density (26,36). By adopting this concept to aid our analysis, we were able to divide the targeted genes of RNAPII into four groups according to their PI values. Upon plotting RNAPII signals around the TSS in WT and spt6l, we found that mutation of SPT6L led to decreased TSS occupancy of RNAPII in all pausing groups (Figure 2J), pointing to a role for SPT6L at early transcription stage. More importantly, the introduction of SPT6LΔtSH2 can partially or even completely rescue the TSS occupancy of RNAPII in higher pausing groups (Figure 2J). In addition, ChIP signals of SPT6LΔtSH2 in all of the pausing groups peaked immediately upstream of TSS followed by RNAPII signals (Figure 2J), suggesting a possible scenario where the presence of SPT6LΔSH2 may help the entry of RNAPII during transcription initiation.
The HtH and YqgF domains are required for the TSS association
We next tried to determine which domain(s) of SPT6L is(are) required for its TSS enrichment. In Arabidopsis, SPT6L contains all five conserved SPT6 domains plus the plant-specific GW/WG domain (Figure 3A) (14). Since the tSH2-deleted version of SPT6L could partially rescue the spt6l phenotype and showed clear enrichment around the TSS, we generated five ‘double-deletion’ constructs by individually deleting each of the five other domains on top of the tSH2 deletion and introduced them into spt6l+/− (Figure 3A). To assess the impacts of domain deletions on the subcellular localization of the proteins, we examined the GFP signals in the transgenic root tips and found that all versions of the domain-deleted SPT6L were still localized in the nuclei (Supplementary Figures S4A–E). Different from the spt6l SPT6LΔtSH2 seedlings, both the spt6l SPT6LΔtSH2ΔHtH and spt6l SPT6LΔtSH2ΔYqgF seedlings failed to develop true leaves and showed similar phenotypes to spt6l (Figures 2D, 3B–D). The other three double-deletion mutants displayed the same phenotypes as the spt6l SPT6LΔtSH2 single mutant (Figure 3E–G). This observation suggests that the HtH and YqgF domains may be required for the TSS enrichment of SPT6LΔtSH2. In addition, we also examined the protein levels of the mutants and observed comparable levels of the mutated SPT6Ls (Figure 3H, Supplementary Figure S4F), indicating that the compromised phenotype was not due to altered protein levels. Finally, we performed ChIP-seq experiments with the spt6l+/− SPT6LΔtSH2ΔHtH and spt6l+/− SPT6LΔtSH2ΔYqgF seedlings and found that signals around the TSS were markedly reduced in spt6l+/− SPT6LΔtSH2ΔHtH and undetectable in spt6l+/− SPT6LΔtSH2ΔYqgF (Figure 3I). This result suggests that the HtH and YqgF domains are required for the TSS enrichment of SPT6LΔtSH2.
Figure 3.

The HtH or YqgF domain is required for TSS enrichment of SPT6LΔtSH2. (A) Diagrams of the protein domains of Arabidopsis SPT6L and the domain deletion versions. (B–G) 30-day old seedlings of spt6l SPT6LΔtSH2 (B), spt6l SPT6LΔtSH2ΔHtH (C), spt6l SPT6LΔtSH2ΔYqgF (D), spt6l SPT6LΔtSH2ΔHhH (E), spt6l SPT6LΔtSH2ΔS1 (F) and spt6l SPT6LΔtSH2ΔGW/WG (G). Bar = 0.5 mm. (H) Immunoblot assessing the levels of the domain-deleted SPT6L proteins in the genetic backgrounds as indicated. H3 levels served as loading controls. Two other biological replicates can be found in Supplementary Figure S4F. (I) Heatmaps of domain-deleted SPT6L reads measured by ChIP-seq, over the same regions and order shown in Figure 2H. The plotted values are the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapped bins, averaged over two replicates. The signals of input sample (input for spt6l+/− SPT6LΔtSH2ΔYqgF) were obtained from one replicate.
The HtH and YqgF domains are indispensable for the distribution of SPT6L along genes
We next asked whether the HtH and YqgF domains also contribute to the distribution of SPT6L along transcribed regions. Two new constructs, with single deletion of either the HtH or YqgF domain, were generated and introduced into spt6l+/− plants. We observed that even though the tSH2 domain was intact in these plants, both of the domain-deleted SPT6Ls failed to rescue the spt6l mutant phenotype (Figure 4A–D), suggesting the critical role of HtH and YqgF in maintaining the normal function of SPT6L. To find out how the deletions affect the function of SPT6L in transcription, we first tested whether these two mutated proteins could still interact with RNAPII by performing Co-IP experiments. As shown in Figure 4E, both the mutated versions of SPT6L could still interact with RNAPIIS2P, but at reduced levels. We then performed ChIP-seq analysis to examine their association with chromatin and observed that the two versions of mutated SPT6L could still weakly associate with RNAPIIS2P, but they were no longer enriched over gene bodies (Figure 4F). These results indicate that the YqgF and HtH domains are also required for the distribution of SPT6L along genes.
Figure 4.

The HtH or YqgF domain is required for SPT6L function (A) to (D) 10-day old seedlings of spt6l (A), spt6l SPT6LΔtSH2 (B), spt6l SPT6LΔHtH (C) and spt6l SPT6ΔYqgF (D). White arrows indicate developed true leaves. Bar = 0.5 mm. (E) Co-immunoprecipitation performed on SPT6L-GFP and domain-deleted SPT6Ls. Immunoprecipitation (IP) and Western blot (WB) were performed using specified antibodies. (F) Heatmaps of domain-deleted SPT6Ls reads as measured by ChIP-seq, over the same regions and order shown in Figure 2H. The plotted values are the means of normalized reads (1× sequencing depth normalization) per 10 bp non-overlapped bins, averaged over two replicates. The signals of input sample (input of spt6l+/− SPT6LΔYqgF) was obtained from one replicate. (G–H) 10-day old F1 progenies of the cross between spt6l+/− SPT6LΔtSH2 and spt6l+/− SPT6LΔHtH (G) or spt6l+/− SPT6LΔYqgF (H). White arrows indicate developed true leaves. Bar = 0.5 mm. (I) Segregation ratio of F1 progenies.
We next took a genetic approach to examine the functional linkage between the tSH2 and HtH/YqgF domains. We reasoned that if the functions of tSH2 and HtH/YqgF could be genetically separated, the spt6l phenotype should be rescued in the co-presence of SPT6LΔtSH2 and SPT6LΔHtH/SPT6LΔYqgF. Toward that end, we crossed spt6l+/−SPT6LΔtSH2+/+ with either spt6l+/−SPT6LΔHtH+/+ or spt6l+/−SPT6LΔYqgF+/+ and examined the phenotypes of the F1 progenies. Approximately 25% of the F1 progenies (spt6l−/−SPT6LΔtSH2+/−SPT6LΔHtH+/- or spt6l−/−SPT6LΔtSH2+/−SPT6LΔYqgF+/−) showed SPT6LΔtSH2-like phenotype (Figure 4G–I). This result indicates that the functional domains of HtH/YqgF and tSH2 have to be preserved in the same SPT6L protein.
Temporal dynamic analysis of SPT6L recruitment to the TSS at heat shock response genes
Our genetic and molecular evidence presented above imply that the TSS recruitment of SPT6L may occur prior to its spreading over the gene bodies. To test this hypothesis, we monitored the recruitment of SPT6L and RNAPII at HEAT SHOCK PROTEIN 70 (HSP70, At3g12580) after heat shock (HS) treatment. As previously reported, its transcription is maintained at a relatively low level at 17 °C and drastically upregulated when the temperature is elevated to 27 °C within 1 h (37). These features make it an ideal model for investigating the temporal recruitment of SPT6L and RNAPII after HS. To optimize the HS condition, we first examined the transcript levels of HSP70 after HS at 5-minute intervals during one hour. Although the transcript level of HSP70 kept increasing throughout the course of the HS treatment (Supplementary Figure S5A), a significant change can be detected within the first 15 min (Figure 5A). Thus, we assessed the occupancy of SPT6L, RNAPII, and RNAPIIS2P at HSP70 within the first 15 mins after HS. The IgG signals served as negative control (Supplementary Figure S5B). After 7.5 min HS, the occupancies of SPT6L, RNAPII and RNAPIIS2P were significantly increased at the HSP70 locus (Supplementary Figure S5B). As the increased expression of HSP70 was detected 5 mins after HS (Figure 5A), this first wave of HS-induced transcription should be accompanied by the enrichment of transcription machinery within the same time frame. Thus, we normalized the ChIP signals within 5 min to that of untreated samples (0 min HS) and found that the peak signal of SPT6L was first detected at the TSS after 5 min HS, while both RNAPII and RNAPIIS2P peaked downstream of the TSS (Figure 5B). To further confirm this recruiting pattern of SPT6L, we also analyzed two other HS responsive genes: HEAT SHOCK TRANSCRIPTION FACTOR A7A (HSFA7A, At3g51910) and HEAT SHOCK TRANSCRIPTION FACTOR B2B (HSFB2B, At4g11660). Different from the continuous induction of HSP70, the two transcription factors were quickly induced within the first 30 mins, followed by a gradual decrease (Supplementary Figure S5A). Compared with 0 min HS, the increased transcription of both HSFA7A and HSFB2B were found at 10 mins after HS (Figure 5A). In ChIP assays, the peak signals of SPT6L were initially observed at TSS after 5 and 7.5 min HS for HSFA7A and HSFB2B, respectively, while both RNAPII and RNAPIIS2P peaked downstream of the TSS at the corresponding time points (Figure 5B). This recruiting pattern is similar to the one that we captured for HSP70. These observations suggest a possible scenario that SPT6L was first recruited to the TSS and that the recruitment was independent of RNAPII, in the first wave of transcription, at these heat shock response genes after HS.
Figure 5.

Temporal occupancies of SPT6L and RNAPII after heat shock treatment at three heat shock response genes. (A) Relative expression of HSP70, HSFA7A and HSFB2B within 15 min after heat shock (HS). The plotted values are the means ± S.D. of three biological replicates. Two tailed student t test. P values are indicated.N.S.: no significant difference. *P < 0.05, **P < 0.01. (B) Occupancies of SPT6L, RNAPII and RNAPS2P at HSP70, HSFA7A and HSFB2B after heat shock (HS). Y-axis presents the fold change of ChIP signals at 2.5, 5 and 7.5 min HS relative to 0 minutes HS. Each of the indicated point represents the middle of PCR fragment and the schematics of gene structures are shown at the bottom. The plotted values are the means ± S.D. of three biological replicates. Numbers on the x-axes are distances to the TSS (TSS = 0).
DISCUSSION
The recognition of tSH2 by phosphorylated RNAPII is a well-established mechanism for the recruitment of SPT6 during transcription (6–8,38). For this reason, SPT6 has been extensively studied for its role in transcription elongation. In this study, we first profiled the genome-wide occupancy of SPT6L and confirmed its conserved function in transcription elongation in plants. Further, we showed that, at both genomic scale and individual loci (three heat shock response genes), SPT6L could target to TSS of transcribing genes in an RNAPII-independent manner and that this targeting is indispensable for the loading of SPT6L into the transcription machinery and distribution along gene bodies. Our findings, thus, reveal a new recruitment mechanism for SPT6L during transcription and shed light on the roles of SPT6 in transcription initiation.
As an elongation factor, SPT6 prefers to associate with highly transcribed genes (30) and improves the elongation rate of RNAPII (5). Interestingly, loss of SPT6 not only reduces the RNAPII levels on transcribed genes (9), but also affects transcription initiation in yeasts (13). Consistently, in plants, we have shown here that decreased occupancies of RNAPII in spt6l were also found at both gene bodies (Figure 1F) and TSS (Figure 2J). In Drosophila, promoter-proximal pausing is considered as a regulatory point for coordinating transcription initiation and elongation (39,40). In plants, the apparent lack of homologs of pausing factors (41) suggests that plants might employ a different strategy to manage the two processes during transcription. The physical presence of SPT6L around the TSS and the partially recovered RNAPII occupancy by SPT6LΔtSH2 (Figure 2H–J) point to a possibility that SPT6L may play roles in both transcription initiation and elongation.
How SPT6L is recruited to the TSS? Structurally, the SPT6 protein can be simply divided into three parts: N-terminal acidic region, core region, and C-terminal region (38,42). The N-terminal region of SPT6 interacts with histones and is involved in the maintenance of genome integrity during transcription (30,31,43,44). The C-terminal region of SPT6, including the tSH2 domain, binds to the phosphorylated linker region of RNAPII during transcription (6,38). In Arabidopsis, the tSH2 domain is followed by a plant-specific GW/WG domain (14), which was proposed to interact with the Argonaute proteins, and is involved in small RNA-mediated gene silencing (45). However, our double-deletion experiments suggest that both the N-terminal and GW/WG domains are less likely to contribute to the TSS enrichment (Figure 3H). The core region of SPT6 consists of the HtH, YqgF, HhH, and S1 domains (38,42). In an activated elongation complex, the core region of SPT6 interacts with the KOWx-KOW4 domain of SPT5 and RNAPII in vitro (38). Consistent with this observation, we found that the interaction between SPT6L and RNAPII was reduced after deleting the HtH or YqgF domain (Figure 4E). Importantly, based on the crystal structure of SPT6 (42), it was proposed that the HtH domain may interact with DNA or proteins (42). These observations point to a possibility that the TSS enrichment of SPT6L might result from interaction(s) between the HtH/YqgF domains and initiation/early elongation factors. Future works are warranted to identify these initiation/early elongation factors.
In our ChIP-seq results, the TSS enrichment and gene body occupancy of SPT6L were both impaired by deletion of the HtH/YqgF domains (Figure 4F). Two possible scenarios could be postulated. One possibility is that the loss of HtH/YqgF may completely disrupt the protein structure, and consequently, the function of SPT6L. The other possibility is that the TSS enrichment may be required for the deposition of SPT6L over gene bodies. The fact that RNAPII and HtH/YqgF-deleted versions of SPT6L could still interact, albeit weakly (Figure 4E), implies that the structures of the mutated proteins were not completely disrupted. On the other hand, the presence of SPT6L at the TSS, rather than the same peak position as RNAPII/RNAPIIS2P after induction (Figure 5B), of the heat shock response genes, suggests that the TSS recruitment of SPT6L may precede the recognition of tSH2 by phosphorylated RNAPII during transcription.
In summary, by integrating genetic and molecular evidence, our work reveals the RNAPII independent recruitment of SPT6L around the TSS region in plants, implying that this recruitment may precede its spreading over the gene body (Figure 5B). Future work on the identification of recruiters of SPT6L at the TSS will certainly provide new insights into the involvement of SPT6L in transcription initiation and how initiation and elongation are coordinated to ensure a productive transcription.
DATA AVAILABILITY
The data, supporting the findings of this study, are available, from the corresponding authors upon request. The ChIP-seq data have been deposited in Gene Expression Omnibus with the accession code GSE108673.
The secure token for reviewing the GSE108673 is qnafmmeinvkrxqd.
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108673
We have upload our data tracks into ENPG (http://www.plantseq.org/) and reviewers can check our data tracks with the following link (http://www.plantseq.org/jbrowse/?data=data%2FArabidopsis&tracks=3329%2C3327%2C3326%2C3349%2C3340%2C3331%2C3355%2C3346%2C3337%2C3343%2C3334%2CTAIR10&loc=1%3A2852616..2889066&highlight=).
The work doesn’t include specialized or in-house scripts and all the parameters for data analyzing were provided in Materials and Methods section.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Arabidopsis Biological Resource Centre for providing the mutant seeds used in this study. Author contributions: C.C. and Y.C. conceived and designed the experiments; J.S. performed immunoblot and Co-immunoprecipitation; R.K.T. performed Real time-qPCR to quantify HSP70 expression level after heat shock; C.L. obtained and validated the spt6l mutant. C.C. generated all the constructs, performed ChIP-seq and bioinformatics analysis; V.N., K.Y., Z.Y., S.E.K., J.L., F.M. and S.H. contributed to sequencing, critical reagents, and data analysis. C.C. and Y.C. wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Science and Engineering Research Council of Canada [RGPIN/04625-561 2017 to Y.C.]; Agriculture and Agri-Food Canada (to Y.C.); Natural Science Foundation of China [31170286 and 31470287 to S.H.]; The Specialized Research Fund for the Doctoral Program of Higher Education [20100171110034 to S.H.]; National Natural Science Foundation of China [31640059, to J.L.]. C.C. and J.S. were supported by a graduate fellowship from the China Scholarship Council. Funding for open access charge: Natural Science and Engineering Research Council of Canada.
Conflict of interest statement. None declared.
REFERENCES
- 1. Andrulis E.D., Guzman E., Doring P., Werner J., Lis J.T.. High-resolution localization of Drosophila Spt5 and Spt6 at heat shock genes in vivo: roles in promoter proximal pausing and transcription elongation. Genes Dev. 2000; 14:2635–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaplan C.D., Morris J.R., Wu C., Winston F.. Spt5 and spt6 are associated with active transcription and have characteristics of general elongation factors in D. melanogaster. Genes Dev. 2000; 14:2623–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mayer A., Lidschreiber M., Siebert M., Leike K., Soding J., Cramer P.. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 2010; 17:1272–1278. [DOI] [PubMed] [Google Scholar]
- 4. Endoh M., Zhu W., Hasegawa J., Watanabe H., Kim D.K., Aida M., Inukai N., Narita T., Yamada T., Furuya A. et al.. Human Spt6 stimulates transcription elongation by RNA polymerase II in vitro. Mol. Cell Biol. 2004; 24:3324–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ardehali M.B., Yao J., Adelman K., Fuda N.J., Petesch S.J., Webb W.W., Lis J.T.. Spt6 enhances the elongation rate of RNA polymerase II in vivo. EMBO J. 2009; 28:1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sdano M.A., Fulcher J.M., Palani S., Chandrasekharan M.B., Parnell T.J., Whitby F.G., Formosa T., Hill C.P.. A novel SH2 recognition mechanism recruits Spt6 to the doubly phosphorylated RNA polymerase II linker at sites of transcription. ELife. 2017; 6:e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun M., Lariviere L., Dengl S., Mayer A., Cramer P.. A tandem SH2 domain in transcription elongation factor Spt6 binds the phosphorylated RNA polymerase II C-terminal repeat domain (CTD). J. Biol. Chem. 2010; 285:41597–41603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoh S.M., Cho H., Pickle L., Evans R.M., Jones K.A.. The Spt6 SH2 domain binds Ser2-P RNAPII to direct Iws1-dependent mRNA splicing and export. Genes Dev. 2007; 21:160–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dronamraju R., Hepperla A.J., Shibata Y., Adams A.T., Magnuson T., Davis I.J., Strahl B.D.. Spt6 association with RNA polymerase II directs mRNA turnover during transcription. Mol. Cell. 2018; 70:1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheung V., Chua G., Batada N.N., Landry C.R., Michnick S.W., Hughes T.R., Winston F.. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 2008; 6:e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hennig B.P., Fischer T.. The great repression: chromatin and cryptic transcription. Transcription. 2013; 4:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaplan C.D., Laprade L., Winston F.. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003; 301:1096–1099. [DOI] [PubMed] [Google Scholar]
- 13. Doris S.M., Chuang J., Viktorovskaya O., Murawska M., Spatt D., Churchman L.S., Winston F.. Spt6 is required for the fidelity of promoter selection. Mol. Cell. 2018; 72:687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gu X.L., Wang H., Huang H., Cui X.F.. SPT6L encoding a putative WG/GW-repeat protein regulates apical-basal polarity of embryo in Arabidopsis. Mol. Plant. 2012; 5:249–259. [DOI] [PubMed] [Google Scholar]
- 15. Antosz W., Pfab A., Ehrnsberger H.F., Holzinger P., Kollen K., Mortensen S.A., Bruckmann A., Schubert T., Langst G., Griesenbeck J. et al.. The composition of the arabidopsis RNA polymerase II transcript elongation complex reveals the interplay between elongation and mRNA processing factors. Plant Cell. 2017; 29:854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li C., Gu L., Gao L., Chen C., Wei C.Q., Qiu Q., Chien C.W., Wang S., Jiang L., Ai L.F. et al.. Concerted genomic targeting of H3K27 demethylase REF6 and chromatin-remodeling ATPase BRM in Arabidopsis. Nat. Genet. 2016; 48:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen C., Li C., Wang Y., Renaud J., Tian G., Kambhampati S., Saatian B., Nguyen V., Hannoufa A., Marsolais F. et al.. Cytosolic acetyl-CoA promotes histone acetylation predominantly at H3K27 in Arabidopsis. Nat. Plants. 2017; 3:814–824. [DOI] [PubMed] [Google Scholar]
- 18. Curtis M.D., Grossniklaus U.. A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol. 2003; 133:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang X., Henriques R., Lin S.S., Niu Q.W., Chua N.H.. Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 2006; 1:641–646. [DOI] [PubMed] [Google Scholar]
- 20. Shu J., Chen C., Thapa R.K., Bian S., Nguyen V., Yu K., Yuan Z.-C., Liu J., Kohalmi S.E., Li C. et al.. Genome-wide occupancy of histone H3K27 methyltransferases CURLY LEAF and SWINGER in Arabidopsis seedlings. Plant Direct. 2019; 3:e00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salmon-Divon M., Dvinge H., Tammoja K., Bertone P.. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics. 2010; 11:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo C., Sidote D.J., Zhang Y., Kerstetter R.A., Michael T.P., Lam E.. Integrative analysis of chromatin states in Arabidopsis identified potential regulatory mechanisms for natural antisense transcript production. Plant J. 2013; 73:77–90. [DOI] [PubMed] [Google Scholar]
- 26. Gilchrist D.A., Dos Santos G., Fargo D.C., Xie B., Gao Y., Li L., Adelman K.. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell. 2010; 143:540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu J., Liu M., Liu X., Dong Z.. RNA polymerase II activity revealed by GRO-seq and pNET-seq in Arabidopsis. Nat. Plants. 2018; 4:1112–1123. [DOI] [PubMed] [Google Scholar]
- 28. DeGennaro C.M., Alver B.H., Marguerat S., Stepanova E., Davis C.P., Bahler J., Park P.J., Winston F.. Spt6 regulates intragenic and antisense transcription, nucleosome positioning, and histone modifications genome-wide in fission yeast. Mol. Cell Biol. 2013; 33:4779–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeronimo C., Watanabe S., Kaplan C.D., Peterson C.L., Robert F.. The histone chaperones FACT and Spt6 restrict H2A.Z from intragenic locations. Mol. Cell. 2015; 58:1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ivanovska I., Jacques P.E., Rando O.J., Robert F., Winston F.. Control of chromatin structure by spt6: different consequences in coding and regulatory regions. Mol. Cell Biol. 2011; 31:531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Bakel H., Tsui K., Gebbia M., Mnaimneh S., Hughes T.R., Nislow C.. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLos Genet. 2013; 9:e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bensaude O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity. Transcription. 2011; 2:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chao S.H., Price D.H.. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001; 276:31793–31799. [DOI] [PubMed] [Google Scholar]
- 34. Adelman K., Lis J.T.. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 2012; 13:720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Min I.M., Waterfall J.J., Core L.J., Munroe R.J., Schimenti J., Lis J.T.. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev. 2011; 25:742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar S.V., Wigge P.A.. H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell. 2010; 140:136–147. [DOI] [PubMed] [Google Scholar]
- 38. Vos S.M., Farnung L., Boehning M., Wigge C., Linden A., Urlaub H., Cramer P.. Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature. 2018; 560:607–612. [DOI] [PubMed] [Google Scholar]
- 39. Krebs A.R., Imanci D., Hoerner L., Gaidatzis D., Burger L., Schubeler D.. Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters. Mol. Cell. 2017; 67:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shao W., Zeitlinger J.. Paused RNA polymerase II inhibits new transcriptional initiation. Nat. Genet. 2017; 49:1045–1051. [DOI] [PubMed] [Google Scholar]
- 41. Hajheidari M., Koncz C., Eick D.. Emerging roles for RNA polymerase II CTD in Arabidopsis. Trends Plant Sci. 2013; 18:633–643. [DOI] [PubMed] [Google Scholar]
- 42. Close D., Johnson S.J., Sdano M.A., McDonald S.M., Robinson H., Formosa T., Hill C.P.. Crystal structures of the S. cerevisiae Spt6 core and C-terminal tandem SH2 domain. J. Mol. Biol. 2011; 408:697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bortvin A., Winston F.. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science. 1996; 272:1473–1476. [DOI] [PubMed] [Google Scholar]
- 44. Farnung L., Vos S.M., Cramer P.. Structure of transcribing RNA polymerase II-nucleosome complex. Nat. Commun. 2018; 9:5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karlowski W.M., Zielezinski A., Carrere J., Pontier D., Lagrange T., Cooke R.. Genome-wide computational identification of WG/GW Argonaute-binding proteins in Arabidopsis. Nucleic Acids Res. 2010; 38:4231–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data, supporting the findings of this study, are available, from the corresponding authors upon request. The ChIP-seq data have been deposited in Gene Expression Omnibus with the accession code GSE108673.
The secure token for reviewing the GSE108673 is qnafmmeinvkrxqd.
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108673
We have upload our data tracks into ENPG (http://www.plantseq.org/) and reviewers can check our data tracks with the following link (http://www.plantseq.org/jbrowse/?data=data%2FArabidopsis&tracks=3329%2C3327%2C3326%2C3349%2C3340%2C3331%2C3355%2C3346%2C3337%2C3343%2C3334%2CTAIR10&loc=1%3A2852616..2889066&highlight=).
The work doesn’t include specialized or in-house scripts and all the parameters for data analyzing were provided in Materials and Methods section.