


Abstract
Regions of genomic instability are not random and often co-localize with DNA sequences that can adopt alternative DNA structures (i.e. non-B DNA, such as H-DNA). Non-B DNA-forming sequences are highly enriched at translocation breakpoints in human cancer genomes, representing an endogenous source of genetic instability. However, a further understanding of the mechanisms involved in non-B DNA-induced genetic instability is needed. Small molecules that can modulate the formation/stability of non-B DNA structures, and therefore the subsequent mutagenic outcome, represent valuable tools to study DNA structure-induced genetic instability. To this end, we have developed a tunable Förster resonance energy transfer (FRET)-based assay to detect triplex/H-DNA-destabilizing and -stabilizing ligands. The assay was designed by incorporating a fluorophore-quencher pair in a naturally-occurring H-DNA-forming sequence from a chromosomal breakpoint hotspot in the human c-MYC oncogene. By tuning triplex stability via buffer composition, the assay functions as a dual-reporter that can identify stabilizers and destabilizers, simultaneously. The assay principle was demonstrated using known triplex stabilizers, BePI and coralyne, and a complementary oligonucleotide to mimic a destabilizer, MCRa2. The potential of the assay was validated in a 384-well plate with 320 custom-assembled compounds. The discovery of novel triplex stabilizers/destabilizers may allow the regulation of genetic instability in human genomes.
INTRODUCTION
Genetic instability that underlies many diseases is characterized by high mutation frequencies at certain ‘mutation hotspot’ regions. These hotspots of genetic instability, including point mutations, deletions, translocations and rearrangements are not random; however, the mechanisms involved are yet to be fully clarified. Recent studies have demonstrated that mutation hotspots often co-localize with naturally-occurring repetitive sequences that can adopt alternatively structured DNA (i.e. non-B DNA, e.g. H-DNA), implicating non-B DNA in disease etiology (1–3). Further, it has been shown that these non-B DNA-forming sequences can induce genetic instability in mammalian cells and in mice (4–6).
H-DNA is an intramolecular triplex DNA structure that forms at polypurine-polypyrimidine mirror-repeat sequences. With the energy provided by negative supercoiling (e.g. during DNA replication, transcription, or repair), a single strand from one half of the mirror symmetry can fold back and bind in the major groove of the duplex across the symmetry plane (7,8). This binding occurs via Hoogsteen or reverse Hoogsteen hydrogen bonding through the major groove of the underlying duplex, thereby forming an intramolecular triplex structure while leaving the complementary strand unpaired. There are two major triads formed: the R*RY triad (R: purine, Y: pyrimidine, *reverse Hoogsteen H-bonds), which can be stabilized by divalent cations (e.g. Mg2+), such as the H-DNA-forming sequence used in this study that co-localizes with a common translocation breakpoint in the human c-MYC gene in Burkitt's lymphoma (Figure 1A) (9–11); and an acidic pH-dependent Y*RY (*Hoogsteen H-bonds) type, when the pyrimidine-rich strand serves as the third strand (7,8,12).
Figure 1.
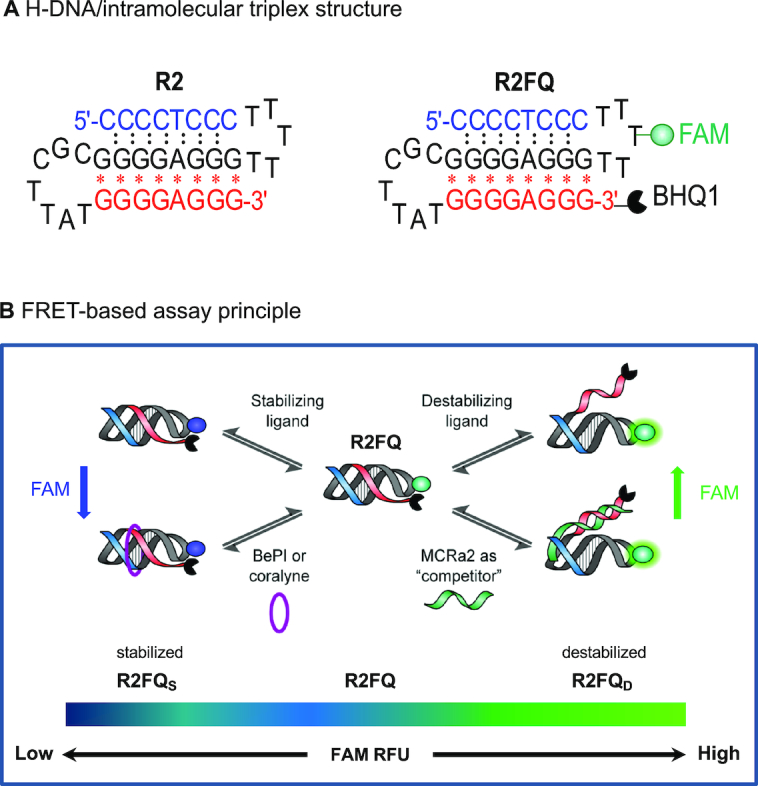
Schematic illustrations of (A) the H-DNA or intramolecular triplex structure used in this study; (B) the FRET-based assay to identify H-DNA/triplex ligands. R2FQ, R2FQS and R2FQD represent R2FQ in solution alone, in the presence of a stabilizer, and in the presence of a destabilizer, respectively.
With evidence linking the DNA structure itself to disease etiology, non-B DNA structure formation and stability are paramount to the mutagenic process. One of the challenges in this area of research, particularly that of H-DNA, is the demonstration that small molecule ligands can modulate non-B DNA structure formation and cause subsequent modulation of the mutagenic outcome of these structures. Further, effective structure-specific, fluorescent H-DNA recognition agents that can serve as real-time probes in the visualization of H-DNA loci in vivo are warranted. The most well-characterized triplex binding ligands, BePI (13) and coralyne (14), lose their fluorescence upon binding to their triplex substrates (15). Other compounds such as the rationally designed triplex ligand BQQ (16,17), YOYO (18), pyrene (19), thiazole orange (20), Cyan 40, (21), and DMT (22) were reported to fluoresce when bound to triplex DNA, but these can be non-specific. This is in contrast to a plethora of G4-DNA-specific fluorescent probes (23–25).
While several triplex stabilizers have been characterized (15,26–29), the identification of triplex destabilizers, on the other hand, is lacking. The few identified destabilizers are mostly limited to minor groove binders, which destabilize the triplex because of their preference for the duplex structures (15,28,30). This bottleneck is in part a result of the shortage of efficient methods to assay for triplex-destabilizing molecules. Of the published assays, most have been designed to identify intercalators or groove-binders, which typically stabilize the triplex structures (15,30,31). The standard method of determining stabilizing or destabilizing effects of a ligand on nucleic acid structures has largely been based on thermal melting measurements (32–34). The limited sampling throughput using this approach has been alleviated through the use of fluorophore–quencher (FQ) combinations (35,36), allowing high-throughput FRET-based thermal melting assays (37–39). However, the thermal melting and monitoring takes time and is sensitive to the nature of the buffers and fluorophores used. Additionally, heating of the samples can complicate the reading/interpretation of the binding process. Further, most ligands identified to date have been tested against triplex homopolymers rather than naturally-occurring H-DNA-forming sequences. The use of high throughput screens (HTS) to identify triplex intercalators has also been rather limited to virtual screening platforms (40). Thus, the development of facile, high-throughput assays that can identify triplex stabilizers and destabilizers using naturally-occurring structure-forming sequences is warranted. The goal of this study was to address this gap.
Thus, in this study, we developed an assay to detect both triplex/H-DNA-stabilizing and -destabilizing ligands and applied the assay in a screen for small-molecule binders using an HTS platform. We utilized the sequence from a chromosomal breakpoint hotspot in the human c-MYC oncogene, which we have shown to form intramolecular triplex/H-DNA (41) (Figure 1A, denoted as ‘R2’). The FRET-based assay employed a FAM–BHQ1 pair covalently linked to the R2 sequence (Figure 1A, denoted as ‘R2FQ’). It is a dual-reporter system, which is the first of its kind that can identify both triplex stabilizers and destabilizers, simultaneously, in a facile HTS-compatible approach (Figure 1B). The assay principle was demonstrated using known triplex stabilizers/intercalators, BePI (13,42,43) and coralyne (14,44–47), and a triplex destabilizer in the form of a complementary sequence to the third strand, MCRa2. The potential of the FRET-based assay to identify triplex binders in a high throughput fashion was tested using 320 custom-assembled compounds in a 384-well plate format.
MATERIALS AND METHODS
Materials
Unless otherwise stated, all chemicals were purchased from Sigma (St. Louis, MO, USA) and were of the highest purity available. Oligonucleotides (Supplementary Table S1) were obtained from Midland Certified Reagent Company, Inc (Midland, TX, USA) and purified by polyacrylamide gel electrophoresis (PAGE), or by reverse-phase high-performance liquid chromatography (HPLC). Oligonucleotides were stored at -20°C until resuspended. Fluorescence assays were performed in a Nunc black 384-well plate obtained from Thermo Scientific (Waltham, MA, catalog # 262260). Coralyne and BePI, used as controls, were purchased from Sigma (Catalog #R278122 and #B7417, respectively) and used as received without further purification.
We determined that the full-length oligonucleotides migrated as single bands by using denaturing PAGE [8 M urea, 89 mM Tris-borate, 2 mM EDTA (TBE)] on 20% acrylamide gels stained with SYBR Gold (Thermo Fisher, Waltham, MA, USA). Concentrations of oligonucleotide stock solutions prepared in nuclease-free water were calculated using their molar extinction coefficient, ϵ (Supplementary Table S1) and absorbances measured at 260 nm using a NanoDrop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). During assay optimization, we tested two buffer conditions: a ‘sodium cacodylate’ buffer (20 mM sodium cacodylate, pH 7.0, 100 mM NaCl, 0.1 mM EDTA, 10 mM MgCl2), which we previously used in characterizing triplex formation of the R2 substrate (41); or a ‘Tris buffer’ (20 mM Tris–HCl, pH 7.4), which has also been used for triplex formation (45,48). In some instances (e.g. all fluorescence-based assays, and CD analyses), 0.01% Tween 20 was added to the buffer.
Chemical compounds
A custom-assembled library containing 320 compounds with known bioactivity against diverse therapeutic targets was acquired from Selleckchem (Houston, TX, USA). Their biological and pharmacological activities have been validated by preclinical research and clinical trials. Also, some compounds in this library were approved by the FDA. Compounds were acquired as a liquid form dissolved in 100% DMSO at 10 mM concentrations and were used as received. All compounds were subjected to rigorous quality control using LC–MS and/or NMR to meet the requirement of >98% purity by the vendor with few exceptions. Details of the compounds are summarized in Supplementary Table S2. For the in-house chemical characterization of doxorubicin, high-resolution mass and liquid chromatography mass spectral data were obtained at the University of Texas at Austin (Austin, TX, USA).
Gel mobility assays using native PAGE
To determine the buffer-specific mobility of the oligonucleotides as an assessment of intramolecular structure formation, oligonucleotides were subjected to native PAGE on 20% native mini gels (19:1 acrylamide/bisacrylamide). DNA at a concentration of 3–5 μM was annealed, then ∼100 pg was mixed with Bio-Rad Nucleic Acid Loading Buffer (15 μl loading volume) and electrophoresed on a 20% native polyacrylamide gel in a cold room at 40–50 V using 1x Tris-Borate (89 mM) in the presence or absence of Mg2+ (at 10 mM final concentration). DNA mobility was visualized on a Chemidoc XRS+ Gel Imaging System (Bio-Rad, Hercules, CA) using Image Quant TL v7 software after SYBR Gold (emission filter: 520–550 nm) staining for 30–45 min. Native gels also included 10- and 20-base pair markers and T30/T40 single-strand polydT markers.
Circular dichroism (CD) spectroscopy
As an additional technique to determine DNA secondary structure formation, circular dichroism (CD) was used. The CD spectra of annealed DNA (10 μM, 300 μl) in 1 cm quartz cuvettes were recorded at 25°C using a Jasco J-815 (Jasco Inc., Easton, MD, USA) CD spectrometer equipped with a Peltier temperature controller. The 200–350 nm range was scanned three times at a rate of 100 nm/min with a 2 s response time. Buffer-corrected CD data, collected as mdeg as a function of wavelength, were processed using the Jasco Spectra Manager software and exported to Graph Pad Prism (La Jolla, CA, USA). As 0.01% Tween 20 was used in the FRET experiments (see below) we routinely included it in the CD experiments when assessing DNA secondary structure.
Development of a FRET-based intramolecular triplex assay
Oligonucleotide solutions were re-suspended in the stated buffer and annealed by heating to 95°C for 5 min followed by slow cooling to room temperature. The annealed DNA solutions were kept at 4°C prior to their use in experiments. Assays were conducted in the presence of sodium cacodylate or Tris buffers in 20 μl volumes at room temperature. We found that the addition of 0.01% Tween 20 maintained the fluorescence signal in all the FRET experiments reported here and was thus used routinely. The final concentration of R2FQ was set to 20 nM in all experiments. The assay was first examined in a 20 μl volume of R2FQ solution by adding either buffer, BePI (7.5 μM final) or MCRa2 (0.5 μM final). The FAM fluorescence was monitored hourly on a Synergy H4 multi-functional plate reader (BioTek, Winooski, VT) for up to 16 h with excitation and emission at 495 and 520 nm, respectively. For experiments examining the effect of Na+ and Mg2+ ions, R2FQ (20 nM final) was mixed with an equal volume of buffer containing either Na+ ions (0–100 mM final) or Mg2+ ions (0–10 mM final). After optimizing the buffer composition, assays were conducted in Tris buffer for further validation. Dose-response assays with BePI, coralyne, or MCRa2 were conducted by mixing R2FQ (20 nM final) with equal volumes of buffer containing ligands. The solutions were incubated at room temperature for 16 h and the fluorescence was recorded on a plate reader. Apparent dissociation constants (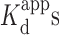 ) were calculated by fitting the experimental data to a one-site ligand binding saturation model using Sigmaplot 13.0 (Systat, San Jose, CA, USA).
) were calculated by fitting the experimental data to a one-site ligand binding saturation model using Sigmaplot 13.0 (Systat, San Jose, CA, USA).
Application to compound screening and ‘hit’ confirmation
The FRET-based assay described above was used to screen for triplex stabilizers or destabilizers in a custom-assembled library containing 320 compounds with known bioactivities (Supplementary Table S2). Because the compounds were dissolved in DMSO, we performed initial tests for DMSO tolerance by mixing a solution of R2FQ (20 nM final) with a DMSO solution (0–2.5%, v/v final). To validate the assay for automation, a 20 μl solution of R2FQ dissolved in Tris assay buffer (20 nM) was dispensed into an assay plate using a Microflo select bulk dispenser (BioTek, Winooski, VT). Twenty μl of buffer-only solution was dispensed to wells as a negative control. Then 100% DMSO (20 nL) was distributed into each well using an Echo 550 Acoustic liquid handler (San Jose, CA, USA). Plates were then incubated for 16 h and fluorescence readings recorded on a plate reader with excitation and emission at 495 and 520 nm, respectively. As internal controls, known triplex binders BePI/coralyne (7.5 μM final) as stabilizers, and MCRa2 (0.5 μM final) as a destabilizer were added manually to designated wells in duplicate. The z’ value was calculated using an equation reported by Zhang et al. (49). The compound screening was conducted similarly to the assay validation method, where 20 nl volumes of each compound dissolved in 100% DMSO (10 μM final) were dispensed. To confirm doxorubicin binding via the FRET assay, 10 μl of a serial dilution of doxorubicin (0–10 μM) was dissolved in assay buffer with an equal volume of 40 nM R2FQ solution. After 16 h incubation, the fluorescence was recorded on a plate reader with excitation and emission at 495 and 520 nm, respectively.
UV thermal denaturation experiments
To determine the thermal melting temperature of R2 (0.5 μM) in the presence of coralyne, DNA solutions were annealed in the presence of Tris buffer without Tween 20. Thermal denaturation of the DNA solutions in 1 cm quartz cuvettes was monitored from 30 to 100°C at 260 nm using a Cary 4000 UV–Vis equipped with a Peltier temperature controller (Santa Clara, CA, USA) using a ramp rate of 0.4°C/min. The end point of the melting curves was normalized to Abs = 1.0 and the thermal melting temperature (Tm) calculated as the maximum value of the first derivative of the absorbance versus temperature using the thermal application of Cary WinUV Ver6.2 software (Santa Clara, CA, USA). The thermal melting assays of the R2 and GT triplex substrates (0.5 μM), in the absence and presence of 0.5 μM of doxorubicin, were conducted as described above.
RESULTS
In this study, we aimed to develop a facile HTS-compatible, fluorescence-based assay to screen for H-DNA/triplex ligands using a naturally-occurring H-DNA/triplex structure-forming sequence from a translocation breakpoint hotspot in the human c-MYC oncogene (R2, Figure 1A). Unlike many homopolymeric DNA sequences, this sequence and its structure-formation is of high biological relevance because of its implication in cancers such as leukemia and lymphoma (1,4,50–53). Previously, we have demonstrated that R2 forms an H-DNA/intramolecular triplex structure both in plasmid (6,50) and synthetic oligonucleotide DNA substrates (4,41). We have also shown that this H-DNA-forming sequence induces genetic instability in mammalian cells and on chromosomes in mouse tissues (5,6), largely resulting in the formation of DNA double-strand breaks, and subsequent deletions, rearrangements and translocations. Inspired by the molecular beacon technology (54,55), and other published assays utilizing fluorophore-quencher (FQ) combinations (35–39,56), this FRET-based assay (Figure 1B) was designed by incorporating a fluorophore (FAM) into the R2 loop sequence (41), and a quencher (BHQ1) at its 3′-end (R2FQ, Figure 1A). The FAM fluorescence was shown to be altered in conjunction with triplex structure modulation by small molecule ligands and a destabilizer mimic, demonstrating the ability of the assay to simultaneously detect both triplex stabilizers and destabilizers. As the oligonucleotide folded into a weakly formed intramolecular triplex structure (depending on buffer composition), the resulting close proximity of the fluorophore-quencher pair resulted in partial quenching of FAM fluorescence (R2FQ, Figure 1B). Notably, the RFU approached background levels when R2FQ was stabilized (R2FQS, Figure 1B), while it was similar to that of free FAM when the R2FQ was destabilized (R2FQD, Figure 1B). Thus, the relative fluorescence intensity (RFU) of FAM can serve as an indicator of triplex stability.
The triplex-structure determination in the presence of the FAM–BHQ1 pair
We tested two buffer conditions: a ‘sodium cacodylate’ triplex-forming buffer (20 mM sodium cacodylate, pH 7.0, 100 mM NaCl, 0.1 mM EDTA, 10 mM MgCl2), which we previously used in characterizing the triplex formation of the R2 sequence (41); or a ‘Tris buffer’ (20 mM Tris–HCl pH 7.4), which has also been used for triplex formation (45,48). In some instances (e.g. all fluorescence-based assays, and CD analyses), 0.01% Tween 20 was added to the buffer. Despite its lower pH (pH 7 compared to Tris at pH 7.4), sodium cacodylate buffer may support more stable triplex formation than the Tris buffer due to the presence of ions (Na+ and Mg2+). These ions, in particular the Mg2+ ions, help neutralize the negatively charged DNA phosphate backbone, resulting in a tighter interaction of the three strands of a triplex structure (30,57,58). We hypothesized that the absence of Na+ or Mg2+ ions in the Tris buffer solution would support a less stable triplex structure, amenable for analysis of structure modulation by the ligands.
To optimize conditions for screening, we assessed triplex formation in both sodium cacodylate and Tris buffers. To characterize triplex formation of R2 and R2FQ under Tris buffer conditions, with and without Mg2+, we used gel mobility assays using native PAGE, thermal melting, and CD spectroscopy as we have described (41). The additional molecular weight from FAM and BHQ caused a slight decrease in electrophoretic mobility on the polyacrylamide gels but did not appear to indicate aggregation or further secondary structure formation (Supplementary Figure S1A). The mobilities of R2 and R2FQ were similar when incubated in Tris or sodium cacodylate buffers, and the presence of Mg2+ had little effect on the mobilities of R2 and R2FQ in the Tris buffer, (compare Supplementary Figure S1A versus Supplementary Figure S1B). These results indicated that secondary structure formation still occurred even in the absence of Mg2+, consistent with our previous observations (41). We found that the presence of FAM and BHQ1 did not negatively affect triplex formation in Tris buffer, as evidenced by the presence of a triplex CD signature, i.e., a negative peak at 210–220 nm (41,59,60) (Supplementary Figure S1C, D). In addition, the predicted ‘weak’ folding of the triplex structure under Tris buffer conditions in the absence of Mg2+ [‘Tris no Mg2+ condition’ (Supplementary Figure S1D)] was indicated by the weaker triplex CD signature, relative to that in the presence of Mg2+. In summary, the use of Tris buffer (20 mM Tris buffer, pH 7.4) supported triplex structure formation of R2FQ, albeit only weakly in the absence of Mg2+ ions. Based on these preliminary investigations, we explored the potential of the FRET-based assay as a dual reporting platform in Tris buffer.
Development of a FRET-based intramolecular triplex assay
We initially examined the FRET-based assay using Tris buffer, containing 0.01% Tween 20 (which stabilized the fluorescence) to screen for triplex destabilizers only, using a mimic in the form of the DNA sequence, MCRa2, which is complementary to the reverse Hoogsteen H-bonded strand (‘third strand’) of the R2FQ sequence. MCRa2 binding to the third strand facilitated the separation of the FAM/BHQ1 pair (by duplex formation, and subsequent loss of the triplex structure), resulting in an increase in FAM fluorescence. As demonstrated in Figure 2, addition of MCRa2 (2.5 μM) to R2FQ (20 nM) resulted in a ∼5-fold increase in fluorescence intensity compared to R2FQ alone. We also observed that the fluorescence of R2FQ was ∼4-fold higher than background, suggesting that the R2 sequence was weakly folded, and thus could be further stabilized or folded. This was then demonstrated by the addition of the triplex intercalator, BePI (7.5 μM), which further reduced the fluorescence intensity of R2FQ (by ∼2-fold Figure 2). To eliminate the possibility that this fluorescence decrease was due to FAM quenching by BePI, a separate titration of BePI and fluorescein alone was performed. We did not observe any fluorescein quenching in the presence of BePI alone (Supplementary Figure S2), thus, the relative fluorescence signal appeared to reflect the stability of the triplex structure, such that the R2FQ fluorescence was near background levels when the intramolecular triplex was near optimal stabilization in the presence of an intercalator (R2FQS, Figure 1B). In addition, the FAM fluorescence was similar to that of the free fluorophore for the destabilized or unfolded (R2FQD, Figure 1B) triplex in the presence of MCRa2, which prevented triplex formation by binding to the third strand. These preliminary results indicated that in Tris buffer, this assay was able to detect both triplex stabilizers and destabilizers in a single platform.
Figure 2.

Initial evaluation of the FRET-based assay in the presence of both a triplex stabilizer and a destabilizer-mimic in Tris buffer. Fluorescence intensity change of R2FQ compared to samples with BePI (stabilizer, 7.5 μM), MCRa2 (destabilizer-mimic, 2.5 μM) and that of background (Buffer only).
Optimizing the dynamic range
We sought buffer conditions that provided robust sensitivity for both triplex stabilization and destabilization. In order to finely tune the stability of R2 in Tris buffer, we explored whether Na+ or Mg2+ ions modulated the stability of the R2FQ triplex (free R2FQ, in the absence of ligands). We expected to effectively modulate the stability of R2FQ and therefore, the dynamic range of the fluorescent signal by varying the concentrations of Na+ (0–100 mM) and Mg2+ (0–10 mM) ions. Although the RFUs of R2FQ were relatively consistent, spanning a range of 1800–2300 RFU, there was a trend of increased fluorescence with decreasing Mg2+ concentration (Figure 3A, left panel). This indicated that the presence of Mg2+ resulted in more efficient quenching, reflecting a more stable triplex-forming condition for R2. Accordingly, the intramolecular triplex was consistently more fluorescent (i.e. less stable) in the absence of Mg2+ ions (Figure 3A, left panel). We hypothesized that this high fluorescence/structural instability in the absence of Mg2+ ions would result in a greater change in fluorescence upon triplex stabilization. It should be noted that the presence or absence of Na+ had little effect on R2FQ fluorescence (Figure 3A, right panel).
Figure 3.

Optimization of buffer conditions and effects of Mg2+ (left panel) and Na+ (right panel) ions in Tris buffer. (A) R2FQ fluorescence alone. Fluorescence signal changes by (B) BePI (7.5 μM)-induced stabilization (ΔRFU = RFUwo/BePI - RFUw/BePI) and (C) MCRa2 (500 nM)-induced destabilization (ΔRFU = RFUw/MCRa2 – RFUwo/MCRa2).
Next, we tested the assay conditions in the presence of the triplex intercalator/stabilizer, BePI, or the triplex destabilizer mimic, MCRa2 (Figures 3B and 3C). When the assay was subjected to an excess concentration of BePI (7.5 μM), and compared to R2FQ (20 nM) alone, the signal change spanned a range of 300–2,000 RFU, depending on the Mg2+ and Na+ levels, with the greatest change in signal achieved in the absence of Mg2+ or Na+ ions (Figure 3B). It was suspected that the presence of Mg2+ or Na+ reduced the affinity of BePI for R2FQ by competing for triplex DNA binding. In the presence of MCRa2 (500 nM), the change in signal ranged from 7500–15 000 RFU and was slightly attenuated at lower concentrations of Mg2+ or Na+ ions. This is likely because MCRa2 is a short DNA sequence and cations may promote its complementary binding to the third strand of the triplex substrate. Accordingly, the observed destabilization by MCRa2 was most dramatic (∼15 000 RFU increase) when R2FQ was incubated in the presence of either 10 mM Mg2+ or 100 mM of Na+ (Figure 3C). In the absence of Mg2+ or Na2+ ions a smaller signal change was observed, which likely reflected the incomplete binding of MCRa2 to R2FQ under these conditions. In summary, we found that a wide dynamic range can be achieved in both directions (triplex stabilization and destabilization) in Tris buffer via control of Mg2+ levels and that the dynamic range for stabilization is much narrower than that of destabilization (2000 versus 15 000 ΔRFU).
Dose-response assay validation
Using Tris buffer in the absence and presence of 10 mM Mg2+, respectively, we performed a dose-dependent validation for triplex stabilizers using coralyne and BePI (15,26). The percent of triplex stabilization was calculated by normalizing the fluorescence in the presence of stabilizers against its fluorescence in the absence of the stabilizers. Addition of increasing concentrations of coralyne with R2FQ resulted in a dose-dependent decrease of FAM fluorescence, confirming the ability of coralyne to stabilize the triplex structure (Figure 4A). The calculated apparent dissociation constants (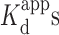 ) of coralyne in the absence or presence of 10 mM Mg2+ were 0.51 ± 0.06 (r2 = 0.992) and 0.92 ± 0.14 μM (r2 = 0.985), respectively (Figure 4B), comparable to published data using homopolymeric DNA sequences (14,45). Its lower affinity (higher
) of coralyne in the absence or presence of 10 mM Mg2+ were 0.51 ± 0.06 (r2 = 0.992) and 0.92 ± 0.14 μM (r2 = 0.985), respectively (Figure 4B), comparable to published data using homopolymeric DNA sequences (14,45). Its lower affinity (higher 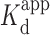 ) in the presence of Mg2+ indicated that the binding affinity of coralyne to the triplex DNA structure was reduced, perhaps by competition with Mg2+ ions in interacting with the triplex structure, which has been observed previously (61).
) in the presence of Mg2+ indicated that the binding affinity of coralyne to the triplex DNA structure was reduced, perhaps by competition with Mg2+ ions in interacting with the triplex structure, which has been observed previously (61).
Figure 4.

Dose-dependent binding characteristics of a known stabilizer, coralyne, and a destabilizer mimic, MCRa2, in the presence and absence of Mg2+ in Tris buffer. Dose-dependent signal change in the presence of (A) coralyne, and its (B) plot of % stabilization. Dose-dependent signal change in the presence of (C) MCRa2, and its (D) plot of % destabilization.
Our results with coralyne were similar to those with BePI in the presence of 10 mM Mg2+ (Supplementary Figure S3), demonstrating the potential of the FRET-based assay to screen for and measure binding affinities of potential triplex interacting ligands. BePI, in the presence of 10 mM Mg2+, affected a dose-dependent decrease in the FAM RFU, based on its ability to stabilize the triplex (Supplementary Figure S3) with a 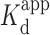 of 4.3 ± 1.1 μM (r2 = 0.979). However, in the absence of Mg2+, the R2FQ fluorescence initially increased up to 1.3 μM, and then decreased in a dose-dependent fashion until saturation (Supplementary Figure S3). These results suggested that in the absence of Mg2+, the presence of BePI at low concentrations (<1.3 μM) appeared to have destabilizing effects on triplex formation, whereas at higher concentrations (>1.3 μM), BePI appeared to have triplex-stabilizing effects. We believe that this concentration-dependent effect of BePI on triplex formation has not previously been reported and further studies are necessary to explain the mechanistic details. However, there have been previous reports of triplex ligands that can either stabilize or destabilize a triplex, depending on the concentration of bound ligand (62–64), ionic condition (63) and even triad sequence (65,66). Evaluation of dose-dependent triplex destabilization by MCRa2 in the presence of Mg2+ (Figures 4C and D) revealed that the binding was saturated above 124 nM MCRa2, with a calculated
of 4.3 ± 1.1 μM (r2 = 0.979). However, in the absence of Mg2+, the R2FQ fluorescence initially increased up to 1.3 μM, and then decreased in a dose-dependent fashion until saturation (Supplementary Figure S3). These results suggested that in the absence of Mg2+, the presence of BePI at low concentrations (<1.3 μM) appeared to have destabilizing effects on triplex formation, whereas at higher concentrations (>1.3 μM), BePI appeared to have triplex-stabilizing effects. We believe that this concentration-dependent effect of BePI on triplex formation has not previously been reported and further studies are necessary to explain the mechanistic details. However, there have been previous reports of triplex ligands that can either stabilize or destabilize a triplex, depending on the concentration of bound ligand (62–64), ionic condition (63) and even triad sequence (65,66). Evaluation of dose-dependent triplex destabilization by MCRa2 in the presence of Mg2+ (Figures 4C and D) revealed that the binding was saturated above 124 nM MCRa2, with a calculated 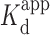 of 21 ± 3.6 nM (r2 = 0.974). In the absence of Mg2+, significant destabilization was observed up to 250 nM MCRa2, however saturation was not achieved. As noted above, this is consistent with the need for cations (e.g. Mg2+) to effectively promote Watson-Crick complementary pairing between MCRa2 and the third strand of the triplex substrate.
of 21 ± 3.6 nM (r2 = 0.974). In the absence of Mg2+, significant destabilization was observed up to 250 nM MCRa2, however saturation was not achieved. As noted above, this is consistent with the need for cations (e.g. Mg2+) to effectively promote Watson-Crick complementary pairing between MCRa2 and the third strand of the triplex substrate.
To correlate the fluorescence changes with the binding interactions between a stabilizer and a destabilizer of R2 triplex formation in Tris buffer, we performed CD experiments in the presence of coralyne or MCRa2 (Figure 5). The weak signature for triplex formation (a negative peak at 220 nm) was strengthened in the presence of coralyne. In addition, a positive CD signal appeared at 330–340 nm, which is characteristic of the intercalative binding interaction of coralyne with DNA (45,47,67,68). Combined, these results indicated that stabilization by intercalation took place upon coralyne binding with R2. Thermal melting experiments of 0.5 μM R2 in the presence of 0.5 μM coralyne revealed an ∼4°C increase in the thermal melting temperature (data not shown). This is similar to the increase in the thermal melting of a T*AT-containing short intramolecular triplex in the presence of coralyne (45). On the other hand, in the presence of MCRa2, there was complete disappearance of the triplex CD signature with the concomitant appearance of a positive peak in its place. There was also a shift of the positive peak toward ∼280 nm, with a crossover at ∼260 nm, characteristic of duplex DNA structures (69). These changes in CD signals indicated the dissolution of the triplex structure when MCRa2 hybridized with the third strand of the triplex to form a duplex DNA structure.
Figure 5.

CD spectroscopy changes accompanying binding interactions of R2 (3.3. μM) with a stabilizer (coralyne, 16.5 μM) and a destabilizer-mimic (MCRa2, 16.5 μM) in Tris buffer.
Application toward library compound screening and ‘hit’ confirmation
Taken together, our results described above provided a ‘proof-of-principle’ of the utility of the FRET-based assay for identifying both triplex stabilizers and destabilizers simultaneously from a single assay platform. To apply the assay toward the screening of libraries of compounds, a pilot screen was conducted using a plate containing a library of 320 custom-assembled compounds with known biological activities (Supplementary Table S2). Tris buffer, in the absence of Mg2+ and Na+, was selected for the screen, as it provided a good optimal dynamic range for the identification of both triplex stabilizers and destabilizers. Because most small molecules were dissolved in 100% DMSO as 10 mM stocks, the effect of DMSO on the fluorescence readings was evaluated. Fluorescence readings of FAM were reproducible to within 5% in a solution containing 0.5% DMSO (Supplementary Figure S4A), which corresponded to a compound concentration of 50 μM if diluted from a 10 mM stock in 100% DMSO. The fluorescence intensity was also monitored hourly to assess assay stability and to determine the optimal time for assay incubation. We found that the signal stabilized after a 2-hour incubation at room temperature and it remained stable for up to 20 h (Supplementary Figure S4B). Thus, we chose a 16-hr incubation at room temperature as the optimized time for incubation.
The assay was also initially validated for robustness using an automated procedure with 0.5% DMSO (to mimic compound levels in an actual assay). As illustrated in Figure 6A, the solutions of R2FQ and buffer alone were plated as positive and negative controls, respectively. In addition, the known triplex binders/stabilizers (BePI and coralyne at 7.5 μM) and MCRa2 (at 500 nM as a destabilizer) were plated as internal controls. The scheme shown in Figure 6B illustrates a very simple mix-and-read and automated assay workflow, where an acoustic dispenser and Microflo bulk dispenser were utilized for compound and assay mix dispensing, respectively. The technologies/equipment employed omitted compound dilution, eliminated use of disposable tips, minimized reagent use and increased the efficiency of the overall high-throughput screen. Statistics obtained from a validation test for stabilizers were 0.81 and 88 for z’ and S/B, respectively, confirming that the assay was robust for automated and high throughput applications. A greater z’ was expected for a destabilizer screen, given the wider dynamic range of the assay.
Figure 6.

Schematic illustrations of (A) the assay plate format and (B) the automation workflow.
The potential of the FRET-based assay for triplex-specific compound screening was then demonstrated by testing a 320 custom-assembled compound set with known bioactivities, at 10 μM each. After correction for inherent fluorescence, we defined a molecule to be a triplex stabilizer if it enhanced the triplex folding and afforded a >2-fold signal reduction compared to the R2FQ control. Whereas we defined a triplex destabilizer as a molecule that induced unfolding of the triplex substrate and afforded a >2-fold signal increase, compared to the R2FQ control. Figure 7 shows the distribution of the activities of the 320 compounds, represented as a percentage of triplex stabilization or destabilization, compared to control compounds. While several compounds showed weak-to-moderate stabilizing effects on the triplex structure, one compound (doxorubicin, Figures 7 and 8A) demonstrated strong activity, comparable to BePI and coralyne.
Figure 7.
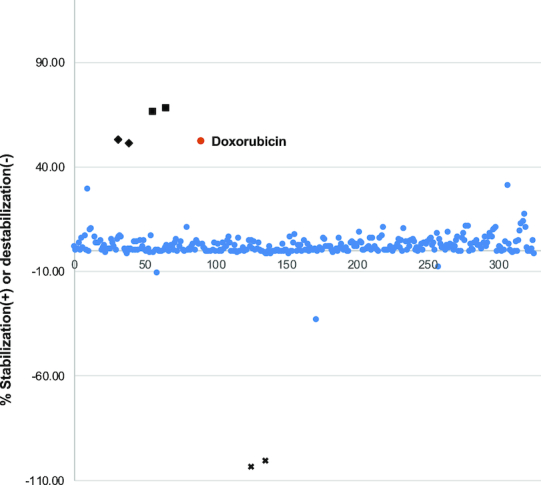
Distribution of compound activities. Percent stabilization or destabilization was normalized with RFUs of R2FQ control or MCR2a. Doxorubicin ( ) is highlighted as a ‘hit’. Controls are marked as BePI (✦), coralyne (▪), and MCRa2 (x).
) is highlighted as a ‘hit’. Controls are marked as BePI (✦), coralyne (▪), and MCRa2 (x).
Figure 8.

Validation of doxorubicin as a triplex stabilizer. (A) Chemical structure; (B) dose-dependent triplex stabilization in Tris buffer and (C) thermal melting profiles of R2 (red lines) and GT (blue lines) in the absence (solid lines) and presence (dotted lines) of 0.5 μM of doxorubicin.
While doxorubicin is known to intercalate in DNA (70,71), this is to our knowledge, the first demonstration implicating it in triplex DNA binding, and so we further validated its potency (Figure 8B). Doxorubicin showed dose-dependent activity with a calculated 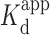 of 0.13 ± 0.04 μM (r2 = 0.915). Of note, an HPLC-UV chromatogram (Supplementary Figure S5A) of the doxorubicin stock solution showed a major single peak indicating its purity. Moreover, mass spectra (Supplementary Figure S5B) with a peak at m/z 544 as the parent ion corresponded with the molecular mass of 543 Da for doxorubicin. These results confirmed the identification of doxorubicin and eliminated any potential of impurity-driven activity. The effect of doxorubicin on triplex stabilization was further validated via thermal melting experiments. In addition to the R2 sequence, another triplex-forming sequence characterized in our buffer conditions, GT (72,73), was employed to examine the general triplex-stabilizing activity of doxorubicin. Thermal melting measurements of R2 and GT (at 0.5 μM) in the absence and presence of 0.5 μM doxorubicin revealed ∼5°C and 4°C Tm increases in the presence of doxorubicin, respectively, as shown in Figure 8C. These increases were similar to that of coralyne, confirming the triplex-stabilizing effects of doxorubicin. On the other hand, none of the compounds showed as strong a destabilizing effect as MCRa2, although one exhibited roughly half the destabilizing effect.
of 0.13 ± 0.04 μM (r2 = 0.915). Of note, an HPLC-UV chromatogram (Supplementary Figure S5A) of the doxorubicin stock solution showed a major single peak indicating its purity. Moreover, mass spectra (Supplementary Figure S5B) with a peak at m/z 544 as the parent ion corresponded with the molecular mass of 543 Da for doxorubicin. These results confirmed the identification of doxorubicin and eliminated any potential of impurity-driven activity. The effect of doxorubicin on triplex stabilization was further validated via thermal melting experiments. In addition to the R2 sequence, another triplex-forming sequence characterized in our buffer conditions, GT (72,73), was employed to examine the general triplex-stabilizing activity of doxorubicin. Thermal melting measurements of R2 and GT (at 0.5 μM) in the absence and presence of 0.5 μM doxorubicin revealed ∼5°C and 4°C Tm increases in the presence of doxorubicin, respectively, as shown in Figure 8C. These increases were similar to that of coralyne, confirming the triplex-stabilizing effects of doxorubicin. On the other hand, none of the compounds showed as strong a destabilizing effect as MCRa2, although one exhibited roughly half the destabilizing effect.
DISCUSSION
By incorporating a FAM–BHQ1 fluorophore-quencher pair into an intramolecular triplex-forming sequence from a breakpoint hotspot in the human c-Myc oncogene, we developed a FRET-based assay that can be used to screen for triplex-destabilizing ligands. Remarkably, the assay was also able to detect triplex-stabilizing ligands. This combination makes this assay the first of its kind to simultaneously detect triplex stabilizers and destabilizers in a single, facile, HTS-compatible platform. We identified Tris buffer (see Methods) as an ideal buffer to support ‘weak’ triplex formation and to enable the identification of triplex-interacting molecules through the induction of robust changes in the fluorescence signal, characteristic of either triplex stabilization or destabilization.
The potential of the design was initially validated through dose-dependent modulation of the FRET signal in the presence of known triplex intercalators/stabilizers (i.e. coralyne and BePI) and MCRa2 (a destabilizer that forms a complementary strand to the third strand of the triplex structure), which were confirmed by structural and thermal melting studies. The assay demonstrated robustness when utilized in an automated procedure and applied to a pilot screen of 320 compounds with known biological activities. Using this screen, we identified several potential triplex stabilizers, with doxorubicin showing a comparable triplex stabilizing effect to that of the positive controls. The assay was automation-friendly and utilized a simple mix-and-read system, which increased the efficiency of the overall high-throughput screen. Further, its use with an acoustic dispenser and Microflo bulk dispenser unit presented cost savings through elimination of disposable tips, and prevention of reagent loss by up to ∼100-fold.
Designed to serve as a preliminary screen for ‘hits’, subsequent validation will be needed using orthogonal assays, such as the native PAGE, CD, and thermal melting approaches employed in this study. Depending on the number of small molecule libraries screened, the possibility of identifying novel and specific triplex-interacting compounds will increase. Thus, we will expand our screening efforts to additional libraries in future efforts. Subsequent screens will also include other nucleic acid structures to further probe for specificity (i.e. duplex DNA, G4-DNA structures) and incorporation of Mg2+ in the workflow, to further parse the binding activities of ligands.
A drawback of our approach, which is typical of fluorescence assays, is the overlap of the fluorescence of some small molecules with the fluorescence of the fluorophore (FAM). For those compounds exhibiting relatively weak fluorescence, this can be addressed by assessing compound fluorescence in control wells.
Of biological significance, we developed these assays using a disease-relevant, H-DNA-forming sequence from the human c-MYC gene that maps to a translocation breakage hotspot in Burkitt lymphoma (50–52,74). Through the use of this newly developed tunable assay, the triplex-specific small molecules identified should allow precise temporal and spatial modulation of the H-DNA structure and its mutagenic outcome. As the occurrence and stability of H-DNA formation, which can be modulated by the identified ligands, is associated with genetic instability, destabilizing ligands are predicted to decrease such genetic instability associated with these mutagenic non-B DNA structures. DNA structure-induced genetic instability has also been implicated in evolution, by providing plasticity to the genome (75). Thus, this approach may provide a valuable path toward considerations for modulating non-B DNA structure-related genetic diseases and evolution. Conversely, triplex-stabilizing ligands, expected to increase the levels of genetic instability, will be useful tools in delineating mechanisms of error-prone processing of non-B DNA structures. Further, such small molecules may be relevant as H-DNA-specific fluorescent ligands that can serve as real-time probes in the visualization of H-DNA loci in vivo, which are still yet to be developed. Thus, structural modulation afforded by ligands from our assay can be tested for their effects on modulating genetic instability. Such insight should assist in delineating the mechanisms involved in genetic instabilities at mutation hotspots in human genetic disease, including cancer, leading to novel therapeutic approaches. Further, such molecules can provide valuable insights toward experimental paths relevant to drug discovery.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Tyler Lieberthal of Imperial College London for assistance with the illustrations, the members of the Vasquez laboratory for their valuable discussions and Dr. Ramakrishna Edupuganti at The University of Texas at Austin for conducting the HPLC-MS for compound characterization.
SUPPLEMENTARY DATA
Supplementary data are available at NAR online.
FUNDING
National Institutes of Health/National Cancer Institute (NIH/NCI) [CA093729 to K.M.V.]; Cancer Prevention Research Institute of Texas (CPRIT) [RP140108 to I.M.A.d.M.]; Cancer Prevention Research Institute of Texas (CPRIT) [RP160657 to K.N.D.]; Welch Foundation [F-1390 to K.N.D.]. Funding for open access charge: NIH/NCI [CA093729].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bacolla A., Tainer J.A., Vasquez K.M., Cooper D.N.. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016; 44:5673–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao J., Bacolla A., Wang G., Vasquez K.M.. Non-B DNA structure-induced genetic instability and evolution. Cell Mol. Life Sci. 2010; 67:43–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wells R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007; 32:271–278. [DOI] [PubMed] [Google Scholar]
- 4. Zhao J., Wang G., Del Mundo I.M., McKinney J.A., Lu X., Bacolla A., Boulware S.B., Zhang C., Zhang H., Ren P. et al.. Distinct mechanisms of Nuclease-Directed DNA-Structure-Induced genetic instability in cancer genomes. Cell Rep. 2018; 22:1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang G., Carbajal S., Vijg J., DiGiovanni J., Vasquez K.M.. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008; 100:1815–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang G., Vasquez K.M.. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13448–13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mirkin S.M., Frank-Kamenetskii M.D.. H-DNA and related structures. Annu. Rev. Biophys. Biomol. Struct. 1994; 23:541–576. [DOI] [PubMed] [Google Scholar]
- 8. Jain A., Wang G., Vasquez K.M.. DNA triple helices: biological consequences and therapeutic potential. Biochimie. 2008; 90:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilda M., Busch K., Klose I., Keller T., Woessmann W., Kreuder J., Harbott J., Borkhardt A.. Level of MYC overexpression in pediatric Burkitt's lymphoma is strongly dependent on genomic breakpoint location within the MYC locus. Genes Chromosomes Cancer. 2004; 41:178–182. [DOI] [PubMed] [Google Scholar]
- 10. Akasaka T., Akasaka H., Ueda C., Yonetani N., Maesako Y., Shimizu A., Yamabe H., Fukuhara S., Uchiyama T., Ohno H.. Molecular and clinical features of non-Burkitt's, diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J. Clin. Oncol. 2000; 18:510–518. [DOI] [PubMed] [Google Scholar]
- 11. Saglio G., Grazia Borrello M., Guerrasio A., Sozzi G., Serra A., di Celle P.F., Foa R., Ferrarini M., Roncella S., Borgna Pignatti C. et al.. Preferential clustering of chromosomal breakpoints in Burkitt's lymphomas and L3 type acute lymphoblastic leukemias with a t(8;14) translocation. Genes Chromosomes Cancer. 1993; 8:1–7. [DOI] [PubMed] [Google Scholar]
- 12. Mukherjee A., Vasquez K.M.. Triplex technology in studies of DNA damage, DNA repair, and mutagenesis. Biochimie. 2011; 93:1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pilch D.S., Waring M.J., Sun J.S., Rougee M., Nguyen C.H., Bisagni E., Garestier T., Helene C.. Characterization of a triple helix-specific ligand. BePI (3-methoxy-7H-8-methyl-11- [(3′-amino)propylamino]-benzo[e]pyrido[4,3-b]indole) intercalates into both double-helical and triple-helical DNA. J. Mol. Biol. 1993; 232:926–946. [DOI] [PubMed] [Google Scholar]
- 14. Lee J.S., Latimer L.J., Hampel K.J.. Coralyne binds tightly to both T.A.T- and C.G.C(+)-containing DNA triplexes. Biochemistry. 1993; 32:5591–5597. [DOI] [PubMed] [Google Scholar]
- 15. Escude C., Garestier T., Sun J.S.. Drug interaction with triple-helical nucleic acids. Methods Enzymol. 2001; 340:340–357. [DOI] [PubMed] [Google Scholar]
- 16. Escude C., Nguyen C.H., Kukreti S., Janin Y., Sun J.S., Bisagni E., Garestier T., Helene C.. Rational design of a triple helix-specific intercalating ligand. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amiri H., Nekhotiaeva N., Sun J.S., Nguyen C.H., Grierson D.S., Good L., Zain R.. Benzoquinoquinoxaline derivatives stabilize and cleave H-DNA and repress transcription downstream of a triplex-forming sequence. J. Mol. Biol. 2005; 351:776–783. [DOI] [PubMed] [Google Scholar]
- 18. Daksis J.I., Erikson G.H.. Specific triplex binding capacity of mixed base sequence duplex nucleic acids used for single-nucleotide polymorphism detection. Genet Test. 2005; 9:111–120. [DOI] [PubMed] [Google Scholar]
- 19. Van Daele I., Bomholt N., Filichev V.V., Van Calenbergh S., Pedersen E.B.. Triplex formation by Pyrene-Labelled probes for nucleic acid detection in fluorescence assays. ChemBioChem. 2008; 9:791–801. [DOI] [PubMed] [Google Scholar]
- 20. Nygren J., Svanvik N., Kubista M.. The interactions between the fluorescent dye thiazole orange and DNA. Biopolymers. 1998; 46:39–51. [DOI] [PubMed] [Google Scholar]
- 21. Kovalska V.B., Losytskyy M.Y., Yarmoluk S.M., Lubitz I., Kotlyar A.B.. Mono and trimethine cyanines Cyan 40 and Cyan 2 as probes for highly selective fluorescent detection of non-canonical DNA structures. J. Fluoresc. 2011; 21:223–230. [DOI] [PubMed] [Google Scholar]
- 22. Chen Z., Zhang H., Ma X., Lin Z., Zhang L., Chen G.. A novel fluorescent reagent for recognition of triplex DNA with high specificity and selectivity. Analyst. 2015; 140:7742–7747. [DOI] [PubMed] [Google Scholar]
- 23. Laguerre A., Wong J.M., Monchaud D.. Direct visualization of both DNA and RNA quadruplexes in human cells via an uncommon spectroscopic method. Sci. Rep. 2016; 6:32141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma D.L., Zhang Z., Wang M., Lu L., Zhong H.J., Leung C.H.. Recent Developments in G-Quadruplex Probes. Chem. Biol. 2015; 22:812–828. [DOI] [PubMed] [Google Scholar]
- 25. Muller S., Rodriguez R.. G-quadruplex interacting small molecules and drugs: from bench toward bedside. Expert Rev. Clin. Pharmacol. 2014; 7:663–679. [DOI] [PubMed] [Google Scholar]
- 26. Strekowski L.W., E. Mojzych M.. Lee MSL. Synthetic and Biophysical Studies of DNA Binding Compounds. 2007; Kerala: Transworld Research Network; 263–278. [Google Scholar]
- 27. Escudé C., Sun J.-S.. Waring MJ, Chaires JB. DNA Binders and Related Subjects. 2005; Berlin, Heidelberg: Springer; 109–148. [Google Scholar]
- 28. Jain A.K., Bhattacharya S.. Groove binding ligands for the interaction with parallel-stranded ps-duplex DNA and triplex DNA. Bioconjug. Chem. 2010; 21:1389–1403. [DOI] [PubMed] [Google Scholar]
- 29. Jenkins T.C. Targeting multi-stranded DNA structures. Curr. Med. Chem. 2000; 7:99–115. [DOI] [PubMed] [Google Scholar]
- 30. Soyfer V.N., Potaman V.N.. Triple-Helical Nucleic Acids. 1995; NY: Springer-Verlag. [Google Scholar]
- 31. Rehman S.U., Sarwar T., Husain M.A., Ishqi H.M., Tabish M.. Studying non-covalent drug-DNA interactions. Arch. Biochem. Biophys. 2015; 576:49–60. [DOI] [PubMed] [Google Scholar]
- 32. Wilson W.D., Tanious F.A., Fernandez-Saiz M., Rigl C.T.. Evaluation of drug-nucleic acid interactions by thermal melting curves. Methods Mol. Biol. 1997; 90:219–240. [DOI] [PubMed] [Google Scholar]
- 33. Schroeder S.J., Turner D.H.. Optical melting measurements of nucleic acid thermodynamics. Methods Enzymol. 2009; 468:371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mergny J.L., Lacroix L.. Analysis of thermal melting curves. Oligonucleotides. 2003; 13:515–537. [DOI] [PubMed] [Google Scholar]
- 35. You Y., Tataurov A.V., Owczarzy R.. Measuring thermodynamic details of DNA hybridization using fluorescence. Biopolymers. 2011; 95:472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mendoza O., Gueddouda N.M., Boule J.B., Bourdoncle A., Mergny J.L.. A fluorescence-based helicase assay: application to the screening of G-quadruplex ligands. Nucleic Acids Res. 2015; 43:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Darby R.A., Sollogoub M., McKeen C., Brown L., Risitano A., Brown N., Barton C., Brown T., Fox K.R.. High throughput measurement of duplex, triplex and quadruplex melting curves using molecular beacons and a LightCycler. Nucleic Acids Res. 2002; 30:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schneider U.V., Severinsen J.K., Geci I., Okkels L.M., Johnk N., Mikkelsen N.D., Klinge T., Pedersen E.B., Westh H., Lisby G.. A novel FRET pair for detection of parallel DNA triplexes by the LightCycler. BMC Biotechnol. 2010; 10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Renciuk D., Zhou J., Beaurepaire L., Guedin A., Bourdoncle A., Mergny J.L.. A FRET-based screening assay for nucleic acid ligands. Methods. 2012; 57:122–128. [DOI] [PubMed] [Google Scholar]
- 40. Holt P.A., Ragazzon P., Strekowski L., Chaires J.B., Trent J.O.. Discovery of novel triple helical DNA intercalators by an integrated virtual and actual screening platform. Nucleic Acids Res. 2009; 37:1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Del Mundo I.M.A., Zewail-Foote M., Kerwin S.M., Vasquez K.M.. Alternative DNA structure formation in the mutagenic human c-MYC promoter. Nucleic Acids Res. 2017; 45:4929–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mergny J.L., Duval-Valentin G., Nguyen C.H., Perrouault L., Faucon B., Rougee M., Montenay-Garestier T., Bisagni E., Helene C.. Triple helix-specific ligands. Science. 1992; 256:1681–1684. [DOI] [PubMed] [Google Scholar]
- 43. Keppler M.D., James P.L., Neidle S., Brown T., Fox K.R.. DNA sequence specificity of triplex-binding ligands. Eur. J. Biochem. 2003; 270:4982–4992. [DOI] [PubMed] [Google Scholar]
- 44. Wilson W.D., Mizan S., Tanious F.A., Yao S., Zon G.. The interaction of intercalators and groove-binding agents with DNA triple-helical structures: the influence of ligand structure, DNA backbone modifications and sequence. J. Mol. Recognit. 1994; 7:89–98. [DOI] [PubMed] [Google Scholar]
- 45. Moraru-Allen A.A., Cassidy S., Asensio Alvarez J.L., Fox K.R., Lane A.N.. Coralyne has a preference for intercalation between TA.T triples in intramolecular DNA triple helices. Nucleic Acids Res. 1997; 25:1890–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xing F., Song G., Ren J., Chaires J.B., Qu X.. Molecular recognition of nucleic acids: coralyne binds strongly to poly(A). FEBS Lett. 2005; 579:5035–5039. [DOI] [PubMed] [Google Scholar]
- 47. Polak M., Hud N.V.. Complete disproportionation of duplex poly(dT)*poly(dA) into triplex poly(dT)*poly(dA)*poly(dT) and poly(dA) by coralyne. Nucleic Acids Res. 2002; 30:983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Christensen L.A., Finch R.A., Booker A.J., Vasquez K.M.. Targeting oncogenes to improve breast cancer chemotherapy. Cancer Res. 2006; 66:4089–4094. [DOI] [PubMed] [Google Scholar]
- 49. Zhang J.H., Chung T.D., Oldenburg K.R.. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999; 4:67–73. [DOI] [PubMed] [Google Scholar]
- 50. Belotserkovskii B.P., De Silva E., Tornaletti S., Wang G., Vasquez K.M., Hanawalt P.C.. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J. Biol. Chem. 2007; 282:32433–32441. [DOI] [PubMed] [Google Scholar]
- 51. Wang G., Vasquez K.M.. Non-B DNA structure-induced genetic instability. Mutat. Res. 2006; 598:103–119. [DOI] [PubMed] [Google Scholar]
- 52. Kinniburgh A.J. A cis-acting transcription element of the c-myc gene can assume an H-DNA conformation. Nucleic Acids Res. 1989; 17:7771–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hoover R.G., Kaushal V., Lary C., Travis P., Sneed T.. c-myc transcription is initiated from P0 in 70% of patients with multiple myeloma. Curr. Top. Microbiol. Immunol. 1995; 194:257–264. [DOI] [PubMed] [Google Scholar]
- 54. Guo J., Ju J., Turro N.J.. Fluorescent hybridization probes for nucleic acid detection. Anal. Bioanal. Chem. 2012; 402:3115–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Biner S.M., Haner R.. A two-color, self-controlled molecular beacon. ChemBioChem. 2011; 12:2733–2736. [DOI] [PubMed] [Google Scholar]
- 56. James P.L., Brown T., Fox K.R.. Thermodynamic and kinetic stability of intermolecular triple helices containing different proportions of C+*GC and T*AT triplets. Nucleic Acids Res. 2003; 31:5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frank-Kamenetskii M.D., Mirkin S.M.. Triplex DNA structures. Annu. Rev. Biochem. 1995; 64:65–95. [DOI] [PubMed] [Google Scholar]
- 58. Mirkin S.M., Frank-Kamenetskii M.D.. H-DNA and related structures. Annu. Rev. Biophys. Biomol. Struct. 1994; 23:541–576. [DOI] [PubMed] [Google Scholar]
- 59. Kaushik S., Kaushik M., Svinarchuk F., Malvy C., Fermandjian S., Kukreti S.. Presence of divalent cation is not mandatory for the formation of intramolecular purine-motif triplex containing human c-jun protooncogene target. Biochemistry. 2011; 50:4132–4142. [DOI] [PubMed] [Google Scholar]
- 60. Xodo L.E., Manzini G., Quadrifoglio F.. Spectroscopic and calorimetric investigation on the DNA triplex formed by d(CTCTTCTTTCTTTTCTTTCTTCTC) and d(GAGAAGAAAGA) at acidic pH. Nucleic Acids Res. 1990; 18:3557–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Keppler M.D., James P.L., Neidle S., Brown T., Fox K.R.. DNA sequence specificity of triplex-binding ligands. Eur. J. Biochem. 2003; 270:4982–4992. [DOI] [PubMed] [Google Scholar]
- 62. Shchyolkina A.K., Borisova O.F.. Stabilizing and destabilizing effects of intercalators on DNA triplexes. FEBS Lett. 1997; 419:27–31. [DOI] [PubMed] [Google Scholar]
- 63. Pilch D.S., Kirolos M.A., Breslauer K.J.. Berenil binding to higher ordered nucleic acid structures: complexation with a DNA and RNA triple helix. Biochemistry. 1995; 34:16107–16124. [DOI] [PubMed] [Google Scholar]
- 64. Boehm B.J., Whidborne C., Button A.L., Pukala T.L., Huang D.M.. DNA triplex structure, thermodynamics, and destabilisation: insight from molecular simulations. Phys. Chem. Chem. Phys. 2018; 20:14013–14023. [DOI] [PubMed] [Google Scholar]
- 65. Scaria P.V., Shafer R.H.. Binding of ethidium bromide to a DNA triple helix. Evidence for intercalation. J. Biol. Chem. 1991; 266:5417–5423. [PubMed] [Google Scholar]
- 66. Vigneswaran N., Mayfield C.A., Rodu B., James R., Kim H.G., Miller D.M.. Influence of GC and AT specific DNA minor groove binding drugs on intermolecular triplex formation in the human c-Ki-ras promoter. Biochemistry. 1996; 35:1106–1114. [DOI] [PubMed] [Google Scholar]
- 67. Feng L., Li X., Peng Y., Geng J., Ren J., Qu X.. Spectral and electrochemical detection of protonated triplex formation by a small-molecule anticancer agent. Chem. Phys. Lett. 2009; 480:309–312. [Google Scholar]
- 68. Eriksson M., Norden B.. Linear and circular dichroism of drug-nucleic acid complexes. Methods Enzymol. 2001; 340:68–98. [DOI] [PubMed] [Google Scholar]
- 69. Nakamoto K., Tsuboi M., Strahan D.. Drug-DNA Interactions: Structures and Spectra. 2008; Hoboken: John Wiley and Sons. [PubMed] [Google Scholar]
- 70. Perez-Arnaiz C., Busto N., Leal J.M., Garcia B.. New insights into the mechanism of the DNA/doxorubicin interaction. J. Phys. Chem. B. 2014; 118:1288–1295. [DOI] [PubMed] [Google Scholar]
- 71. Aubel-Sadron G., Londos-Gagliardi D.. Daunorubicin and doxorubicin, anthracycline antibiotics, a physicochemical and biological review. Biochimie. 1984; 66:333–352. [DOI] [PubMed] [Google Scholar]
- 72. Gondeau C., Maurizot J.C., Durand M.. Spectroscopic investigation of an intramolecular DNA triplex containing both G.G:C and T.A:T triads and its complex with netropsin. J. Biomol. Struct. Dyn. 1998; 15:1133–1145. [DOI] [PubMed] [Google Scholar]
- 73. Gondeau C., Maurizot J.C., Durand M.. Circular dichroism and UV melting studies on formation of an intramolecular triplex containing parallel T*A:T and G*G:C triplets: netropsin complexation with the triplex. Nucleic Acids Res. 1998; 26:4996–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Marcu K.B., Bossone S.A., Patel A.J.. myc function and regulation. Annu. Rev. Biochem. 1992; 61:809–860. [DOI] [PubMed] [Google Scholar]
- 75. Xie K.T., Wang G., Thompson A.C., Wucherpfennig J.I., Reimchen T.E., MacColl A.D.C., Schluter D., Bell M.A., Vasquez K.M., Kingsley D.M.. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science. 2019; 363:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.